
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CO2 electroreduction activity and dynamic structural evolution of in situ reduced nickel-indium mixed oxides†
Laura C.
Pardo Pérez
 *a,
Zora
Chalkley
ab,
Robert
Wendt
c,
Ibbi Y.
Ahmet
c,
Markus
Wollgarten
c and
Matthew T.
Mayer
*a
*a,
Zora
Chalkley
ab,
Robert
Wendt
c,
Ibbi Y.
Ahmet
c,
Markus
Wollgarten
c and
Matthew T.
Mayer
*a
aYoung Investigator Group Electrochemical Conversion of CO2, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Hahn-Meitner-Platz 1, 14109 Berlin, Germany. E-mail: lcpardop@gmail.com; m.mayer@helmholtz-berlin.de
bInstitut für Chemie & Biochemie, Freie Universität Berlin, Berlin 14195, Germany
cInstitute for Solar Fuels, Helmholtz-Zentrum Berlin für Materialien und Energie GmbH, Hahn-Meitner-Platz 1, 14109 Berlin, Germany
First published on 5th September 2022
Abstract
In the field of CO2 electroreduction (CO2ER), tuning the selectivity among diverse products remains a major challenge. Mixed metal catalysts offer possible synergetic effects which can be exploited for tuning product selectivity. We present a simple wet chemical approach to synthesize a range of nickel-indium mixed oxide (NiAInBOx) thin films with homogeneous metal distribution. CO2 electroreduction results indicate that the NiAInBOx mixed oxide thin films can achieve high CO selectivity (>70%) in contrast with the single metal oxides NiO (H2 >90%) and In2O3 (formate >80%). The relative composition Ni40In60Ox attained the best CO selectivity of 71% at moderate cathodic bias of −0.8 VRHE, while a higher cathodic bias (E < −0.9 V) promoted a decrease of CO in favor of formate. A detailed investigation of the Ni40In60Ox thin films following progressive stages of reduction during CO2ER revealed dynamic structural transformations strongly dependent on applied bias and electrolysis time. For the CO-selective catalyst composition, at moderate cathodic bias (E < −0.8 V) and short electrolysis times (1 h), the catalyst is composed of nickel-indium alloy grains embedded in amorphous Ni–In mixed oxide as observed by electron microscopy. Extending electrolysis time at −0.8 V for 10 h, or increasing the applied reductive bias to −1.0 V, result in a complete reduction of the residual oxide film into an interconnected array of multicomponent (In, Ni, Ni3In7) nanoparticles which display significantly lower CO selectivity (<50%). Our results indicate that the persistent amorphous NiInOx oxide/alloy composite material preserved in the early stages of reduction at −0.8 V plays a key role in CO selectivity. The highly dynamic structure observed in this catalytic system demonstrates the importance of conducting detailed structural characterization at various applied potentials to understand the impact of structural changes on the observed CO2ER selectivity trends; and thus be able to distinguish structural effects from mechanistic effects triggered by increasing the reductive bias.
1 Introduction
The electrochemical reduction of carbon dioxide (CO2ER) has gained increasing attention as a sustainable pathway for CO2 reutilization and upcycling into added-value chemicals. As many as 16 products have been observed and identified during CO2ER in aqueous media,1 such as formate, CO, hydrocarbons, and alcohols. Tuning the selectivity of CO2ER catalysts remains a major challenge in the field. The initial 2 e− reduction of CO2 can yield formate or CO. While formate is commonly accepted as a terminal product that cannot be further reduced, CO has been demonstrated to be a key intermediate in the formation of higher (>2 e−) reduction products such as hydrocarbons and alcohols.2,3 The pioneering work on CO2ER activity of different metal electrodes in aqueous media established an empirical correlation between the CO binding strength of metals and the main observed CO2ER product.4–9 The empirical trends were further supported in more recent DFT studies concluding that interfacial CO2 and CO binding modes and energetics are key descriptors determining the CO2ER selectivity.3,10,11Among single metal surfaces, Cu is unique in exhibiting an intermediate CO binding strength that favors further reduction to hydrocarbons or alcohols.1,12 Other early transition metals such as Ni bind CO too strongly leading to surface poisoning and are unable to reduce CO further. On these metal surfaces, CO2ER is suppressed and instead hydrogen evolution reaction (HER) is the dominant process in aqueous media.13 On the other hand, post-transition metals like In exhibit a weak interaction with CO2 and are known to favor formate production.4,6,14–17 Au and Ag adsorb CO weakly and release it as the final product.3,9,11
Bimetallic catalysts offer possible synergetic effects among different metals to yield optimal binding of CO2 and the key intermediate CO and have been demonstrated as a successful strategy for tuning selectivity in Cu-based catalysts.18–22 Such an approach can be adapted for the modulation of CO binding on other early transition metals; for instance, the strong CO binding on Ni can be modulated by combination with a weak CO2/CO binding surface like In.9,11 Nickel-indium composites were previously reported by He and coworkers23 as a potential bimetallic composition with decreased CO binding energy capable of producing CO with appreciable selectivity. In that study, a Ni25In75 composite gave a CO faradaic efficiency (FE) of less than 30%, while a sample with low Ni content Ni2In98 was reported to produce 60% CO at −0.7 VRHE. In contrast, In2O3 nanocrystals doped with 6 at% Ni were shown to produce predominantly formate with 93% faradaic efficiency at −0.9 VRHE.
Herein we present a detailed investigation of CO2ER activity and associated structural changes of oxide-derived Ni–In composite catalysts. A facile synthetic approach by spin coating of metal precursors in solution followed by a thermal treatment at 400 °C in air was employed to produce NiAInBOx mixed oxide thin films with homogenous distribution of both metals. The mixed oxide thin films are reduced in situ during CO2ER. The structural changes induced during reduction are investigated by electron microscopy, EDS, and XPS. The investigation of various nickel-indium metal compositions revealed that Ni40In60Ox delivers the best CO selectivity of 71% at moderate cathodic bias of −0.8 VRHE, while the increase of applied cathodic bias to −1.0 V leads to a change in selectivity towards formate (62%). The selectivity change from CO to formate is accompanied by strong structural transformations of the pristine Ni40In60Ox mixed oxide. The structural and morphological changes observed in the composites during in situ reduction and their correlation with the CO2ER activity will be discussed in detail below.
2 Experimental methods
2.1 Synthesis
The NiAInBOx mixed oxides were synthesized by a simple wet chemical method by deposition of a solution of metal precursors, NiCl2·6H2O (CAS: 7791-20-0) and anhydrous InCl3 (CAS: 10025-82-8) in the desired molar Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :In ratio, in a solvent mixture composed of dimethyl-ethanolamine (DMAE) and methanol (MeOH) in a 1:3 ratio (by volume). Each metal was dissolved separately in a single solvent. NiCl2·6H2O is added to 0.5 mL of DMAE and stirred for 1.5 h until all solid was dissolved to yield homogenous green solution. Anhydrous InCl3 was promptly dissolved in 1.5 mL of methanol. The solutions were then mixed by adding the InCl3/MeOH slowly to the NiCl2/DMAE solution shortly before film preparation by spin coating. The obtained solution was deposited onto Si or glassy carbon substrates (GCS) by spin coating of 25 μL cm−2 at 2000 rpms for 23 s and 28 s for Si and GCS, respectively, to achieve a homogenous thin film coating on the substrate. The wet film is set by a drying step for 10 s at 100 °C. The film deposition is followed by a thermal decomposition step by annealing at 400 °C for 30 min to transform the metal precursor solution film into a mixed oxide thin film. Representative SEM images of the obtained films are presented in Fig. S1.†
:3 ratio by volume of concentrated acids HNO3:HCl) over three days to induce surface oxidation and improve adhesion of the coating solution. After aqua regia treatment, the Si substrates were rinsed and sonicated for 10 minutes in fresh Milli-Q water (resistivity 18.2 MΩ cm), followed by rinsing and sonication twice in fresh ethanol and isopropyl alcohol. Chips were then blown dry with a N2(g) gun and dried in an oven at 80 °C. Glassy carbon substrates (GCS) are reused. Old films were removed by placing the GCS in a test tube with a small amount (∼4 mL) of freshly prepared aqua regia overnight to dissolve the existing film. The clean GCS were then rinsed and sonicated twice in fresh Milli-Q water twice and polished with 3.0 μm and 0.05 μm alumina suspension, rinsing and sonicating in fresh mQ water in between and after polishing steps to remove alumina particle residues. The GCS were then blown dry with a N2(g) gun and dried in an oven at 100 °C.
:In ratio, in a solvent mixture composed of dimethyl-ethanolamine (DMAE) and methanol (MeOH) in a 1:3 ratio (by volume). Each metal was dissolved separately in a single solvent. NiCl2·6H2O is added to 0.5 mL of DMAE and stirred for 1.5 h until all solid was dissolved to yield homogenous green solution. Anhydrous InCl3 was promptly dissolved in 1.5 mL of methanol. The solutions were then mixed by adding the InCl3/MeOH slowly to the NiCl2/DMAE solution shortly before film preparation by spin coating. The obtained solution was deposited onto Si or glassy carbon substrates (GCS) by spin coating of 25 μL cm−2 at 2000 rpms for 23 s and 28 s for Si and GCS, respectively, to achieve a homogenous thin film coating on the substrate. The wet film is set by a drying step for 10 s at 100 °C. The film deposition is followed by a thermal decomposition step by annealing at 400 °C for 30 min to transform the metal precursor solution film into a mixed oxide thin film. Representative SEM images of the obtained films are presented in Fig. S1.†
:3 ratio by volume of concentrated acids HNO3:HCl) over three days to induce surface oxidation and improve adhesion of the coating solution. After aqua regia treatment, the Si substrates were rinsed and sonicated for 10 minutes in fresh Milli-Q water (resistivity 18.2 MΩ cm), followed by rinsing and sonication twice in fresh ethanol and isopropyl alcohol. Chips were then blown dry with a N2(g) gun and dried in an oven at 80 °C. Glassy carbon substrates (GCS) are reused. Old films were removed by placing the GCS in a test tube with a small amount (∼4 mL) of freshly prepared aqua regia overnight to dissolve the existing film. The clean GCS were then rinsed and sonicated twice in fresh Milli-Q water twice and polished with 3.0 μm and 0.05 μm alumina suspension, rinsing and sonicating in fresh mQ water in between and after polishing steps to remove alumina particle residues. The GCS were then blown dry with a N2(g) gun and dried in an oven at 100 °C.
Synthesis of Ni2In3 alloy nanoparticles was carried out by heat treatment of Ni25In75Ox mixed oxide film in forming gas with 2.4 vol% H2 in Ar at 500 °C for 1 h. Note that the precursor mixed oxide film employed for alloy synthesis is enriched in In fraction in order to compensate for some In loss observed during forming gas heat treatment. The formation of the Ni2In3 alloy was verified by GI-XRD measurement on a sample prepared on a Si substrate (Fig. 7b) and the alloy composition was further verified by ICP-OES (Table S2†).
2.2 CO2 electroreduction testing
Electrolysis tests were carried out in a custom two-compartment cell separated by a Nafion 115 membrane, in CO2 saturated 0.1 M KHCO3 (pH 6.8) under a constant CO2 flow of 20 mL min−1. A BioLogic SP-200 potentiostat was used in a three-electrode configuration with Ag/AgCl in 3 M KCl (Dri-ref. 2 mm WPI) and Pt mesh as reference and counter electrodes, respectively. All potentials presented through the manuscript are referenced to the reversible hydrogen electrode (RHE, ERHE = EAg/AgCl + 0.211 + 0.0591 × pH). The CO2ER test typically comprised two steps: (1) a linear sweep voltammetry (LSV) step from open circuit potential (OCP) to −0.7 V vs. RHE and (2) a chronoamperometry (CA) step with increasingly reductive potential between −0.7 V and −1.0 V at 0.1 V intervals holding each potential for 1 h. Representative behavior is displayed in Fig. S2.†The gas products were quantified by in-line gas chromatography with a Thermo Scientific TRACE 1310 gas chromatograph using He as a carrier gas. The gas chromatograph is equipped with two columns – HayeSepS column (1 mm ID, Restek) and molecular sieve MS5a column (1 mm ID, Restek) – as well as two detectors – pulse discharge detector (PDD) and flame ionization detector (FID). Representative chromatograms and calibration curves of different products are presented in Fig. S11.† The outlet flow of the headspace in the cathode compartment of the two-compartment cell was fed into GC through a transfer line (heated at 60 °C). The product concentration was measured every 15 minutes for the duration of the CO2ER catalytic test. The faradaic efficiency (FE) was calculated according to eqn (1), where [X] is the concentration measure for gas X determined by gas chromatography, flowrate corresponds to the constant inlet rate of CO2 into the cell (20 mL min−1), z the number of mol of electrons required to produce 1 mol of gas X, i is the recorded experimental current, and F is the Faraday constant.
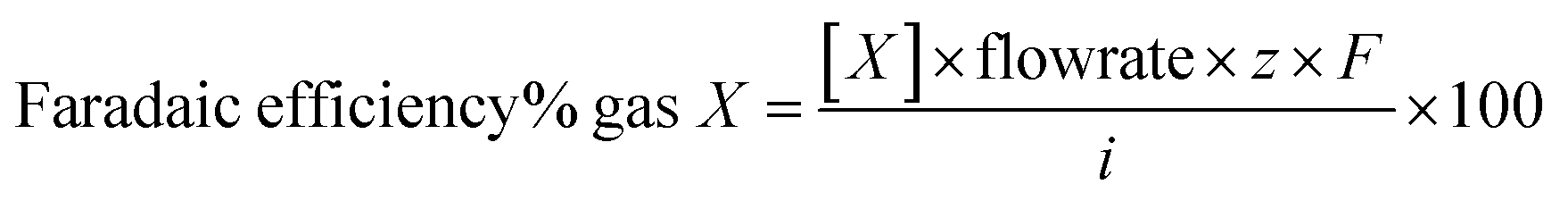 | (1) |
Liquid products are analyzed post-electrolysis by high performance liquid chromatography (HPLC), using a Thermo Scientific Dionex UltiMate 3000 UHPLC instrument equipped with UV (UltiMate 3000, Dionex) and RI (RefractoMax 520, ERC) detectors and an autosampler unit. The separation is achieved with a HyperREZ XP H+ column using 5 mM H2SO4(aq) as eluent. The electrolyte was sampled at the end of each electrolysis test by taking a 1 mL aliquot. Formate faradaic efficiency was determined based on the experimentally determined formate concentration [HCOO−], the total amount of charge passed (q) and the total volume of electrolyte (Velectrolyte), as shown in eqn (2). Representative chromatograms and calibration curves of formate are presented in Fig. S12.†
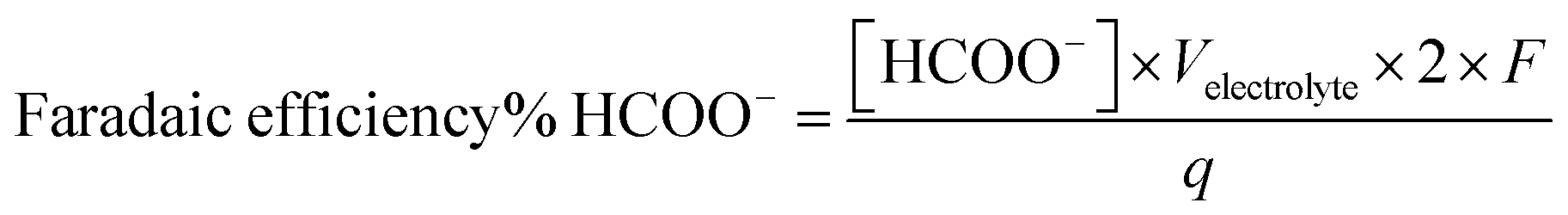 | (2) |
2.3 Materials characterization
Scanning Electron Microscopy (SEM) and associated energy-dispersive X-ray spectroscopy (EDS) spectra were collected on a Zeiss LEO 1530 Gemini SEM system with a ThermoFisher UltraDry EDS detector. The SEM images were acquired at 3 kV using the in-lens secondary electron detector and EDS spectra were collected at an acceleration voltage of 15 kV. Transmission electron microscopy (TEM), high angle annular dark field (HAADF), scanning transmission electron microscopy (STEM), selected area electron diffraction (SAED) and energy-dispersive X-ray spectroscopy (EDS) micrographs and spectra were collected on a TEM Zeiss LIBRA 200FE instrument operated at 200 kV accelerating voltage equipped with a Thermo Scientific Noran System Six X-ray spectrometer. The thin film samples in as-synthesized condition or after electrolysis testing were scraped off the GCS and transferred to carbon coated Cu or SiNx TEM grids. X-ray photoelectron spectroscopy (XPS) measurements were carried out in a SPECS PHOIBOS 100 analyzer using Monochromatic Al Kα radiation (hν = 1486.74 eV, SPECS FOCUS 500 monochromator). The pass energy was set to 10 eV with step size of 0.05 eV. The spectra were fitted in CasaXPS software using a Shirley background subtraction. The as-synthesized samples were calibrated with respect to the adventitious carbon C 1s peak at 285 eV. The XPS analysis of post-electrolysis samples was conducted on samples first subjected to CO2ER electrolysis tests inside a O2-free glovebox (O2 < 1 ppm) using the typical two-compartment cell and experimental conditions described in CO2ER testing details (Section 2.2). The post-electrolysis samples were washed in de-aerated mQ water, dried, and stored under N2 atmosphere inside the glovebox prior to XPS measurements. In order to avoid any exposure to air, the samples were transferred from the glovebox to the XPS vacuum chamber under inert atmosphere using a transfer arm module. Inductively Coupled Plasma – Optical Emission Spectrometry (ICP-OES) measurements were conducted in an iCAP 7400 Duo MFC ICP-OES Analyzer system (Thermo Scientific) in axial Ar (5.0 purity, Air Liquide) plasma mode. Each sample was digested in 4 mL of fresh aqua regia overnight to dissolve the Ni–In composite. The resulting solution was then quantitatively transferred to a 50 mL volumetric flask and completed to volume for ICP-OES analysis. Grazing incidence X-ray diffraction (GIXRD) was performed on a PANalytical X'Pert Pro MPD diffractometer for thin film analysis, using a Cu Kα radiation (λ = 1.5418 Å), and a Xenon scintillation counter detector with parallel plate collimator. The diffractograms were collected at an incidence angle of 0.3° with a step size of 0.006 and collection time of 10 s.3 Results & discussion
3.1 Effect of In–Ni composition on CO2ER activity
A range of NiAInBOx films were studied. Herein, A and B denote the nominal Ni and In fraction respectively, as defined by the composition of the precursor solution used for film coating; Ox refers to the mixed oxide obtained. The fractions A and B are expressed as percentage referred to total atomic composition of Ni and In; i.e. (A = [Ni/(Ni + In)] × 100). NiAInBOx films with Ni fraction between 10 and 75 at% were synthesized to evaluate the effect of Ni–In composition on the CO2ER activity. Single metal oxide thin films of NiO and In2O3 were synthesized by an analogous method for comparison. The synthesis method yields NiAInBOx mixed oxide thin films with homogenous distribution of both metals and oxygen composed of small crystallites (with diameter <5 nm) as can be seen in Fig. S1 and S3.† The CO2ER activity of the various NiAInBOx samples was evaluated in CO2 saturated 0.1 M KHCO3. The typical test comprises a reductive LSV followed by electrolysis at constant potential in the range of −0.7 V to −1.0 V vs. RHE (Fig. S2†) with quantification of gaseous and liquid products at each tested potential. Note that all potentials are referred to RHE unless otherwise specified. The electrocatalysis results are summarized in Fig. 1, comparing CO2ER product distributions observed from electrodes with a range of compositions, tested at different applied potentials. It should be noted that under CO2ER conditions, the NiAInBOx tend to become reduced in situ due to the applied reductive bias. Such catalysts are often described as “oxide-derived” catalysts which we shall refer to as being “in situ reduced”. Comparing the selectivity trends as a function of composition, it is clear that the mixed oxides NiAInBOx exhibit distinctly different CO2ER activity as compared to their single metal oxide counterparts NiO and In2O3. Over the potential range evaluated, in situ reduced NiO produces over 90% H2 faradaic efficiency, while In2O3 exhibits high faradaic efficiency towards formate >80%. Uniquely, the mixed oxides NiAInBOx exhibit high selectivity towards CO, particularly at moderate cathodic bias E ≥ −0.8 V (Fig. 1a and b), reaching maximum FECO of ∼71–73% for a nominal Ni fraction between 25 to 40 at%. This points to a synergy between Ni and In under these conditions. Interestingly, at more cathodic bias E < −0.9 V, a shift in selectivity to favor HCOO− is observed.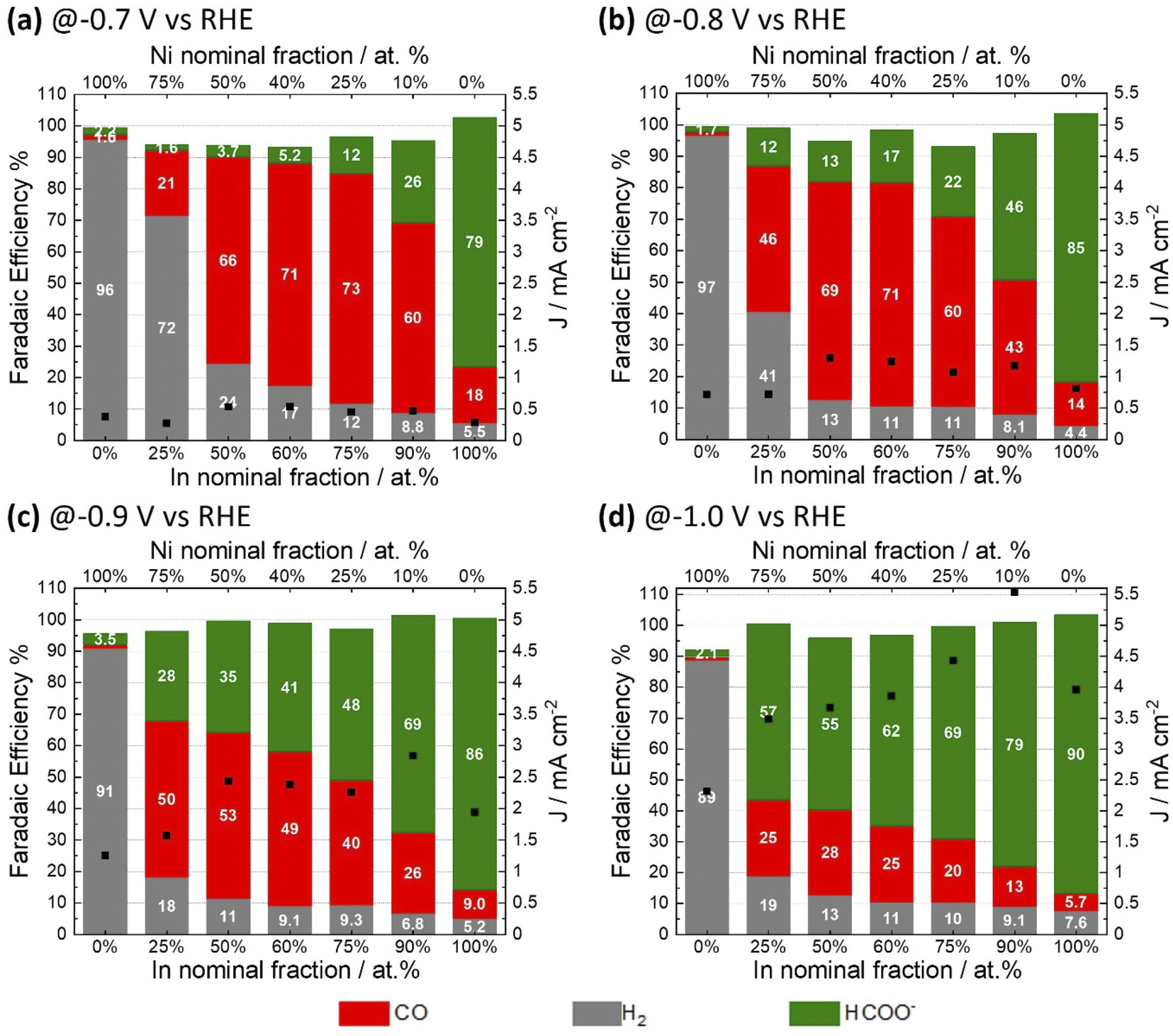 | ||
| Fig. 1 Summary of CO2ER activity for different NiAInBOx mixed oxides tested in CO2 saturated 0.1 M KHCO3 at different applied electrolysis potentials of −0.7 V (a), −0.8 V (b), −0.9 V (c) and −1.0 V (d). Stacked bars correspond to faradaic efficiencies of H2 (grey), CO (red), and formate (green); the total current density is indicated by black squares. | ||
Nickel-indium composites have been previously reported as potential CO2ER catalysts. He et al. reported mixed oxides with composition Ni0.5In0.5 and Ni0.25In0.75 (Ni fraction between 25 to 50 at%) yielding a CO faradaic selectivity of 10 and 25% at −0.7 V.23 In the present study, similar compositions of Ni40In60Ox and Ni25In75Ox display a significantly improved CO selectivity of 71 and 73% at the same potential (Fig. 1a). Interestingly, the best performance in the report by He et al. was observed for a composite with a low Ni fraction of 2 at% (Ni0.02In0.98)23 which exhibited a CO faradaic selectivity of 60% at −0.7 V similar to that observed for the Ni10In90Ox catalyst in the present study (Fig. 1a). However, the Ni10In90Ox catalyst shifts its selectivity to produce predominantly formate with FE of 79% at higher cathodic bias of −1.0 V (Fig. 1d) in agreement with the behavior expected for such In-rich composite. Similar observations have been reported for In2O3 nanocrystals doped with Ni (6 at%) which yield 93% formate FE at −0.9 V. Further comparison of CO2ER activity with literature reports for nickel-indium composites and other catalysts based on earth-abundant elements is presented in Table S7.†
The CO2ER activity of the NiAInBOx samples was also evaluated as product generation rates (partial current densities) as a function of potential and composition (Fig. 2), the results show that the mixed oxides NiAInBOx with nominal Ni fraction between 25 and 50 at% exhibit similar CO rates (−0.4 mA cm−2) at low cathodic bias (−0.7 V), but the formate rate increases with nominal In fraction. The CO partial current peaks at roughly −1.0 to −1.3 mA cm−2 at −0.9 V and either stabilizes or declines at more reductive bias, where formate selectivity becomes favored. On the other hand, formate and H2 partial current densities grow with increasing cathodic bias. The results indicate that at low cathodic bias (E > −0.8 V) NiAInBOx thin films display improved CO production activity, a behavior markedly different from the CO2ER activity of monometallic Ni-9,13 or In-based catalysts.24,25
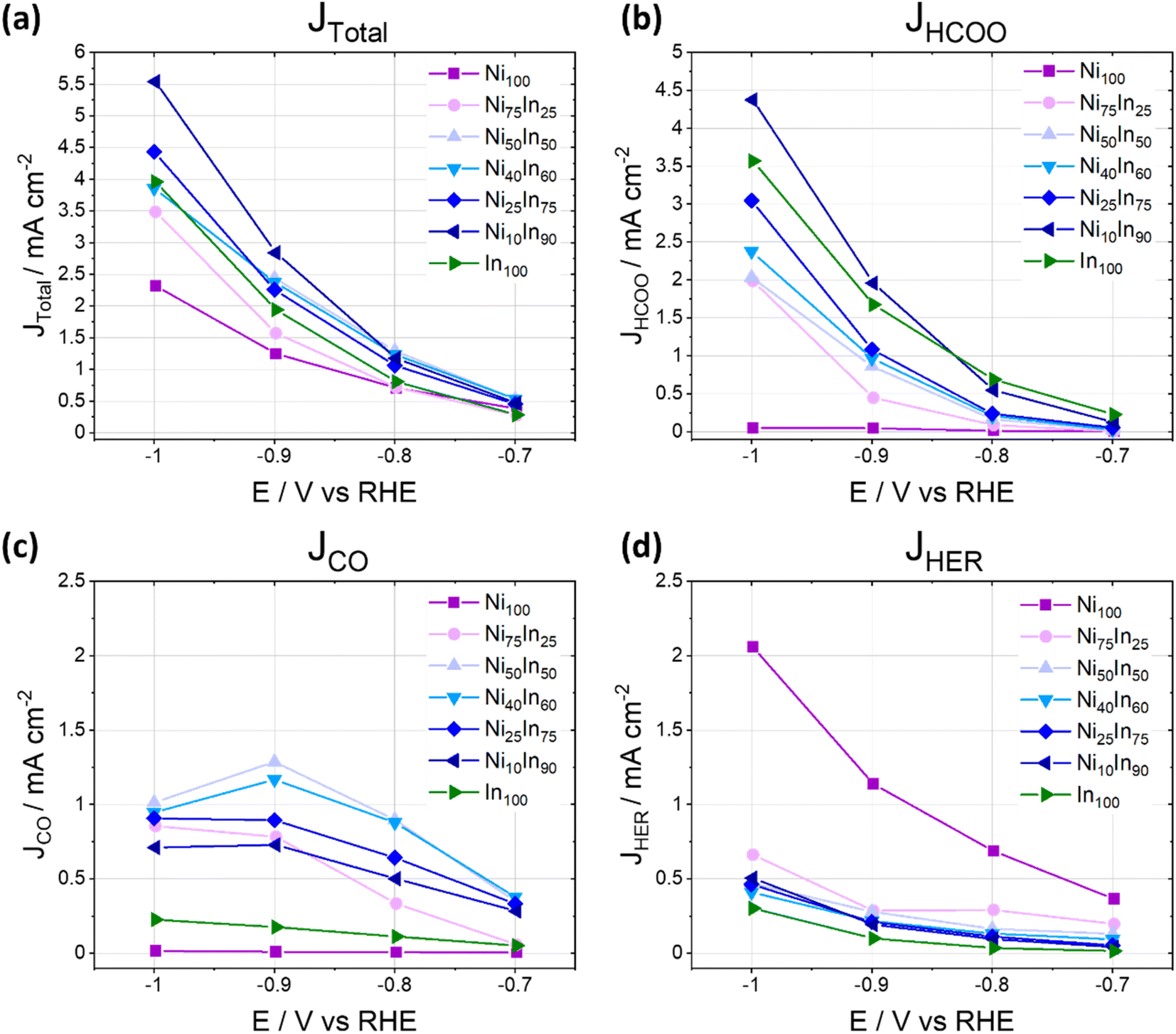 | ||
| Fig. 2 Total and partial current densities as a function of applied potential for different products during CO2ER activity tests for different NiAInBOx mixed oxides tested in CO2 saturated 0.1 M KHCO3 at different applied electrolysis potentials. Note that Fig. 1 and 2 display the same dataset presented as faradaic efficiencies or partial current densities, respectively. | ||
3.2 Structural evolution during CO2 reduction
The structure and composition of NiAInBOx thin films were investigated by SEM and EDS before and after CO2 electrolysis (Fig. S3a–f; and Table S1†). In the as-synthesized condition, the samples have a smooth homogenous thin film morphology with an In:Ni ratio slightly lower than the expected nominal content used in the synthesis. This discrepancy could be attributed to the sublimation of small amounts of In as InCl3(g) during thin film annealing in air.26 After a typical CO2 electrolysis test between up to −1.0 V, the Ni-rich thin films (NiO and Ni75In25Ox) exhibit minor morphological changes (Fig. S3g and h†). For NiAInBOx thin films with Ni fraction ≤50 at%, significant changes in morphology are observed after in situ reduction during CO2ER electrolysis. In general, the transformation from smooth homogenous thin film into an array of nanoparticles is observed (Fig. S3i–l†). For NiAInBOx thin films with (Ni fraction = 25–50 at%) the in situ formed nanoparticles are interconnected in islands with a wide size distribution between 20–200 nm. For In-rich samples (with Ni fraction ≤10 at%), homogenously distributed round particles are formed during CO2ER testing. Interestingly, all NiAInBOx thin films display a decrease in Ni fraction after CO2ER electrolysis, as indicated by the EDS and ICP-OES (Fig. S3 and Tables S1, S2†); these results point to an electrochemically induced leaching of Ni during CO2ER electrolysis.
As discussed above, CO2ER activity tests revealed that NiAInBOx thin films with Ni fraction of 25–50 at% display favored CO selectivity at low bias (E > −0.8 V) while larger bias (E < −0.9 V) favor formate production. In order to investigate the correlation of the selectivity shift with structural changes and Ni leaching, we conducted a detailed investigation of structural changes on a sample with a nominal composition Ni40In60Ox. Note that the following sections of this study focus on samples with nominal composition Ni40In60Ox which display superior CO selectivity and formation rate. Ni40In60Ox films were investigated at different stages of in situ reduction: namely at −0.8 V, where the highest FE towards CO for this electrode (71%) was observed, and −1.0 V, where the highest FE towards formate (62%) was observed. The results are displayed in Fig. 4.
In the as-synthesized condition, the Ni40In60Ox thin films display homogenous coverage of the glassy carbon surface as seen in the SEM image (Fig. 3a). HAADF-STEM-EDS mapping reveals a homogenous distribution of Ni and In across the thin film (Fig. 3g). TEM reveals that the film is composed of small crystallites with diameter <5 nm. SAED analysis displays a wide diffraction ring with d between 2.46–2.90 Å indicating the presence of randomly oriented small crystallites (Fig. 3d and S4†) with lattice spacings typical of NiO d = 2.55 Å and In2O3d = 2.53 and 2.92 Å. Additionally, narrower diffraction rings at d = 2.14, 1.77 and 1.52 Å also assigned to NiO or In2O3 are observed (see further detailed SAED analysis in ESI, Table S3†).
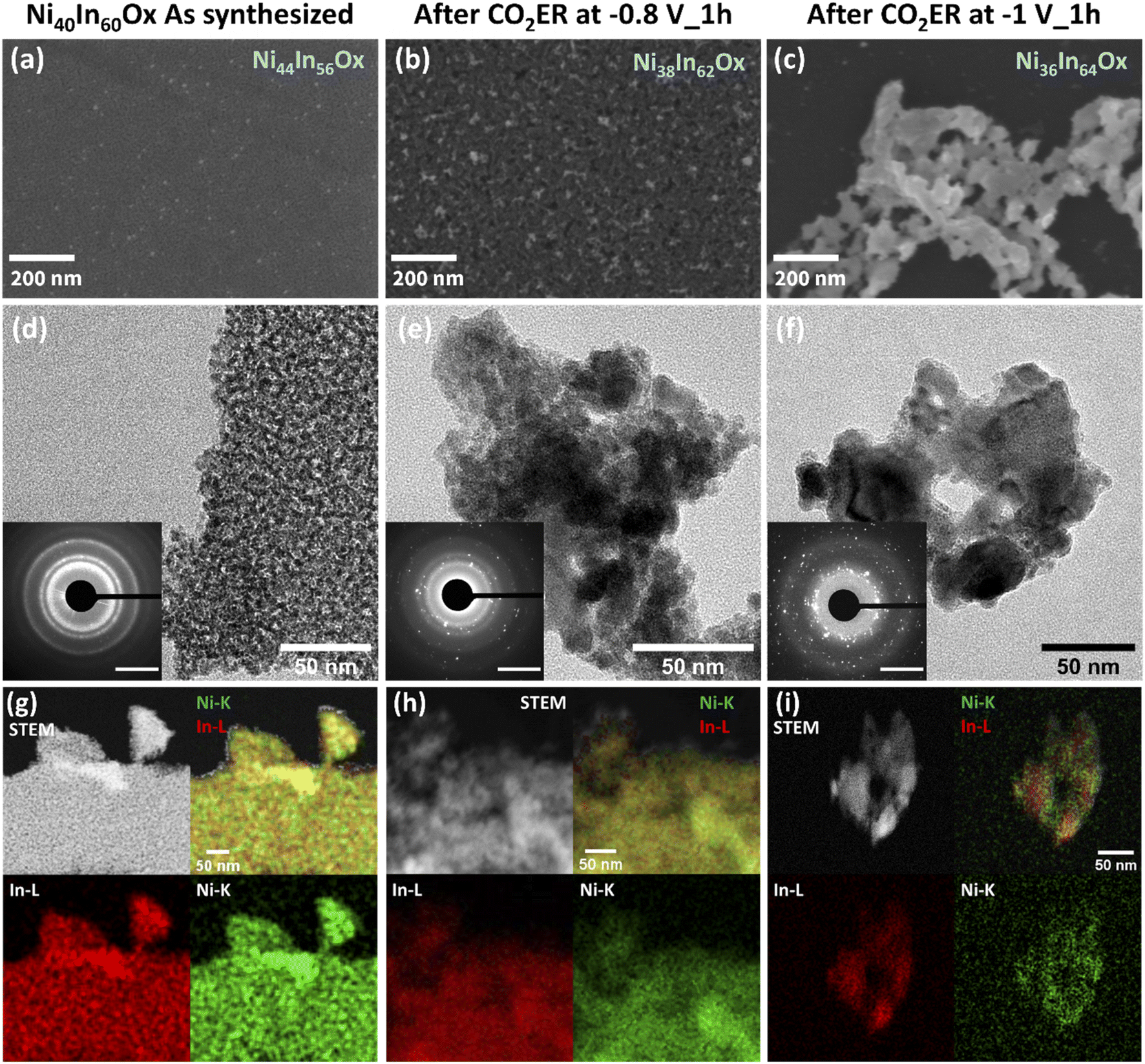 | ||
| Fig. 3 Scanning (SEM) and transmission (TEM) electron microscopy investigation of Ni40In60Ox thin film structural evolution during CO2ER at different applied bias. SEM micrographs of the thin film as-synthesized (a), after CO2ER electrolysis test at −0.8 V (b) and −1.0 V (c). TEM micrographs of the thin film as-synthesized (d), after CO2ER electrolysis test at −0.8 V (e) and −1.0 V (f); SAED analysis is presented as inset (scale bar is 5 nm−1). HAADF-STEM-EDS mapping of the thin film as-synthesized (g), after CO2ER electrolysis test at −0.8 V (h) and −1.0 V (i). Experimental composition as determined by ICP-OES is presented as text inset on the SEM image for each sample. | ||
After CO2ER testing at −0.8 V vs. RHE for 1 h, part of the thin film is preserved decorated with embedded nanoparticles (diameter <50 nm) as seen by SEM and TEM micrographs (Fig. 3b and e). SAED reveals numerous bright diffraction spots together with the diffuse rings of diffraction observed for the pristine sample providing further evidence of the formation of crystalline nanoparticles embedded in persistent amorphous mixed oxide. SAED analysis indicates that the crystalline particles display d spacings which could be assigned to metallic In and Ni, as well as NiXInY alloys matching NiIn and Ni3In7 particularly (Fig. S4†). Note that the overlapping d spacings among these species hinders unequivocal assignment of phases. Further details on possible assigned phases are provided in Table S3.† HRTEM investigation enables identification and clustering of the d spacings observed in different areas of the sample which confirms the presence of small grains of Ni and/or NiXInY alloys embedded in a disordered matrix likely composed of residual mixed oxide or small NiXInY alloys crystallites, denoted as Ni–NiXInY/Ni40−XIn60−YOx. Details on HRTEM analysis, d spacings, and phase assignment are provided in Fig. S5 and Table S4.† The HAADF-STEM-EDS (Fig. 3h) mapping indicates that the amorphous small crystallites and nanoparticles are composed of both In and Ni, though the nanoparticles appear to be enriched in Ni. The persistence of Ni–In oxidized species was further investigated by HAADF-STEM-EDS. A sample Ni40In60Ox was tested for CO2ER at −0.8 V for 1 h inside an O2-free glovebox. To prevent oxidation of the catalysts during handling and thereby evaluate the catalysts as close as possible to the in situ conditions, the sample was then transferred to the TEM under inert atmosphere using a transfer holder. The results are presented in Fig. S9c† and indicate the persistence of O content in the sample, providing further evidence of the persistence of oxidized Ni and In phases.
After CO2ER at a stronger reducing potential of −1.0 V for 1 h (note that the film is sequentially tested at −0.7, −0.8, −0.9 and −1 V for 1 h), the film has completely transformed into an array of nanoparticles of varied dimensions (10–100 of nm) as seen in the SEM and TEM micrographs (Fig. 3c and f). SAED reveals an increase of bright diffraction spots and the disappearance of diffuse diffraction rings associated with the pristine amorphous oxide film in comparison with the catalyst tested at −0.8 V for 1 h (Fig. 3b and e). The d spacings observed in SAED analysis are consistent with the formation of In and Ni3In7 particles (see Fig. S4 and Table S3†). Correlation of local d spacings identified by HRTEM (Fig. S6†) and HAADF-STEM-EDS (Fig. 3i) mapping enables us to establish the formation of In-rich particles composed by In and/or Ni3In7 alloy decorated with smaller Ni-rich crystallites composed of NiIn alloy, metallic Ni, and/or NiO. Further details on HRTEM d spacing assignments are displayed in Table S5.† Note that the HAADF-STEM-EDS was conducted on a sample tested on a O2-free glovebox; handled and transferred to the TEM under inert atmosphere to prevent oxidation in air. The sample displays no discernible O content (Fig. S9d†). These results provide further evidence of a complete bulk reduction of oxide-derived catalyst under these conditions; within the detection limits of the EDS.
The bulk composition analyzed by ICP-OES indicates that the Ni fraction decreases from 44% in the as-synthesized sample to 38 and 36% at −0.8 V and −1.0 V, respectively (Table S2†). These results indicate that the morphology change from amorphous thin film to a composite of crystalline nanoparticles composed of metallic In, Ni, or NiXInY alloys induced by the applied reductive bias is accompanied by a minor loss of Ni. Furthermore, the structural changes lead to a dynamic roughness of the catalytic surface as evidenced by double layer capacitance (CDL) measurements (Fig. 6b and S10†). In the as-synthesized condition Ni40In60Ox the amorphous film displays a capacitance of 244 μF cm−2. After electrolysis at −0.8 V, the Ni–NiXInY/Ni40−XIn60−YOx composite with the highest CO selectivity is formed and the capacitance increases to 319 μF cm−2. Further increase of cathodic bias to −1 V leads to the aggregation of metallic In, Ni, or NiXInY alloys grains which coincides with the decrease of capacitance to 177 μF cm−2. These observations indicate that the optimal CO selectivity observed for the Ni–NiXInY/Ni40−XIn60−YOx composite is favored by the persistent amorphous oxide and the improved catalyst roughness.
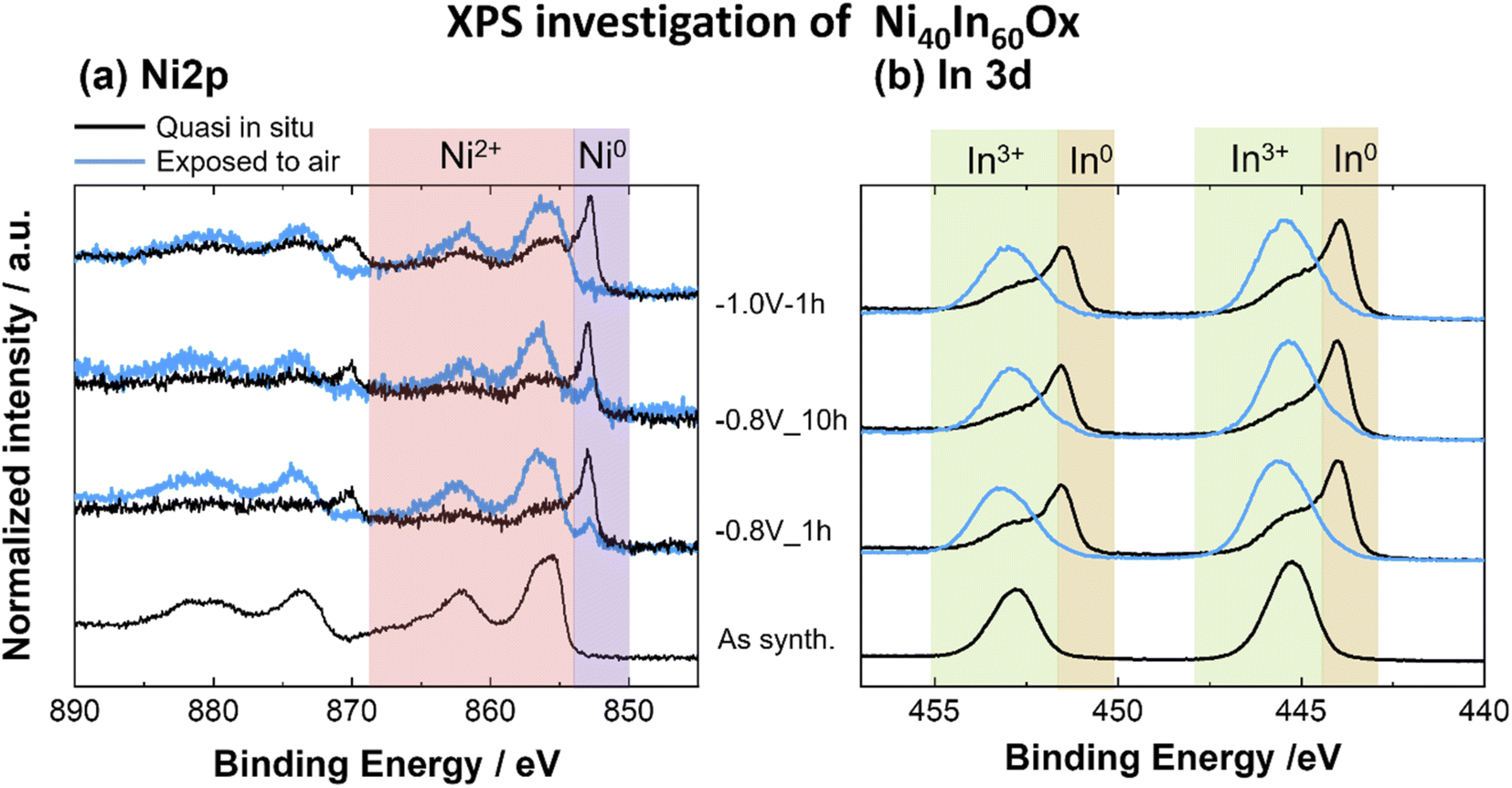 | ||
| Fig. 4 XPS spectra for (a) Ni 2p, (b) In 3d core levels for Ni40In60Ox thin films as-synthesized and after CO2ER electrolysis test at −0.8 V for 1 h, 10 h or −1.0 V for 1 h. | ||
Furthermore, a very dynamic surface composition was observed by XPS (Fig. 6a). In the as-synthesized condition, the sample displays a surface Ni fraction (referred to total Ni + In surface content, [Ni/(Ni + In)] × 100) of 27 at%, a value lower than the bulk Ni fraction determined by ICP-OES, which indicates an enrichment of In at the oxide thin film surface. After CO2ER testing up to −0.8 V, the surface Ni fraction decreases to 20 at% in contrast with the 38 at% bulk Ni fraction determined by ICP-OES. Taken together with the observations of Ni-rich dense particles embedded in amorphous matrix seen by STEM-EDS mapping (Fig. 3h), we conclude that the −0.8 V condition induces Ni depletion from the surface and enrichment in the bulk and increases the relative In fraction at the surface. Similar segregation of In towards the surface has been previously observed in Cu–In21 and Sn–In.27 Interestingly, after CO2ER testing up to higher bias (−1.0 V), the surface Ni fraction is 33 at%, a value closer to the bulk Ni fraction determined by ICP-OES at this potential (36 at%). These observations agree with the formation of In-rich larger particles (In and Ni3In7) decorated with Ni-rich smaller crystallites observed by HRTEM (Fig. S6†) and TEM-EDS mapping (Fig. 3i). The dynamic changes in metal composition observed at the surface (XPS) and bulk (ICP-OES) for the Ni40In60Ox samples after CO2ER testing are summarized in Fig. 6a.
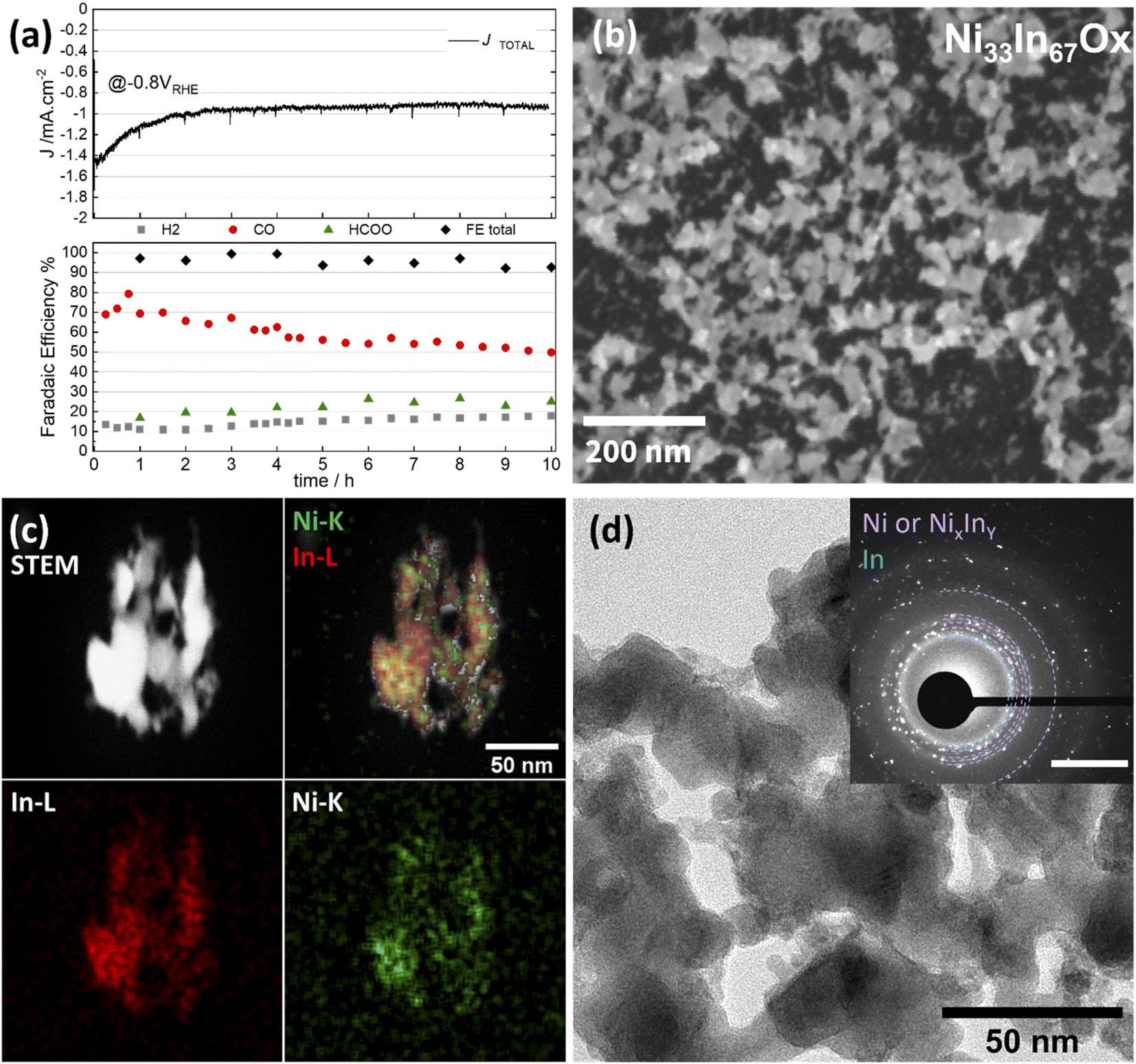 | ||
| Fig. 5 Study of long-term stability of Ni40In60Ox thin films during CO2ER testing at −0.8 V in CO2 sat. 0.1 M KHCO3. (a) CO2ER activity over 10 h test. (Top) Current density profile vs. time. (Bottom) Faradaic efficiencies for H2 (grey squares), CO (red circles), formate (green triangles) as well as added total (black diamonds). Structural characterization of thin film sample after long electrolysis (10 h) test (b) SEM, (c) STEM-EDS mapping and (d) TEM-SAED analysis is displayed as inset (scale bar 5 nm−1). | ||
The changes in metal composition observed before and after the extended electrolysis test at −0.8 V are summarized in Fig. 6a. The bulk Ni fraction determined by ICP-OES decreases from 38 at% to 33 at% after 1 and 10 h of CO2 electrolysis at −0.8 V, respectively, indicating a slow but sustained leaching of Ni. However, the surface Ni fraction determined by quasi in situ XPS remains rather constant at around 20% following the 10 h electrolysis test. This stability indicates that the CO selectivity loss observed in this experiment is not simply attributable to changes in the surface Ni fraction (i.e. Ni:In surface ratio) but rather correlates with the structural changes taking place: progressive reduction of the residual amorphous oxide to alloy and aggregation of larger alloy grains leading to a decrease in roughness as evidenced by decrease of capacitance to 74 μF cm−2 (Fig. 6d and S10†). The CO selectivity decrease in favor of formate and H2 occurring during long electrolysis test at −0.8 V is likely caused by these structural transformations.
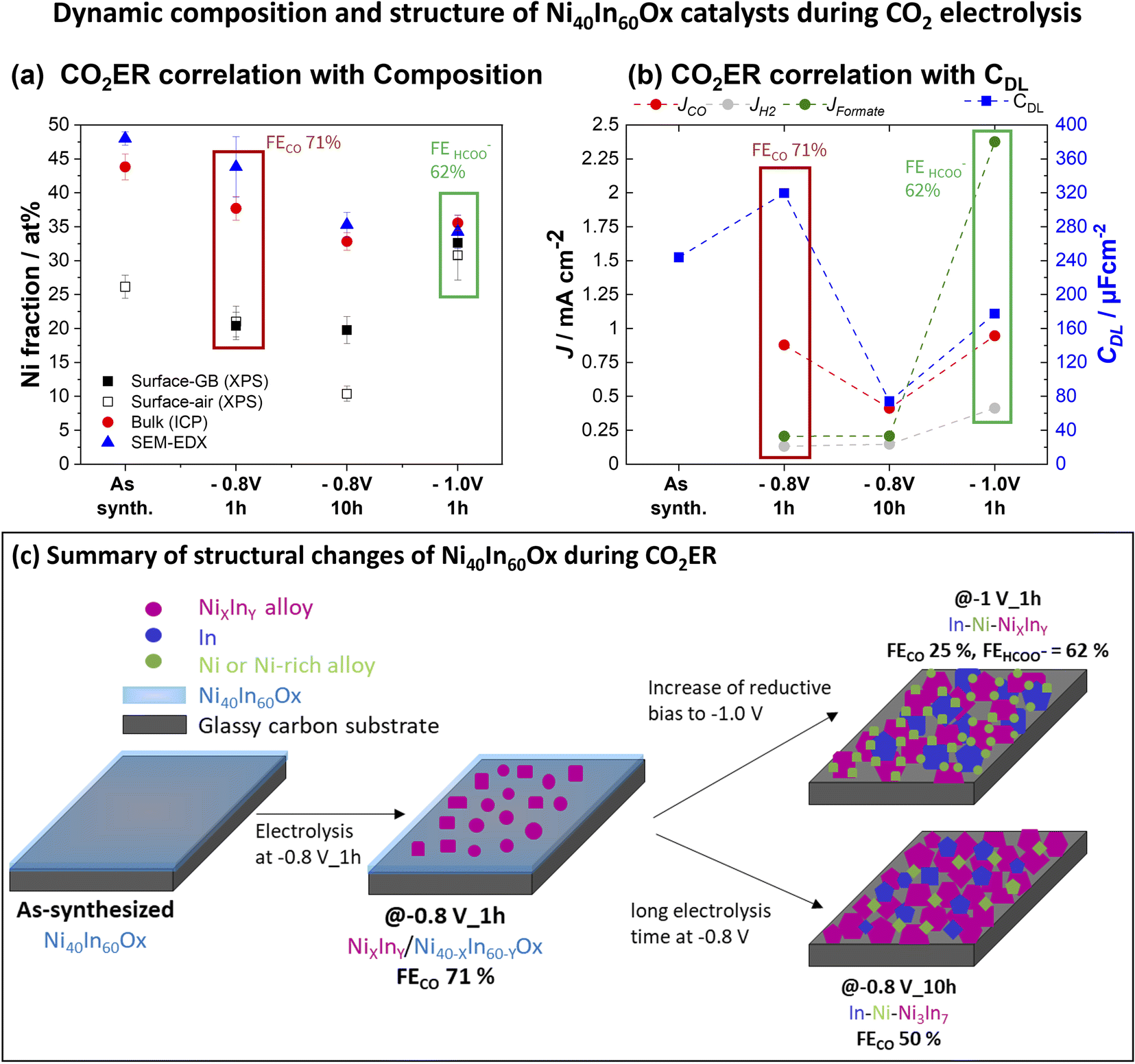 | ||
| Fig. 6 Dynamic composition and structure of Ni40In60Ox catalysts during CO2 electrolysis. (a) Surface and bulk Ni fraction (referred to total metal Ni + In) observed for in Ni40In60Ox samples during CO2ER electrolysis, surface composition was investigated by XPS in samples transferred from the glovebox (filled black squares) and after exposure to air for 24 h (hollow black squares). Bulk composition was investigated by ICP-OES (red circles) and EDS in SEM (blue triangles). Error bars display the standard deviation in quantification results on at least two samples measured 3 different spots. (b) Correlation of surface roughness (as CDL, blue squares) of Ni40In60Ox samples and CO2ER activity. (c) Summary of structural evolution of Ni40In60Ox mixed oxides during CO2ER in 0.1 M KHCO3. | ||
It is worth noting that the surface Ni fraction measured on this sample after exposure to air for 24 h is significantly lower (∼10 at%) than that observed on the sample preserved under inert atmosphere between electrochemical testing and XPS analysis (20 at%). Such an effect is likely the result of a segregation of In towards the surface of Ni3In7 grains upon oxidation in air. This observation highlights the importance of minimizing or excluding air exposure before the analysis of the surface composition, as it leads not only to changes in surface speciation (i.e. oxidation of the majority of metal surface sites), but also to changes in the relative metal surface composition.
3.3 Structure–activity correlation
The detailed structural investigation after diverse electrochemical testing conditions reveals that the Ni40In60Ox mixed oxides act as a pre-catalyst material which undergoes complex structural transformations during in situ reduction under CO2ER conditions. The structural changes are strongly dependent on applied bias and electrolysis time. Our conclusions are summarized in Fig. 6c and as follows: At low bias E < −0.8 V during short electrolysis times (1 h), an early stage of material reduction is observed in which NiXInY alloy grains are formed embedded in amorphous Ni–In mixed oxide, denoted NiXInY/Ni40−XIn60−YOx, as observed by SEM/TEM (Fig. 3 and S5†). It is at this heterogenous NiXInY/Ni40−XIn60−YOx alloy/oxide composite that the highest roughness and CO selectivity is observed (71%). Further increase of reductive bias to −1.0 V (Fig. S6†) or prolonged electrolysis for 10 h at −0.8 V (Fig. S7†) results in complete reduction of the residual Ni40In60Ox film into interconnected array of In–Ni–NiXInY nanoparticles, which display significantly lower CO selectivity (<50%) at −0.8 V. Two main structural changes are triggered at high reductive bias (E < −0.9 V) or over extended electrolysis at −0.8 V: (1) the reduction of the residual amorphous oxide to alloy and (2) the aggregation of larger metal/alloy grains leading to a decrease in roughness as evidenced in the CDL measurements (Fig. 6b and S10†). The CO selectivity decrease from 71% to 50% at −0.8 V during 10 h or the increase in formate selectivity (62%) at −1.0 V could therefore be correlated to these structural transformations. However, the experiments presented so far do not allow us to conclude if the selectivity changes are only directed by the changes in catalyst composition/morphology, or if they are governed by a change in bias-dependent CO2ER activation mechanism as well.The increase in formate selectivity at higher reductive bias (E < −0.9 V) is likely to be combined effect of both processes: structural changes and activation of a favorable formate pathway in this potential range. Several formate selective CO2ER electrocatalysts composed of Sn,28–30 In15–17 as well as bimetallic compositions such as Cu–Sn31,32 and Cu–In33 reach their best formate selectivity at medium–high reductive bias ranges (E < −0.9 V).
In order to distinguish the effects, a sample was tested up to −1.0 V to ensure material transformation from amorphous oxide film into In–Ni–Ni3In7 nanoparticles composite and subsequently tested again for CO2ER at −0.8 V, the potential at which CO production is typically favored. The results are displayed in Fig. S8.† The FECO observed at −0.8 V for this sample (46%) is very similar to the FECO observed on the nanoparticle network formed after 10 h of electrolysis at −0.8 V (50%). Two conclusions can be drawn from this experiment. The first is that the catalyst fully transformed into In–Ni–Ni3In7 nanoparticles composite displays a shift in selectivity from CO (46%) at −0.8 V to formate (62%) at −1.0 V, indicating that increasing bias does favor a formate production pathway in this catalytic system, even if structural changes have already been completed. The second is that in the early stage of structural transformation from mixed oxide to In–Ni–Ni3In7 nanoparticle composite catalyst, the amorphous materials composed of oxide and small crystallite NiXInY play a key role in the high CO selectivity (70%) observed during the first hours of catalytic test at −0.8 V (Fig. 5). To summarize, the CO selectivity loss observed during CO2ER tests at −0.8 V from 71% (1 h) to 50% (10 h) is associated with the slow transformation of amorphous oxide/alloy composite into aggregated In–Ni–Ni3In7 nanoparticles composite at this potential. The increase in formate selectivity at E < −0.9 V appears to be triggered, at least partially, to the activation of favorable formate pathway at this potential.15–17,28–33
The observation of these highly dynamic structural transformations emphasizes the necessity to investigate catalyst materials in detail not only before catalytic testing in the as-synthesized condition, but also during and after CO2ER electrolysis. Such detailed characterization allows evaluation of the impact of structural changes on the observed CO2ER selectivity trends while recognizing that often both structure and CO2ER selectivity are dependent on applied potential and experiment runtime. In the specific case of the Ni40In60Ox mixed oxide catalyst in this study, the identification of the residual mixed oxide film as an important aspect in the observed CO selectivity provides a catalyst design goal to stabilize the oxidized phases during catalysis. The CO selectivity of oxidized Ni(II) sites has already been demonstrated for Ni “single atom” sites on N-doped carbon materials,34–38 pointing to a possible common feature favoring CO selectivity on Ni-based catalysts when comparing to our results. While in our case the progressive in situ metallization and loss of oxide is sensitive to potential and time under steady-state operation, we note that innovative strategies to stabilize metastable oxidized species – such as electrochemical pulsing involving periodic surface re-oxidation, which has been demonstrated for tuning selectivity and improving stability on copper12 – could be pursued for mixed-metal systems such as those shown here toward the goal of further controlling selectivity and stability.
3.4 Oxide-derived vs. alloy catalyst
To further investigate if the original Ni40In60Ox mixed oxide is necessary to attain high CO selectivity at low bias (E > −0.8 V), an alloy of similar metal composition, Ni2In3, was synthesized by thermal treatment of a mixed oxide thin film under forming gas atmosphere (see Experimental section for details). This method yields an array of Ni2In3 alloy nanoparticles as seen in the SEM micrographs and GI-XRD characterization (Fig. 7a and b). The comparison of CO2ER activity between mixed oxide Ni40In60Ox thin film and the Ni2In3 alloy nanoparticles is displayed in Fig. 7c. The results show that the CO selectivity for Ni2In3 is significantly poorer than for Ni40In60Ox mixed oxide and provide further evidence that the use of the mixed oxide as a starting material (or pre-catalyst) is essential to the in situ formation of an oxide-derived Ni–In composite with high CO selectivity during CO2ER.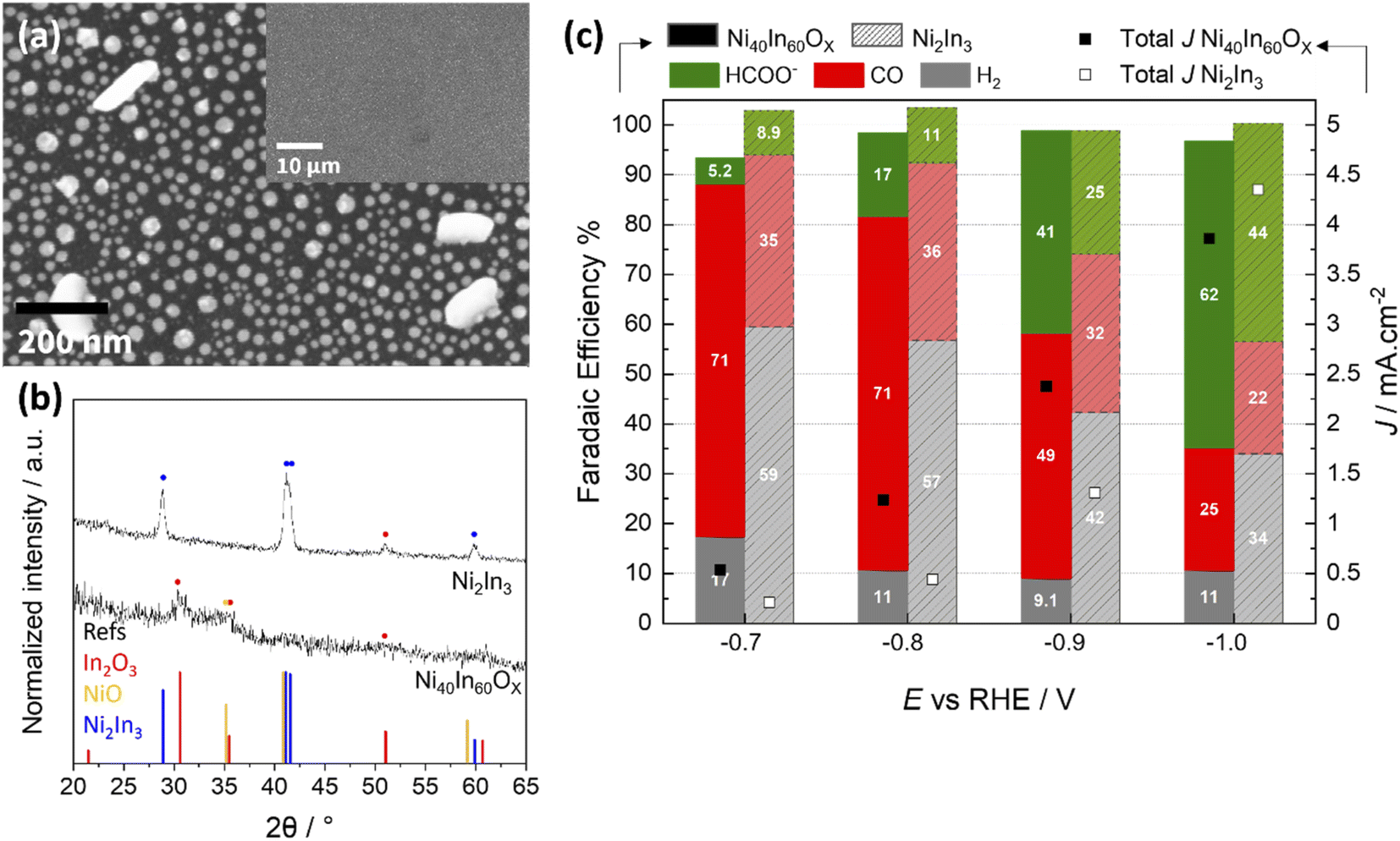 | ||
| Fig. 7 Structural characterization of Ni2In3 alloy synthesized by annealing of mixed oxide film in forming gas at (2.4% H2 in Ar) at 500 °C. (a) SEM micrographs (low magnification image presented as inset) (b) GI-XRD patterns collected on samples deposited on Si wafer; reference XRD patterns for In2O3 (PDF 00-006-0416), NiO (PDF 01-085-5454) and Ni2In3 alloy (PDF 04-003-1606) are displayed for comparison. (c) CO2ER activity of Ni40In60Ox mixed oxide and Ni2In3 alloy in CO2 sat. 0.1 M KHCO3 at various applied bias between −0.7 V and −1.0 V vs. RHE. Note that at each potential, the left components correspond to Ni40In60Ox mixed oxide (FE: solid-colored bars; total current density: black squares), while the right components represent Ni2In3 alloy (FE: lighter, patterned bars; total current density: white squares). | ||
These observations, together with residual amorphous mixed oxide observed for Ni40In60Ox sample in its CO-selective form (CO2ER at −0.8 V for 1 h) by TEM, SAED (Fig. S4, S5†) and quasi in situ XPS (Fig. 5), indicate that residual amorphous oxide favors improved CO selectivity. These persistent Ni–In oxides exhibit poor stability under CO2ER conditions and are slowly reduced over the course of several hours. It is worth noting, however, that the metal In–Ni–NiXInY alloy nanoparticles produced by in situ reduction of the Ni40In60Ox thin film at −0.8 V (10 h) or −1.0 V (1 h) display significantly better CO2ER selectivity (CO + formate) over H2 in comparison with the Ni2In3 alloy nanoparticles (Fig. 7c). These results indicate an advantage of oxide-derived Ni–In composites over metallic/alloy materials as has been previously reported for Cu,39,40 In15,24 based catalysts, other bimetallic composites,27,41 and more recently Ni catalysts.42 In the aforementioned study, the authors found that the persistence of polarized Niδ+ sites in oxide-derived Ni catalysts weakens *CO binding at the catalyst surface facilitating its further reduction to hydrocarbons. Similar effect of weaking of *CO and *H binding has been reported for positively polarized In sites.43 It is likely that for the Ni40In60Ox mixed oxides presented here, the persistence of oxidized Ni and In sites at −0.8 V contribute to the weakening of *CO, facilitating its release as main product.
4 Conclusions
NiAInBOx mixed oxide amorphous thin films with homogeneous metal distribution were successfully synthesized by spin coating of a solution containing metal chloride precursors in desired ratios. The CO2ER activity evaluation of in situ reduced NiAInBOx mixed oxides with Ni fraction between 25–50 at% shows favorability towards CO production (FECO > 70%), in strong contrast with the behavior of the single metal oxides NiO (>90% H2) and In2O3 (FEFORMATE > 80%). The best performance in terms of FECO and CO partial current density was found for the Ni40In60Ox thin film at −0.8 V with 71% FECO and CO partial current density ∼−1.0 mA cm−2. At higher reductive bias of E < −0.9 V, the selectivity shifts to favor formate. This shift in selectivity is accompanied by a structural transformation starting from an amorphous mixed oxide into a composite of interconnected nanoparticles composed of metallic In, Ni and NiXInY alloys denoted In–Ni–NiXInY. The correlation of structural characterization after CO2 electrolysis at different conditions (potential, time) with observed CO2ER catalytic activity reveals that the CO selectivity is favored by the presence of persistent oxidized Ni(II) and In(III) species as well as small NiXInY alloy crystallites in the form of amorphous oxide/alloy composite during the early stages of material transformation at low reductive bias (E ≥ −0.8 V). However, this amorphous oxide/alloy composite material is unstable under operation conditions and transforms – gradually at −0.8 V (10 h) or more readily at −1.0 V (<1 h) – to form an In–Ni–NiXInY nanoparticle composite displaying lower CO faradaic efficiency at a low bias of −0.8 V (FECO 50%) and favored formate selectivity (62%) at −1.0 V. Our experimental results indicate that the CO selectivity loss from 71% to 50% during 10 h of electrolysis at a low bias of −0.8 V is associated with the slow structural transformation of amorphous Ni40In60Ox mixed oxide into In–Ni–NiXInY nanoparticles composite at this potential. The increase in formate selectivity at high reductive bias of E < −0.9 V is likely the result of a more favorable formate pathway in this potential range, as such an effect has been observed in other In and Sn based electrocatalysts.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Helmholtz Association's Initiative and Networking Fund through Helmholtz Young Investigator Group (VH-NG-1225) and the Helmholtz Climate Initiative (Net-Zero-2050). The research utilized instrumentation within the Helmholtz Energy Materials Foundry (HEMF), the HZB corelate Correlative Microscopy and Spectroscopy, the HZB Institute for Solar Fuels and the HZB X-ray core lab. We thank the following colleagues for experimental and technical support: Ursula Michalczik, Álvaro Diaz Duque, Christian Höhn, René Gunder and Michael Tovar.References
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- Y. Hori, A. Murata and R. Takahashi, J. Chem. Soc., Faraday Trans. 1, 1989, 85, 2309 RSC.
- A. A. Peterson, J. K. Nørskov and J. K. Norskov, J. Phys. Chem. Lett., 2012, 3, 251–258 CrossRef CAS.
- M. Azuma, J. Electrochem. Soc., 1990, 137, 1772 CrossRef CAS.
- K. Ito, S. Ikeda, N. Yamauchi, T. Iida and T. Takagi, Bull. Chem. Soc. Jpn., 1985, 58, 3027–3028 CrossRef CAS.
- S. Ikeda, T. Takagi and K. Ito, Bull. Chem. Soc. Jpn., 1987, 60, 2517–2522 CrossRef CAS.
- H. Noda, S. Ikeda, Y. Oda, K. Imai, M. Maeda, K. Ito, S. Ideka, Y. Oda, K. Imai, M. Maeda and I. Kaname, Bull. Chem. Soc. Jpn., 1990, 63, 2459–2462 CrossRef CAS.
- H. Yoshio, K. Katsuhei and S. Shin, Chem. Lett., 1985, 14, 1695–1698 CrossRef.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Electrochim. Acta, 1994, 39, 1833–1839 CrossRef CAS.
- J. T. Feaster, C. Shi, E. R. Cave, T. Hatsukade, D. N. Abram, K. P. Kuhl, C. Hahn, J. K. Nørskov and T. F. Jaramillo, ACS Catal., 2017, 7, 4822–4827 CrossRef CAS.
- A. Bagger, W. Ju, A. S. Varela, P. Strasser and J. Rossmeisl, ChemPhysChem, 2017, 18, 3266–3273 CrossRef CAS PubMed.
- R. M. Arán-Ais, F. Scholten, S. Kunze, R. Rizo and B. Roldan Cuenya, Nat. Energy, 2020, 5, 317–325 CrossRef.
- E. R. Cave, C. Shi, K. P. Kuhl, T. Hatsukade, D. N. Abram, C. Hahn, K. Chan and T. F. Jaramillo, ACS Catal., 2018, 3035–3040 CrossRef CAS.
- M. N. Mahmood, D. MAsheder and C. J. Harty, J. Appl. Electrochem., 1987, 17, 1159–1170 CrossRef CAS.
- Z. M. Detweiler, J. L. White, S. L. Bernasek and A. B. Bocarsly, Langmuir, 2014, 30, 7593–7600 CrossRef CAS PubMed.
- Z. Xia, M. Freeman, D. Zhang, B. Yang, L. Lei, Z. Li and Y. Hou, ChemElectroChem, 2018, 5, 253–259 CrossRef CAS.
- J. Zhang, R. Yin, Q. Shao, T. Zhu and X. Huang, Angew. Chem., Int. Ed., 2019, 58, 5609–5613 CrossRef CAS PubMed.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S. Z. Qiao, Chem, 2018, 1–23 Search PubMed.
- M. K. Birhanu, M.-C. Tsai, A. W. Kahsay, C.-T. Chen, T. S. Zeleke, K. B. Ibrahim, C.-J. Huang, W.-N. Su and B.-J. Hwang, Adv. Mater. Interfaces, 2018, 5, 1800919 CrossRef.
- L. C. Pardo Pérez, A. Arndt, S. Stojkovikj, I. Y. Ahmet, J. T. Arens, F. Dattila, R. Wendt, A. Guilherme Buzanich, M. Radtke, V. Davies, K. Höflich, E. Köhnen, P. Tockhorn, R. Golnak, J. Xiao, G. Schuck, M. Wollgarten, N. López and M. T. Mayer, Adv. Energy Mater., 2022, 12, 2103328 CrossRef.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2016, 6, 6265–6274 CrossRef.
- J. Monzó, Y. Malewski, R. Kortlever, F. J. Vidal-Iglesias, J. Solla-Gullón, M. T. M. Koper and P. Rodriguez, J. Mater. Chem. A, 2015, 3, 23690–23698 RSC.
- J. He, K. E. Dettelbach, D. A. Salvatore, T. Li and C. P. Berlinguette, Angew. Chem., Int. Ed., 2017, 56, 6068–6072 CrossRef CAS PubMed.
- J. L. White and A. B. Bocarsly, J. Electrochem. Soc., 2016, 163, H410–H416 CrossRef CAS.
- J. Li, M. Zhu and Y. F. Han, ChemCatChem, 2021, 13, 514–531 CrossRef CAS.
- C. Karakaya, S. Ricote, D. Albin, E. Sánchez-Cortezón, B. Linares-Zea and R. J. Kee, Thermochim. Acta, 2015, 622, 55–63 CrossRef CAS.
- L. C. Pardo Pérez, D. Teschner, E. Willinger, A. Guiet, M. Driess, P. Strasser and A. Fischer, Adv. Funct. Mater., 2021, 31, 2103601 CrossRef.
- L. Fan, Z. Xia, M. Xu, Y. Lu and Z. Li, Adv. Funct. Mater., 2018, 28, 1706289 CrossRef.
- B. Kumar, V. Atla, J. P. Brian, S. Kumari, T. Q. Nguyen, M. Sunkara and J. M. Spurgeon, Angew. Chem., Int. Ed., 2017, 56, 3645–3649 CrossRef CAS PubMed.
- S. Zhang, P. Kang and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 1734–1737 CrossRef CAS.
- C. Chen, Y. Pang, F. Zhang, J. Zhong, B. Zhang and Z. Cheng, J. Mater. Chem. A, 2018, 6, 19621–19630 RSC.
- X. Hou, Y. Cai, D. Zhang, L. Li, X. Zhang, Z. Zhu, L. Peng, Y. Liu and J. Qiao, J. Mater. Chem. A, 2019, 7, 3197–3205 RSC.
- Q. Xie, G. O. Larrazábal, M. Ma, I. Chorkendorff, B. Seger and J. Luo, J. Energy Chem., 2021, 63, 278–284 CrossRef.
- T. N. Nguyen, M. Salehi, Q. Van Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- K. Jiang, S. Siahrostami, T. Zheng, Y. Hu, S. Hwang, E. Stavitski, Y. Peng, J. Dynes, M. Gangisetty, D. Su, K. Attenkofer and H. Wang, Energy Environ. Sci., 2018, 11, 893–903 RSC.
- T. Zheng, K. Jiang, N. Ta, Y. Hu, J. Zeng, J. Liu and H. Wang, Joule, 2019, 3, 265–278 CrossRef CAS.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang, J. Liu and C. He, Nat. Commun., 2020, 11, 1–8 CrossRef PubMed.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- J. E. Pander, D. Ren, Y. Huang, N. W. X. Loo, S. H. L. Hong and B. S. Yeo, ChemElectroChem, 2018, 5, 219–237 CrossRef CAS.
- Y. Lum, B. Yue, P. Lobaccaro, A. T. Bell and J. W. Ager, J. Phys. Chem. C, 2017, 121, 14191–14203 CrossRef CAS.
- G. O. Larrazábal, A. J. Martín, S. Mitchell, R. Hauert and J. Pérez-Ramírez, ACS Catal., 2016, 6, 6265–6274 CrossRef.
- Y. Zhou, A. J. Martín, F. Dattila, S. Xi, N. López, J. Pérez-Ramírez and B. S. Yeo, Nat. Catal., 2022, 5, 545–554 CrossRef CAS.
- D. Pavesi, F. Dattila, R. C. J. Van de Poll, D. Anastasiadou, R. García-Muelas, M. Figueiredo, G. J. M. Gruter, N. López, M. T. M. Koper and K. J. P. Schouten, J. Catal., 2021, 402, 229–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta05214h |
| This journal is © The Royal Society of Chemistry 2022 |