
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantifying triplet formation in conjugated polymer/non-fullerene acceptor blends†
Junjun
Guo
 a,
Benjamin
Moss
b and
Tracey M.
Clarke
*a
a,
Benjamin
Moss
b and
Tracey M.
Clarke
*a
aDepartment of Chemistry, University College London, Christopher Ingold Building, London WC1H 0AJ, UK. E-mail: tracey.clarke@ucl.ac.uk
bDepartment of Chemistry, Molecular Science Research Hub, White City Campus, Imperial College London, London W12 0BZ, UK
First published on 8th September 2022
Abstract
Triplet formation is generally regarded as an energy loss process in organic photovoltaics. Understanding charge photogeneration and triplet formation mechanisms in non-fullerene acceptor blends is essential for deepening understanding of photophysics in these important organic photovoltaic materials. Here, we present a comprehensive spectroscopy and morphology study on non-fullerene acceptors ITIC, ITIC-Th, ITIC-2F and Y6, both pristine and blended with reference polymer PffBT4T-C9C13. Atomic force microscopy and grazing-incidence X-ray diffraction provided information regarding the morphology of the films while spectroelectrochemistry combined with microsecond transient absorption spectroscopy allowed triplets and charge carriers to be investigated in detail. Crucially, we used triplet sensitisation to determine molar extinction coefficients of the non-fullerene acceptor triplets (2.7–6.5 × 104 L mol−1 cm−1), allowing triplet populations to be quantified in the blends. Intriguingly, no consistent trends were found in the photophysics of the studied blend systems, with each presenting its own unique mechanism. PffBT4T-C9C13:Y6 showed no triplet formation, only charge carriers that decayed rapidly in a relatively crystalline environment, consistent with the observed highly segregated morphology. In contrast, all blends in the ITIC series produced evidence of considerable triplet formation in addition to charge carriers. PffBT4T-C9C13:ITIC-Th blend produced acceptor triplets irrespective of excitation wavelength, and these were formed via intersystem crossing and/or energy transfer. Conversely, both ITIC and ITIC-2F blends displayed triplet formation via non-geminate recombination of charge carriers, with both NFA and polymer triplets observed. However, PffBT4T-C9C13:ITIC-2F produced a substantially higher charge carrier population than the ITIC blend. Because its triplet formation mechanism relies on the presence of charge carriers, PffBT4T-C9C13:ITIC-2F, with the highest charge carrier population, also had the highest triplet population. These results exemplify the prevalence of triplet states across a range of NFA blend systems, despite the varying formation mechanisms. Furthermore, they showcase that triplet populations can reach very high levels, particularly in cases of concomitantly high charge populations. Since high charge carrier densities correlate with large short circuit currents, this has significant ramifications for organic photovoltaic performance.
Introduction
The past several years have witnessed a dramatic increase in the power conversion efficiency of solution-processed organic photovoltaic (OPV) devices. The efficiency for a single junction organic solar cell can now achieve 19%.1 The improvement is largely a consequence of newly designed non-fullerene acceptors (NFAs), providing a series of options to match with the donor and covering a broader spectral range than fullerenes. A key advantage of the most efficient NFA organic solar cells is their ability to achieve a relative high charge generation yield with a small (or negligible) driving force for charge separation.2–4It has recently been reported that triplet states appear prominently in highly efficient OPV blends, including those based on NFAs.5,6 Considering that triplet formation is traditionally seen as a loss mechanism in OPV, these are very surprising results. Since the T1 state is often one of the lowest energy states of the system, for example, its formation and subsequent relaxation back to the ground state entails a non-radiative voltage loss.7 An important aspect to consider is the multiple mechanisms by which donor or acceptor triplets can be created in an organic solar cell. There are three primary mechanisms: intersystem crossing (ISC), back electron transfer from a spin-mixing charge transfer (CT) state, and via bimolecular recombination of free carriers. The rate of ISC is dictated by the size of the spin–orbit coupling and the energy gap between the singlet and triplet states involved, ΔEST. The latter two mechanisms both proceed via a CT state and are dependent on the energetics of the system, kinetic competition, spin statistics, and the nature of the spin-mixing.
Triplet formation can also depend on the morphology of the active layer. Rao et al. have shown, for example, that the charge delocalisation enabled by ordered fullerene domains promotes CT state separation over the competing process of back electron transfer to create triplets.8 To optimise OPV devices further, it is essential to better understand energy alignment in polymer:NFA blends together with morphology variation to suppress non-radiative recombination losses.9
In this paper, triplet formation and behaviour are studied in a series of NFAs: ITIC, ITIC-Th, ITIC-2F (also called ITIC-4F, IT-4F), and Y6. The chemical structures for these molecules are shown in Fig. 1, pertinent characteristics such as energy levels presented in Table 1, and cyclic voltammetry (CV) in Fig. S1 (ESI).† The thienyl side chain in ITIC-Th deepens molecular energy levels relative to the phenyl group in ITIC, providing a strong sulphur–sulphur interaction that facilitates π-stacking and charge transfer.10 For ITIC-2F, fluorination enhances the inter/intramolecular interactions because of non-covalent F–H and S–F interactions, improving crystallinity and facilitating charge transfer.11 In addition, ITIC-2F shows a high polarisation and reduced Coulombic interaction between electrons and holes.12 Fluorinated NFAs have been designed to suppress triplet formation and improve device efficiency by optimising triplet energy level alignment.7 Y6 is the prototypical high-achiever NFA, capable of OPV device efficiencies of 18%. NFA photophysics are also examined in blends with a reference polymer, poly [(5,6-difluoro-2,1,3-benzothiadiazol-4,7-diyl)-alt-(3,3′′′-di (2-nonyltridecyl)-2,2′; 5′,2′′; 5′′,2′′′ -quaterthiophen-5,5′′′-diyl)] (PffBT4T-C9C13). PffBT4T-C9C13 is capable of device efficiencies of over 12% with PC70BM.13 Blends of PffBT4T-C9C13 with fullerenes were extensively studied in our previous work and this provides the tools to facilitate analysis of the selected NFAs blended with this polymer.14 Notably, PffBT4T-C9C13 blends with fullerenes exhibited strong tendencies to triplet formation, despite its high performance in OPV devices, and morphology-dependent bimodal polaron formation.
 | ||
| Fig. 1 The structure of polymer donor PffBT4T-C9C13 and non-fullerene acceptors ITIC, ITIC-Th, ITIC-2F and Y6. | ||
| S1 energy (eV) | HOMO (eV) | LUMO (eV) | PLQE (%) | 3NFA* ε (x 104 L mol−1 cm−1) | Triplet τ solution (μs) | 3NFA* τ in blend (μs) | 3P* τ in blend (μs) | |
|---|---|---|---|---|---|---|---|---|
| PffBT4T-C9C13 | 1.71 | −5.34 | −3.63 | — | 12 | |||
| ITIC | 1.66 | −5.68 | −4.02 | 77 | 2.7 | 1.6 | 23 | 33 |
| ITIC-Th | 1.67 | −5.75 | −4.08 | 84 | 5.7 | 6.0 | 8.5 | — |
| ITIC-2F | 1.61 | −5.79 | −4.18 | 68 | 4.1 | 6.0 | >6 | 40 |
| Y6 | 1.48 | −4.99 | −3.94 | 95 | 6.5 | 1.1 | — | — |
In this work we employ μs transient absorption spectroscopy (TAS) and spectroelectrochemistry (SEC) to study relevant transient species in different pristine NFAs, with a particular emphasis on the anions and triplet states. The SEC data shows the ITIC series anions to absorb at ∼800 nm, while the Y6 anion is located at 925 nm. The ITIC-Th and ITIC triplets absorb at 1100 and 1130 nm respectively, ITIC-2F triplet at 1210 nm, and the Y6 triplet absorbs even further to the red at 1360 nm. Importantly, we used a sensitisation procedure to estimate the molar extinction coefficient of each triplet, allowing triplet populations in the blends to be determined from the TA data. Indeed, each blend with the reference polymer presented very different photophysical behaviour. PffBT4T-C9C13:Y6 showed no triplet formation, only charge carriers that decayed rapidly in a relatively crystalline environment, consistent with the highly segregated morphology observed using atomic force microscopy. In contrast, all blends in the ITIC series produced evidence of considerable triplet formation in addition to charge carriers. PffBT4T-C9C13:ITIC-Th blend produced NFA triplets irrespective of excitation wavelength, indicative of an energy transfer process, and also no evidence of non-geminate triplet formation. In contrast, both ITIC and ITIC-2F blends displayed non-geminate triplet formation, and both NFA and polymer triplets were observed. Because this triplet formation mechanism relies on the presence of charge carriers, the blend system with the highest charge carrier population (PffBT4T-C9C13:ITIC-2F) also had the highest triplet population. These results exemplify the prevalence of triplet states across a range of NFA blend systems, despite the varying formation mechanisms. Furthermore, they showcase that triplet populations can reach very high levels, particularly in cases of concomitantly high charge populations. Since high charge carrier densities correlate with large short circuit currents, this has significant ramifications for organic photovoltaic performance.
Results
Steady state absorption spectra and photoluminescence spectra
The normalised ground state absorption spectra for non-fullerene acceptors ITIC, ITIC-Th, ITIC-2F, and Y6 blended with polystyrene (PS), pristine donor PffBT4T-C9C13, and each blend are shown in Fig. 2a–d. The 0–0 vibronic transition is at approximately 700 nm for the pristine polymer PffBT4T-C9C13 and the NFAs ITIC and ITIC-Th, while it is red-shifted by 20 nm for ITIC-2F. The low bandgap Y6 absorbs across a wide wavelength range with the 0–0 vibronic transition at 810 nm. The ITIC blends with PffBT4T-C9C13 reveal some interesting behaviour. For ITIC, the breadth of the blend absorption is less than either of the individual components. PffBT4T-C9C13:ITIC has a FWHM of 0.35 eV, demonstrably less than pristine PffBT4T-C9C13 (0.52 eV) or ITIC (0.41 eV). Furthermore, the intensity ratio of the 0–0 to 0–1 bands is greater in the ITIC and ITIC-Th blends compared to the individual components. Both observations indicate an increased crystallinity in the blend films, noting that acceptor-induced ordering has previously been reported for PffBT4T-C9C13.14 This is less obviously the case for the ITIC-2F blend, which shows a smaller change in intensity ratio and an intermediate FHWM of 0.48 eV (with pristine ITIC-2F having a FWHM of 0.41 eV).15 The absorption spectrum for the PffBT4T-C9C13:Y6 blend film shows polymer and NFA contributions, with both components showing clear red-shifts relative to the pristine films, potentially indicating an enhanced crystallinity for both the donor and acceptor regions. | ||
Fig. 2 Normalised steady state absorption spectra (solid lines) and photoluminescence spectra (dashed lines) of (a) PffBT4T-C9C13:ITIC film, (b) PffBT4T-C9C13:ITIC-Th film, (c) PffBT4T-C9C13:ITIC-2F film, and (d) PffBT4T-C9C13:Y6 film compared to the individual components. All blend films are made with D/A ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2. The excitation wavelength 650 nm is used for all film PL measurements. PS represents the polystyrene. :1.2. The excitation wavelength 650 nm is used for all film PL measurements. PS represents the polystyrene. | ||
Photoluminescence spectroscopy is used to analyse the role of non-fullerene acceptors in the PffBT4T-C9C13 blend films. The normalised PL emission spectra for the individual materials and their blends are shown in Fig. 2a–d, with excitation wavelength 650 nm. Because both donor and acceptor components absorb at 650 nm, the PL spectra of the blends are a combination of both components. For PffBT4T-C9C13:ITIC, for example, the shoulder at 740 nm is unshifted polymer PL, while the main peak at 798 nm appears to be red-shifted ITIC PL. This red-shift cannot be attributed to dispersion of the ITIC in the blend film, because PS:ITIC exhibits a blue shift (765 nm) compared to pristine ITIC (780 nm), and thus the red-shift is likely due to the donor/acceptor interaction in the blend. Conversely, the PL spectrum of PffBT4T-C9C13:ITIC-Th appears identical to the spectrum of PS:ITIC-Th. Although all pristine NFA PL spectra display aggregation-induced quenching compared to their corresponding PS:NFA spectra (Fig. S2†), it is noted that this is considerably less prominent for ITIC-Th. This implies that even in a dispersing medium, ITIC-Th forms clusters. As such, even in the blend with the polymer, these clusters are likely also present and thus comprise the remaining PL. An additional possibility that cannot be discounted is singlet energy transfer from the polymer to the NFA, noting that their very similar S1 energy levels could facilitate coupling between the two states at the donor/acceptor interface. The PL peak of PffBT4T-C9C13:ITIC-2F appears intermediate between the polymer and NFA, but the high PL quantum yield of ITIC-2F makes it difficult to assess relative contributions.16,17 The PffBT4T-C9C13:Y6 PL is dominated by the polymer component, and this can be attributed to the greater proportion of light being absorbed by the polymer at the excitation wavelength 650 nm.
To estimate the polymer PL quenching yield (Fig. S3†), an excitation wavelength of 455 nm was used, such that NFA absorbance was minimised (<0.1). Care was taken to ensure the polymer in both pristine and blend samples absorbed the same number of photons. To further minimise contribution from the NFA, the PL amplitude at 700 nm was used for the estimation, noting that NFA PL at this wavelength was ≤ 10% of the polymer's. The uncertainty in the estimated polymer PL quenching yields, mostly arising from the small NFA contribution, is ±5%. This analysis showed that PffBT4T-C9C13:ITIC-Th displayed the least PL quenching (68%), consistent with its hypothesised clustering, while PffBT4T-C9C13:Y6 showed the greatest (92%). The high quenching in the Y6 blend, indicating efficient exciton dissociation at the D/A interface, is likely due to the long exciton lifetime and consequently long exciton diffusion length of Y6.18,19 PffBT4T-C9C13:ITIC-2F also exhibited polymer PL quenching of over 90%, potentially assisted by its greatest driving force for electron transfer and a favourable morphology.
Morphology
Atomic force microscopy was used to understand morphology of the PffBT4T-C9C13:NFA blend films (Fig. 3). From the ITIC series' blend height images in Fig. 3a–c, the root mean square (Rq) ranged from the ITIC (4.1 nm) to the ITIC-Th blend (5.6 nm) to ITIC-2F blend (5.1 nm), all of which are rougher than the pristine PffBT4T-C9C13 film (2.4 nm, Fig. S4†). The increased roughness upon blending shows that phase segregation is induced with addition of different acceptors, with the ITIC-Th blend potentially having the largest domain size of the ITIC series (as hypothesised above). However, the roughness of PffBT4T-C9C13:Y6 (Fig. 3d) is substantially larger than the other blends, with Rq approximately 17 nm. This large surface roughness after blending with PffBT4T-C9C13 contrasts with smooth films reported for PM6:Y6, which show similar morphology as the pristine PM6 film.20 To check if such high roughness is inherent to Y6, both pristine Y6 and PS:Y6 films were investigated (Fig. S5†). Pristine Y6 films are relatively smooth (2.2 nm), with an increased roughness in the PS:Y6 film (6.6 nm). As such, the substantial roughness of the PffBT4T-C9C13:Y6 film is due to considerable Y6 phase segregation, suggesting low miscibility between the two components. In contrast, the pristine ITIC series' films are very smooth, with Rq values of less than 1 nm (Fig. S5†). | ||
| Fig. 3 AFM images of topography for (a)PffBT4T-C9C13:ITIC, (b)PffBT4T-C9C13:ITIC-Th, (c) PffBT4T-C9C13:ITIC-2F, and (d)PffBT4T-C:Y6. Scale bars are 400 nm. GIXRD (normalised per absorption) for pristine PffBT4T-C9C13 film and (e) ITIC and PffBT4T-C9C13: ITIC (1:1.2) film (f) ITIC-Th and PffBT4T-C9C13: ITIC-Th (1:1.2) film (g) ITIC-2F and PffBT4T-C9C13: ITIC-2F (1:1.2) film (h) Y6 and PffBT4T-C9C13: Y6 (1:1.2) film. | ||
The molecular order packing structure for pure polymer, NFAs and their blend films are investigated by GIXRD measurements, as shown in Fig. 3e–h. Previous GIXRD measurements have shown an amorphous morphology for pristine polymer PffBT4T-C9C13, with the weak, broad [010] π–π stacking peak at q = 16 nm−1 (stacking distance d = 0.393 nm).14,21 Compared to the polymer, the [010] feature is also exhibited in the NFAs but with slightly different peak scattering position and intensity, occurring at ∼17 nm−1 and consistent with the previously reported values.12,22 The GIXRD for pristine ITIC shows a relatively broad and weaker diffraction peak compared to the other NFAs, indicating lower crystallinity. In the polymer:NFA blends, the [010] peak of the polymer becomes increasingly prominent (with the NFA peak still visible as a shoulder), suggesting an enhanced crystallinity in the polymer domains. Similar to the pristine ITIC, the blend PffBT4T-C9C13:ITIC has the lowest integrated peak area, suggesting the lowest crystallinity of all blends studied. Conversely, PffBT4T-C9C13:Y6 shows the greatest integrated peak area and the Y6 shoulder is particularly visible, suggesting both components exhibit an enhanced crystallinity, a possible result of the low miscibility observed in the AFM.
Pristine NFAs
A primary reason for the success of NFAs in OPV is their ability to absorb more light than fullerenes. However, this presents a potential issue in spectroscopic analysis, as spectral data is no longer dominated by polymer features. NFA species will also contribute, causing possible spectral congestion and difficulties in analysis. To overcome this, we examined each NFA in its pristine form (solution and film), and also with the inert polymer polystyrene (PS) to simulate the blend environment. The NFA species of interest are anions and triplets, and these will be examined using SEC and TAS, respectively.SEC results for the ITIC film are shown in Fig. 4a as a function of voltage; the other NFA films are shown in Fig. S6.† Care was taken to keep the voltage below the first reduction peak of the NFA, in order to avoid double reduction. The ITIC anion peak is at 780 nm, ITIC-Th anion at 795 nm, and ITIC-2F anion at 793 nm. Although weak, the Y6 anion can be observed at 925 nm (Fig. S6† and the inset in Fig. 4a). Additional confidence in these results arises from the good correspondence between peak amplitude and the total charge extracted as a function of voltage (Fig. S7†). The NIR region was also checked, but no additional peaks were observed.
 | ||
| Fig. 4 SEC absorption spectra for (a) ITIC film and (b) polymer PffBT4T-C9C13 film in 0.1 M TBAP/acetonitrile with Ag/AgNO3 electrode, plotted versus applied potential from 0.4 V to 0.8 V for the polymer and 0 V to −0.8 V for ITIC film. The inset is the normalised SEC absorption spectra for ITIC (−0.8 V), ITIC-Th (−0.9 V), ITIC-2F (−0.5 V), and Y6 (−0.8 V) films. (c) Normalised (to 1) TA spectra for PffBT4T-C9C13, ITIC, ITIC-Th, ITIC-2F and Y6 solutions in chlorobenzene. (d) Normalised (to 1) triplet decay dynamics for PffBT4T-C9C13 (probe at 1100 nm), ITIC (probe at 1100 nm), ITIC-Th (probe at 1100 nm), ITIC-2F (probe at 1200 nm), Y6 (probe at 1400 nm) solutions in chlorobenzene. The excitation energies used for the above solutions is 16 to 26 μJ cm−2. | ||
SEC was also employed to determine the PffBT4T-C9C13 cation (positive polaron) absorption spectrum; the results are shown in Fig. 4b. The two expected polymer cation bands are measured at 760 nm and 1600 nm. An apparent shift in band position with voltage is observed for the 760 nm band, which is superseded by a broad 850 nm absorption. Given that neither the ground-state bleach nor 1600 nm band shift, this apparent shift of the 760 nm band may relate to the growth of the dication, noting that the dication absorption of a conjugated polymer is typically intermediate between the two cation absorptions.23
TAS of the NFA solutions showed solely triplets in each case, as shown in Fig. 4c. Strong TA signal quenching in an oxygen environment was observed for all NFAs tested, even the very low-bandgap Y6 (Fig. S8†). Critically, the original TA signal amplitude and lifetime was recovered after the sample was returned to an inert environment, a clear hallmark of a triplet state. ITIC-Th and ITIC triplets absorb similarly at 1100 and 1130 nm respectively while the ITIC-2F triplet absorbs slightly further to the red at 1210 nm, reflecting the differences in their ground-state absorbance spectra. The Y6 triplet absorbs even further to the red, at 1360 nm. The triplet decay lifetimes (Fig. 4d) in solution are all mono-exponential and in the early μs range, with τ = 1.1 μs for Y6, τ = 1.6 μs for ITIC, and τ = 6.0 μs for both ITIC-2F and ITIC-Th.
Because condensed phases can often incur substantial red-shifts relative to the solution phase, PS:NFA films were also investigated (Fig. S9†). Triplet formation in PS:Y6 is switched off, and only anion formation is observed (Fig. S9d†). Triplets are still observed in the ITIC series blended with PS. PS:ITIC and PS:ITIC-Th triplets display 0.05 eV red-shifts compared to the solution, while the PS:ITIC-2F shows a significant broadening, indicating numerous triplet environments (Fig. S9a to S9c†). This hypothesis can be verified by an examination of the triplet spectrum of a pristine ITIC-2F film, which shows the triplet red-shifted to 1300 nm. Unusually, the lifetimes of the ITIC series triplets in PS blends are longer than in solution, with mono-exponential lifetimes in the range of 120–160 μs (Fig. S10†). Since the triplet decays in both phases are mono-exponential, this observation cannot be accounted for by annihilation or any other second-order loss process.
To quantify the NFA triplet formation, we assessed the triplet molar extinction coefficient in solution. This was accomplished using a sensitisation process with well-known triplet sensitiser ZnTAPP (zinc tetraphenyl-porphyrin). By selectively exciting at the porphyrin's strong Soret band, efficient triplet formation and transfer onto each NFA enabled its triplet extinction coefficient to be determined. We used the estimation method outlined by Land et al.,24 including their suggested corrections for situations when the acceptor triplet decays during its formation, and when the donor triplet decays by means other than triplet transfer. Full calculation details are given in the SI and all NFA triplet extinction coefficients were estimated to be of the order of 104 L mol−1 cm−1, with a range of 2.7–6.5 × 104 L mol−1 cm−1 (Table 1). We estimate the uncertainty in these values, based on signal-to-noise of the ΔOD amplitude and fitting errors, to be approximately ± 0.2 × 104 L mol−1 cm−1.
NFAs in polymer blends
Microsecond TAS was employed to study the photophysics of the selected NFAs blended with reference polymer PffBT4T-C9C13. PffBT4T-C9C13 has been well-characterised in blends with fullerenes, and thus its polaron and triplet spectral features are known. Normalised transient absorption spectra of each NFA with PffBT4T-C9C13 at two pump wavelengths are shown in Fig. 5, and spectral evolutions are shown in Fig. S11.† 455 nm primarily excites the polymer, 700 nm is able to excite both the NFA and polymer (709 nm was used for the lower band-gap Y6). The observed photophysics are summarised in Jablonski diagrams in Fig. 6.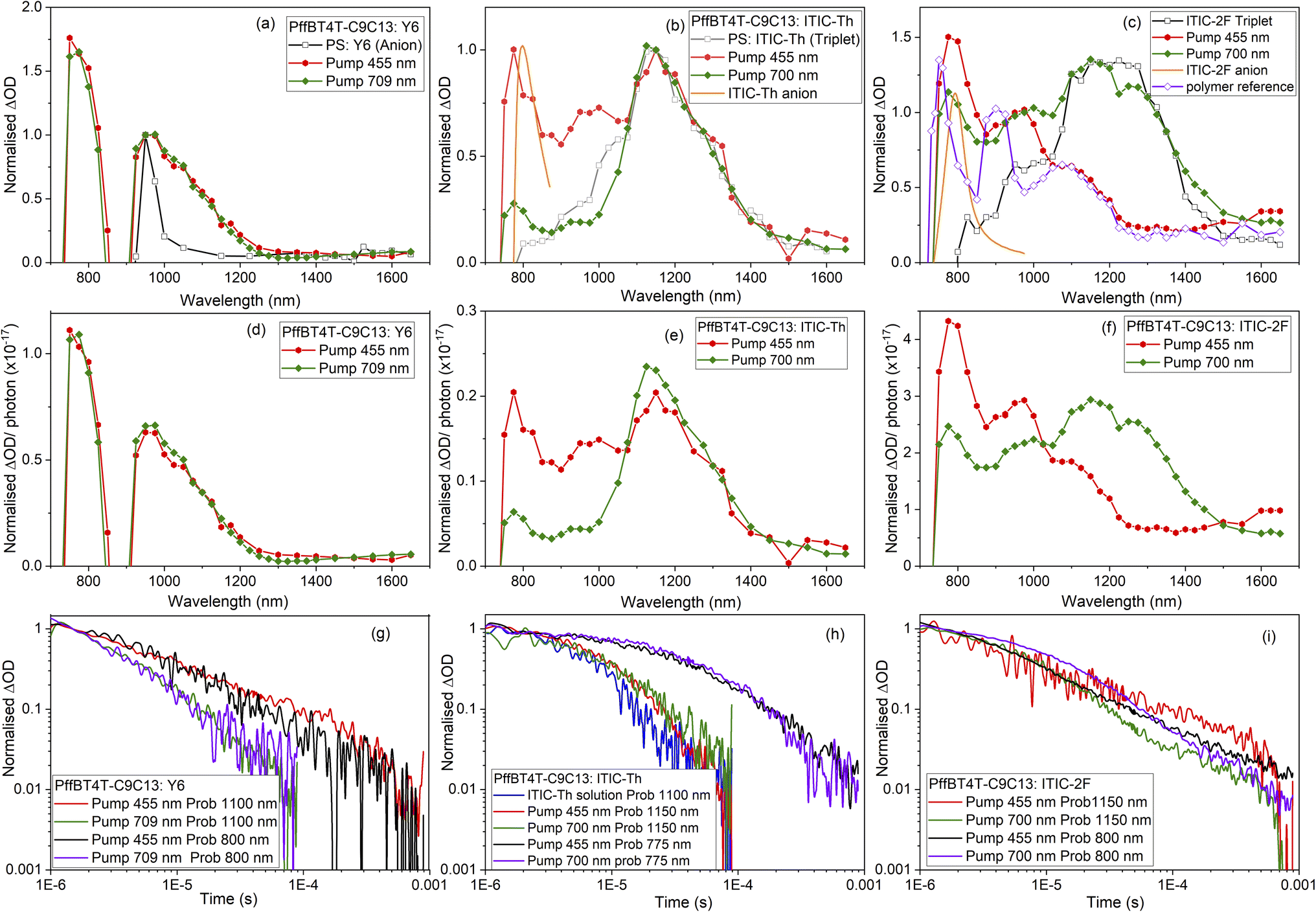 | ||
| Fig. 5 Normalised (to 1) and (per photon) transient absorption spectra of (a) and (d) PffBT4T-C9C13:Y6, (b) and (e) PffBT4T-C9C13:ITIC-Th, (c) and (f) PffBT4T-C9C13:ITIC-2F films, with excitation pump wavelengths of 455 and 700/709 nm. Excitation densities used were in the range 10–30 μJ cm−2. Also shown are reference spectra including the Y6 anion (from PS:Y6 film), relevant NFA triplet (NFA:PS film), relevant NFA anion (from SEC), and polymer polaron and triplet (“polymer reference”: PffBT4T-C9C13:PC70BM14). Corresponding normalised decay dynamics for (g) PffBT4T-C9C13:Y6 film probing at 800 nm and 1100 nm, (h) PffBT4T-C9C13: ITIC-Th film probing at 775 nm and 1150 nm, and (i) PffBT4T-C9C13: ITIC-2F film probing at 800 nm and 1150 nm. | ||
 | ||
| Fig. 6 Photophysical mechanisms for the polymer:NFA blends studied here. Blue arrows denote polymer (P) excitation with 455 nm pump, while red arrows denote NFA excitation with 700 nm excitation (although some polymer excitation is also present with this pump wavelength). Green arrows denote processes that occur irrespective of excitation wavelength. The excited species highlighted in yellow show those present on μs timescales in each blend. Note that we do not observe charge transfer (CT) states in μs-TAS, and therefore focus only on the charge-separated (CS) states; however, it should be noted that CT states will certainly contribute during both charge photogeneration and recombination processes. | ||
PffBT4T-C9C13:Y6 shows the most straight-forward TAS behaviour: neither polymer nor NFA triplet is observed, and the standard power law decay kinetics are consistent with charge carriers (Fig. 5g). The spectrum is dominated by charge carriers at both excitation wavelengths (Fig. 5a and d), consistent with previous reports of ultrafast energy transfer in Y6 blends.25 The precise wavelength of the charges cannot be determined from TAS in this case due to the Y6 ground-state bleach bisecting the photoinduced absorption peak, but the position is consistent with the SEC results.
In contrast, PffBT4T-C9C13:ITIC-Th displays evidence of both charge carrier and triplet formation (Fig. 5b). The ITIC-Th triplet is seen very prominently at 1140 nm for both excitation wavelengths. Despite the closeness in spectral position between the PffBT4T-C9C13 triplet (1080 nm) and ITIC-Th triplet (1140 nm), the triplet identified in this case is the clearly NFA triplet due to the identical match to the PS:ITIC-Th spectrum in terms of spectral position, shape, and breadth. Furthermore, the considerable increase in relative intensity of the 1140 nm peak when exciting the NFA directly is strongly indicative of an NFA species (Fig. 5e). The ITIC-Th triplet decays rapidly in the blend with a mono-exponential lifetime of 6 μs and reveals more of the charge carrier spectral features at longer times (Fig. 5h). Intriguingly, this ITIC-Th triplet lifetime is identical to that found in solution, suggesting that the ITIC-Th triplet is not being regenerated via charge recombination. Furthermore, the appearance of similar populations of NFA triplets irrespective of excitation wavelength suggests the presence of an energy transfer mechanism (either singlet or triplet), with the NFA triplet being the lowest energy triplet in the system. The PffBT4T-C9C13:ITIC-Th charge carriers are characterised by two peaks at 1000 nm and <800 nm, with the latter being most intense. These two bands are similar to that seen previously for PffBT4T-C9C13:PC70BM and were attributed to polymer polarons in mixed donor/acceptor domains and relatively pure polymer domains respectively. For PffBT4T-C9C13:ITIC-Th, however, the ITIC-Th anion also absorbs strongly at 800 nm. The additive effect of both cation and anion absorbing at 800 nm lead this band to dominate the TA spectrum, particularly at late times once the NFA triplet has decayed (Fig. S11c and d†). The 1600 nm polymer polaron band is therefore only weakly apparent.
PffBT4T-C9C13:ITIC shows strong excitation wavelength dependent TA spectra (Fig. S12a and S12b†). As with the ITIC-Th blend, two longer-lived polymer polaron peaks are evident at <800 nm and 920 nm at 1 μs for PffBT4T-C9C13:ITIC, with the former peak also having a contribution from the NFA anion at 780 nm. However, the third band is very different for the two excitation wavelengths: appearing at 1080 nm with polymer excitation and 1150 nm with NFA excitation. These match well with the corresponding triplets; for example, the red edge of the 455 nm PffBT4T-C9C13:ITIC TA spectrum matches perfectly with the PffBT4T-C9C13:PC70BM spectrum, indicating polymer triplets. In contrast the 700 nm PffBT4T-C9C13:ITIC TA spectrum's 1150 nm band is very similar to the ITIC solution and PS:ITIC TA spectra (Fig. S12a†). These observations suggest that polymer triplets dominate with polymer excitation and NFA triplets dominate with NFA excitation. This could occur, for example, if the triplet energy levels of both components are very similar (which is feasible, given their similar S1 energies) and thus there is no impetus for triplet energy transfer, or the triplets are trapped in their individual component domains. In contrast to the ITIC-Th blend, the triplets in the PffBT4T-C9C13:ITIC blend (Fig. S12c†) have a slightly longer lifetime than either individual component in solution. While the ITIC triplets in solution have a lifetime of 1.6 μs and the PffBT4T-C9C13 triplets have a lifetime of 12 μs, the ITIC and PffBT4T-C9C13 triplets in the blend have mono-exponential lifetimes of 23 μs and 33 μs respectively. The prolonged lifetime may have its origin in charge recombination repopulating the triplet state on longer timescales.
PffBT4T-C9C13:ITIC-2F (Fig. 5c and f) also exhibits both triplets and charge carriers, with strong excitation wavelength dependence. The same dual polaron peaks are present at 950 and <800 nm and, like the other blends in the ITIC series, are particularly prominent with the polymer 455 nm excitation. Once again it appears that at early times of 1 μs, polymer triplets are present with the 455 nm excitation while NFA triplets are present with 700 nm excitation. However (Fig. S11e, f and S13c†), at late times there is a strong contribution to the TA spectrum by triplets, not just polarons. Furthermore, the late-time spectrum is roughly the same for both excitation wavelengths and the triplet region matches the polymer triplet. This is similar to what was observed by the ITIC blend, but considerably more prominent. An examination of the triplet kinetics with NFA excitation at 700 nm shows a biphasic decay consistent with this observation (Fig. 5i). The NFA triplet decay is observed until approximately 80 μs, after which time the polymer triplet is more apparent and decays at the same rate as the 455 nm excitation decay. The polymer triplet lifetime contains a longer-lived component of ∼40 μs in the blend, three times longer than the lifetime in solution, indicative of non-geminate recombination. Indeed, the polymer triplet lifetime is of the same order of magnitude as the polymer polarons, consistent with this hypothesis. Another striking observation for the PffBT4T-C9C13:ITIC-2F system is that both the charge photogeneration yield and triplet population are substantially higher than in the other ITIC-based series' blends.
Discussion
Our results have shown that triplet formation is particularly prevalent in polymer:NFA blends. Since triplet formation is generally regarded as an energy loss pathway, it is critical to understand not only how the triplets are formed, but also their population in the blend film. As such, we employed a sensitisation procedure to estimate triplet extinction coefficient for each NFA and thus estimate NFA triplet populations for both solution and blend film (Fig. 7). Note that for this quantitative analysis, we only consider NFA excitation due to the spectral proximity of the polymer triplet upon 455 nm excitation at early times. In solution, it is observed that Y6 has the highest triplet population by a factor of almost two (Fig. 7a). It has been previously reported by Ohkita et al. that pristine Y6 can generate triplets via singlet fission in addition to ISC, and this could account for the higher triplet population on these later timescales.26 ITIC-2F has the second highest triplet population, an unusual result given its high PL quantum yield: a low ISC efficiency would therefore be expected. However, this could be compensated for by its relative long triplet lifetime.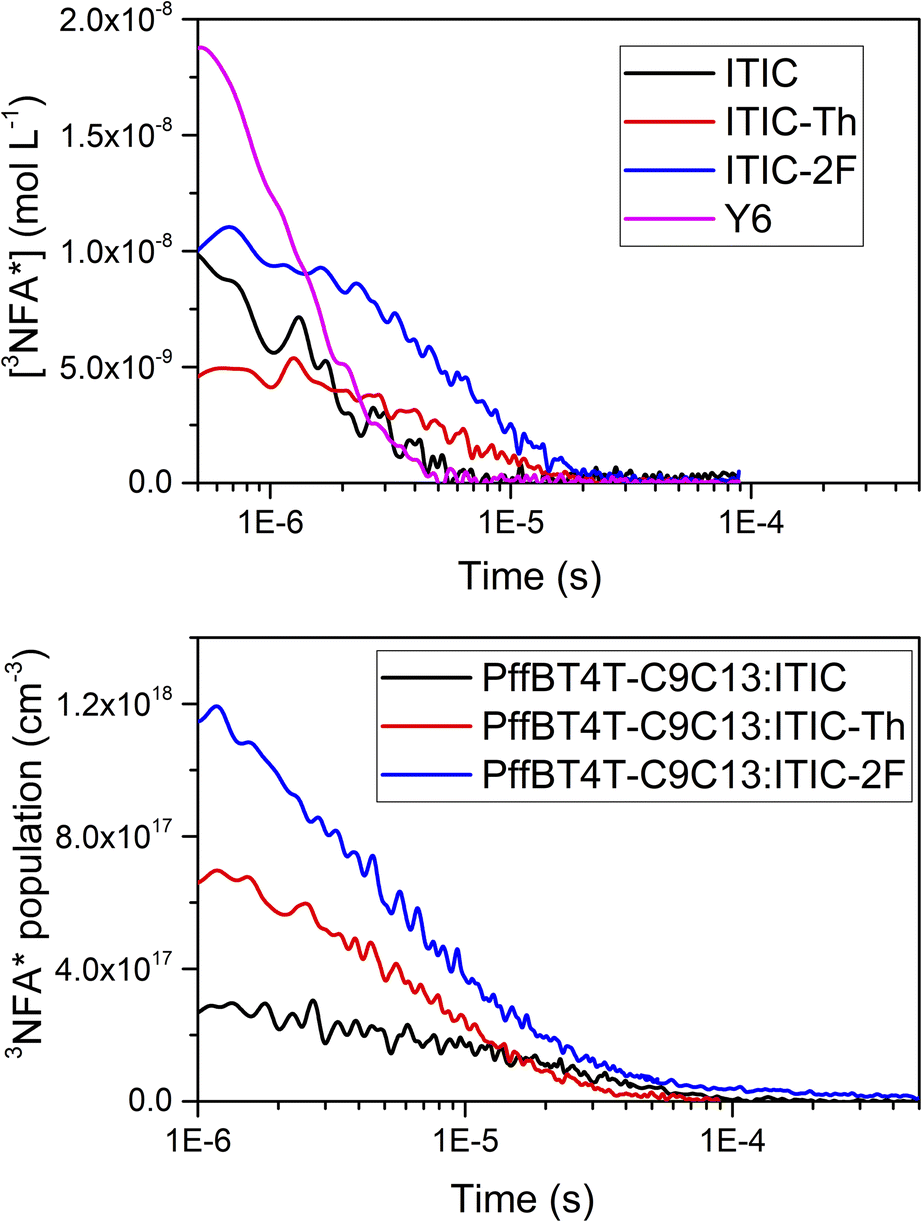 | ||
| Fig. 7 The triplet yields calculated (a) in pristine NFA solutions in chlorobenzene and (b) in blend PffBT4T-C9C13:NFA films using 700 nm excitation. | ||
It is important to note that the extinction coefficient of a material can vary between solution and solid phases. A quantitative triplet sensitisation procedure in the solid phase is very difficult due to the lack of standards with known triplet absorption coefficients in the solid phase and the high likelihood of excited state annihilation processes leading to inaccurate ε values. As such, we have addressed this issue by obtaining correction factors for the change in absorption cross-section between solution and film for the ground state of each NFA, and then applying these correction factors to the solution phase triplet ε values we have already determined (details in the ESI†). Even accurately establishing ground state absorption coefficients for film samples is non-trivial, particularly for dispersive materials, but this has been achieved for ITIC, ITIC-2F, and Y6 by Armin et al.27 Our estimated film triplet extinction coefficients are in the range of 2–3 × 104 L mol−1 cm−1. However, applying a correction factor does introduce an additional uncertainty, but this is likely to be a systematic error and thus will not significantly affect the results discussed below.
An examination of the triplet populations in the PffBT4T-C9C13:NFA blends (using the estimated film triplet extinction coefficients) is shown in Fig. 7b, noting that the PffBT4T-C9C13:Y6 film showed little evidence of triplets on these ns–μs timescales. PffBT4T-C9C13:ITIC-2F has a very high NFA triplet population of 1.2 × 1018 cm−3, greater by a factor of 1.7 than PffBT4T-C9C13:ITIC-Th. The likely origin of this is the non-geminate recombination of charges to create triplets, as also evidenced by the substantially longer triplet lifetimes in the blend, thereby providing an additional avenue for populating the triplet state. Previous works have shown fluorinated NFAs have supressed non-geminate triplet formation,7 for example by stabilising the 3CT state below the local NFA triplet. Indeed, although PffBT4T-C9C13:ITIC-2F has the highest triplet population, this supressed triplet formation is also evident here. This is because PffBT4T-C9C13:ITIC-2F also has the highest charge carrier population and thus more charges are available to recombine to create triplets. Instead, we can correct the triplet population for charge yield for PffBT4T-C9C13:ITIC-2F and PffBT4T-C9C13:ITIC (noting that PffBT4T-C9C13:ITIC-Th exhibited no evidence of non-geminate triplet formation). PffBT4T-C9C13:ITIC-2F produces 40 times more charge carriers but only 4 times more triplets compared to the ITIC blend, thus we estimate that PffBT4T-C9C13:ITIC-2F produces only one-tenth the number of triplets per charge carrier compared to the ITIC blend.
This therefore raises the question as to why PffBT4T-C9C13:ITIC-2F blend produces the highest charge carrier yield, even more so than the high-performing Y6 and despite their similar polymer PL quenching yields. Both excitation wavelengths produce this highest charge carrier yield. It is evident from the energetics that PffBT4T-C9C13:ITIC-2F has both the largest ΔHOMO and ΔLUMO, and it is well-known that greater driving forces for charge separation can induce larger charge carrier populations.3 Furthermore, a higher dielectric constant has been reported for ITIC-2F (5.83) compared to Y6 (5.73) and ITIC (4.3).28 The higher dielectric constant of ITIC-2F indicates a smaller binding energy between electrons and holes which can facilitate the charge separation process.
It was also observed that there are considerable differences in the charge carrier decay dynamics between the blends upon polymer excitation (Fig. S14†), with PffBT4T-C9C13:Y6 and PffBT4T-C9C13:ITIC-2F both exhibiting distinct power law kinetics with α = 0.70 while PffBT4T-C9C13:ITIC and PffBT4T-C9C13:ITIC-Th display stretched exponential decay kinetics. This can be correlated with the observation that the first two blends also have the highest charge generation yields by an order of magnitude. Stretched exponential kinetics are typically associated with multiple decay pathways in a dispersive environment.29,30 Conversely, an α of 0.70 is very high compared to most other polymer:acceptor blends,31,32 and indicates a crystalline, relatively trap-free environment. This can lead to higher charge mobilities and greater CT state delocalisation, both of which can facilitate the higher charge carrier photogeneration yields observed for the ITIC-2F and Y6 blends.
To explore the intriguingly high charge carrier and triplet populations of PffBT4T-C9C13:ITIC-2F further, we examined its spectroscopic and morphological behaviour as a function of blend ratio. The μs-TA data for 1:3 and 3:1 ratio blends are compared to the original 1:1.2 blend in Fig. 8a. It is immediately evident that the 1:1.2 blend produces the highest charge carrier and triplet populations. With excess NFA in the blend (1:3), or 700 nm excitation (for either 1:3 or 3:1 blends), very few excited state species are still present on these μs timescales, indicating rapid relaxation processes back to the ground state. Indeed, the populations of triplets and charges under these conditions are very similar to pristine or PS blend samples. A particularly interesting observation is that the NFA triplet is only observed for the 1:1.2 blend. Examination of the morphology of these 1:3 and 3:1 blend ratios (Fig. 8b and c) reveals strong phase segregation and very rough films (Rq values of 11.9 and 7.3 nm respectively), in contrast to the smoother 1:1.2 film (Rq 5.1 nm). The implication here is that the charge recombination to form NFA triplets, which is mainly apparent for the 1:1.2 blend, primarily occurs in well-dispersed, intermixed domains. In contrast, charge recombination to form NFA triplets is considerably less apparent when large, segregated, possibly crystalline, domains are present.
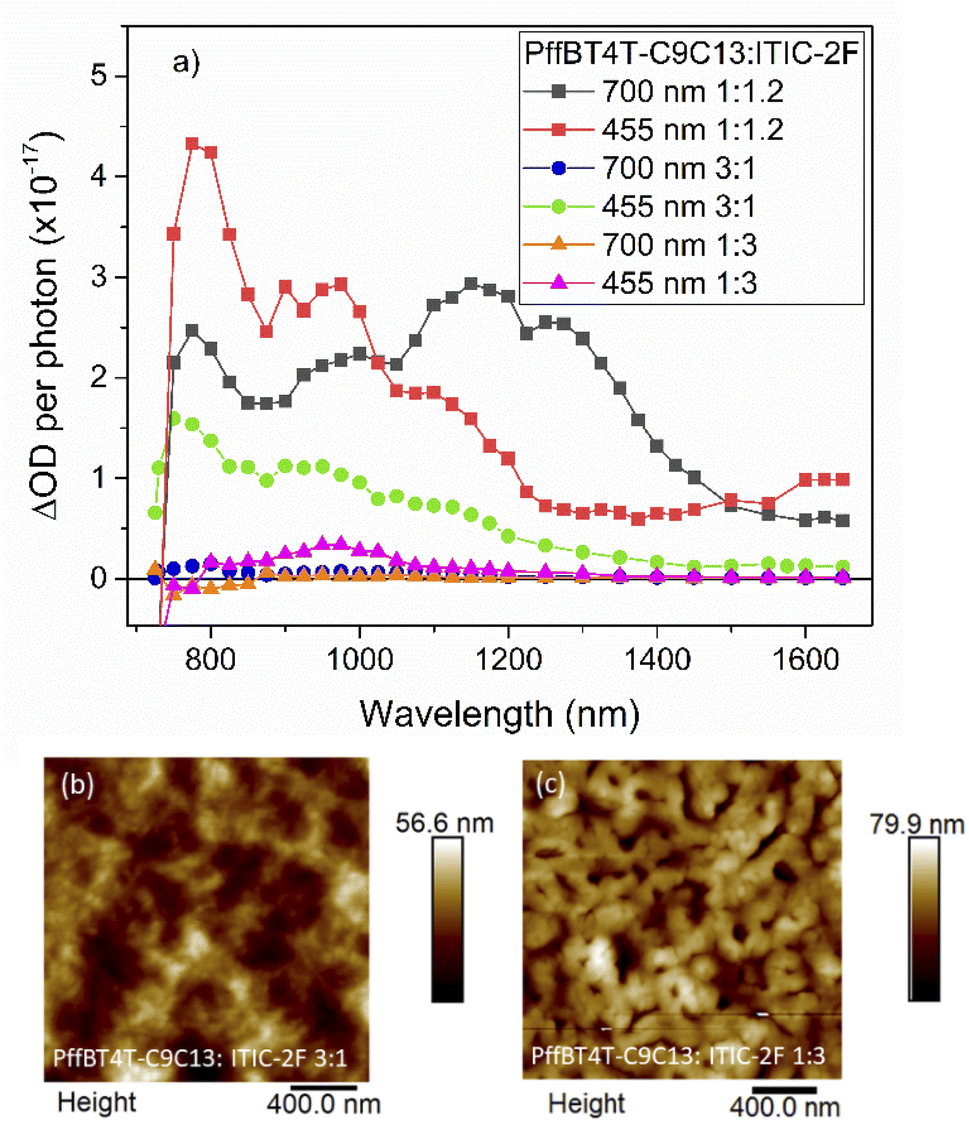 | ||
| Fig. 8 The μs-TA spectra, corrected for photons absorbed, for PffBT4T-C9C13:ITIC-2F blends with weight ratios 3:1, 1:1.2, and 1:3 with excitation wavelengths of 455 and 700 nm (a). AFM images for PffBT4T-C9C13:ITIC-2F 3:1 (b) and 1:3 (c) ratios, noting that the AFM data for the 1:1.2 ratio are displayed in Fig. 3. | ||
Interestingly, only PffBT4T-C9C13:ITIC-2F and PffBT4T-C9C13:ITIC share common behaviour in terms of the excited states formed and their mechanisms. In contrast, PffBT4T-C9C13:Y6 produces negligible evidence of triplets while PffBT4T-C9C13:ITIC-Th produces only NFA triplets and these decay extremely quickly in the blend. The lack of Y6 triplets on ns–μs timescales is likely related to the longer lifetime of the Y6 singlet exciton and ultrafast energy transfer19 enabling efficient exciton quenching despite the large domain sizes observed. The Y6 blend also has the largest GIXRD integrating factor of all the blends studied, which is consistent with lower energetic disorder compared to the ITIC series, as reported by Brédas et al.33 The ordered domains and fast charge carrier transfer rates34 enable charge carrier generation to dominate in PffBT4T-C9C13:Y6 films. However, non-geminate triplet formation has been previously observed in PM6:Y6,9 but it should be noted the PM6:Y6 also presents a very smooth morphology with the two components highly miscible with one another,35 in contrast to the strongly segregated PffBT4T-C9C13:Y6. It has been recently reported that Y6 is capable of efficient intrinsic charge photogeneration,36 and this could contribute to the lack of non-geminate triplet formation: if charges are generated in large, relatively pure domains, then donor/acceptor non-geminate recombination is less likely to occur, as observed in the above morphology study for PffBT4T-C9C13:ITIC-2F.
The prevalence of NFA triplets and their short lifetime in PffBT4T-C9C13:ITIC-Th is likely partially related to ITIC-Th's greater propensity to cluster, as indicated by the morphology and PL results. Furthermore, the similarity in NFA triplet population irrespective of excitation wavelength is strongly indicative of an energy transfer process. In contrast, the others in the ITIC series show distinct excitation wavelength dependence. 455 nm primarily excites the polymer and PffBT4T-C9C13:ITIC and PffBT4T-C9C13:ITIC-2F both show polymer triplet formation under these conditions, with no evidence of NFA triplets at all. The energetics of energy transfer do not demonstrably vary across the polymer:ITIC series, where the S1 energies of PffBT4T-C9C13 and the ITICs are very similar, and the T1 energy levels of the ITIC series are calculated to be invariant.37 However, it has been previously reported by Marina et al. that the polymorphism and phase behaviour of ITIC-Th are different to the others in the ITIC series due to differing chemical moiety occurring in the central building block rather than the terminal groups, thereby altering intermolecular interactions.38 The presence of energy transfer in PffBT4T-C9C13:ITIC-Th could therefore be related to differences in the nanomorphology at the donor/acceptor interface, possibly enabling a smaller reorganisation energy and thus facilitating energy transfer.
Finally, we consider the ramifications of these results for OPV device performance. Firstly, the variation in photophysical mechanisms across the different NFA blends suggests a complex interplay between intrinsic behaviour, energetics, and morphology that dictates that new OPV materials must be considered on their own merits. Our results suggest that charge recombination to form triplets is significantly enhanced when intermixed, well-dispersed domains are present. However, the clear observation that triplet populations also scale with charge carrier populations implies that future research directions must attempt to decouple the two, or alternatively employ strategies to harness triplet states to benefit device performance.
Conclusions
In summary, the triplet formation and behavior are studied in several NFAs (ITIC, ITIC-Th, ITIC-2F and Y6), both in pristine and blend films with the reference polymer PffBT4T-C9C13. We also performed SEC to acquire the NFA anion spectra to assist in understanding the more complex μs transient absorption spectral data, and triplet sensitisation measurements to quantify NFA triplet populations. While PffBT4T-C9C13:Y6 presents only charge carriers, NFA and/or polymer triplets are also observed for all the ITIC series blends. Interestingly, triplet identity, formation mechanism, and population vary across the ITIC series. PffBT4T-C9C13:ITIC-Th presents NFA triplets generated by both ISC and energy transfer process from the polymer excited states. Both polymer and NFA triplets are present for PffBT4T-C9C13:ITIC-Th and PffBT4T-C9C13:ITIC-2F, and the triplet lifetime extension in the blends suggests triplet formation via non-geminate recombination. Although triplet formation is suppressed in the fluorinated ITIC-2F relative to ITIC, the extraordinarily high charge carrier population observed in PffBT4T-C9C13:ITIC-2F coupled with the non-geminate recombination formation mechanism also resulted in a very high triplet population. These results show that while there is no consistent pattern with regard to triplet behaviour in NFA blends, triplets are extremely prevalent and can reach very high populations.Experimental
Materials
The polymer PffBT4T-C9C13 was sourced from Ossila (Mw = 123796, PDI = 1.68). The NFAs ITIC, ITIC-Th, ITIC-2F, and Y6 were all purchased from Ossila. The control material polystyrene (PS), the triplet sensitiser zinc tetraphenyl-porphyrin (ZnTpp), the solvent chlorobenzene (CB, 99.9%), and the additive diiodooctane (DIO) were purchased from Sigma-Aldrich.
Sample preparation
All the solutions were prepared outside the glove box and then transferred inside the glove box with stirring rate 450 rpm overnight. All the pristine acceptors' solutions for spectroscopy (ITIC, ITIC-Th, ITIC-2F, and Y6) were made with concentration 0.01 mg mL−1 in CB. Freeze–pump–thaw procedure was used to extract air from solutions for TAS measurements. Thin films were made by spin coating inside the glove box onto 1 cm2 glass substrates under nitrogen atmosphere. The pristine acceptors' films were fabricated from 10 mg mL−1 in CB solutions, heated overnight at 40 °C and spin coated at 900 rpm. All polystyrene (PS):acceptor blend films were made with weight ratio 1:1.2 from 12 mg mL−1 in CB solution, heated at 40 °C and spin coated with spin rate 900 rpm. The glass substrates and glass pipette to make blend films PffBT4T-C9C13 with non-fullerene acceptors were pre-heated at 100 °C for 20 minutes and blend films were made by spin coating from hot solution at a spin rate 900 rpm for 60 seconds. All PffBT4T-C9C13/acceptor blend films were made with weight ratio 1:1.2 from 10 mg mL−1 in CB and 3% DIO at a spin rate at 900 rpm and heating with temperature 100 °C. For the sensitisation experiment, both ZnTpp and non-fullerene acceptors solution were made with concentration 7 × 10−6 M in CB, and the blend solution ZnTpp with non-fullerene acceptors were made with molar ratio 1:1.
Ground state absorption spectroscopy
Ground state absorption spectra of all samples were recorded with a PerkinElmer Lambda 365 from 300 nm to 1100 nm at ambient atmosphere.Photoluminescence (PL) spectroscopy
Photoluminescence emission and excitation spectra were obtained with a Fluorolog-3 Spectrometer (Horiba). The 450 W xenon short arc lamp provided the excitation. The spectra were measured at 100 accumulated exposures of 0.1 s. Corrections were made by subtracting the background and accounting for the detector and lamp response. PL spectra were normalised per sample absorption at the excitation wavelength.Microsecond to millisecond transient absorption spectroscopy (TAS)
A pump-probe micro-millisecond TA spectroscopy set-up was used to measure the TA spectra and kinetics. Laser pulses (repetition rate 10 Hz, pulse duration 6 ns) were generated by a Nd:YAG laser (Spectra Physics, INDI-40-10). Excitation wavelengths were selected by a versaScan L-532 OPO and the excitation density was set in the range between 0.3 and 120 μJ cm−2 using neutral density filters, measured by a ES111C power meter (Thorlabs). The probe light was provided by a quartz tungsten halogen lamp (IL1, Bentham). Probe wavelength selectivity was achieved using bandpass filters and a Cornerstone 130 monochromator (Oriel Instrument) before the detector. The TA signals were recorded with Si and InGaAs photodiodes. The signal from the photodiodes was preamplified and sent to the main amplification system with an electronic filter (Costronic Electronics), which was connected to an oscilloscope (Tektronics, DPO4034 B) and PC.Atomic force microscopy (AFM)
AFM images were recorded by Bruker Dimension Icon in ScanAsyst peak force mode. Cantilevers used for AFM were Bruker scanasyst-Air with a nominal radius of 4 nm.Grazing incidence X-ray diffraction (GIXRD)
X-ray diffraction analysis was carried out using a Bruker AXS D8 Advanced X-ray diffractometer in grazing-incidence mode with parallel beam optics equipped with a LynxEye silicon strip detector and copper source (Cu Kα1, 1.54056 Å and Kα2, 1.54439 Å) run at 40 kV, 40 mA. The angular range was 4° to 30° 2θ counted at 0.05°/sec with data-step of 0.05° using a 1° grazing incidence angle (θ) on the films. All the GIXRD samples were spin cast on glass substrates.Spectroelectrochemistry (SEC)
A collimated beam 10 mW W halogen lamp (Ocean) was used as a light source, passing through the sample which was placed in an airtight glass three electrode cell. After passing through the cell, the beam was re-collimated and focused into an optical fibre used for detection. Two CCD cameras were used, enabling spectra to be captured from in the visible region from 650 nm to 950 nm (Ocean Maya 2000) and from 1000 to 1700 nm (Ocean Insight NIR quest). Potentials were controlled in a three-electrode configuration using an IVIUM VERTEX potentiostat using custom built LABVIEW software. Pristine films coated on ITO were used as the working electrode, Ag/AgNO3 was used as a reference electrode and platinum mesh as the counter electrode. The electrolyte used was 0.1 M tetrabutylammonium hexaflurophosphate in acetonitrile.Author contributions
JG performed all spectroscopic and morphology experiments and analysis, with BM assisting in performing and analysing the SEC. TMC conceptualised, supervised, and administered the project. All authors contributed to writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
TMC would like to acknowledge support from EPSRC project EP/N026411/1. Thank you to Professor James Durrant for access to SEC equipment and the EPSRC project EP/TO28513/1 for funding. We acknowledge Dr Ardalan Armin for helpful discussions.References
- L. Zhu, M. Zhang, J. Xu, C. Li, J. Yan, G. Zhou, W. Zhong, T. Hao, J. Song, X. Xue, Z. Zhou, R. Zeng, H. Zhu, C. C. Chen, R. C. I. MacKenzie, Y. Zou, J. Nelson, Y. Zhang, Y. Sun and F. Liu, Nat. Mater., 2022, 21, 656–663 CrossRef CAS PubMed.
- J. Liu, S. Chen, D. Qian, B. Gautam, G. Yang, J. Zhao, J. Bergqvist, F. Zhang, W. Ma, H. Ade, O. Inganäs, K. Gundogdu, F. Gao and H. Yan, Nat. Energy, 2016, 1, 1–7 Search PubMed.
- C. Yang, J. Zhang, N. Liang, H. Yao, Z. Wei, C. He, X. Yuan and J. Hou, J. Mater. Chem. A, 2019, 7, 18889–18897 RSC.
- S. Chen, Y. Wang, L. Zhang, J. Zhao, Y. Chen, D. Zhu, H. Yao, G. Zhang, W. Ma, R. H. Friend, P. C. Y. Chow, F. Gao and H. Yan, Adv. Mater., 2018, 30, 1804215 CrossRef PubMed.
- Z. Chen, X. Chen, Z. Jia, G. Zhou, J. Xu, Y. Wu, X. Xia, X. Li, X. Zhang, C. Deng, Y. Zhang, X. Lu, W. Liu, C. Zhang, Y. Michael Yang and H. Zhu, Joule, 2021, 5, 1832–1844 CrossRef CAS.
- S. Karuthedath, J. Gorenflot, A. Melianas, Z. Kan, M. Kemerink and F. Laquai, J. Phys. Chem. Lett., 2020, 11, 2838–2845 CrossRef CAS PubMed.
- R. Wang, J. Xu, L. Fu, C. Zhang, Q. Li, J. Yao, X. Li, C. Sun, Z. G. Zhang, X. Wang, Y. Li, J. Ma and M. Xiao, J. Am. Chem. Soc., 2021, 143, 4359–4366 CrossRef CAS.
- A. Rao, P. C. Y. Chow, S. Gélinas, C. W. Schlenker, C. Z. Li, H. L. Yip, A. K. Y. Jen, D. S. Ginger and R. H. Friend, Nature, 2013, 500, 435–439 CrossRef CAS.
- A. J. Gillett, A. Privitera, R. Dilmurat, A. Karki, D. Qian, A. Pershin, G. Londi, W. K. Myers, J. Lee, J. Yuan, S. J. Ko, M. K. Riede, F. Gao, G. C. Bazan, A. Rao, T. Q. Nguyen, D. Beljonne and R. H. Friend, Nature, 2021, 597, 666–671 CrossRef CAS.
- Y. Lin, F. Zhao, Q. He, L. Huo, Y. Wu, T. C. Parker, W. Ma, Y. Sun, C. Wang, D. Zhu, A. J. Heeger, S. R. Marder and X. Zhan, J. Am. Chem. Soc., 2016, 138, 4955–4961 CrossRef CAS PubMed.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- A. C. Stuart, J. R. Tumbleston, H. Zhou, W. Li, S. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- Z. Li, K. Jiang, G. Yang, J. Y. L. Lai, T. Ma, J. Zhao, W. Ma and H. Yan, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- J. Guo, J. M. Marin-Beloqui and T. M. Clarke, J. Phys Mater, 2021, 4, 044009 CrossRef CAS.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- M. T. Sajjad, A. Ruseckas, L. K. Jagadamma, Y. Zhang and I. D. W. Samuel, J. Mater. Chem. A, 2020, 8, 15687–15694 RSC.
- C. Im, S. Kang, J. Choi and J. An, Polymer, 2021, 13, 1770 CAS.
- Y. Firdaus, V. M. Le Corre, S. Karuthedath, W. Liu, A. Markina, W. Huang, S. Chattopadhyay, M. M. Nahid, M. I. Nugraha, Y. Lin, A. Seitkhan, A. Basu, W. Zhang, I. McCulloch, H. Ade, J. Labram, F. Laquai, D. Andrienko, L. J. A. Koster and T. D. Anthopoulos, Nat. Commun., 2020, 11, 5220 CrossRef CAS PubMed.
- A. Classen, C. L. Chochos, L. Lüer, V. G. Gregoriou, J. Wortmann, A. Osvet, K. Forberich, I. McCulloch, T. Heumüller and C. J. Brabec, Nat. Energy, 2020, 5, 711–719 CrossRef CAS.
- L. Zhu, M. Zhang, G. Zhou, T. Hao, J. Xu, J. Wang, C. Qiu, N. Prine, J. Ali, W. Feng, X. Gu, Z. Ma, Z. Tang, H. Zhu, L. Ying, Y. Zhang and F. Liu, Adv. Energy Mater., 2020, 10, 1–9 Search PubMed.
- Z. Chen, P. Cai, J. Chen, X. Liu, L. Zhang, L. Lan, J. Peng, Y. Ma and Y. Cao, Adv. Mater., 2014, 26, 2586–2591 CrossRef CAS PubMed.
- H. Zhang, H. Yao, J. Hou, J. Zhu, J. Zhang, W. Li, R. Yu, B. Gao, S. Zhang and J. Hou, Adv. Mater., 2018, 30, 1–7 CAS.
- R. Dhanker, C. L. Gray, S. Mukhopadhyay, S. Nunez, C. Y. Cheng, A. N. Sokolov and N. C. Giebink, Nat. Commun., 2017, 8, 2252 CrossRef PubMed.
- E. Amouyal, R. Bensasson and E. J. Land, Photochem. Photobiol., 1974, 20, 415–422 CrossRef CAS.
- S. Karuthedath, J. Gorenflot, Y. Firdaus, N. Chaturvedi, C. S. P. De Castro, G. T. Harrison, J. I. Khan, A. Markina, A. H. Balawi, T. A. Dela Peña, W. Liu, R. Z. Liang, A. Sharma, S. H. K. Paleti, W. Zhang, Y. Lin, E. Alarousu, D. H. Anjum, P. M. Beaujuge, S. De Wolf, I. McCulloch, T. D. Anthopoulos, D. Baran, D. Andrienko and F. Laquai, Nat. Mater., 2021, 20, 378–384 CrossRef CAS PubMed.
- S. I. Natsuda, Y. Sakamoto, T. Takeyama, R. Shirouchi, T. Saito, Y. Tamai and H. Ohkita, J. Phys. Chem. C, 2021, 125, 20806–20813 CrossRef CAS.
- R. Kerremans, C. Kaiser, W. Li, N. Zarrabi, P. Meredith and A. Armin, Adv. Opt. Mater, 2020, 8, 202000319 Search PubMed.
- P. Li, J. Fang, Y. Wang, S. Manzhos, L. Cai, Z. Song, Y. Li, T. Song, X. Wang, X. Guo, M. Zhang, D. Ma and B. Sun, Angew. Chem., Int. Ed., 2021, 60, 15054–15062 CrossRef CAS PubMed.
- J. Albero, Y. Zhou, M. Eck, F. Rauscher, P. Niyamakom, I. Dumsch, S. Allard, U. Scherf, M. Krüger and E. Palomares, Chem. Sci., 2011, 2, 2396–2401 RSC.
- J. Nelson, Phys. Rev. B - Condens. Matter Mater. Phys, 1999, 59, 15374–15380 CrossRef CAS.
- J. Guo, H. Ohkita, S. Yokoya and H. Benten, J. Am. Chem. Soc., 2010, 132, 9631–9637 CrossRef CAS.
- T. M. Clarke, F. C. Jamieson and J. R. Durrant, J. Phys. Chem. C, 2009, 113, 20934–20941 CrossRef CAS.
- G. Kupgan, X. K. Chen and J. L. Brédas, Mater. Today Adv., 2021, 11, 7 Search PubMed.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS PubMed.
- J. Kosco, S. Gonzalez-Carrero, C. T. Howells, T. Fei, Y. Dong, R. Sougrat, G. T. Harrison, Y. Firdaus, R. Sheelamanthula, B. Purushothaman, F. Moruzzi, W. Xu, L. Zhao, A. Basu, S. De Wolf, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Nat. Energy, 2022, 7, 340–351 CrossRef CAS.
- M. B. Price, P. A. Hume, A. Ilina, I. Wagner, R. Ronnie, K. E. Thorn, W. Jiao, A. Campbell, P. J. Conaghan, G. Lakhwani, N. J. L. K. Davis, Y. Wang, P. Xue, H. Lu, K. Chen, X. Zhan and J. M. Hodgkiss, Nat. Commun., 2022, 13, 2827 CrossRef CAS PubMed.
- M. S. Kotova, G. Londi, J. Junker, S. Dietz, A. Privitera, K. Tvingstedt, D. Beljonne, A. Sperlich and V. Dyakonov, Mater. Horiz., 2020, 7, 1641–1649 RSC.
- S. Marina, A. D. Scaccabarozzi, E. Gutierrez-Fernandez, E. Solano, A. Khirbat, L. Ciammaruchi, A. Iturrospe, A. Balzer, L. Yu, E. Gabirondo, X. Monnier, H. Sardon, T. D. Anthopoulos, M. Caironi, M. Campoy-Quiles, C. Müller, D. Cangialosi, N. Stingelin and J. Martin, Adv. Funct. Mater., 2021, 31, 3103784 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta05172a |
| This journal is © The Royal Society of Chemistry 2022 |