
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effect of caesium alloying on the ultrafast structural dynamics of hybrid organic–inorganic halide perovskites†
Nathaniel P.
Gallop
 *a,
Junzhi
Ye
bc,
Gregory M.
Greetham
d,
Thomas L. C.
Jansen
e,
Linjie
Dai
b,
Szymon J.
Zelewski
bf,
Rakesh
Arul
b,
Jeremy J.
Baumberg
b,
Robert L. Z.
Hoye‡
c and
Artem A.
Bakulin
a
*a,
Junzhi
Ye
bc,
Gregory M.
Greetham
d,
Thomas L. C.
Jansen
e,
Linjie
Dai
b,
Szymon J.
Zelewski
bf,
Rakesh
Arul
b,
Jeremy J.
Baumberg
b,
Robert L. Z.
Hoye‡
c and
Artem A.
Bakulin
a
aDepartment of Chemistry, Imperial College London, Molecular Sciences Research Hub, 83 Wood Lane, London W12 0BZ, UK. E-mail: nathaniel.gallop12@imperial.ac.uk
bCavendish Laboratory, University of Cambridge, JJ Thomson Avenue, Cambridge CB3 0HE, UK
cDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK
dCentral Laser Facility, Rutherford Appleton Laboratory, Harwell Campus, Didcot, OX11 0QX, UK
eZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG, Groningen, Netherlands
fDepartment of Semiconductor Materials Engineering, Faculty of Fundamental Problems of Technology, Wrocław University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland
First published on 4th October 2022
Abstract
Hybrid inorganic–organic perovskites have attracted considerable attention over recent years as promising processable electronic materials. In particular, the rich structural dynamics of these ‘soft’ materials has become a subject of investigation and debate due to their direct influence on the perovskites' optoelectronic properties. Significant effort has focused on understanding the role and behaviour of the organic cations within the perovskite, as their rotational dynamics may be linked to material stability, heterogeneity and performance in (opto)electronic devices. To this end, we use two-dimensional IR spectroscopy (2DIR) to understand the effect of partial caesium alloying on the rotational dynamics of the methylammonium cation in the archetypal hybrid perovskite CH3NH3PbI3. We find that caesium incorporation primarily inhibits the slower ‘reorientational jump’ modes of the organic cation, whilst a smaller effect on the fast ‘wobbling time’ may be due to distortions and rigidisation of the inorganic cuboctahedral cage. 2DIR centre-line-slope analysis further reveals that while static disorder increases with caesium substitution, the dynamic disorder (reflected in the phase memory of the N–H stretching mode of methylammonium) is largely independent of caesium addition. Our results contribute to the development of a unified model of cation dynamics within organohalide perovskites.
1. Introduction
Hybrid inorganic-organic perovskites (HOIPs) are presently attracting considerable academic and industrial interest.1–3 Since their first use as light-harvesting materials in 2009, the certified photovoltaic power conversion efficiencies of HOIPs have climbed to in excess of 25%,4 the highest for any emerging PV technology. These record efficiencies, combined with their ease and low cost of production, broad material tuneability, and potential use in flexible PV modules, have resulted in applications as diverse as thermoelectric materials,5 LEDs,1,6,7 lasers,8 media for non-volatile memory,9 and many others. Consequently, developing a comprehensive understanding of their macroscopic and microscopic structural behaviours is essential for understanding the unique optoelectronic properties of HOIPs. In particular, HOIPs typically comprise an ensemble of organic cations within the cuboctahedral voids of an inorganic crystal lattice,10 as can be seen in Fig. 1(a). The inorganic lattice—consisting of covalently bonded PbX6 octahedra (here, X refers to any combination of halide ions)—creates a cage whose cavities are occupied by the organic (or inorganic) so-called A-site cations. These organic A-site cations have been shown to be highly rotationally mobile at conditions relevant to device operation. Moreover, their motions and interplay with the electronic dynamics of their parent material is a point of considerable interest as researchers aim to develop a comprehensive understanding of the molecular origins of the remarkable optoelectronic properties of HOIPs.11–16 As such, the rotational properties of the A-site cations have been extensively studied using a battery of experimental techniques, including steady-state IR17 and millimetre wave18 spectroscopy, calorimetry,17 solid-state NMR,19–21 neutron scattering,22–24 and most recently time-resolved ultrafast spectroscopy.10,14,25–27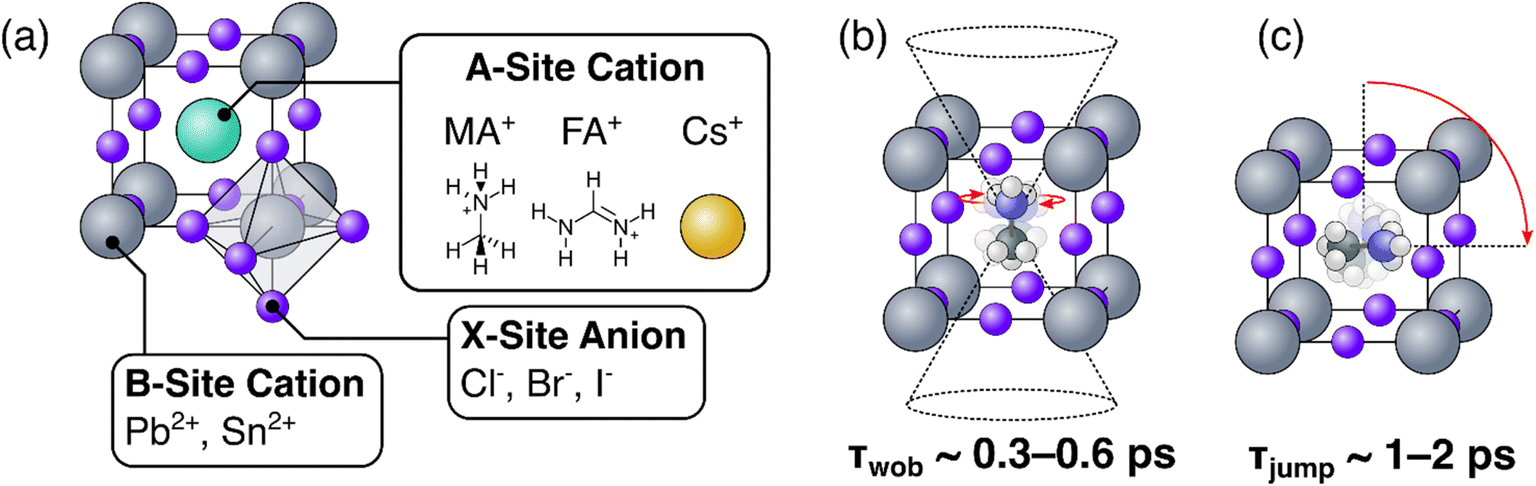 | ||
| Fig. 1 (a) Generalized crystal structure of a metal-halide perovskite; (b) and (c) illustration of wobbling (b) and jump (c) motions of methylammonium in a lead halide perovskite. | ||
From these extensive investigations, a picture of the rotational dynamics of the A-site cation within HOIPs has emerged.10,28 In the case of the common organic cation methylammonium (MA), the long (C–N) axis of the molecule is known to preferentially orient itself towards the ‘faces’ of the unit cell (see Fig. 1(b)). Within this preferential orientation, we distinguish two regimes of rotational motion: a rapid (but small angle) ‘wobbling in a cone’ motion in which the organic cation rapidly librates around its local equilibrium but does not change its gross orientation (Fig. 1(b)), and wide angle (but slower) thermally activated reorientational ‘jumps’, where the organic cation rotates towards a different face of its inorganic cage.10,14,25,26 These wobble and jump mechanisms dominate at timescales of 300–600 fs, and ∼1–2 ps respectively. Conversely, for the popular organic formamidinium (FA) cation, the lack of axial symmetry means that whilst coexisting fast and slow rotational processes occur, they cannot be easily ascribed to a single rotational mode.24,27 Regardless of the A-site species however, these rotational dynamics have been found to be highly sensitive to the chemical structure, phase, and other factors intrinsic to the material.10
Studies on mixed-halide perovskites have found that partial substitution of the X-site anions results in a substantial slowdown in the rotational dynamics of the MA ion and has been ascribed to the formation of an anisotropic potential within the octahedral cavity.10,29,30 Here, the potential anisotropy could be understood as arising from variations in the hydrogen bonds between the ammonium group of MA and the halide ions. However, it has been noted that such a static picture would severely underestimate the ease with which organic cations rotate within HOIPs.10 An alternative (and potentially complementary) explanation has been put forward, in which the [PbX6]4− phonon frequencies—which will change with differing halide composition—dictate the rotational mobility of the cation.26,31 Here, the dynamic fluctuations in the PbX3 sub-lattice could be understood as modulating the energy barriers between adjacent equilibrium sites in a way that provides the organic cation with ‘windows of opportunity’ in which they can reorient themselves. Understanding the interplay of these two effects is essential to developing a more unified model of cation dynamics in MHPs.
Recent experimental work has focused on the effect of cation rotations upon changing the halide content of the perovskite. This makes it difficult to distinguish the effect of cation–halide interactions from other effects (e.g.: rigidisation of the inorganic lattice, changing symmetry of the material unit cell) which could give rise to changes in cation dynamics. In this paper we aim to understand the effect of partial caesium substitution on the rotational dynamics of methylammonium in the popular organohalide perovskite MAPbI3. This approach allows introduction of distortions in the perovskite lattice without directly affecting the cation–halide interactions. Using 2DIR transient anisotropy, we monitor the rotational dynamics of methylammonium in CsxMA1−xPbI3 (x = 0–0.3) with sub-ps time resolution. We find that addition of caesium strongly inhibits the slower reorientation process of the organic cation, with this inhibition becoming more pronounced as the proportion of caesium increases; the corresponding effect on the angular orientation and fast ‘wobbling time’ of MA is significantly less pronounced. Centre–Line–Slope (CLS) analysis of the 2DIR data suggests that the phase memory of the perovskite is on the order of 300–600 fs, similar in magnitude to the ‘wobbling time’ of the organic cation. We suggest therefore that the observed memory loss is driven by the fast librational motion of the organic cation, and that the slower reorientation is driven instead by other processes. Our results implicate the dynamical motion of the inorganic sublattice as responsible for the change in jump time of the organic cations within the perovskite.
2. Experimental
2.1 Material synthesis
All films were prepared on either pre-cleaned quartz or CaF2 substrates. Prior to film deposition, the substrates were sonicated for 15 min, first in acetone and then subsequently in IPA. The substrates were then dried with compressed N2 and plasma treated with O2 under vacuum for 10 min. The MAPbI3 precursors were prepared by dissolving 158.97 mg MAI (GreatCell Solar, >99.99%) and 461.01 mg PbI2 (TCI, 99.99%, trace metals basis) in 200 μL DMSO and 800 μL DMF under 60 °C at 1000 rpm for 1 hour inside an N2 filled glovebox. The CsPbI3 precursors were prepared by dissolving 259.81 mg CsI (Sigma-Aldrich, 99.999% trace metals basis) and 461.01 mg PbI2 (TCI) in 200 μL DMSO and 800 μL DMF under 60 °C at 1000 rpm for 1 hour inside the N2 filled glovebox. Thereafter, the two precursor solutions were left under room temperature for 30 min and were then separately passed through 0.45 μm PTFE filters. The two precursors were then mixed according to the required volume ratio to achieve desirable MA1−xCsxPbI3 compositions. After mixing the two precursor solutions, the perovskite films were prepared by spin-coating (40 μL of solution was dropped on the substrates and spun, initially at 1000 rpm for 30 s and thereafter at 4000 rpm for 60 s, with 75 μL of chlorobenzene added 30 s before the spin finished). The substrates were then annealed initially at 60 °C for 2 s and thereafter at 100 °C for 30 min.2.2 2DIR transient anisotropy
For our 2DIR experiments, we employed MAPbI3 alongside three other derivatives (MA0.9Cs0.1PbI3, MA0.8Cs0.2PbI3, and MA0.7Cs0.3PbI3). The four perovskite samples were synthesised according to the protocol outlined above and subsequently spin-cast from solution onto 1 mm thick CaF2 windows (EKSMA Optics). The resulting thin films were then dried, annealed and stored in the dark under vacuum until needed.To monitor the rotational dynamics of the organic cation in our perovskites, we employed the 2DIR transient anisotropy approach, which has been applied previously to study the rotational dynamics of the organic cation in other perovskites.10,26,27,32 To acquire the 2DIR spectra, we made use of the LIFETime experimental system at the Central Laser Facility (Rutherford Appleton Laboratory).33 Detailed descriptions of the system, its capabilities, and its applications to the study of rotational dynamics in perovskites have been described elsewhere.27 Briefly, a 100 kHz Yb:KGW pulsed laser (PHAROS, Light Conversion Ltd.) was used to pump two separate OPAs capable of operation in the mid-IR to produce pump and probe pulse trains both centred at ∼1470 cm−1, which are resonant with the N–H stretching mode of methylammonium.10,25,26,31 The N–H stretching mode of methylammonium was chosen as its transition dipole is collinear with the molecule's C–N axis.10,23,24 To produce pairs of phase-locked, collinear pump pulses, a programmable acousto-optic modulator (Phasetech Spectroscopy Inc.) was employed. The polarisation of the pump and probe pulses were set separately using a combination half waveplate and linear polariser. The polarisation of the pump pulse was kept constant throughout the experiments. The probe pulse was polarised at an angle of 45° relative to the pump. The pump and probe pulses were thereafter focused to a single point on the sample in a partially collinear geometry. The delay between the pump and probe pulses (i.e.: the population time, t3) was achieved via a mechanical delay stage. At each population time, the probe spectrum was measured as a function of the time delay between the pump pulse pairs (t1). At each value of t1, the probe spectrum was averaged over 10 seconds, with phase cycling employed to remove background pump-probe scatter signals. After passing through the sample, the probe beam was recollimated, the parallel and orthogonally polarised components separated and, and the two resulting beams dispersed spectrally and imaged side-by-side onto a 128 element HgCdTe mid-IR array detector (Infrared Associates Inc.).
Analysis of the 2DIR spectra was performed using a suite of custom analysis software. In all experiments, both the pump and probe pulse centroid frequencies are identical, meaning that it is possible for any scattered light from the pump pulse to contaminate the probe spectrum, resulting in a broad and strongly negative feature localised to the diagonal of the resulting 2DIR spectrum (Fig. S4†). Prior to all subsequent analysis, these scattering effects were suppressed as much as possible by averaging the 2DIR data obtained prior to the time-zero of the t3 axis and subtracting this from all subsequent spectra. Moreover, the off-diagonal excited state absorption (ESA) bands were used for calculation of the transient anisotropy and the centre line slope. For the transient anisotropy measurements, the intensities of the ESA bands were estimated by averaging the amplitudes of the 9 pixels of the peak and its immediate neighbours (i.e.: the peak and the 8 neighbouring pixels). The 1/e population lifetime of the ESA signals for all samples ranged from 2 to 3 ps, with detectable signal observed over the full 50 ps measurement range (see ESI, Fig. S5†).
2.3 Structural/optical characterization
In addition to the 2DIR measurements outlined above, the perovskite films were characterised using X-Ray Diffraction (XRD), Steady-State and Time Resolved Photoluminescence, and Raman spectroscopy. Details of these experimental techniques can be found in the ESI.†To characterise the absorption of the perovskite at the band edge, we employed Photothermal Deflection Spectroscopy (PDS), which enables the detection of absorbance signals that are 5–6 orders of magnitude weaker than the band edge absorption.34,35 For the measurements, the samples (film on quartz substrate) were illuminated with a monochromatic pump beam that is modulated mechanically at 13 Hz. Light absorption then leads to a thermal gradient near the sample surface via non-radiative relaxation induced heating, resulting in a refractive index gradient in the area surrounding the sample surface. This is further enhanced by immersing the sample in the inert liquid FC-72 Fluorinert® (3M Inc.) which has a high refractive index change per unit change in temperature. A fixed wavelength CW laser probe beam was passed through the refractive index gradient close to the sample surface (i.e.: in a transverse configuration), producing a deflection proportional to the absorbed light at a particular wavelength, which is detected using a quadrant photodiode and lock-in amplifier. Scanning through different wavelengths yields the complete absorption spectra. Because this technique makes use of the intrinsic non-radiative relaxation processes in the sample, it is immune to other optical effects, such as interference and scattering.
3. Results
3.1 Characterisation of CsxMA1−xPbI3 films
CsxMA1−xPbI3 (x = 0–30%) thin films were prepared using methods described in the experimental section. X-Ray Diffraction (XRD) patterns of the films in Fig. 2(a) show that the films remain purely in their black, photoactive alpha (α) phase for Cs substitution ratios of ≤30%. For Cs ratios above 30%, photo-inactive delta (δ) phase and PbI2 peaks are observed, in agreement with previous studies.36–39 We therefore consider only substitution ratios of 0–30% in this study. Successful doping of Cs into the MAPbI3 matrix is evidenced by the shift in 2θ values, as well as by the shift in the photoluminescence peak positions and UV-Vis absorption spectra (ESI, Fig. S1†). The 2θ value shifts from 14.13° for pure MAPbI3 to 14.18° for Cs0.3 MA0.7PbI3 (Fig. 2(a)), indicating a reduction in the lattice parameter as smaller A-site cations (Cs) partially substitute MA cations. Concomitantly, the PL emission maxima and absorption onsets are blue shifted as the Cs content is increased from 10% to 30%.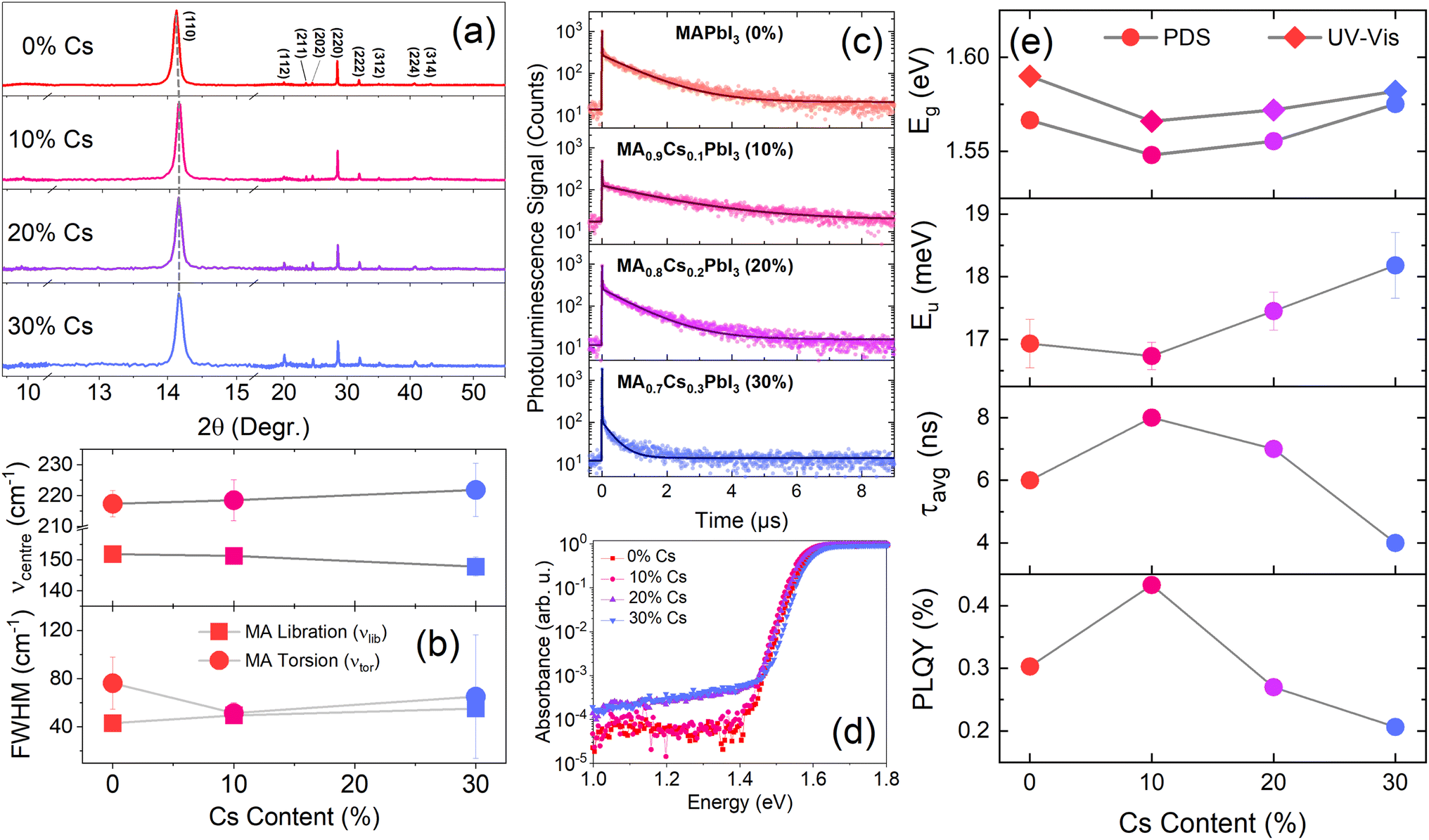 | ||
| Fig. 2 Optical and structural characterization of MA1−xCsxPbI3. (a) X-Ray Diffraction (XRD) of CsxMA1−xPbI3 (x = 0–30%) films on quartz substrates; the 2θ = 12.5–15.5° region is expanded for clarity. (b) Characteristics of the Raman peaks of the torsional and librational modes of MA with increasing Cs content. (c) and (d) Time-resolved photoluminescence (c) and photothermal deflection spectra (d) of MA1−xCsxPbI3 (x = 0–30%) films. (e) Effect of caesium alloying on the optical and photoluminescence properties of MA1−xCsxPbI3. | ||
To understand the effect of Cs doping on the optical properties of MAPbI3, photoluminescence decay and photoluminescence quantum yield (PLQY) measurements were taken (Fig. 2(c) and (e), ESI, Fig. S1, and Table S2†). The PLQY of the film increases from 0.30% for pure MAPbI3 to a maximum of 0.43% for Cs0.1MA0.9MAPbI3; further increasing the Cs content to 20% and 30% decreases the PLQY to 0.27% and 0.21%, respectively. The average PL lifetime—obtained using an amplitude-weighted average of lifetimes from a bi-exponential fit40 (see ESI, Section 1.1.2†)—yields a similar trend (Fig. 2(c)), with the average carrier lifetime increasing from 6 ns to a maximum of 8 ns at a Cs content of 10% before decreasing again. The decrease in the PLQY and lifetime at Cs contents of >10% can be attributed to the increase in the sub-bandgap absorption and Urbach energy (EU), shown in Fig. 2(d) (see ESI, Section 1.2.1† for a description of the fitting method and results). The increase in the sub-bandgap absorption measured using PDS indicates an increase in sub-bandgap/defect states when increasing the Cs content beyond 10% (Fig. 2(d)); these states act as non-radiative recombination centres which reduce the carrier lifetime via trapping. Moreover, the increase in EU also suggests that a higher doping concentration increases the system's disorder, which is detrimental to carrier lifetime. However, we do observe an increase in PLQY and PL lifetime at low Cs doping concentration (10%), and a decrease in Urbach energy (Fig. 2(d) and (e)). This observation is consistent with previous studies, which demonstrate that incorporating small (∼10%) amounts of Cs into MAPbI3 increases the carrier lifetime and the power conversion efficiency of the corresponding solar cell.36–38,41,42
To further understand the reason behind the improvement in optical properties when doping smaller amount of Cs, we performed Raman spectral mapping on pure MAPbI3, as well as on Cs0.1MA0.9MAPbI3 and Cs0.3MA0.7MAPbI3. The resulting spectra are shown in Fig. S3,† in which two clear peaks can be identified at ∼150 cm−1 and ∼218 cm−1. The fitted average peak positions and peak full width at half maxima (FWHM) are listed in Table S3.† The Raman band at 150 cm−1 can be assigned to a librational mode of the MA cations (νlib) and the band at ∼218 cm−1 can be assigned to a torsional mode of MA (νtor).37,43 Upon addition of caesium, we observe a red shift in the position of νlib, from 152 ± 2 cm−1 (0% Cs) to 151 ± 0.6 cm−1 (10% Cs) and 148 ± 3 cm−1 (30% Cs), indicating a continuous tilting of PbI2 octahedral framework and lattice shrinking.43 This is consistent with the blue-shifted PL peaks and increasing XRD 2θ value, which are also indicating the lattice or unit cell size is shrinking upon Cs doping. The incorporation of the smaller Cs cations also increases the rigidity of the lattice, which constrains the libration of the MA ions, in agreement with previous reports.43 In particular, the large increase in the νlib and νtor linewidth at 30% Cs substitution, compared with those at 0% and 10% indicates an increase in lattice distortion, which matches our observations in Fig. 2(a). In contrast, the position of νtor redshifts upon Cs doping, and the linewidth νtor decreases first when adding 10% Cs, and then increases when more Cs is added. The sharpening of νtor upon addition of 10 mol% Cs suggests that the torsional vibrations are first suppressed then recovers upon further addition of Cs. The initial reduction in the torsional mode could potentially reduce the degree of carrier-phonon coupling, thus explaining the increase in PLQY and charge-carrier lifetime44 when adding a tiny amount of Cs, as observed in Fig. 2(c) and (e).
3.2 Rotational dynamics of methylammonium in CsxMA1−xPbI3
To further investigate the relationship between MA cation reorientation and the optical properties of our films, we employed 2DIR transient anisotropy, which has been used previously to understand the rotational dynamics of organic A-site cations in other hybrid perovskite materials.10,26,27,32Representative 2DIR spectra of the four material systems under study are displayed in Fig. 3. In all cases, we observe a well-separated ground state bleach and excited state absorption (ESA) bands with minimal on-diagonal broadening. The degree of on-diagonal broadening and its change over time provides useful information about the local structural dynamics of the cation's surroundings and will be discussed quantitatively in subsequent sections. To extract the transient anisotropy decay curve for each perovskite, we first obtain the intensity of the 2DIR ESA band for both parallel and orthogonal polarisations of the probe pulse. For each of the four samples, between 7 and 10 individual 2DIR spectra were obtained for different points across two separately fabricated films. For each individual spectrum, the intensities of ESA bands for both the parallel and orthogonally polarised pump/probe combination were extracted and converted to the dimensionless transient rotational anisotropy via:
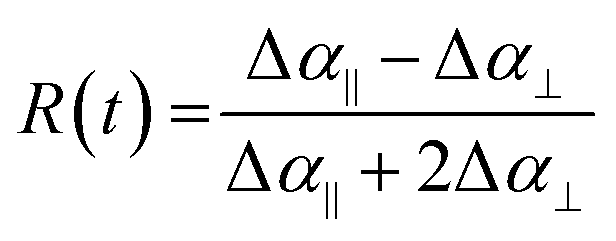 | (1) |
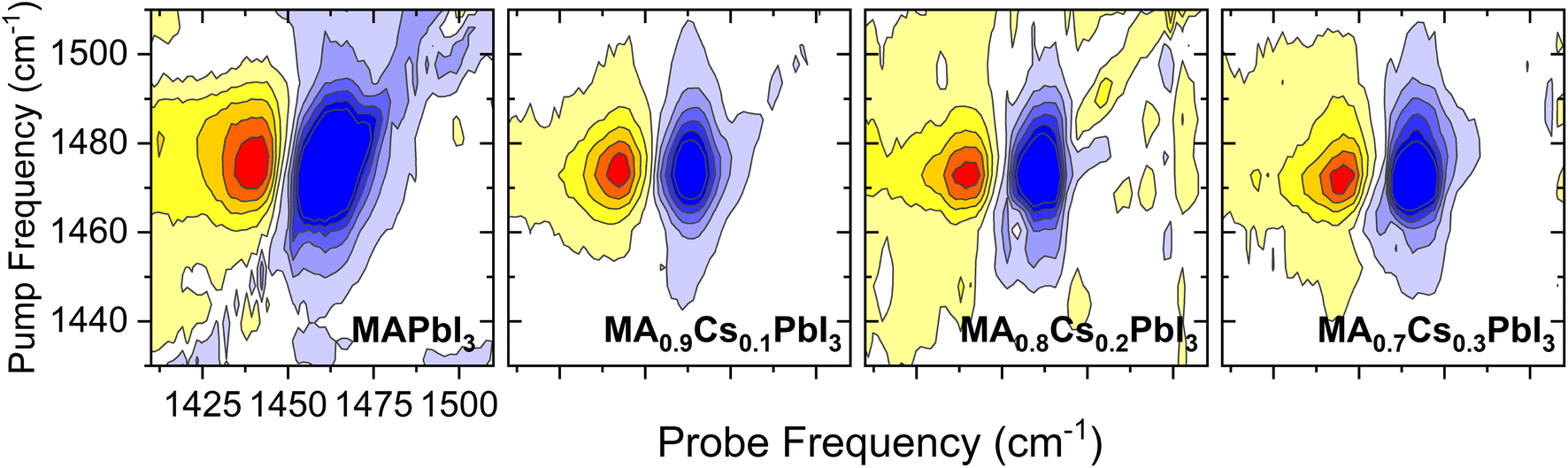 | ||
| Fig. 3 Exemplar 2DIR spectra for the four MA1−xCsxPbI3 systems studied, obtained at a population time of 300 fs. Red (blue) regions are given in bins of 20% of the positive (negative) maxima at 300 fs. | ||
The resulting transient anisotropy traces of the four samples are given in Fig. 4. Pulse-induced heating of samples is known to generate highly persistent 2DIR features through modification of the 0 → 1, 1 → 2 etc. vibrational transitions and so may result in erroneous decay characteristics. However, previous studies on MAPbI3 and other perovskite systems have demonstrated that the transient anisotropy response of the ESA signal for our chosen mode is insensitive to thermal effects and can thus be wholly ascribed to differing rotational dynamics across the material series.25,27 In all cases, addition of caesium results in a slowing of the anisotropy decay: for example, the transient anisotropy of MAPbI3 (i.e.: no caesium addition) effectively decays to 0 within the total measurement time, whilst at the same time range the transient anisotropy decays of all other perovskites in the series have only decreased by ∼75%. We quantify the differences in these anisotropy dynamics using the 2-timescale model put forward by the Fayer group:26,32
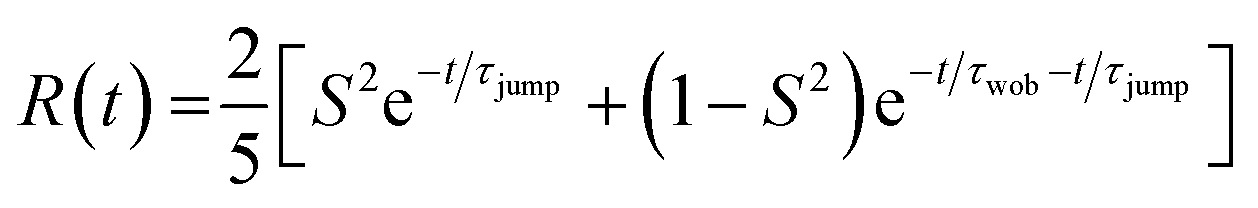 | (2) |
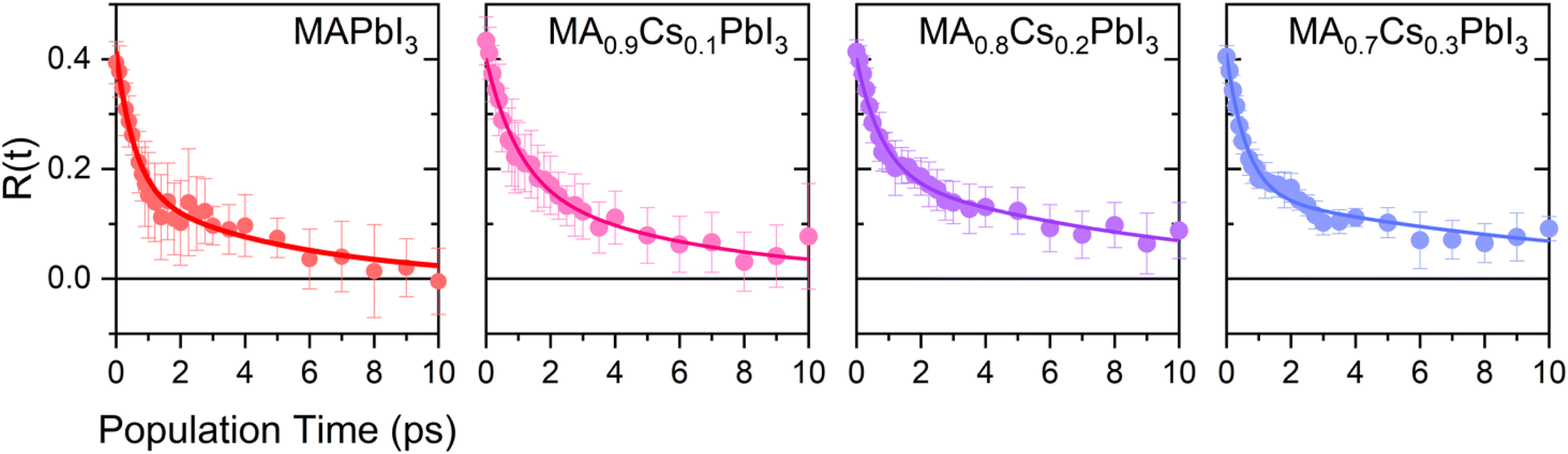 | ||
| Fig. 4 Rotational anisotropy decays of CsxMA1−xPbI3 (x = 0.0–0.3). The solid points represent averages across all runs whilst the solid lines represent a fit of this data using eqn (2). | ||
where τjump and τwob are respectively the timescales of the two rotational processes within the perovskite: a high-amplitude, low-frequency ‘jump’ between adjacent equilibrium positions, and a low-amplitude, high-frequency ‘wobbling’ around the local equilibrium position. The parameter S denotes a dimensionless constant related to the solid angle associated with the wobbling motion. The extracted values of τjump and τwob are plotted in Fig. 5. Across all samples, the addition of caesium is associated with an increase in both τjump and τwob. However, the sensitivity of τjump and τwob to the caesium content of the perovskite varies substantially; for example, across the range of samples studied, τjump increases by a factor of 3, whilst only increasing by roughly 30% for τwob in the same range. Moreover, whilst no significant trend is seen for τwob, τjump increases approximately linearly with the degree of caesium alloying, with this increase occurring in concert with an increase in the cone half-angle. This is suggestive of a picture in which the organic cation is freer to move around its local equilibrium position, whilst its ability to move between adjacent equilibria is inhibited. This is consistent with the relative insensitivity of τwob to the caesium content within the perovskite, as well as with other studies which demonstrated the insensitivity of τwob to the halide content of differing perovskites.26 Interestingly however, we note that the (∼4–12 ps) increase in jump time upon addition of caesium is dramatic in comparison to previous reports (1.5–1.8 ps).26 The structural characterisation data discussed previously—as well as ab initio studies by Ghosh et al. on FAxCs1−xPbI3—suggest that the octahedral cavity distorts in response to caesium addition,45,46 whilst Raman spectra of our samples suggest that the inorganic cage rigidizes in response to caesium. Interestingly, the trend in lattice distortion obtained by Ghosh et al. is similar to that observed for τwob, but differs substantially from the observed values of τjump. Notably however, both distortion of the cuboctahedral cavity and rigidisation of the inorganic sub-lattice contribute to the slowdown of the organic cation's rotational behaviour. This would suggest that the two rotational processes within the perovskite are controlled by different properties of the material that nevertheless work in tandem: distortions of the octahedral cavity create an anisotropic electrostatic potential that will favour certain cation orientations, whilst the rigidized lattice precludes modulations of the energetic barrier which would otherwise facilitate reorientation.
 | ||
| Fig. 5 Extracted wobbling and jump time parameters, obtained by fitting the four anisotropy decays in Fig. 4 to eqn (2). The orange line in (a) is digitally extracted from ref. 45. | ||
We now attempt to better rationalise the effect of caesium alloying on MAPbI3. Initially, to better link our 2DIR results to the atomic-scale dynamics of the A-site cations' surroundings, we performed classical molecular dynamics simulations on a 6 × 6 × 6 MAxCs1−xPbI3 supercell containing a total of 216 A-site cations (194 MA cations and 22 Cs cations), corresponding to a total caesium content of approximately 10%. The polarizable force field employed for these simulations is identical to that used previously in ref. 10; further details of the molecular dynamics simulations are given in ESI, Section 4.† Two separate simulations were considered: one in which the a, b, and c lattice parameters were kept fixed and equal (here termed “isotropic”), and another in which the a/c lattice ratio was allowed to vary (“anisotropic”). The average rotational anisotropy is obtained through application of the ensemble averaged rotational autocorrelation function:
R(t) = 2/5〈L2(![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) (0)·(t))〉 (0)·(t))〉 | (3) |
The two transient anisotropy curves obtained from the simulation are given in Fig. 6. Allowing the a/c lattice ratio to vary has a dramatic effect of the dynamics of the organic cations within the perovskite, resulting in a slowdown of the transient anisotropy decay that becomes particularly pronounced at later times. The simulated transient anisotropy curves are poorly modelled by the 2-timescale model expressed in eqn (2), potentially due to the relatively low number of organic cations included in the simulation and the use of a classical forcefield. Therefore, whilst it is not possible to link the distortion of the inorganic lattice to a specific regime of motion of the organic cation, we are nevertheless able to demonstrate that distortion of the octahedral cage is of crucial importance in determining the dynamics of the organic cation in caesium substituted perovskites.
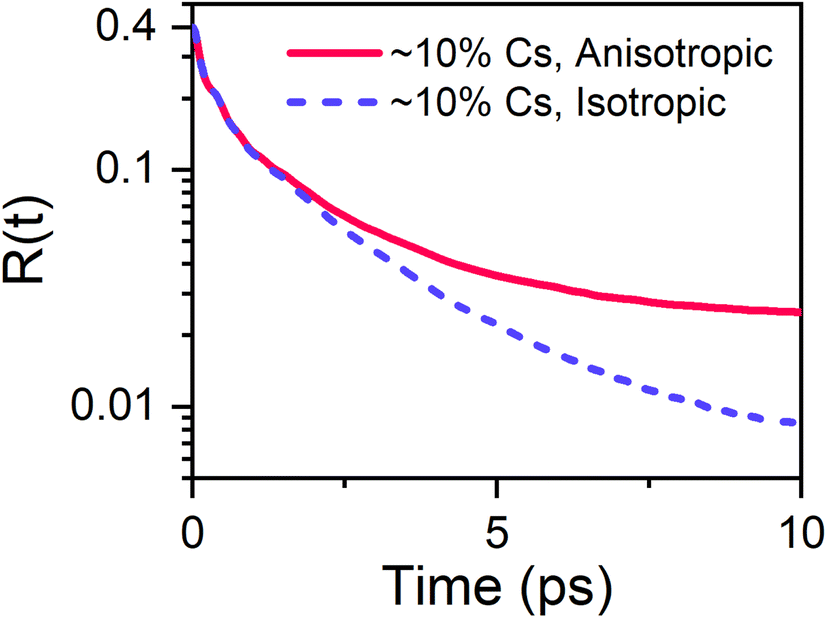 | ||
| Fig. 6 Effect of constraining lattice constants on the rotational dynamics of the organic cation, obtained from molecular dynamics simulations. | ||
Another important means of gleaning atomistic insight from 2DIR data is through analysis of the 2D spectral lineshape. Whilst anisotropy dynamics provide information about the molecular rotations, 2DIR spectral lineshapes are linked to the dynamics of the local environment. In the hybrid perovskites studied herein, the A-site organic ions are surrounded by the inorganic PbI6 octahedral framework, and thus the obtained 2DIR spectral lineshapes should exhibit a dependence on the interaction between the organic cations and the surrounding inorganic sublattice. Of particular interest is the time dependence of changes to the spectral lineshape, which provides a measure of the speed with which fluctuations of the organic cations' surroundings cause the cations to ‘forget’ their initial local configurations (the so-called phase memory). Thus, through analysis of the dynamics of the 2DIR spectral lineshapes, we can better understand the coupling between the organic cation and its surrounding inorganic cage, and thus elucidate the effect that this has on their rotational dynamics. Analysis of 2DIR spectral lineshapes is typically conducted through calculation and analysis of the so-called frequency–frequency correlation function (FFCF).52 Whilst direct extraction of the FFCF from experimental data is known to be computationally intensive and sensitive to instrumental artefacts, there now exist several computationally simple techniques to estimate the FFCF, including nodal line analysis, lineshape ellipticity, 2D Gaussian correlation, and centre–line–slope (CLS)47,53 analysis. Of these techniques, CLS analysis is both relatively straightforward to implement and has been shown to be robust against a variety of experimental imperfections in comparison to other methods, including phase twisting, anharmonicity, apodization, and low signal-to-noise ratios.47
The extracted CLS dynamics for the four perovskite systems are given in Fig. 7(a)–(d). As with the transient anisotropy experiments discussed above, the CLS of the ESA band is taken to minimise any artefacts from pump-induced scattering. In all cases, the CLS attains a maximum value of 0.2–0.5, before decaying away within 2–3 ps. In keeping with previous reports, we quantify these timescales by fitting the data with a simple single exponential decay (τr). The values of τr for our four samples, along with the initial CLS values (CLS0) are given in Fig. 7(e) and (f). Considering first the CLS0 values, a clear positive correlation between the caesium content and CLS0 can be seen. Importantly, whilst absolute FFCF amplitudes estimated using CLS analysis are sensitive to apodization, relative amplitudes are not,47 meaning that the observed trend can be directly ascribed to the effect of caesium addition. Moreover, the observed positive trend—in which CLS0 increases with increasing Cs substitution—is broadly consistent with the expected increase in structural inhomogeneity brought about by A-site ion alloying. By contrast, no clear trend can be observed for the relaxation time τr. However, of particular interest is the fact that the range of extracted relaxation times (0.2–0.6 ps) are between 1 and 2 orders of magnitude smaller than the ‘jump times’ of the four perovskites but are similar in magnitude to the vales of τwob extracted from the transient anisotropy response (Fig. 4). This is most striking for pure MAPbI3 and MA0.7Cs0.3PbI3, where quantitative agreement between τr (0.49 ± 0.13 and 0.57 ± 0.13 respectively), and τwob (0.52 ± 0.06 and 0.65 ± 0.07 respectively) is obtained. Such a result suggests that the fast ‘wobbling’ motion of the organic cation determines the phase memory of the N–H ion. Regardless of the observed origin, our results suggest that the phase memory of the perovskite is too short for τjump to exhibit a dependence on local effects, such as hydrogen bonding and further implies that τjump and τwob are dependent on different factors within the material.
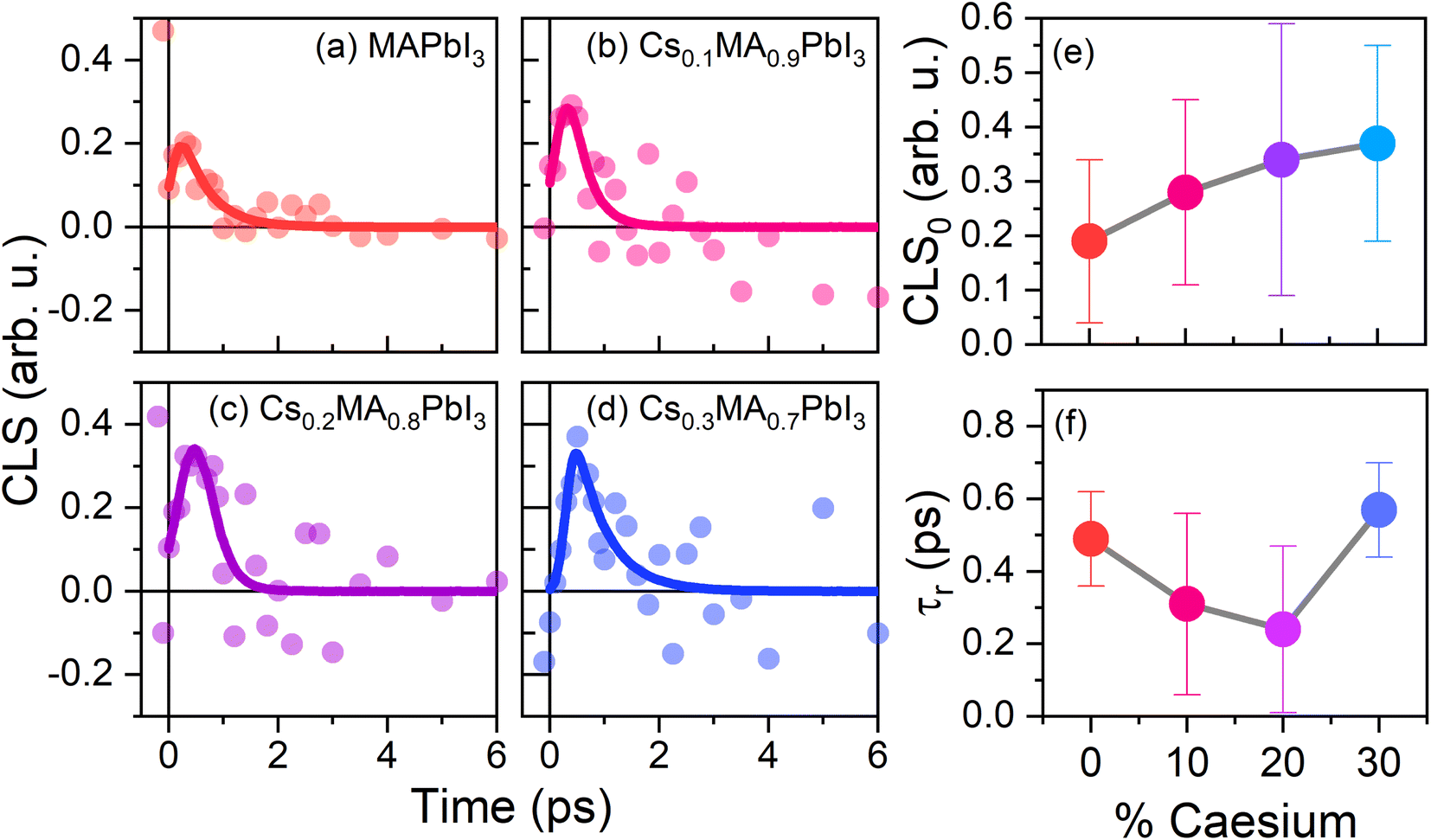 | ||
| Fig. 7 Results of CLS analysis. (a–d) CLS values of the four perovskite systems along with the corresponding fits. Solid points represent the average CLS at each population time, whilst the solid lines correspond to the fit of the CLS using a single exponential function; (e) and (f) extracted fitting parameters for the CLS curves given in (a–d). | ||
4. Discussion
Despite the considerable amount of effort that has been directed to understanding cation rotations in perovskites, there still exists a significant amount of ambiguity regarding the physical origins of differences in the rotational dynamics of the organic cations. To date, several phenomena have been proposed to explain the experimental observations, particularly the observed slowdown in cation dynamics upon alteration of the halide and cation composition of the perovskite. Firstly, the alteration of the hydrogen bonding should directly affect the rotational mobility of the cation.10,26 Secondly, changes to frequencies of the inorganic cage modes may affect both wobbling amplitude and rotational-jump motions.22,26,48 Thirdly, ab initio calculations on a variety of perovskite systems have suggested that distortion of the octahedral cage plays a substantial role. Finally, the structural dynamics may be affected by the inorganic cage rigidising in response to caesium (or other cation) addition.43 Developing a comprehensive and unified picture that accounts for these varied phenomena is important for the future development of HOIPs as both solar absorbers, as well as in other applications.The results of our 2DIR studies presented herein, alongside the findings of previous studies, suggest that the contributions discussed above can build up a comprehensive picture of cation behaviour in HOIPs. As we use a single halide species in this study, the hydrogen bond strengths between the ammonium group and the halide atoms only vary as a result of distortions of the cuboctahedral cavity. Previous ab initio studies—as well as some experimental evidence—have demonstrated that caesium alloying both deforms and shrinks the cuboctahedral cavity in which the organic cations are located. The effect that these distortions have on the interactions between the organic cations and the inorganic sublattice can be seen through the increase in the inhomogeneous linewidth broadening (quantified by the CLS0 values given in Fig. 7(e)) with increasing caesium content. Increased inhomogeneous broadening implies stronger static disorder originating from a stronger degree of distortion in the octahedral cavities and a greater distribution of octahedral cavity environments. It also indicates that these changes significantly affect the interactions between the organic cation and the inorganic sublattice. As a result, the anisotropy curves of caesium doped MAPbI3 represent an ensemble average of octahedral cavity dynamics with differing degrees of lattice–cation interactions, depending on both distortion and contraction of the local inorganic lattice.49
Also of interest is the substantial difference in the response of the two regimes of organic cation motion to caesium addition. In particular, the close agreement between distortion of the inorganic lattice and the fast dynamics of the organic cation strongly implies a correlation between them. This also accounts for the larger effect of caesium substitution on the fast rotational dynamics of MA+versus halide substitution: whilst the difference in ionic radii between I− (220 pm) and Br− (196 pm) is significant, it is substantially smaller than between Cs+ (167 pm) and MA+ (217 pm).49–51 By contrast, the observed trend for the ‘jump’ process is entirely different and does not correlate with changes to the distortion of the octahedral cage. To explain the trends seen for this slower rotational regime of the organic cation, we consider instead the dynamics of the inorganic sublattice. The Raman studies provided herein identified two important changes upon addition of caesium: a rigidisation of the inorganic sub-lattice and a slowdown in the lattice dynamics, respectively. Thus, alongside distortion of the octahedral cavity, changes to the phonon structure and rigidity of the organic lattice also occur in response to caesium alloying. Both phenomena could be expected to have a significant effect on the rotational dynamics of the organic cation. This is best understood by considering the reorientation of the organic cation as being an activated process whose energetic barrier is dictated by cation–lattice interactions. Here, the low-frequency fluctuations of the inorganic lattice dynamically modulate the energetic barrier between adjacent equilibrium positions, effectively providing ‘windows of opportunity’, during which the energetic barrier is sufficiently low for large-angle rotational diffusion to become facile. In this case, caesium incorporation reduces both the amplitude (through rigidisation of the inorganic cage) and frequency of inorganic cage fluctuations, in effect creating both fewer and smaller ‘windows’ of opportunity for the cation to reorient itself within the octahedral cavity. Within the two-timescale model used to rationalise cation rotations, we would expect this to most strongly affect the large-angle, low-frequency jumps (quantified by the ‘jump time’ τjump) of the cation. This is supported by the relatively close agreement between the timescales of spectral diffusion (quantified by τr, see Fig. 7(f)) and the ‘wobbling time’ of the organic cation (τwob, Fig. 5(b)), which suggests that the organic cations are relatively free to move around their local equilibrium orientations. Conversely, if one imagines a scenario in which the organic cation was tightly bound to the inorganic lattice, one expects the rigidisation and slowdown of the lattice dynamics to result in slower relaxation of the inhomogeneous linewidth, yet no such trend is observed. This suggests that rather than a single dominant phenomenon influencing the dynamics of the organic cation, the two timescales of motion are instead affected by a confluence of differing phenomena: the fast ‘wobbling’ dynamics of the organic cation are dictated by the size and shape of the octahedral cavity, whilst the slower ‘jump’ process is primarily dependent on lattice dynamics.
5. Conclusions
We have applied 2DIR spectroscopy to explicate the relationship between the interaction of the inorganic lattices and the dynamics of the organic A-site ions in HIOPs. In contrast to previous 2DIR experiments, we have focused on the effect of A-site cation alloying on the HOIPs, which has not previously been experimentally studied in this manner. In particular, we have supplemented our 2DIR transient anisotropy methods with 2DIR spectral lineshape analysis, which enables the interaction between the organic A-site ion and its surroundings to be more clearly ascertained. Collectively, our results suggest that the two regimes of motion of the organic cation exhibit dependencies on different aspects of the material; the fast ‘wobbling’ motion is weakly dependent on the shape of the octahedral cavity, whilst the slower reorientation of the cation depends on fluctuations of the inorganic lattice, in particular the rigidity of the inorganic cage and the inorganic lattice modes. Our findings will help to further guide the rational design of mixed-cation perovskite solar cells.Conflicts of interest
There are no conflicts to declare.Acknowledgements
NPG and AAB thank Maxim S. Pshenichnikov for helpful discussions and feedback over the course of this study. NPG and AAB gratefully acknowledge support from the ERC (grant no. 639750) under their Horizon2020 framework. RLZH thanks the Royal Academy of Engineering for support under the Research Fellowships scheme (no.: RF\201718\1701). LD thanks the Chinese Scholarship Council and Cambridge Trust for funding. AAB is a Royal Society University Research Fellow. Access to the Central Laser Facility's LIFEtime 2DIR instrument was provided by STFC. RA acknowledges support from the Rutherford Foundation of the Royal Society Te Apārangi of New Zealand, the Winton Program for the Physics of Sustainability, and Trinity College Cambridge. SJZ acknowledges support from the Polish National Agency for Academic Exchange within the Bekker programme (grant no. PPN/BEK/2020/1/00264/U/00001). RLZH and JY thank the EPSRC Centre of Advanced Materials for Integrated Energy Systems (CAM-IES, grant no. EP/P007767/1) for financial support.References
- S. D. Stranks and H. J. Snaith, Metal-Halide Perovskites for Photovoltaic and Light-Emitting Devices, Nat. Nanotechnol., 2015, 10(5), 391–402, DOI:10.1038/nnano.2015.90.
- D. A. Egger, A. Bera, D. Cahen, G. Hodes, T. Kirchartz, L. Kronik, R. Lovrincic, A. M. Rappe, D. R. Reichman and O. Yaffe, What Remains Unexplained about the Properties of Halide Perovskites?, Adv. Mater., 2018, 30(20), 1800691, DOI:10.1002/adma.201800691.
- T. M. Brenner, D. A. Egger, L. Kronik, G. Hodes and D. Cahen, Hybrid Organic - Inorganic Perovskites: Low-Cost Semiconductors with Intriguing Charge-Transport Properties, Nat. Rev. Mater., 2016, 15007, DOI:10.1038/natrevmats.2015.7.
- Best Research-Cell Efficiency Chart | Photovoltaic Research | NRELhttps://www.nrel.gov/pv/cell-efficiency.html, (accessed Oct 26, 2020).
- Y. He and G. Galli, Perovskites for Solar Thermoelectric Applications: A First Principle Study of CH3NH3AI3 (A = Pb and Sn), Chem. Mater., 2014, 26(18), 5394–5400, DOI:10.1021/cm5026766.
- N. Yantara, S. Bhaumik, F. Yan, D. Sabba, H. A. Dewi, N. Mathews, P. P. Boix, H. V. Demir and S. Mhaisalkar, Inorganic Halide Perovskites for Efficient Light-Emitting Diodes, J. Phys. Chem. Lett., 2015, 6(21), 4360–4364, DOI:10.1021/acs.jpclett.5b02011.
- H. Cho, S. H. Jeong, M. H. Park, Y. H. Kim, C. Wolf, C. L. Lee, J. H. Heo, A. Sadhanala, N. S. Myoung, S. Yoo, S. H. Im, R. H. Friend and T. W. Lee, Overcoming the Electroluminescence Efficiency Limitations of Perovskite Light-Emitting Diodes, Science, 2015, 350(6265), 1222–1225, DOI:10.1126/science.aad1818.
- Q. Zhang, R. Su, W. Du, X. Liu, L. Zhao, S. T. Ha and Q. Xiong, Advances in Small Perovskite-Based Lasers, Small Methods, 2017, 1(9), 1700163, DOI:10.1002/smtd.201700163.
- D. Liu, Q. Lin, Z. Zang, M. Wang, P. Wangyang, X. Tang, M. Zhou and W. Hu, Flexible All-Inorganic Perovskite CsPbBr3 Nonvolatile Memory Device, ACS Appl. Mater. Interfaces, 2017, 9(7), 6171–6176, DOI:10.1021/acsami.6b15149.
- N. P. Gallop, O. Selig, G. Giubertoni, H. J. Bakker, Y. L. A. Rezus, J. M. Frost, T. L. C. Jansen, R. Lovrincic and A. A. Bakulin, Rotational Cation Dynamics in Metal Halide Perovskites: Effect on Phonons and Material Properties, J. Phys. Chem. Lett., 2018, 9(20), 5987–5997, DOI:10.1021/acs.jpclett.8b02227.
- J. Gong, M. Yang, X. Ma, R. D. Schaller, G. Liu, L. Kong, Y. Yang, M. C. Beard, M. Lesslie, Y. Dai, B. Huang, K. Zhu and T. Xu, Electron-Rotor Interaction in Organic-Inorganic Lead Iodide Perovskites Discovered by Isotope Effects, J. Phys. Chem. Lett., 2016, 7(15), 2879–2887, DOI:10.1021/acs.jpclett.6b01199.
- M. Bonn, K. Miyata, E. Hendry and X. Y. Zhu, Role of Dielectric Drag in Polaron Mobility in Lead Halide Perovskites, ACS Energy Lett., 2017, 2555–2562, DOI:10.1021/acsenergylett.7b00717.
- X. Y. Zhu and V. Podzorov, Charge Carriers in Hybrid Organic-Inorganic Lead Halide Perovskites Might Be Protected as Large Polarons, J. Phys. Chem. Lett., 2015, 4758–4761, DOI:10.1021/acs.jpclett.5b02462.
- H. Zhu, K. Miyata, Y. Fu, J. Wang, P. P. Joshi, D. Niesner, K. W. Williams, S. Jin and X. Y. Zhu, Screening in Crystalline Liquids Protects Energetic Carriers in Hybrid Perovskites, Science, 2016, 353(6306), 1409–1413, DOI:10.1126/science.aaf9570.
- A. Johnston, G. Walters, M. I. Saidaminov, Z. Huang, K. Bertens, N. Jalarvo and E. H. Sargent, Bromine Incorporation and Suppressed Cation Rotation in Mixed-Halide Perovskites, ACS Nano, 2020, 14(11), 15107–15118, DOI:10.1021/acsnano.0c05179.
- M. M. Byranvand, C. Otero-Martínez, J. Ye, W. Zuo, L. Manna, M. Saliba, R. L. Z. Hoye and L. Polavarapu, Recent Progress in Mixed A-Site Cation Halide Perovskite Thin-Films and Nanocrystals for Solar Cells and Light-Emitting Diodes, Adv. Opt. Mater., 2022, 2200423, DOI:10.1002/adom.202200423.
- N. Onoda-Yamamuro, T. Matsuo and H. Suga, Calorimetric and IR Spectroscopic Studies of Phase Transitions in Methylammonium Trihalogenoplumbates (II), J. Phys. Chem. Solids, 1990, 51(12), 1383–1395, DOI:10.1016/0022-3697(90)90021-7.
- A. Poglitsch and D. Weber, Dynamic Disorder in Methylammoniumtrihalogenoplumbates (II) Observed by Millimeter-Wave Spectroscopy, J. Chem. Phys., 1987, 87(11), 6373–6378, DOI:10.1063/1.453467.
- R. E. Wasylishen, O. Knop and J. B. Macdonald, Cation Rotation in Methylammonium Lead Halides, Solid State Commun., 1985, 56(7), 581–582, DOI:10.1016/0038-1098(85)90959-7.
- G. M. Bernard, R. E. Wasylishen, C. I. Ratcliffe, V. Terskikh, Q. Wu, J. M. Buriak and T. Hauger, Methylammonium Cation Dynamics in Methylammonium Lead Halide Perovskites: A Solid-State NMR Perspective, J. Phys. Chem. A, 2018, 122(6), 1560–1573, DOI:10.1021/acs.jpca.7b11558.
- D. J. Kubicki, D. Prochowicz, A. Hofstetter, P. Péchy, S. M. Zakeeruddin, M. Grätzel and L. Emsley, Cation Dynamics in Mixed-Cation (MA)x(FA)1−XPbI3 Hybrid Perovskites from Solid-State NMR, J. Am. Chem. Soc., 2017, 139(29), 10055–10061, DOI:10.1021/jacs.7b04930.
- A. M. A. Leguy, J. M. Frost, A. P. McMahon, V. G. Sakai, W. Kochelmann, C. Law, X. Li, F. Foglia, A. Walsh, B. C. O'Regan, J. Nelson, J. T. Cabral and P. R. F. Barnes, The Dynamics of Methylammonium Ions in Hybrid Organic-Inorganic Perovskite Solar Cells, Nat. Commun., 2015, 6(1), 7124, DOI:10.1038/ncomms8124.
- T. Chen, B. J. Foley, B. Ipek, M. Tyagi, J. R. D. Copley, C. M. Brown, J. J. Choi and S. H. Lee, Rotational Dynamics of Organic Cations in the CH3NH3PbI3 Perovskite, Phys. Chem. Chem. Phys., 2015, 17(46), 31278–31286, 10.1039/c5cp05348j.
- D. H. Fabini, T. A. Siaw, C. C. Stoumpos, G. Laurita, D. Olds, K. Page, J. G. Hu, M. G. Kanatzidis, S. Han and R. Seshadri, Universal Dynamics of Molecular Reorientation in Hybrid Lead Iodide Perovskites, J. Am. Chem. Soc., 2017, 139(46), 16875–16884, DOI:10.1021/jacs.7b09536.
- A. A. Bakulin, O. Selig, H. J. Bakker, Y. L. A. Rezus, C. Müller, T. Glaser, R. Lovrincic, Z. Sun, Z. Chen, A. Walsh, J. M. Frost and T. L. C. Jansen, Real-Time Observation of Organic Cation Reorientation in Methylammonium Lead Iodide Perovskites, J. Phys. Chem. Lett., 2015, 6(18), 3663–3669, DOI:10.1021/acs.jpclett.5b01555.
- O. Selig, A. Sadhanala, C. Müller, R. Lovrincic, Z. Chen, Y. L. A. Rezus, J. M. Frost, T. L. C. Jansen and A. A. Bakulin, Organic Cation Rotation and Immobilization in Pure and Mixed Methylammonium Lead-Halide Perovskites, J. Am. Chem. Soc., 2017, 139(11), 4068–4074, DOI:10.1021/jacs.6b12239.
- V. C. A. Taylor, D. Tiwari, M. Duchi, P. M. Donaldson, I. P. Clark, D. J. Fermin and T. A. A. Oliver, Investigating the Role of the Organic Cation in Formamidinium Lead Iodide Perovskite Using Ultrafast Spectroscopy, J. Phys. Chem. Lett., 2018, 9(4), 895–901, DOI:10.1021/acs.jpclett.7b03296.
- E. M. Mozur and J. R. Neilson, Cation Dynamics in Hybrid Halide Perovskites, Annu. Rev. Mater. Res., 2021, 269–291, DOI:10.1146/annurev-matsci-080819-012808.
- O. Selig, A. Sadhanala, C. Müller, R. Lovrincic, Z. Chen, Y. L. A. Rezus, J. M. Frost, T. L. C. Jansen and A. A. Bakulin, Organic Cation Rotation and Immobilization in Pure and Mixed Methylammonium Lead-Halide Perovskites, J. Am. Chem. Soc., 2017, 139(11), 4068–4074, DOI:10.1021/jacs.6b12239.
- H. Grüninger, M. Bokdam, N. Leupold, P. Tinnemans, R. Moos, G. A. De Wijs, F. Panzer and A. P. M. Kentgens, Microscopic (Dis)Order and Dynamics of Cations in Mixed FA/MA Lead Halide Perovskites, J. Phys. Chem. C, 2021, 125, 54, DOI:10.1021/acs.jpcc.0c10042.
- A. M. A. Leguy, A. R. Goñi, J. M. Frost, J. Skelton, F. Brivio, X. Rodríguez-Martínez, O. J. Weber, A. Pallipurath, M. I. Alonso, M. Campoy-Quiles, M. T. Weller, J. Nelson, A. Walsh and P. R. F. Barnes, Dynamic Disorder, Phonon Lifetimes, and the Assignment of Modes to the Vibrational Spectra of Methylammonium Lead Halide Perovskites, Phys. Chem. Chem. Phys., 2016, 18(39), 27051–27066, 10.1039/c6cp03474h.
- A. A. Bakulin, O. Selig, H. J. Bakker, Y. L. A. Rezus, C. Müller, T. Glaser, R. Lovrincic, Z. Sun, Z. Chen, A. Walsh, J. M. Frost and T. L. C. Jansen, Real-Time Observation of Organic Cation Reorientation in Methylammonium Lead Iodide Perovskites, J. Phys. Chem. Lett., 2015, 6(18), 3663–3669, DOI:10.1021/acs.jpclett.5b01555.
- P. M. Donaldson, G. M. Greetham, D. J. Shaw, A. W. Parker and M. A. Towrie, 100 KHz Pulse Shaping 2D-IR Spectrometer Based on Dual Yb:KGW Amplifiers, J. Phys. Chem. A, 2018, 122(3), 780–787, DOI:10.1021/acs.jpca.7b10259.
- T. A. S. Doherty, A. J. Winchester, S. Macpherson, D. N. Johnstone, V. Pareek, E. M. Tennyson, S. Kosar, F. U. Kosasih, M. Anaya, M. Abdi-Jalebi, Z. Andaji-Garmaroudi, E. L. Wong, J. Madéo, Y. H. Chiang, J. S. Park, Y. K. Jung, C. E. Petoukhoff, G. Divitini, M. K. L. Man, C. Ducati, A. Walsh, P. A. Midgley, K. M. Dani and S. D. Stranks, Performance-Limiting Nanoscale Trap Clusters at Grain Junctions in Halide Perovskites, Nature, 2020, 580(7803), 360–366, DOI:10.1038/s41586-020-2184-1.
- A. Sadhanala, F. Deschler, T. H. Thomas, S. E. Dutton, K. C. Goedel, F. C. Hanusch, M. L. Lai, U. Steiner, T. Bein, P. Docampo, D. Cahen and R. H. Friend, Preparation of Single-Phase Films of CH3NH3Pb(I1−xBrx)3 with Sharp Optical Band Edges, J. Phys. Chem. Lett., 2014, 5(15), 2501–2505, DOI:10.1021/jz501332v.
- M. Wang, F. Cao, K. Deng and L. Li, Adduct Phases Induced Controlled Crystallization for Mixed-Cation Perovskite Solar Cells with Efficiency over 21, Nano Energy, 2019, 63, 103867, DOI:10.1016/j.nanoen.2019.10.
- V. Pawar, M. Kumar, P. A. Jha, S. K. Gupta, P. K. Jha and P. Singh, Cs/MAPbI3 Composite Formation and Its Influence on Optical Properties, J. Alloys Compd., 2019, 783, 935–942, DOI:10.1016/j.jallcom.2018.12.333.
- G. Murugadoss, R. Thangamuthu, S. Vijayaraghavan, H. Kanda and S. Ito, Caesium−Methyl Ammonium Mixed-Cation Lead Iodide Perovskite Crystals: Analysis and Application for Perovskite Solar Cells, Electrochim. Acta, 2017, 257, 267–280, DOI:10.1016/j.electacta.2017.10.092.
- X. Guo, C. McCleese, C. Kolodziej, A. C. S. Samia, Y. Zhao and C. Burda, Identification and Characterization of the Intermediate Phase in Hybrid Organic-Inorganic MAPbI3 Perovskite, Dalton Trans., 2016, 45(9), 3806–3813, 10.1039/c5dt04420k.
- G. Zatryb and M. M. Klak, On the Choice of Proper Average Lifetime Formula for an Ensemble of Emitters Showing Non-Single Exponential Photoluminescence Decay, J. Phys.: Condens. Matter, 2020, 32(41), 11, DOI:10.1088/1361-648X/ab9bcc.
- M. Imran, A. Saleem, N. A. Khan and A. H. Kamboh, Enhanced Efficiency and Stability of Perovskite Solar Cells by Partial Replacement of CH3NH3+ with Inorganic Cs+ in CH3NH3PbI3 Perovskite Absorber Layer, Phys. Rev. B: Condens. Matter Mater. Phys., 2019, 572, 1–11, DOI:10.1016/J.PHYSB.2019.07.041.
- X. Liu, M. Chen, Y. Zhang, J. Xia, J. Yin, M. Li, K. G. Brooks, R. Hu, X. Gao, Y. H. Kim, A. Züttel, J. M. Luther, S. Kinge, Y. Feng and M. K. Nazeeruddin, High-Efficiency Perovskite Photovoltaic Modules Achieved via Cesium Doping, Chem. Eng. J., 2022, 431, 133713, DOI:10.1016/J.CEJ.2021.133713.
- V. H. Damle, L. Gouda, S. Tirosh and Y. R. Tischler, Structural Characterization and Room Temperature Low-Frequency Raman Scattering from MAPbI3 Halide Perovskite Films Rigidized by Cesium Incorporation, ACS Appl. Energy Mater., 2018, 1(12), 6707–6713, DOI:10.1021/acsaem.8b01539.
- D. Ghosh, C. M. Perez, O. Prezhdo, W. Nie, S. Tretiak and A. J. Neukirch, Impact of Composition Engineering on Charge Carrier Cooling in Hybrid Perovskites: Computational Insights, J. Mater. Chem. C, 2022, 10(25), 9563–9572, 10.1039/d2tc01413k.
- D. Ghosh, A. R. Smith, A. B. Walker and M. S. Islam, Mixed A-Cation Perovskites for Solar Cells: Atomic-Scale Insights into Structural Distortion, Hydrogen Bonding, and Electronic Properties, Chem. Mater., 2018, 30(15), 5194–5204, DOI:10.1021/acs.chemmater.8b01851.
- D. Ghosh, P. Walsh Atkins, M. S. Islam, A. B. Walker and C. Eames, Good Vibrations: Locking of Octahedral Tilting in Mixed-Cation Iodide Perovskites for Solar Cells, ACS Energy Lett., 2017, 2(10), 2424–2429, DOI:10.1021/acsenergylett.7b00729.
- Q. Guo, P. Pagano, Y. L. Li, A. Kohen and C. M. Cheatum, Line Shape Analysis of Two-Dimensional Infrared Spectra, J. Chem. Phys., 2015, 142(21), 212427, DOI:10.1063/1.4918350.
- W. Shockley and H. J. Queisser, Detailed Balance Limit of Efficiency of P-n Junction Solar Cells, J. Appl. Phys., 1961, 32(3), 510–519, DOI:10.1063/1.1736034.
- J. Nishida, A. H. Alfaifi, T. P. Gray, S. E. Shaheen and M. B. Raschke, Heterogeneous Cation–Lattice Interaction and Dynamics in Triple-Cation Perovskites Revealed by Infrared Vibrational Nanoscopy, ACS Energy Lett., 2020, 5(5), 1636–1643, DOI:10.1021/acsenergylett.0c00522.
- G. Kieslich, S. Sun and A. K. Cheetham, An Extended Tolerance Factor Approach for Organic-Inorganic Perovskites, Chem. Sci., 2015, 6(6), 3430–3433, 10.1039/c5sc00961h.
- G. Kieslich, S. Sun and A. K. Cheetham, Solid-State Principles Applied to Organic–Inorganic Perovskites: New Tricks for an Old Dog, Chem. Sci., 2014, 5(12), 4712–4715, 10.1039/c4sc02211d.
- Kees Lazonder, Maxim S Pshenichnikov and Douwe A Wiersma, Easy interpretation of optical two-dimensional correlation spectra., Opt Lett, 2006, 31(22), 3354–3356, DOI:10.1364/ol.31.003354.
- Evgeniia Salamatova, Ana V Cunha, Keisuke Shinokita, Thomas L C Jansen and Maxim S Pshenichnikov, Hydrogen bond and lifetime dynamics in diluted alcohols., Phys Chem Chem Phys, 2017, 19(41), 27960–27967, 10.1039/c7cp03222f.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta05207e |
| ‡ Present address: Inorganic Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QR, UK. |
| This journal is © The Royal Society of Chemistry 2022 |