
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thin film nanocomposite membranes of superglassy PIM-1 and amine-functionalised 2D fillers for gas separation†
Faiz
Almansour
 a,
Monica
Alberto
a,
Andrew B.
Foster
b,
Sajjad
Mohsenpour
a,
Peter M.
Budd
b and
Patricia
Gorgojo
*acd
a,
Monica
Alberto
a,
Andrew B.
Foster
b,
Sajjad
Mohsenpour
a,
Peter M.
Budd
b and
Patricia
Gorgojo
*acd
aDepartment of Chemical Engineering, Faculty of Science and Engineering, The University of Manchester, M13 9PL, UK. E-mail: p.gorgojo@manchester.ac.uk
bDepartment of Chemistry, The University of Manchester, M13 9PL, UK
cInstituto de Nanociencia y Materiales de Aragón (INMA) CSIC-Universidad de Zaragoza, C/Mariano Esquillor s/n, 50018 Zaragoza, Spain
dDepartamento de Ingeniería Química y Tecnologías del Medio Ambiente, Universidad de Zaragoza, C/Pedro Cerbuna 12, 50009 Zaragoza, Spain. E-mail: pgorgojo@unizar.es
First published on 14th October 2022
Abstract
Loss of free volume over time (i.e. aging) is the main hurdle towards the commercial use of super glassy polymers for gas separation membranes. Aging takes place at a much faster rate in polymeric thin films, with permeability reductions of over 50% in only a few days. In this work 2D reduced holey graphene oxide (rHGO) nanosheets containing amine groups were added into thin films of the super-glassy polymer of intrinsic microporosity PIM-1. At filler loadings of 1 wt% of rHGO-tris(4-aminophenyl)amine, the CO2 permeance after 1 year of physical aging was 846 ± 37 GPU, which remained very close to that of the fresh membrane tested right after preparation (1050 ± 70 GPU), and was double that of 1 year-aged purely PIM-1 thin film composite membranes (432 ± 4 GPU). Membranes with lower filler concentrations of 0.1 wt% showed CO2 permeance values of 604 ± 34 GPU after 1 year of aging, but they aged quite rapidly; the initial CO2 permeance values of the fresh thin film nanocomposite (TFN) membrane at filler loading of 0.1 wt% was 3351 ± 662 GPU. The aging behaviour was also investigated in several tens of micrometres thick membranes (up to 2 years) for filler loadings of 0.1 wt% and the gas separation performance showed similar tendencies to that of thin films; leading to higher CO2 permeability without sacrificing CO2/CH4 selectivity.
1. Introduction
The demand for new technologies to mitigate high atmospheric levels of carbon dioxide (CO2) is experiencing an unprecedented growth. Membrane-based processes can contribute to the reduction of greenhouse gases emissions,1 as they feature minimal operating cost, relatively small footprint, and are less energy-intensive than conventional CO2 separation technologies such as amine absorption.2 Polymers are by far the most commonly used materials in membranes for gas separation applications. However, despite the extensive research in recent years, only a handful of them are commercially available. The need to find new materials that can overcome current long-term stability issues is crucial. Alcohol vapour and liquid treatments,3 as well as blending with organic polymers and fillers to obtain mixed matrix membranes (MMMs)4,5 have been shown to be efficient strategies to slow down physical aging, or to recover from it. It is also important to bear in mind that physical aging (i.e. densification due to polymer chain relaxation) and plasticisation (i.e. swelling at high CO2 partial pressures) effects are more pronounced in polymeric thin films, which are the preferred configurations for commercial membranes due to their higher flux. Thin films can be produced by depositing a thin selective layer (typically only a few μm thick) on top of a previously prepared porous support, giving so-called thin film composite (TFC) membranes. Another fabrication approach is the one-step immersion-precipitation technique, where a thin polymer solution is cast and introduced into a coagulation bath to obtain integrally skinned asymmetric membranes. In both techniques, nanofillers may be introduced in the selective top layer to render the membranes with superior properties. TFCs containing disperse nanomaterials are known as thin film nanocomposite (TFN) membranes.Over recent years, two-dimensional materials, including graphene oxide (GO)6–8 and other GO derivatives such as porous holey graphene oxide (HGO),9,10 have arisen as promising materials for membrane technology. Like GO, HGO possesses oxygen functional groups that result from the oxidation of graphite and can act as anchoring sites for a range of molecules and nanoparticles to tailor their properties. As the name suggests, HGO can be described as a GO sheet with some carbon atoms vacancies (holes/pores) in its lattice. When used as a filler, membranes can benefit from the structural changes it induces in the polymeric matrix and also from the additional pores in its lattice, creating additional diffusion paths with low resistance for the gas molecules, enhancing the membrane permeability.11 Moreover, it is hypothesised that the incorporation of graphene can affect the chain packing and restrict the mobility of the polymer chains, consequently reducing the membrane swelling and physical aging.12–14 Most of the research on the use of HGO in membrane technology is based on simulations15–17 and only a few experimental works are available in the literature. Li et al. reported an improvement in CO2 permeability and CO2/N2 selectivity of 21% (70 GPU) and 20.8% (130), respectively, by incorporating porous graphene oxide into thin-film composite (TFC) polyimide membranes through interfacial polymerization.10 Recently, Luque-Alled and co-workers18 reported mitigation of physical aging of MMMs when incorporating 10 wt% (PIM-1)-functionalised HGO into PIM-1 polymeric matrix. These retained 70% of the initial CO2 permeability after 150 days. Zhang and co-workers9 studied Pebax@1657 membranes that were functionalised by o-hydroxyazo-hierarchical porous organic polymers (o-POPs) and PGO. The gas separation performance of these membranes surpassed the Robeson’s upper bounds (2008), exhibiting a CO2 permeability and CO2/N2 ideal selectivity of 232.7 barrer and 80.7, respectively. Chen et al.19 reported a method to improve the permeability and CO2 selectivity of PIM-1 by incorporating GO nanosheets. The combination of the hydrophilic/hydrophobic microphase segregation in the membrane from the uniform assembly of GO nanosheets and the very porous MMMs with pore size of about 0.78 nm significantly improved the gas separation performance of PIM-1 membrane with an excellent CO2 permeability of up to 6169 barrer as well as a high CO2/N2 selectivity of 123.5. Tamaddondar et al.20 showed that the incorporation of a low cross-link density (LCD) network PIM-1 into a PIM-1 matrix is an effective way to reduce the physical aging effects of the membrane performance.
In this work, supported TFN and freestanding mixed matrix membranes were fabricated by incorporating amine-functionalised reduced graphene oxide into polymer of intrinsic microporosity PIM-1. PIM-1 possesses high CO2 permeability and relative high selectivity for important gas-pairs, such as CO2/CH4, which contributed to the revision of the Robeson’s upper bound in 2008.21 Two different amines were investigated (N,N,N′,N′-tetrakis(4-aminophenyl)-1,4-phenylenediamine (tetrakis) and tris(4-aminophenyl)-amine (TAPA)). The performance of all membranes was investigated for CO2/CH4 gas separation, as well as the effect of filler loading on the physical aging.
2. Experimental part
2.1. Materials
Hydrogen peroxide (H2O2, 30% w/v) was purchased from Acros Organics. L-Ascorbic acid (AA, 99%), anhydrous dimethylacetamide (DMAc), anhydrous dichlorobenzene (DCB), methanol, ethanol, 1,4-dioxane and chloroform were purchased from Sigma Aldrich (UK) and used as received. Anhydrous potassium carbonate (K2CO3, 99%) from Fisher Scientific Ltd (UK) was ground into a fine powder and dried in a vacuum oven at 110 °C overnight before use. Tetrachloroterephthalonitrile (TCTPN, ≥99%) was purchased from Jinan Finer Chemical Co. Ltd (China). Aqueous GO at a concentration of 10 mg mL−1 was kindly provided by William Blythe (UK). N,N,N′,N′-tetrakis(4-aminophenyl)-1,4-phenylenediamine (tetrakis) (C30H28N6, ≥95%) was purchased from Carbosynth (UK). Tris(4-aminophenyl)-amine (TAPA) (C18H18N4, 97%) and 3,3,3′,3′-tetramethyl-1,1′-spirobisindane-5,5′,6,6′-tetraol (TTSBI, ≥96.0%) were purchased from Alfa Aesar. Both TTSBI and TCTPN were dried in a vacuum overnight at room temperature before use. Polyacrylonitrile (PAN) supports were supplied by Saudi Aramco (Saudi Arabia). The chemical structures of TAPA and tetrakis are shown in Fig. 1(a) and (b), respectively. | ||
| Fig. 1 (a) Tris(4-aminophenyl)-amine (TAPA) and (b) N,N,N′,N′-tetrakis(4-aminophenyl)-1,4-phenylenediamine (tetrakis). | ||
2.2. Synthesis and characterisation of PIM-1
The PIM-1 synthesis was modified from the procedure described by Foster et al.,22 with 51.06 g of TCTPN (0.15 mol), 39.89 g of TTSBI (0.15 mol) and 62.19 g of K2CO3 (0.45 mol) reacted in a solvent mixture of 300 mL of DMAc and 150 mL of DCB under N2 at 120 °C (Fig. 2). The polymerization was carried out under mechanical stirring with the addition of the base in the monomers-containing solution after 25 min, when the internal temperature of the reaction mixture had reached 60 °C. The reaction mixture was brought up to 120 °C and maintained at that temperature for 2.5 h (Fig. S1†). These temperature conditions were selected to provide a topologically-rich PIM-1 polymer which has been shown in membranes to give improved gas separation performance. The stirring rate was increased throughout the reaction time as the viscosity of the solution increased. The viscous solution was then quenched into excess methanol. The yellow crude product was collected by vacuum filtration, redissolved in chloroform (5 g/120 mL), and then reprecipitated by pouring slowly into excess methanol. The polymer was again collected via filtration and then refluxed in deionized water for 16 h. The recovered product was immersed in a minimal amount of 1,4-dioxane for 15 min, before being washed with copious amounts of acetone during filtration. The polymer sample was left to soak in methanol overnight. It was then finally vacuum-filtered using a sintered glass funnel and dried in a vacuum oven at 120 °C for 2 days. 67 g (97% yield) was obtained after purification. A Viscotek gel permeation chromatography (GPC) max VE 2001 instrument (Malvern, UK) equipped with two PL mixed B columns and a Viscotek TDA302 triple detector array were used to determine the weight-average molecular weight (Mw), number-average molecular weight (Mn) and dispersity (Đ) of PIM-1. PIM-1 was dissolved in chloroform to obtain a concentration of 1 mg mL−1. Analysis was performed in chloroform at a flow rate of 1 mL min−1 and an injection volume of 100 μL. OmniSEC software (Malvern, UK) was used to analyze the data. GPC results: Mw = 74![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 100, Mn = 49500, Đ = 1.5. A Flash 2000 organic elemental analyser (Thermo Scientific, The Netherlands) was employed to obtain elemental analysis data. 5 mg of powder was used for each experiment. Elemental analysis results (wt%): C = 72.32%, H = 4.33%, N = 5.75%, Cl = 1.42% (theoretical content for C29H20N2O4: C = 75.64%, H = 4.37%, N = 6.08%). 1H nuclear magnetic resonance (NMR) spectrum of PIM-1 polymer was recorded using a Bruker Avance II 500 MHz instrument. 50 mg mL−1 polymer solutions in CDCl3 were prepared for the NMR analysis. 1H NMR spectrum indicated a branched PIM-1 is formed, this is shown in the ESI, Fig. S2.†
100, Mn = 49500, Đ = 1.5. A Flash 2000 organic elemental analyser (Thermo Scientific, The Netherlands) was employed to obtain elemental analysis data. 5 mg of powder was used for each experiment. Elemental analysis results (wt%): C = 72.32%, H = 4.33%, N = 5.75%, Cl = 1.42% (theoretical content for C29H20N2O4: C = 75.64%, H = 4.37%, N = 6.08%). 1H nuclear magnetic resonance (NMR) spectrum of PIM-1 polymer was recorded using a Bruker Avance II 500 MHz instrument. 50 mg mL−1 polymer solutions in CDCl3 were prepared for the NMR analysis. 1H NMR spectrum indicated a branched PIM-1 is formed, this is shown in the ESI, Fig. S2.†
 | ||
| Fig. 2 PIM-1 polymerization. | ||
2.3. Synthesis of holey graphene oxide (HGO)
Holey graphene oxide (HGO) was prepared by gradually adding 5 mL of H2O2 into a 200 mL GO aqueous dispersion (1 mg mL−1). The mixture was constantly stirred and refluxed for 10 h at 80 °C. After that, part of the HGO suspension (120 mL) was centrifuged and washed with deionized (DI) water to remove the remaining H2O2 and debris, and finally dried in a vacuum oven for 24 h at room temperature. The remaining HGO suspension (80 mL) was functionalised with two different amines as described in the next Section 2.4.2.4. Synthesis of reduced amine-functionalised graphene oxide
Amine-functionalisation of HGO was carried out following the procedure described by Alberto et al.23 The concentration ratio of HGO to amines (tetrakis and TAPA) was fixed at 1.0 mg of HGO to 0.01 mmol of amine. Firstly, amine was dissolved in ethanol up to a concentration of 5 mg mL−1. Then, the amine solution was slowly added into the HGO aqueous dispersion and stirred for approximately 24 h at 60 °C under reflux. The reaction suspension was filtered with ethanol to remove the unreacted amine followed by a final wash with chloroform.The obtained functionalised HGOs were chemically reduced to improve their dispersion in chloroform for membrane preparation. For that, 0.992 g of AA were added to 140 mg of amine-functionalised HGO (1 mg mL−1) and the mixture was allowed to stir for 2 h at 90 °C. The resulting suspension was filtered and washed with ethanol and chloroform. The final product was collected and dispersed in chloroform (2.86 mg mL−1) and labelled as rHGO-TAPA or rHGO-tetrakis.
2.5. Membrane fabrication
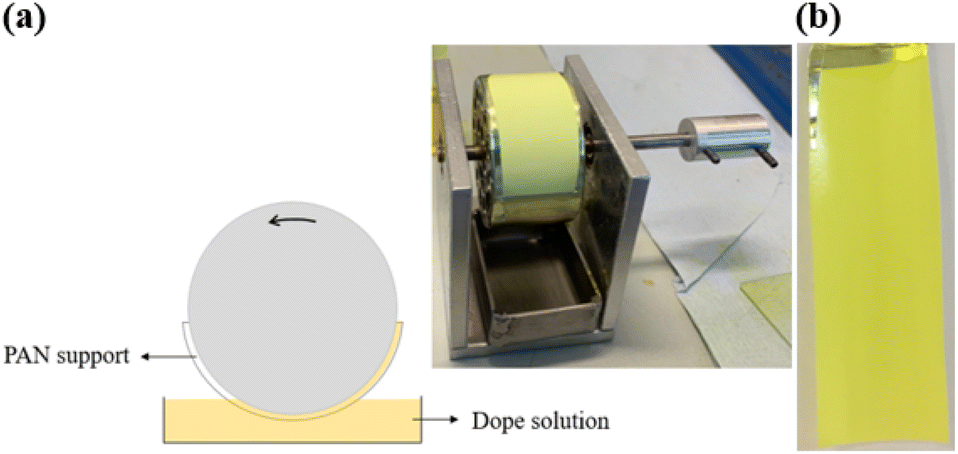 | ||
| Fig. 3 (a) Diagram and picture of the apparatus used for the fabrication of TFN membranes via kiss coating, and (b) representative TFN PIM-1 membrane supported on a PAN porous support. | ||
2.6. Characterisation of HGO fillers and membranes
Attenuated total reflection fourier transform infrared spectroscopy (ATR-FTIR) was used to analyse the structure of the graphene-based materials, pristine PIM-1 membranes (freestanding and TFC), MMMs, and TFN membranes. The spectra were acquired with an iDS Nicolet iS5 instrument (Thermo Scientific, UK), equipped with a Ge crystal as background over the wavenumber range of 500–4000 cm−1, and step size of 0.5 cm−1.Thermogravimetric analysis (TGA) was used to study the thermal stability of the synthesized graphene-like fillers, MMMs and TFC membranes. A TGA 550 thermal analyser (TA instruments, USA) was used, with a heating rate of 10 °C min−1 and under 60 mL min−1 nitrogen flow rate. The analysis was carried out from 50 to 800 °C.
A scanning electron microscope (SEM) FEI Quanta 650 FEG SEM (FEI, USA) was used to study the morphology of the membranes and the lateral size distribution of the GO nanosheets. Cross sectional samples were prepared by cryo-fracturing in liquid nitrogen. Initially, the sample membranes were immersed in ethanol for about 30 s, and subsequently introduced into liquid nitrogen for another 30 s, where the membranes were fractured. Also, the samples were sputtered with platinum nanoparticles using an Emitech sputter coater (Quorum Technologies, UK) before imaging under SEM. To obtain the lateral size of the prepared GO nanosheets, a silicon wafer was dipped in a GO dispersion (0.05 mg mL−1), dried at room temperature under vacuum and imaged. The processing software ImageJ® was used to perform the flake size analysis.
2.7. Gas permeation measurements
The gas permeability P for each gas was calculated using eqn (1).
 | (1) |
 | (2) |
All membrane samples were sandwiched between two aluminum tape rings and sealed with epoxy resin to prevent breakage and cracking when handling and during testing. The effective membrane area ranged from 0.18 to 0.48 cm2. At least two membranes of each composition were tested.
 | (3) |
 | (4) |
 | (5) |
400 cm3 mol−1), R is the gas constant (cmHg cm3 K−1 mol−1), T is the absolute temperature (K), dp/dt is the pressure build-up of permeate side (cmHg s−1), Δp is the average pressure difference between feed and permeate sides (cmHg), and l is the membrane thickness (cm).
The diffusion coefficient (D) was obtained using eqn (6). The solubility coefficient (S) was calculated from the permeability (P) and diffusion (D) coefficients using eqn (7); the time-lag (θ) was obtained from the graph of permeate pressure against time, and l is the membrane thickness.
 | (6) |
| P = DS | (7) |
The permeability of the membrane can be modelled using Maxwell–Wagner–Sillars equation. Eqn (8),25 which is an adjusted function of the Maxwell equation, includes a parameter n that can be related to the orientation and shape of the holey GO fillers in the membranes
 | (8) |
3. Results and discussion
The lateral size distribution of the GO nanosheets was obtained from several SEM images; a representative SEM image is shown in Fig. S4(a).† According to the normal distribution curve-fitting used, the mean value of the lateral GO flake size is 3.5 ± 2.5 μm (Fig. S4(b)†).Fig. S3a and b† show the porosity of the surface of PAN supports at low and high magnifications, respectively. From the acquired images, the obtained average pore size and surface porosity are 45.6 nm and 11.7%, respectively. Fig. 4 shows SEM images of the surface and cross-sections of pristine PIM-1 TFC membranes and TFN-1.0rHGO-TAPA-TFC membranes. From these images, it can be observed that the kiss-coating method followed in this study led to the production of defect-free TFN membranes and no agglomerates were visible, suggesting that the fillers were uniformly dispersed in the polymeric matrix. Generally, the membrane thickness for thin membranes was about 2–5 μm. More SEM images of the surface and cross sections of the TFC prepared in this study are shown in Fig. S5.†
 | ||
| Fig. 4 SEM images of surface and cross sections of (a and c) PIM-1 TFC, and (b and d) TFN-1.0rHGO-TAPA membranes. | ||
FTIR spectra of rHGO, rHGO-TAPA and rHGO-tetrakis samples are shown in Fig. S6(a).† The FTIR spectrum of rHGO showed the oxygen-containing groups in which the main broad absorption band occurs at 3320 cm−1 representing O–H group stretching vibrations of hydroxyl and carboxyl groups. Moreover, the absorption peaks at 1700 cm−1 and 1600 cm−1 correspond to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of carboxylic and CC stretching in the aromatic ring, respectively. The absorption peaks at approximately 1200 cm−1 and 1040 cm−1 represent epoxy C–O stretching vibration and alkoxy C–O stretching vibrations, respectively.28 After functionalisation, the FTIR spectra of TAPA/GO and tetrakis/GO show the N–H wag peaks at 875 cm−1. Additionally, both GO-based materials show two peaks at 1535 and 1571 cm−1 that are attributed to N–H stretching. New medium intensity peaks appeared in these spectra in the 1400–1200 cm−1 region due to antisymmetric C–N stretching vibrations coupled with out-of-plane NH2 and NH modes.29 The peaks in the region from 3400 to 3250 cm−1 correspond to N–H stretching. FTIR was also employed to study the chemical structure of the as-cast free PIM-1 membrane. As shown in Fig. S6(b),† the characteristic peaks from approximately 2800 cm−1 to 3010 cm−1 correspond to the aliphatic and aromatic C–H stretching. In addition to that, the FTIR also showed characteristics peak at ∼2240 cm−1 for C
O stretching of carboxylic and CC stretching in the aromatic ring, respectively. The absorption peaks at approximately 1200 cm−1 and 1040 cm−1 represent epoxy C–O stretching vibration and alkoxy C–O stretching vibrations, respectively.28 After functionalisation, the FTIR spectra of TAPA/GO and tetrakis/GO show the N–H wag peaks at 875 cm−1. Additionally, both GO-based materials show two peaks at 1535 and 1571 cm−1 that are attributed to N–H stretching. New medium intensity peaks appeared in these spectra in the 1400–1200 cm−1 region due to antisymmetric C–N stretching vibrations coupled with out-of-plane NH2 and NH modes.29 The peaks in the region from 3400 to 3250 cm−1 correspond to N–H stretching. FTIR was also employed to study the chemical structure of the as-cast free PIM-1 membrane. As shown in Fig. S6(b),† the characteristic peaks from approximately 2800 cm−1 to 3010 cm−1 correspond to the aliphatic and aromatic C–H stretching. In addition to that, the FTIR also showed characteristics peak at ∼2240 cm−1 for C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, ∼1610 cm−1 for CC aromatic bending and 1000–1300 cm−1 for C–O bending.30 Fig. S6(c) and (d)† show the FTIR spectra of MMMs and TFN membranes, respectively. No significant difference was observed between the FTIR spectrum of pristine PIM-1 membranes (freestanding and TFC) and the spectra of MMMs and TFN membranes, which might be due to the low filler concentration of the prepared membranes.
N, ∼1610 cm−1 for CC aromatic bending and 1000–1300 cm−1 for C–O bending.30 Fig. S6(c) and (d)† show the FTIR spectra of MMMs and TFN membranes, respectively. No significant difference was observed between the FTIR spectrum of pristine PIM-1 membranes (freestanding and TFC) and the spectra of MMMs and TFN membranes, which might be due to the low filler concentration of the prepared membranes.
The thermal stability of TAPA (as received), tetrakis (as received), GO, rHGO-tetrakis and rHGO-TAPA was assessed by TGA and the curves are shown in Fig. S7(a).† For all the samples the weight loss up to approximately 100 °C is attributed to the evaporation of adsorbed water. The weight loss between 100 and 300 °C of GO stems from the decomposition of oxygen-containing functional groups, and at higher temperatures up to 650 °C the pyrolysis of the carbon skeleton takes place.31 At 700 °C, the GO registered a weight loss of 95.4%. TAPA and tetrakis are thermally stable up to approximately 250 °C, showing negligible weight loss in this temperature range, with subsequent degradation at higher temperatures.28 rHGO-TAPA and rHGO-tetrakis are both more hydrophobic than GO due to the amine functionalisation and chemical reduction, as confirmed by the lower weight loss up to 100 °C. The thermal stability of MMMs and TFN membranes was also investigated and the results are shown in Fig. S7(b) and (c),† respectively. Results show that the thermostability of those membranes has not been significantly affected by both the type and loading of the fillers.
Gas permeation tests of PIM-1 TFC membranes and rHGO-tetrakis- and TAPA-based PIM-1 TFN membranes were carried out with single gases (CO2 and CH4) on a fixed pressure cell at 25 °C. The CO2 permeance and the ideal CO2/CH4 selectivity of the rHGO-tetrakis/PIM-1 and rHGO-TAPA/PIM-1 TFNs are plotted in Fig. 5(a) and (b), respectively, and the raw data is provided in ESI (Tables S1 and S2†). As can be observed, the incorporation of the fillers affected the CO2 and CH4 gas permeation of the PIM-1 thin films, with increased selectivity for less permeable membranes (>0.25 wt% of fillers). The TFNs containing a filler loading of 0.1 wt% showed a two-fold increase in CO2 permeances as compared to pristine PIM-1 TFC membranes for both fillers: 3351 ± 662 and 3200 ± 842 GPU for 0.1rHGO-TAPA-TFN and 0.1rHGO-tetrakis-TFN, respectively. The ideal CO2/CH4 selectivity of both sets of membranes, decreases from ∼9 for pure PIM to ∼7. Li et al.10 studied the incorporation of porous graphene nanosheets into polyimide membranes for CO2/N2 separation and reported similar results; at relatively low filler loadings (0.04–0.05 wt%) the CO2 permeation through the membrane increased and it then decreased for higher filler loadings. Similarly, Kim et al. incorporated GO and reduced GO (rGO) into thermally rearranged (TR) polymers to form ultrathin TFN membranes. TFNs containing rGO showed a high initial CO2 permeance of 1784 GPU which corresponds to an increase of 482 times compared to pristine polymer TFC membranes, with a selectivity of 32.4.32 This behaviour can be rationalised considering two competitive factors – disruption of the polymers chains that cause the increase in the gas permeability and the creation of the more tortuous path for the gas transport that leads to a reduction in gas permeation. This phenomenon is also reported in other studies in the literature.33,34 A similar study by Zhao et al. where metallic ion-cross-linked PIM-1 thin film membranes were fabricated, reported high CO2/N2 selectivity (23) and good CO2 permeance (1058 GPU).35 Likewise, PIM-1 based ultrathin membranes containing 2D metal–organic framework (MOF) nanosheets fabricated by Cheng et al. resulted in a CO2/CH4 selectivity of 15.6 and CO2 permeance up to 407 GPU, surpassing other PIM-1 based membranes.36
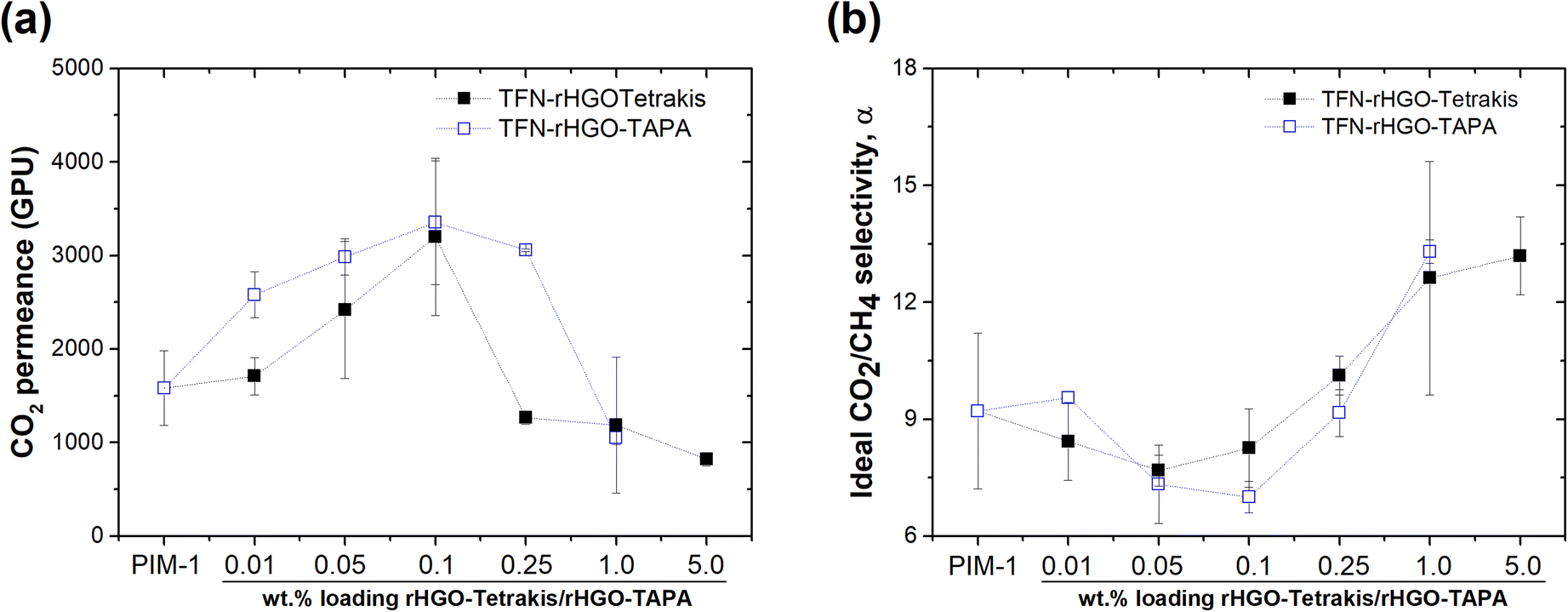 | ||
| Fig. 5 (a) CO2 permeance and (b) CO2/CH4 ideal selectivity of rHGO-tetrakis and rHGO-TAPA-based PIM-1 TFN membranes. ★ represents the performance of TFC PIM-1 membranes (dashed lines are used for guide purposes only). | ||
More important than the initial gas separation values, is the performance of the thin films over time. Stability is crucial in industrial operations, where predictable outputs are required. Due to the intrinsic high free volume of PIM-1, polymer chain relaxation that leads to a lower energy state and thus lower free volume is expected.37,38 The prepared TFNs in this work were kept in plastic petri dishes, sealed with parafilm and monitored up to 1 year and the results are plotted in Fig. 6 (raw data is provided in ESI in Tables S1 and S4†). On day 1, PIM-1 TFC membranes showed CO2 permeance of (1583 ± 400) GPU and ideal CO2/CH4 selectivity of (9.2 ± 2). After 66 days of aging, 58% of the initial CO2 permeance was lost, and after 1 year the loss was 73% ((432 ± 4) GPU). The reduction in CH4 permeance was higher (c.a. 81%) than that for CO2 over the same period, leading to the expected increase in ideal selectivity for the CO2/CH4 mixture upon aging. As explained in the work by Bernardo et al.39 aging in PIM-1 membranes mainly affects the gas diffusivity, which is more pronounced for molecules with larger kinetic diameters. The kinetic diameters of CH4 and CO2 are 3.8 Å and 3.3 Å, respectively. Thus, the physical aging in PIM-1 membranes tends to restrict to a higher degree the diffusion of CH4, improving the CO2/CH4 selectivity, as observed for the prepared TFNs. Moreover, the results also show a more rapid physical aging rate in thin films than in thick films (ESI†). In this study, thick freestanding PIM-1 membranes showed a CO2 permeability reduction of approximately 67% after 457 days. The physical aging rate dependency on the membrane thickness has been reported previously.40 As an example, Borisov et al. showed a drop in CO2 permeance of 3 month-aged PIM-1 TFC membranes between 10 and 40 times (208–297 GPU).41 More recently, Foster et al. reported the effect of blending topologically distinct samples of PIM-1 on the performance and aging behaviour of TFCs.42 Those containing a multi-loop polymeric structure (with 3 small loops) and very few chain ends (0–1 max) showed the best aging behaviour, with CO2 permeance of 671 GPU after 120 days.
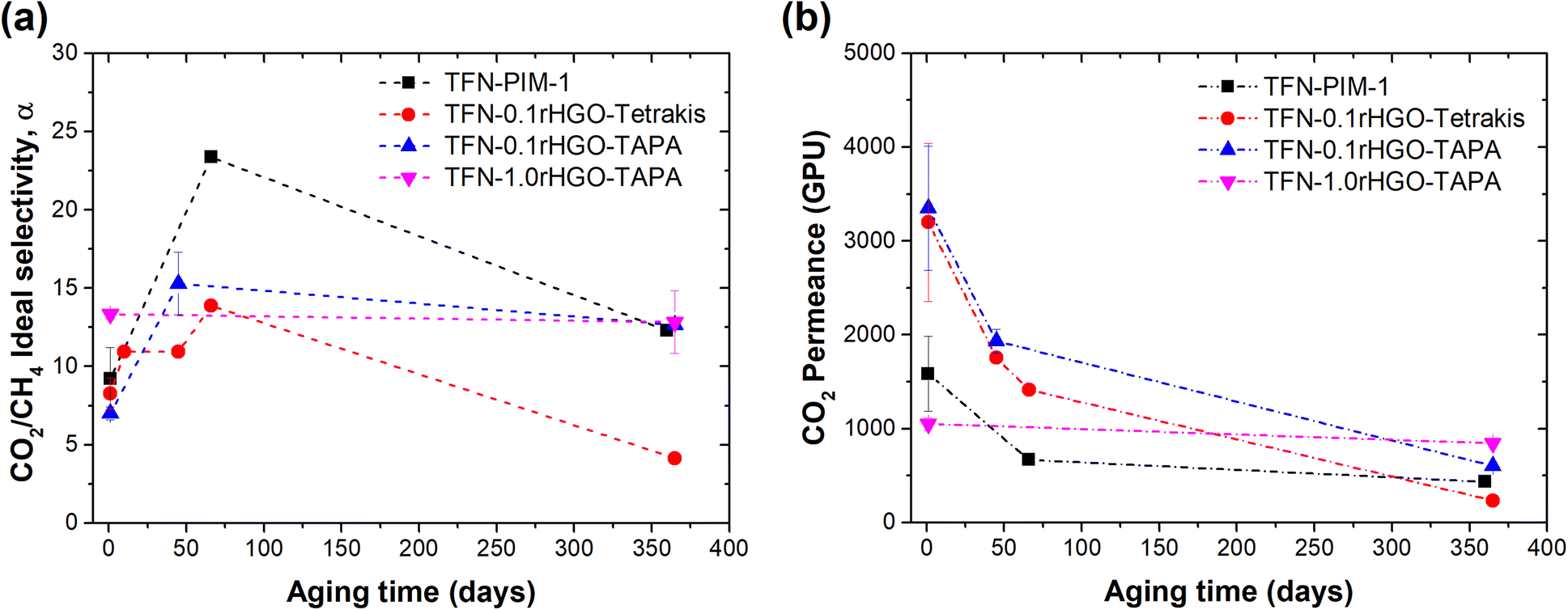 | ||
| Fig. 6 (a) CO2 permeance and (b) ideal CO2/CH4 selectivity of PIM-1 TFC and TFN membranes containing rHGO-tetrakis and rHGO-TAPA as fillers. Pure gas measurements at room temperature and a feed pressure of 2.4 barg. Effective membrane area 2.1 cm2, and samples stored at ambient conditions between testing. (1 GPU = 10−6 cm3[STP] cm−2 s−1 cmHg−1) (dashed lines are used for guide purposes only). | ||
Some of the prepared TFN membranes with amine-functionalised-rHGO were able to reduce the physical aging of the PIM-1 polymer, for example the membrane containing 0.25 wt% of tetrakis (TFN-0.25rHGO-tetrakis) experienced a loss of CO2 permeance of only 14% (from (1265 ± 70) to (1092 ± 108) GPU) over a period of 66 days. Similarly, the membrane containing 1 wt% of TAPA (TFN-1.0rHGO-TAPA) was able to preserve 81% of its initial CO2 permeance after one year from preparation at (846 ± 37) GPU, only a reduction of ∼19.4%. In all cases, CO2/CH4 ideal selectivities increased quite substantially for the first couple of months (Fig. 6b) due to the aforementioned more effective packing of the polymer and, therefore, an increase in the diffusivity-selectivity. However, prolonged aging of the TFNs led to selectivities down to their original values, which could be explained by the formation of small interfacial gaps between the polymer and the filler during the testing period.
The incorporation of fillers into the PIM-1 polymer to obtain gas separation membranes has been previously reported in the literature. However, most studies focus on freestanding thick MMMs, and only a few report the preparation of TFNs. In a recent publication by our research group,43 thin film nanocomposites (TFNs) membranes were prepared by incorporating porous silica nanosheets (SN) functionalized by a sulfonic acid (S-SN) in the PIM-1 polymer matrix. The incorporation of 0.05 wt% of S-SN led to 35% higher initial CO2 permeance (3771 ± 5 GPU) than the TFC PIM-1 (2778 ± 1010 GPU), and enhanced physical aging was obtained. However, the CO2 permeance of 28 days-aged TFC PIM-1 and TFN PIM/S-SN0.05 dropped by 97% (81 ± 10 GPU) and 87% (403 ± 43 GPU), respectively.
Bhavsar et al.44 showed that the incorporation of highly permeable nanoparticulate fillers (carbonized form of hypercrosslinked polystyrene, C-HCP) into PIM-1 TFN membranes drastically reduced the physical aging rate. The incorporation of a very high loading of C-HCP (60 wt%) into the PIM-1 polymeric active layer showed CO2 permeance of >9300 GPU after 90 days.
Tables S3 and S8(a)† show the gas separation performance of freestanding PIM-1 membranes and freestanding MMMs containing rHGO-tetrakis as filler (0.01–5.0 wt%). A 50/50 vol% CO2 and CH4 binary mixture was introduced to the feed side and tests were conducted at 25 °C under a transmembrane pressure of 1 bar. At least two membranes of each composition were tested. Experimental results showed that the incorporation of rHGO-tetrakis into the polymeric matrix has distinct effects of the gas separation performance, depending on the loading of the filler. A significant increase in CO2 permeability of the MMMs was observed with increasing filler loading over the range 0.01–0.1 wt% rHGO-tetrakis, reaching a maximum CO2 permeability of (9760 ± 449) barrer for the sample 0.1rHGO-tetrakis, representing an increase of 31% when compared to pristine PIM-1 membrane. On the other hand, the permeability of CO2 dropped for filler loadings higher than 1.0 wt% rHGO-tetrakis. The membrane 5.0rHGO-tetrakis presented a CO2 permeability of (3393 ± 315) barrer, the lowest amongst all, even lower than a pristine PIM-1 membranes. In addition to that, membrane selectivity increased for this range of filler loadings. These findings are underpinned by both the semi-impermeability of holey graphene and by the creation of a more tortuous path for the gas molecules to cross the membrane. Similar results were obtained by Althumayri et al.45 that reported a two-fold increase in CO2 permeability of MMMs containing a loading of graphene of 0.05 vol% in a PIM-1 polymeric matrix, whereas, the CO2 permeability decreases with the further increasing loading of graphene. Shen et al.34 reported a similar trend when GO was incorporated into Pebax matrix. CO2 permeability increased upon addition of 0.1 wt% GO to the Pebax membrane reaching 100 barrer. Further addition of GO led to a decrease of the CO2 permeability from 100 to 30 Barrer for an addition of 0.5 wt% GO. MMMs based on polyimide with either GO or laminated GO as fillers prepared by Ge et al.46 showed the same behaviour. The addition of both fillers resulted in an increase of CO2 permeability with the increase of filler loading up to 3.0 wt% which was followed by a decrease in CO2/N2 selectivity. Opposite behaviour was shown by both MMMs when the filler loading surpassed 4.0 wt%. Functionalised multi-walled carbon nanotubes (f-MWCNTs) with polyethylene glycol (PEG) in PIM-1 MMMs were investigated by Khan et al. Similar to previous studies, the addition of 0.5 wt% of f-MWCNTs loading to polymer matrix resulted in ∼21% increase in CO2 permeability. Likewise, f-MWCNTs loading between 1 wt% and 2 wt% further enhanced the gas permeability. On the other hand, with the increase of f-MWCNTs loading from 2% to 3%, the CO2 permeability decreased.47 A related study on MMMs incorporating g-C3N4 nanosheets into the PIM-1 matrix reported by Tian and co-workers also showed an enhancement in gas permeabilities with increase filler loadings. At low g-C3N4 loadings (<1.5 wt%), there was an apparent increase in gas permeability until it reached the maximum of 1 wt% g-C3N4 loading, then further loading decreased the permeability. The decrease in permeability was hypothesised to be due to the partial agglomeration of restacked g-C3N4, which blocked the ultramicropores of the PIM-1 matrix.48
Assuming the addition of 2D sheets (rHGO) into PIM-1 follows the ellipsoids theory and knowing that the permeability of the rHGO is way lower than the PIM-1 permeability, hence the Pf can be neglected. The n parameter can be calculated from the experimental gas permeability values for the PIM-1 and the TFN membranes. The calculation of the volume fraction Ø was determined by considering the wt% of each membrane. The density of PIM-1 was considered to be 0.948 g cm−3,12 and the density of GO was calculated as the two-dimensional mass density of a graphene single layer (7.63 × 10−8 g cm−2) divided by the GO flake thickness (1 nm) as previously reported.18 The n parameter was calculated for each TFN membranes using eqn (8) with PM (experimental gas permeability of the TFN membranes) and Pp (experimental gas permeability of pure PIM-1), and the results are shown in Table S5.† The n values results range between 0.943 and 1, which indicates that most of the rHGO flakes are parallel to each other and horizontally orientated to the gas flow. Fig. S8† shows the CO2 permeability prediction for the rHGO-tetrakis-containing membranes based on the Maxwell equation.
The CO2/CH4 separation performance of MMMs containing different loadings of rHGO-TAPA into PIM-1 polymeric matrix was also investigated under the same conditions as the previous rHGO-tetrakis MMMs. The performance of rHGO-TAPA/PIM-1 MMMs is shown in Table S4. Fig. S9(b)† compares the performance of rHGO-TAPA- and rHGO-tetrakis-based PIM-1 MMMs. For filler loading up to 0.1 wt%, both fillers induced an increase in CO2 permeability. Among these membranes, the maximum CO2 permeability was reached by the membrane 0.1rHGO-TAPA ((11077 ± 261) barrer). In terms of CO2/CH4 selectivity, rHGO-TAPA/PIM-1-based membranes showed always higher selectivity than rHGO-tetrakis/PIM-1-based membranes. This might be due to the size of the molecules TAPA and tetrakis. Tetrakis molecules are bigger than TAPA molecules, which can induce a greater disruption of the polymer chains and therefore create more non-selective voids between the filler and the polymer chains. Moreover, bigger molecules may also cause an increase in tortuosity path for the gas molecules and therefore a decrease in membrane permeability.
Physical aging of the membranes was evaluated in terms of CO2 permeability and CO2/CH4 selectivity for 611 days and the results are shown in Fig. S10(a) and (b)†, respectively (Tables S3 and S4 in the ESI†). The CO2 permeability of all MMMs decreased over time, while the CO2/CH4 selectivity is enhanced for the same testing period. After 611 days, 0.05rHGO-TAPA and 0.05rHGO-tetrakis showed the highest ((3839 ± 907) barrer) and lowest ((1999 ± 173) barrer) average CO2 permeability, respectively. The same tendency of highly permeable glassy polymers to show a decrease in CO2 permeability over time has been shown in the literature.13,39,49 The relative CO2 permeability and relative CO2/CH4 selectivity of pristine PIM-1 membranes and MMMs, 611 days after preparation are shown in Fig. S10(c) and (d)†, respectively. Pristine PIM-1 membranes experienced a drop of 66% in CO2 permeability after 611 days. Over the same period, the lowest CO2 permeability loss, 55%, was shown by 0.05rHGO-TAPA. It is worth noting that the membrane 0.05rHGO-tetrakis presented the highest relative CO2 permeability loss (79%) and also the second highest initial CO2 permeability ((9361 ± 374) barrer). This might be due to the greater free volume generated by the introduction of the filler into the polymeric matrix. The higher the initial free volume, the higher the gas permeability, increasing the tendency to raise the physical aging rate. Generally, MMMs presented a lower increase in relative CO2/CH4 selectivity compared to pristine PIM-1 membranes. This might be due to voids still present in the membrane structure upon the disruption of the polymer chains created by the presence of the filler in the membrane matrix.
Fig. S11† displays the gas separation performance of freestanding PIM-1 and rHGO-tetrakis/PIM-1 and rHGO-TAPA/PIM-1 MMMs, the 2008 Robeson and the 2019 Jansen/McKeown upper-bounds. It can be observed that the performance of the membranes moved towards lower CO2 permeability and higher CO2/CH4 selectivity.
To further investigate the effect of the fillers in the MMMs, single gas permeation measurements of aged membranes (aged for 850 days) were undertaken using a time-lag apparatus. The solution-diffusion mechanism is the most widely used transport model for gas permeation in polymer membranes, hence pure gas measurements were obtained by a constant volume instrument to evaluate the solubility (S) and diffusivity (D) coefficients of PIM-1 and rHGO-tetrakis- and TAPA-based PIM-1 membranes (shown in Table 1). The calculated values of S and D in the PIM-1 membrane are similar to those reported by Budd et al.21D and S values for CO2 are both greater than CH4, owing to the smaller kinetic diameter of CO2 and higher polarity, respectively. It has been found that aging has a significant effect on the diffusivity, however, the solubility is almost constant as a function of time. The D values for all MMMs are greater than pristine PIM-1. Therefore, the addition of the fillers leads to higher free volume after a long period of aging. It is worth mentioning that the permeability of all the MMMs was still higher than that of neat PIM-1. Thus, the improvements in permeability are due to faster diffusion of CO2 through the MMMs. This suggests that adding amine-functionalised rHGO helps keep a higher diffusivity, avoiding the collapse of free volume. However, the lower values of solubility suggest some of the polymer sorption sites in PIM-1 are blocked due to interactions with the fillers, as has been reported in the literature.50
| Aged (850) days membrane | P (barrer) | D × 107 (cm2 s−1) | S × 103 (cm3(STP) cm−3 cmHg−1) | Ideal sel CO2/CH4 | Diffusivity selectivity | Solubility selectivity | |||
|---|---|---|---|---|---|---|---|---|---|
| CO2 | CH4 | CO2 | CH4 | CO2 | CH4 | ||||
| PIM-1 | 2195 | 156 | 5 | 1 | 456 | 109 | 14.1 | 3.4 | 4.2 |
| 0.01rHGO-TAPA | 2453 | 154 | 6 | 2 | 384 | 90 | 15.9 | 3.7 | 4.3 |
| 0.05rHGO-TAPA | 3179 | 210 | 7 | 2 | 427 | 107 | 15.1 | 3.8 | 4.0 |
| 0.1rHGO-TAPA | 3245 | 247 | 9 | 4 | 350 | 63 | 13.2 | 2.4 | 5.6 |
| 0.01rHGO-tetrakis | 3062 | 231 | 7 | 3 | 442 | 85 | 13.3 | 2.6 | 5.2 |
| 0.05rHGO-tetrakis | 3088 | 172 | 9 | 2 | 326 | 102 | 17.9 | 5.6 | 3.2 |
| 0.1rHGO-tetrakis | 2738 | 173 | 24 | 5 | 115 | 33 | 15.8 | 4.5 | 3.5 |
For comparison reasons, the gas separation performance of TFC PIM-1, TFN membranes and aged MMMs was plotted in a selectivity-permeability log–log graph with the 2008 Robeson upper-bound (Fig. 7). An average membrane thickness of 3 μm (obtained from the SEM images) was considered. Results show that TFC and TFN films do age more rapidly than MMMs, which is in accordance with the findings reported in the literature.51,52 It can be also seen that fresh MMMs at day 1 are all above the 2008 upper bound and loss in permeability over time makes them go slightly below it. Contrarily, the initial performance of the TFN containing TAPA-functionalised holey GO is below the selectivity-permeability trade-off, which at low filler loadings move parallel to it, and for the 1 wt% membrane (the less permeable TFN at day 1) it remains almost unaltered after 360 days.
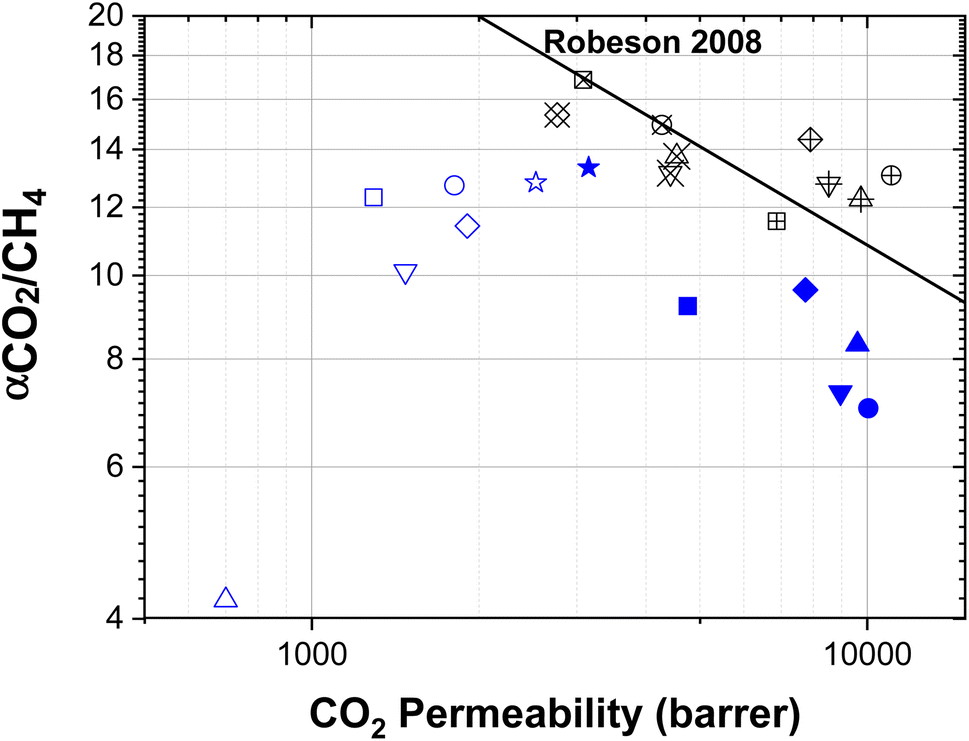 | ||
| Fig. 7 Membrane performance of aged MMMs (457 days), PIM-1 TFC and TFN membranes (360 days) for CO2/CH4 separation with the 2008 Robeson upper-bound. A membrane thickness of 3 μm was considered. ■ PIM-1, • 0.1 TAPA, ▼ 0.05 TAPA, ♦ 0.01 TAPA, ★ 1.0 TAPA and ▲ 0.1 tetrakis. Filled symbols: performance of TFC and TFN membranes at day 1, unfilled symbols: performance of TFC and TFN membranes at day 360, unfilled symbols with a +: performance of MMM at day 1, unfilled symbols with X: performance of MMMs at day 457. | ||
4. Conclusions
Thin film nanocomposite (TFN) membranes of PIM-1 containing amine-functionalised reduced HGO (rHGO-tetrakis and rHGO-TAPA) were prepared and tested for CO2/CH4 separation. Also the effects of filler loadings on physical aging were investigated.After 1 year of physical aging, membranes with filler loading of 1 wt% rHGO-TAPA showed CO2 permeance of (846 ± 37) GPU. The results obtained were close to the permeance value of fresh membranes tested after preparation and double that of 1 year-aged purely PIM-1 thin film composite membranes, (1050 ± 70) GPU and (432 ± 4) GPU, respectively. On the other hand, membranes with lower filler concentrations of 0.1 wt% after 1 year of aging obtained a CO2 permeance of (604 ± 34) GPU, however, they aged faster. Fresh TFN membrane at filler loading of 0.1 wt% showed an initial CO2 permeance value of (3351 ± 662) GPU.
The aging behaviour was also investigated in several tens of micrometres thick membranes for filler loadings of 0.1 wt% and the gas separation performance showed similar tendencies to that of thin films; leading to higher CO2 permeability without sacrificing CO2/CH4 selectivity. 0.1wt% rHGO-tetrakis and 0.1wt% rHGO-TAPA reached an initial CO2 permeability of (9760 ± 449) and (11077 ± 261) barrer, respectively. The CO2/CH4 separation performance outperformed pristine PIM-1 membranes after 611 days, where 0.05rHGO-TAPA showed the highest CO2 permeability of (3839 ± 907) barrer. Gas solubility and diffusivity values were obtained experimentally and confirmed that the addition of amine-functinalised rHGO helped maintain a higher diffusivity, avoiding the collapse of free volume, and suggested some extent of blockage of the polymer sorption sites.
In conclusion, the incorporation of amine-functionalised reduced HGO is an effective strategy to enhance the gas performance of PIM-1 based membranes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
F. Almansour is grateful to the Department of Research & Development, Saudi Aramco for funding and supporting his PhD studies. M. Alberto is grateful to EPSRC for funding under the research grant number EP/S032258/1. A. B. Foster acknowledges the EPSRC for the grant number EP/v047078/1 “SynHiSel”. P. Gorgojo acknowledges the Spanish Ministry of Economy and Competitiveness and the European Social Fund through the Ramon y Cajal programme (RYC2019-027060-I/AEI/10.13039/501100011033).References
- R. W. Baker and K. Lokhandwala, Ind. Eng. Chem. Res., 2008, 47, 2109–2121 CrossRef CAS.
- P. Bernardo, E. Drioli and G. Golemme, Ind. Eng. Chem. Res., 2009, 48, 4638–4663 CrossRef CAS.
- F. Almansour, M. Alberto, R. S. Bhavsar, X. Fan, P. M. Budd and P. Gorgojo, Front. Chem. Sci. Eng., 2021, 15, 872–881 CrossRef CAS.
- M. Rezakazemi, A. Ebadi Amooghin, M. M. Montazer-Rahmati, A. F. Ismail and T. Matsuura, Prog. Polym. Sci., 2014, 39, 817–861 CrossRef CAS.
- S. He, B. Zhu, S. Li, Y. Zhang, X. Jiang, C. Hon Lau and L. Shao, Sep. Purif. Technol., 2022, 284, 120277 CrossRef CAS.
- G. Dong, Y. Zhang, J. Hou, J. Shen and V. Chen, Ind. Eng. Chem. Res., 2016, 55, 5403–5414 CrossRef CAS.
- F. Zhou, H. N. Tien, W. L. Xu, J.-T. Chen, Q. Liu, E. Hicks, M. Fathizadeh, S. Li and M. Yu, Nat. Commun., 2017, 8, 2107 CrossRef.
- J. Zhang, Q. Xin, X. Li, M. Yun, R. Xu, S. Wang, Y. Li, L. Lin, X. Ding, H. Ye and Y. Zhang, J. Membr. Sci., 2019, 570–571, 343–354 CrossRef CAS.
- Y. Li, W. Zhao, M. Weyland, S. Yuan, Y. Xia, H. Liu, M. Jian, J. Yang, C. D. Easton, C. Selomulya and X. Zhang, Environ. Sci. Technol., 2019, 53, 8314–8323 CrossRef CAS PubMed.
- H. Li, X. Ding, Y. Zhang and J. Liu, J. Membr. Sci., 2017, 543, 58–68 CrossRef CAS.
- D. Jang, J.-C. Idrobo, T. Laoui and R. Karnik, ACS Nano, 2017, 11, 10042–10052 CrossRef CAS.
- A. Gonciaruk, K. Althumayri, W. J. Harrison, P. M. Budd and F. R. Siperstein, Microporous Mesoporous Mater., 2015, 209, 126–134 CrossRef CAS.
- M. Alberto, R. Bhavsar, J. M. Luque-Alled, A. Vijayaraghavan, P. M. Budd and P. Gorgojo, J. Membr. Sci., 2018, 563, 513–520 CrossRef CAS.
- J. M. Luque-Alled, A. Ameen, M. Alberto, M. Tamaddondar, A. B. Foster, P. M. Budd, A. Vijayaraghavan and P. Gorgojo, J. Membr. Sci., 2020, 118902, DOI:10.1016/j.memsci.2020.118902.
- S. Wang, S. Dai and D.-e. Jiang, ACS Appl. Nano Mater., 2019, 2, 379–384 CrossRef CAS.
- D.-e. Jiang, V. R. Cooper and S. Dai, Nano Lett., 2009, 9, 4019–4024 CrossRef CAS.
- H. Liu, S. Dai and D.-e. Jiang, Nanoscale, 2013, 5, 9984–9987 RSC.
- J. M. Luque-Alled, M. Tamaddondar, A. B. Foster, P. M. Budd and P. Gorgojo, ACS Appl. Mater. Interfaces, 2021, 13, 55517–55533 CrossRef CAS.
- M. Chen, F. Soyekwo, Q. Zhang, C. Hu, A. Zhu and Q. Liu, J. Ind. Eng. Chem., 2018, 63, 296–302 CrossRef CAS.
- M. Tamaddondar, A. B. Foster, M. Carta, P. Gorgojo, N. B. McKeown and P. M. Budd, ACS Appl. Mater. Interfaces, 2020, 12, 46756–46766 CrossRef CAS.
- P. M. Budd, N. B. McKeown, B. S. Ghanem, K. J. Msayib, D. Fritsch, L. Starannikova, N. Belov, O. Sanfirova, Y. Yampolskii and V. Shantarovich, J. Membr. Sci., 2008, 325, 851–860 CrossRef CAS.
- A. B. Foster, M. Tamaddondar, J. M. Luque-Alled, W. J. Harrison, Z. Li, P. Gorgojo and P. M. Budd, Macromolecules, 2020, 53, 569–583 CrossRef CAS.
- M. Alberto, J. M. Luque-Alled, L. Gao, M. Iliut, E. Prestat, L. Newman, S. J. Haigh, A. Vijayaraghavan, P. M. Budd and P. Gorgojo, J. Membr. Sci., 2017, 526, 437–449 CrossRef CAS.
- S. A. Stern, P. J. Gareis, T. F. Sinclair and P. H. Mohr, J. Appl. Polym. Sci., 1963, 7, 2035–2051 CrossRef CAS.
- R. H. B. Bouma, A. Checchetti, G. Chidichimo and E. Drioli, J. Membr. Sci., 1997, 128, 141–149 CrossRef CAS.
- D. Q. Vu, W. J. Koros and S. J. Miller, J. Membr. Sci., 2003, 211, 335–348 CrossRef CAS.
- S. Mohsenpour, A. W. Ameen, S. Leaper, C. Skuse, F. Almansour, P. M. Budd and P. Gorgojo, Sep. Purif. Technol., 2022, 298, 121447 CrossRef CAS.
- X. Zhao, X. Jiang, D. Peng, J. Teng and J. Yu, J. Rare Earths, 2020, 39, 90–97 CrossRef.
- M. X. Pulikkathara, O. V. Kuznetsov and V. N. Khabashesku, Chem. Mater., 2008, 20, 2685–2695 CrossRef CAS.
- L. Hao, K.-S. Liao and T.-S. Chung, J. Mater. Chemistry A, 2015, 3, 17273–17281 RSC.
- M.-C. Hsiao, C.-C. M. Ma, J.-C. Chiang, K.-K. Ho, T.-Y. Chou, X. Xie, C.-H. Tsai, L.-H. Chang and C.-K. Hsieh, Nanoscale, 2013, 5, 5863–5871 RSC.
- S. Kim, J. Hou, Y. Wang, R. Ou, G. P. Simon, J. G. Seong, Y. M. Lee and H. Wang, J. Mater. Chem. A, 2018, 6, 7668–7674 RSC.
- G. Dong, J. Hou, J. Wang, Y. Zhang, V. Chen and J. Liu, J. Membr. Sci., 2016, 520, 860–868 CrossRef CAS.
- J. Shen, G. Liu, K. Huang, W. Jin, K.-R. Lee and N. Xu, Angew. Chem., Int. Ed., 2015, 54, 578–582 Search PubMed.
- H. Zhao, L. Feng, X. Ding, X. Tan and Y. Zhang, Chin. J. Chem. Eng., 2018, 26, 2477–2486 CrossRef CAS.
- Y. Cheng, X. Wang, C. Jia, Y. Wang, L. Zhai, Q. Wang and D. Zhao, J. Membr. Sci., 2017, 539, 213–223 CrossRef CAS.
- H. W. H. Lai, F. M. Benedetti, J. M. Ahn, A. M. Robinson, Y. Wang, I. Pinnau, Z. P. Smith and Y. Xia, Science, 2022, 375, 1390–1392 CrossRef CAS PubMed.
- P. M. Budd, Science, 2022, 375, 1354–1355 CrossRef CAS.
- P. Bernardo, F. Bazzarelli, F. Tasselli, G. Clarizia, C. R. Mason, L. Maynard-Atem, P. M. Budd, M. Lanč, K. Pilnáček, O. Vopička, K. Friess, D. Fritsch, Y. P. Yampolskii, V. Shantarovich and J. C. Jansen, Polymer, 2017, 113, 283–294 CrossRef CAS.
- S. Harms, K. Rätzke, F. Faupel, N. Chaukura, P. M. Budd, W. Egger and L. Ravelli, J. Adhes., 2012, 88, 608–619 CrossRef CAS.
- I. Borisov, D. Bakhtin, J. M. Luque-Alled, A. Rybakova, V. Makarova, A. B. Foster, W. J. Harrison, V. Volkov, V. Polevaya, P. Gorgojo, E. Prestat, P. M. Budd and A. Volkov, J. Mater. Chem. A, 2019, 7, 6417–6430 RSC.
- A. B. Foster, J. L. Beal, M. Tamaddondar, J. M. Luque-Alled, B. Robertson, M. Mathias, P. Gorgojo and P. M. Budd, J. Mater. Chem. A, 2021, 9, 21807–21823 RSC.
- S. Mohsenpour, Z. Guo, F. Almansour, S. M. Holmes, P. M. Budd and P. Gorgojo, J. Membr. Sci., 2022, 661, 120889 CrossRef CAS.
- R. S. Bhavsar, T. Mitra, D. J. Adams, A. I. Cooper and P. M. Budd, J. Membr. Sci., 2018, 564, 878–886 CrossRef CAS.
- K. Althumayri, W. J. Harrison, Y. Shin, J. M. Gardiner, C. Casiraghi, P. M. Budd, P. Bernardo, G. Clarizia and J. C. Jansen, Philos. Trans. R. Soc., A, 2016, 374, 20150031 CrossRef.
- B.-S. Ge, T. Wang, H.-X. Sun, W. Gao and H.-R. Zhao, Polym. Adv. Technol., 2018, 29, 1334–1343 CrossRef CAS.
- M. M. Khan, V. Filiz, G. Bengtson, S. Shishatskiy, M. M. Rahman, J. Lillepaerg and V. Abetz, J. Membr. Sci., 2013, 436, 109–120 CrossRef CAS.
- Z. Tian, S. Wang, Y. Wang, X. Ma, K. Cao, D. Peng, X. Wu, H. Wu and Z. Jiang, J. Membr. Sci., 2016, 514, 15–24 CrossRef CAS.
- S. Yi, B. Ghanem, Y. Liu, I. Pinnau and W. J. Koros, Sci. Adv., 2019, 5, eaaw5459 CrossRef.
- A. W. Ameen, J. Ji, M. Tamaddondar, S. Moshenpour, A. B. Foster, X. Fan, P. M. Budd, D. Mattia and P. Gorgojo, J. Membr. Sci., 2021, 636, 119527 CrossRef CAS.
- N. R. Horn and D. R. Paul, Polymer, 2011, 52, 5587–5594 CrossRef CAS.
- M. S. McCaig and D. R. Paul, Polymer, 2000, 41, 629–637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta06339e |
| This journal is © The Royal Society of Chemistry 2022 |