
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tailoring charge reconfiguration in dodecahedral Co2P@carbon nanohybrids by triple-doping engineering for promoted reversible oxygen catalysis†
Luhan
Li
a,
Lei
Zhang
 *a,
Zhicheng
Nie
a,
Wenyu
Ma
a,
Nianpeng
Li
ab,
Thomas
Wågberg
*c and
Guangzhi
Hu
*bc
*a,
Zhicheng
Nie
a,
Wenyu
Ma
a,
Nianpeng
Li
ab,
Thomas
Wågberg
*c and
Guangzhi
Hu
*bc
aSchool of Materials Science and Engineering, State Key Laboratory of Mining Response and Disaster Prevention and Control in Deep Coal Mines, Anhui University of Science and Technology, Huainan, Anhui 232001, PR China. E-mail: leizhang@aust.edu.cn
bInstitute for Ecological Research and Pollution Control of Plateau Lakes, School of Ecology and Environmental Science, Yunnan University, Kunming, Yunnan 650504, PR China. E-mail: guangzhihu@ynu.edu.cn
cDepartment of Physics, Umeå University, Umeå S-901 87, Sweden. E-mail: thomas.wagberg@umu.se
First published on 21st September 2022
Abstract
Simultaneously tuning the electronic structure of active sites and the microenvironment of the carbon matrix in metal phosphide/carbon nanohybrids is the most effective way to design and develop bi-functional electrocatalysts for electrochemically related energy storage devices. Inspired by this, a robust and advanced N/P co-doped carbon-based dodecahedron catalyst with confined Fe-doped Co2P particles was successfully prepared through a multi-doping engineering strategy. Phytic acid molecules, which were used in the synthesis of the catalyst, not only contribute to the formation of the porous structure, but also act as a phosphorus source to form the corresponding metal phosphide and the P dopant in the carbon matrix. Thanks to the unique composition and structure-dependent merits, the microenvironment of the electrocatalyst was significantly modulated, thus promoting the advantageous local charge rearrangement and smooth mass/charge transfer processes during the oxygen-related electrocatalytic reactions. As a result, the resultant catalyst exhibited significantly enhanced reversible oxygen activity, as evidenced by an ultra-small potential gap of 0.655 V (half-wave potential of 0.895 V for the oxygen reduction reaction; η10 of 320 mV for the oxygen evolution reaction), a remarkable specific capacity of 762 mA h gZn−1, and high voltaic efficiency, exceeding most previous reports. This study provides a new synthetic approach for fabricating highly efficient bi-functional oxygen catalysts and can be handily extended to the synthesis of other heterogeneous electrocatalysts for sustainable energy storage.
Introduction
Because of the ever-increasing environment-related concerns and explosive growth of worldwide energy demand, developing sustainable energy materials and green nanotechnology to construct fuel cells, metal–air batteries, and electrolytic water systems has attracted global attention.1,2 Particularly, the Zn–air battery (ZAB) has been demonstrated as a promising technology to meet the requirement of future energy conversion equipment owing to its widely recognized advantages of high theoretical energy density, low material cost, environmental friendliness, and reliable security.3–5 As the key reactions in rechargeable ZAB devices, the oxygen reduction reaction (ORR) during discharge and the oxygen evolution reaction (OER) during charge are pivotal for the whole electrocatalytic process.6 Nevertheless, the sluggish dynamics for the ORR and OER cause adverse polarization loss, so an efficient catalyst to speed up these reactions has been a central issue.7,8 Although noble-metal materials, such as Pt-based catalysts and RuO2, have long been regarded as benchmarks for catalyzing the ORR and OER, respectively, they suffer from the disadvantages of single catalytic activity, high cost, and poor cycle stability. Within this context, it is of practical value to design a highly efficient and durable bi-functional ORR–OER catalyst for ZABs.9–11Recently, carbon/transition-metal phosphide (TMP) nanohybrids with nanoconfinement structures have received extensive and continuous interest because of their unique features of spatial confinement and functional integration, thus making them attractive for potential utilization in energy-related electrocatalysis areas.12,13 Many early studies have demonstrated that constructing these N-doped carbon (NDC) frameworks with confined phosphide-based moieties represents a feasible strategy to fine-tune the electronic interactions between the embedded metal phosphide particles and their adjacent carbon-based substrates, thereby enhancing the intrinsic activities of the catalysts.14–16 Taking cobalt phosphide as an example, Liu and co-workers reported a highly efficient and durable tri-functional electrocatalyst comprised of Co2P nanoparticles and the carbon matrix.17 Theoretical simulations showed that Co2P exhibited a favorable density of states around the Fermi level.17 As a consequence, it was concluded that a possible synergetic catalysis effect and promoted electron-transfer at the interface between the Co2P component and carbon skeleton could greatly optimize the activity of this hybrid catalyst.
Despite these breakthroughs, the further improvement of the electrocatalytic ORR–OER efficiency of these TMP-based catalysts is tremendously impeded by their inherent physical and chemical characteristics, including adverse electroconductibility, unsatisfactory local electronic structure, and the consequent undesirable adsorption/desorption energy barriers of reaction intermediates.18 To conquer these bottlenecks, foreign metal elements can be incorporated into the microstructure of TMPs to effectively tailor the electronic structure of the host catalyst, thereby realizing the optimization of catalytic performance at the atomic scale.19,20 On the other hand, it is generally believed that the derived metal–N moiety that existed in the NDC skeleton serves as the true ORR active site due to the strong coordination ability of nitrogen.21–23 However, recent studies have demonstrated that the nitrogen atom around the metal center may lead to unsuitable free energy for the adsorption of intermediates owing to its excessive electronegativity.24 Predictably, this non-optimal adsorption behavior deteriorates the kinetic activity and thus hinders the activity improvement.25 Actually, the adsorption strength of the intermediate product at the catalytic center can be tuned by optimizing the microenvironment of the metal atom to reduce the potential barriers, thereby contributing to elevated catalytic efficiency.26 Because of its relatively weak electronegativity, incorporating phosphorus into the NDC framework seems to be a promising pathway to regulate the electronic structure of the metal–N moiety.27 Theoretically, the alien phosphorus atom can tailor and boost the kinetic activity of the active site by availably regulating the electron-withdrawing and donating properties.28 Expectedly, this unique compositional and structural design, featuring Co2P component intercalation into the N/P co-doped carbon matrix, can provide efficient electrocatalytic activity towards ORR and OER processes.
Encouraged by these features, we herein report the successful synthesis of a N/P co-doped carbon porous dodecahedron catalyst with confined Fe-doped Co2P nanoparticles via the sequential etching–coordination–phosphorization process using the Zn/Co-ZIF dodecahedron as the precursor, iron ion as the dopant, and phytic acid as the etching agent and phosphorus source. Benefitting from the synergistic effect between the Fe-doped Co2P component and porous conductive N/P co-doped carbon dodecahedron, the optimized Fe-Co2P@NPDC nanohybrid delivered excellent oxygen-related catalytic performances in an alkaline medium, with a notable half-wave potential of 0.895 V for ORR and a small OER overpotential of 320 mV for 10 mA cm−2 (here, N/P co-doped carbon is termed as NPDC). Beyond that, the rechargeable ZAB device assembled with the Fe-Co2P@NPDC catalyst exhibited high voltaic efficiency and remarkable long-term durability, exceeding those of the advanced Pt/C-RuO2 pair-based air-electrode. Therefore, our discovery affords a feasible strategy for the controllable synthesis and activity regulation of TMP-based bi-functional ORR–OER electrocatalysts for energy applications.
Experimental section
Preparation of Zn/Co-ZIF dodecahedrons
Typically, 0.2854 g of Zn(CH3COO)2·2H2O, 0.0324 g of Co(CH3COO)2·4H2O, and 1.12 g of 2-methylimidazole were added into 10 mL of deionized water. After 24 h of aging, the product was taken out and washed with deionized water, followed by drying at 55 °C for 12 h. For the preparation of Zn-ZIF dodecahedrons, the synthesis method was the same as above, except that the corresponding cobalt source was missing.Preparation of E-Zn/Co-ZIF dodecahedrons
0.15 g of Zn/Co-ZIF dodecahedron was dispersed into 25 mL of phytic acid solution (0.0006 M), followed by heating at 90 °C for 3 h. Finally, the product was washed with deionized water, and then dried at 55 °C for 12 h. For the preparation of E-Zn-ZIF porous dodecahedrons, the synthesis method was the same as above, except that an equal amount of Zn-ZIF was used instead of Zn/Co-ZIF.Preparation of E-Zn/Co-ZIF-Fe dodecahedrons
10 mg of Fe(NO3)3·9H2O was added into 20 mL of a methanol solution containing 0.087 g of E-Zn/Co-ZIF. After stirring for 24 h, the product was washed with deionized water, and then dried at 55 °C for 12 h. In order to regulate the amount of Fe-doping in the product, three other control samples were prepared using the same synthesis strategy, except that the amount of introduced Fe source was 0, 5, and 15 mg, respectively.Preparation of Fe-Co2P@NPDC dodecahedrons
0.08 g of E-Zn/Co-ZIF-Fe was treated at 600 °C for 1 h and 920 °C for 2 h with a heating rate of 3 °C min−1 under a N2 atmosphere (Anhui Kemi Machinery Technology Co., Ltd). The same pyrolysis process was used for the synthesis of the other samples, except that the precursor to be calcined was different.For other experimental information, such as the used reagents, characterization methods, and electrochemical testing, please see ESI.†
Results and discussion
Composition and morphology characterization of the Fe-Co2P@NPDC catalyst
The brief synthesis strategy of the Fe-Co2P@NPDC catalyst is schematically depicted in Fig. 1a. First, the Zn/Co-ZIF precursor was prepared by self-assembly of metal ions (Co2+ and Zn2+) and the 2-methylimidazole ligand through a mild liquid precipitation method.29 After phytic acid etching, many holes were created on the surface of the Zn/Co-ZIF dodecahedron. In addition, due to the porous feature of the precursor, some of the phytic acid molecules could be captured, which might act as a phosphorus source in the subsequent pyrolysis process.30 Third, the resulting porous Zn/Co-ZIF polyhedron was ultrasonically dispersed in an aqueous solution containing iron ions, followed by a high-temperature calcination treatment. During this process, the P–O chemical bond could be reduced at high temperatures to release phosphorus, thus producing the final product of the Fe-Co2P@NPDC catalyst through an in situ phosphorization and doping process.31 The morphology and composition of the Zn/Co-ZIF dodecahedron were investigated by X-ray diffraction (XRD), energy-dispersive X-ray spectroscopy (EDS), and scanning electron microscopy (SEM). As shown in Fig. S1a†, the XRD pattern of the produced precursor exhibits obvious signals, which is in line with the simulated pattern of pure ZIF-8, thus indicating that the Co-doping process hardly changes the crystal structure of the ZIF-8 material.32 The corresponding EDS spectrum confirms the successful doping of Co ions, as evidenced by the characteristic signals of four compositional elements of Zn, Co, C, and N (Fig. S1b†). The presence of oxygen can be attributed to carbon dioxide adsorbed on the product surface.33 The SEM images displayed in Fig. S1c and d† reveal that the as-prepared product is comprised of many uniform Zn/Co-ZIF dodecahedrons with a size distribution in the range of 400–500 nm. After being etched in phytic acid solution, the product still maintains its original phase and morphology, except that many nanosized holes are formed on the surface of the polyhedron (Fig. S2a–e).† Interestingly, the characteristic signal of the P element can be identified in the EDS spectrum, as demonstrated in Fig. S2f,† presumably due to the substitution of 2-methylimidazole by phytic acid molecules.31 The subsequent Fe-loading step produced a corresponding E-Zn/Co-ZIF-Fe sample. As revealed in Fig. S3,† it is concluded that the expected iron ions are uniformly distributed in the dodecahedron structure, thus creating convenient conditions for subsequent Fe-doping into the crystal structure of the Co2P matrix. | ||
| Fig. 1 (a) The formation procedure, (b, c) XRD patterns, and (d–i) XPS spectra of the Fe-Co2P@NPDC product. | ||
The subsequent calcination procedure converted E-Zn/Co-ZIF-Fe dodecahedrons into the desired Fe-Co2P@NPDC product. The XRD analysis in Fig. 1b shows pure Co2P diffraction peaks with an orthorhombic phase (ICDD 01-089-3030). No other irrelevant characteristic signals of iron- or cobalt-based materials are identified, implying the phase purity of the product. If examined carefully, the diffraction peaks of the Fe-Co2P@NPDC catalyst shift towards lower angles compared to the Co2P@NPDC sample and standard card (Fig. 1c), which may be attributed to the lattice expansion of the Co2P matrix caused by the doping of iron with a slightly larger radius.34–36 Fig. S4a† illustrates the presence of these five compositional elements (e.g., Co, Fe, P, C, and N), and the calculated molar ratio of Co/Fe in the as-developed Fe-Co2P@NPDC is determined to be approximately 4.4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Next, the Raman spectrum was recorded to obtain structural information about the carbon component in the product. As depicted in Fig. S4b,† two dominant signals at 1350 and 1600 cm−1 assignable to the representative D- and G-bands of carbon can be identified.37 The large Id/Ig value of 1.03 reveals abundant structural defects in the carbon matrix, which may be related to heteroatom doping, thus providing rich active sites for the oxygen-involved catalytic reactions through the fine-regulation of the local electronic and geometric structures in the derived carbon frame.38 Moreover, the remaining two small signals at 1190 and 1500 cm−1 correspond to the carbon atoms outside of a perfectly planar sp2 carbon network and heteroatoms in carbon layers or integrated five-number rings, respectively.39 It is noted that the decent Raman peak located at 1500 cm−1 in the Fe-Co2P@NPDC catalyst further confirms the expected hetero-element doping and the derived irregular carbon structure (Fig. S4b–g),† which may in turn significantly enhance the interaction between the oxygen species and the catalyst surface and thereby accelerate the electrocatalytic reaction kinetics, matching well with the previous findings.40
:1. Next, the Raman spectrum was recorded to obtain structural information about the carbon component in the product. As depicted in Fig. S4b,† two dominant signals at 1350 and 1600 cm−1 assignable to the representative D- and G-bands of carbon can be identified.37 The large Id/Ig value of 1.03 reveals abundant structural defects in the carbon matrix, which may be related to heteroatom doping, thus providing rich active sites for the oxygen-involved catalytic reactions through the fine-regulation of the local electronic and geometric structures in the derived carbon frame.38 Moreover, the remaining two small signals at 1190 and 1500 cm−1 correspond to the carbon atoms outside of a perfectly planar sp2 carbon network and heteroatoms in carbon layers or integrated five-number rings, respectively.39 It is noted that the decent Raman peak located at 1500 cm−1 in the Fe-Co2P@NPDC catalyst further confirms the expected hetero-element doping and the derived irregular carbon structure (Fig. S4b–g),† which may in turn significantly enhance the interaction between the oxygen species and the catalyst surface and thereby accelerate the electrocatalytic reaction kinetics, matching well with the previous findings.40
X-ray photoelectron spectroscopy (XPS) was conducted to probe the surface composition and electronic state of the Fe-Co2P@NPDC sample. The full survey displayed in Fig. 1d reveals the presence of carbon, nitrogen, oxygen, iron, cobalt, and phosphorus compositional elements, confirming the doping of iron species into the Fe-Co2P@NPDC system, compatible with the results of XRD and Raman analyses. Fig. 1e illustrates the narrow scan image of the Co 2p3/2 region, and four peaks located at 778.6, 780.0, 782.1 and 785.9 eV can be assigned to the Co–P bond, Co–N moiety, Co2+ oxidized species, and the associated shake-up satellite, respectively.41–43 The formation of the Co–N bond should be related to the nitrogen element with a strong coordination function in the carbon matrix, which can produce the desired Co–N active site with the adjacent metal center to catalyze the ORR process.44 In comparison with the binding energy of the zero-valence state of cobalt (778.1–778.2 eV) reported recently, the cobalt component in the Co–P bond exhibits a partial positive charge (Coδ+, 0 < δ < 2), presumably due to the electron transfer from the Co domain to P or N regions.45 With regard to the Fe 2p3/2 profile, four sub-bands at 707.7, 711.2, 714.3, and 720.2 eV match well with the Fe–N bond, Fe2+, Fe3+, and its satellite peak, respectively, as revealed in Fig. 1f.45–47 The appearance of relatively weak Fe–N signals suggests that nitrogen atoms in the carbon matrix can also form ORR-active metal–N active centers with trace iron, presumably consistent with the formation mechanism of Co–N bonds. No characteristic signal of iron phosphide is observed, thus further demonstrating the successful incorporation of iron ions in the Co2P core of Fe-Co2P@NPDC product, matching well with the previous report.41 The formation of Fe2+-related species can be attributable to the reduction of Fe3+ to Fe2+ during the thermal phosphorization process, consistent with the most recent results recorded in the literature.48 The high-resolution P 2p spectrum can be deconvoluted into three separated signals with binding energies at 129.5 eV for metal–P, 132.1 eV for P–C, and 133.4 eV for the P–O bond (Fig. 1g).49 The obvious metal–P peak signifies the formation of the Co2P phase and the presence of the P–C bond indicates the phosphorus-doped nature of the carbon framework.50 Particularly, the Co signal of Co–P (778.6 eV) in Fe-Co2P@NPDC is shifted slightly towards higher binding energy, compared with that in Co2P@NPDC (778.3 eV in Fig. S7†). As for phosphorus, the movement direction of the P-metal bond is opposite (129.5 eV of Fe-Co2P@NPDC vs. 129.8 eV of Co2P@NPDC). This phenomenon probably originates from the charge redistribution by the introduction of iron ions, which may play a pivotal role in tailoring the electronic environment of catalytic sites.51 Note that the existence of oxidized species in as-developed phosphide-based electrocatalysts is a common phenomenon, perhaps due to the inevitable oxidation of the surface caused by exposure to air,52 which can be further demonstrated by the oxygen signal in Fig. 1d. The high-resolution C 1s scan in Fig. 1h exhibits main peaks at 284.6, 285.4, 285.9, and 286.6 eV, assignable to C–C/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–P, C–N, and C–O bonds,53 respectively, verifying the structural features of N/P co-doped carbon materials. The N 1s spectrum (Fig. 1i) displays several peaks at 398.3 (pyridinic-N, 34.65%), 399.1 (metal–N, 20.13%), 400.4 (pyrrolic-N, 23.40%), 401.0 (graphitic-N, 11.84%) and 402.5 eV (oxidized-N, 9.98%).54 In addition to the high content of metal–N sites for possible ORR activity, it is documented that pyridinic- and graphitic-N species can effectively enhance the conductivity of the electrocatalyst and thus accelerate the involved electrocatalytic process.55 As a consequence, diversified N-doping collectively promotes the electrocatalytic efficiency of the Fe-Co2P@NPDC sample. Besides, the observed metal–N bond as discussed above indicates the positive interactions between the phosphide phase and the NPDC matrix, allowing improved charge-transfer to maintain efficient oxygen-related catalysis.42
C, C–P, C–N, and C–O bonds,53 respectively, verifying the structural features of N/P co-doped carbon materials. The N 1s spectrum (Fig. 1i) displays several peaks at 398.3 (pyridinic-N, 34.65%), 399.1 (metal–N, 20.13%), 400.4 (pyrrolic-N, 23.40%), 401.0 (graphitic-N, 11.84%) and 402.5 eV (oxidized-N, 9.98%).54 In addition to the high content of metal–N sites for possible ORR activity, it is documented that pyridinic- and graphitic-N species can effectively enhance the conductivity of the electrocatalyst and thus accelerate the involved electrocatalytic process.55 As a consequence, diversified N-doping collectively promotes the electrocatalytic efficiency of the Fe-Co2P@NPDC sample. Besides, the observed metal–N bond as discussed above indicates the positive interactions between the phosphide phase and the NPDC matrix, allowing improved charge-transfer to maintain efficient oxygen-related catalysis.42
Fig. 2a and b depict the SEM images of the well-managed Fe-Co2P@NPDC product, which clearly show that the morphology of this sample hardly changes after the thermal phosphorization, and still maintains a satisfactory rhombic dodecahedron geometry similar to that of its precursor. The size of this particle is slightly reduced to approximately 410 nm. Apart from that, careful observation reveals that this polyhedron exhibits a rough surface with many nanoporous structures. Both of these subtle structural evolutions may be ascribed to the phytic acid etching in the preparation steps and abundant gas products generated during the high-temperature treatment.56 Perceivably, this delicate nanoporous structure contributes to the transport kinetics of electrolytes and thus effectively accelerates the mass exchange during electrocatalytic ORR/OER processes.57 For a detailed examination, a transmission electron microscopy (TEM) image of the Fe-Co2P@NPDC demonstrates that this polyhedral nanoarchitecture exhibits loose and porous features with abundant particles embedded in the carbon matrix and some carbon nanotubes decorated on its surface (Fig. 2c). The typical structural characteristic of the nanoparticle embedded in a carbon nanotube indicates that the growth and graphitization process of a carbon nanotube may follow the metal-catalyzed mechanism.58 This in situ grown carbon nanotube can indeed accelerate the mass/charge transport, thus affording efficient ORR/OER performance for reversible ZAB devices.59 The high-resolution TEM image obtained from the nanoparticle domain exhibits a well-resolved fringe spacing of 0.209 nm, which can be assigned to the (211) plane of the orthorhombic Co2P component (Fig. 2d). The slightly increased interplanar spacing may be attributed to the lattice expansion caused by iron doping.35 Elemental mapping images reveal that the embedded nanoparticles are comprised of three constituent elements of Co, Fe, and P, thus demonstrating the Fe-Co2P component (Fig. 2e–h). The remaining C and N elements depict the homogeneous distribution throughout the whole rhombic dodecahedron, as evidenced in Fig. 2i and j. Moreover, the doped P element imparted by phytic acid in the carbon matrix also contributes to the phosphorus signal as desired.60 Based on these findings, it is noted that the Fe-Co2P@NPDC product with a well-maintained dodecahedron shape, porous structure, nano-sized Fe-Co2P active unit, and conductive NPDC frame can be easily fabricated via the sequential etching–coordination–phosphorization process.
 | ||
| Fig. 2 (a and b) SEM, (c) TEM, (d) HRTEM, (e–j) elemental mapping images of the well-regulated Fe-Co2P@NPDC electrocatalyst. | ||
ORR performance of the Fe-Co2P@NPDC catalyst
To disclose the essential factors of the elevated performance of the Fe-Co2P@NPDC catalysts, five other contrast samples were also prepared according to similar synthetic steps. In our case, the Fe-Co2P component was successfully intercalated on the carbon-based support. Therefore, the Zn-ZIF dodecahedron was selected as the precursor to synthesize the corresponding NDC support, which can prove the role of the carbon skeleton in the catalytic activity (Fig. S5†). With this in mind, Zn-ZIF polyhedra were then etched and doped with phytic acid to produce the NPDC matrix, thus elucidating the intrinsic correlation between its missing Fe-Co2P component and electrocatalytic efficiency (Fig S6†). The effect of Fe-doping on the electrochemical performance can be determined by synthesizing the corresponding undoped sample of Co2P@NPDC (Fig. S7†). Lastly, two samples with different Fe contents (Fe-Co2P@NPDC-1 and Fe-Co2P@NPDC-2) were also synthesized accordingly to elucidate the effect of different amounts of Fe-doping (Fig S8–S11†). Relevant experimental results are documented in the ESI.†The porous nanostructure, conductive carbon-based framework, and optimized Fe-Co2P catalytic centers make the as-designed Fe-Co2P@NPDC electrocatalyst a qualified substitute for commercial electrocatalysts. As a proof-of-concept, the ORR activity of the as-fabricated samples was first examined in oxygen-saturated 0.1 M KOH solution through cyclic voltammetry (CV) plots. As stated in Fig. 3a, Fe-Co2P@NPDC displays a positive ORR peak potential at 0.918 V and strong cathodic current, exceeding that of Fe-Co2P@NPDC-1 (0.893 V), Fe-Co2P@NPDC-2 (0.895 V), Co2P@NPDC (0.870 V), NPDC (0.844 V) and NDC (0.813 V), thus implying its optimal catalytic efficiency among all materials.61,62 Linear scan voltammetry (LSV) plots further verify the best ORR catalytic activity of Fe-Co2P@NPDC (Fig. 3b), exhibiting a notable onset potential (Eonset = 1.059 V), excellent half-wave potential (E1/2 = 0.895 V) and boosted diffusion limited current density (jL = 5.79 mA cm−2). These important index parameters have great advantages compared to those of Fe-Co2P@NPDC-1 (Eonset = 1.056 V; E1/2 = 0.889 V; jL = 4.30 mA cm−2), Fe-Co2P@NPDC-2 (Eonset = 1.058 V; E1/2 = 0.890 V; jL = 5.07 mA cm−2), Co2P@NPDC (Eonset = 1.019 V; E1/2 = 0.870 V; jL = 4.12 mA cm−2), NPDC (Eonset = 0.996 V; E1/2 = 0.817 V; jL = 4.03 mA cm−2), NDC (Eonset = 0.966 V; E1/2 = 0.803 V; jL = 3.53 mA cm−2), Pt/C electrocatalysts (Eonset = 1.025 V; jL = 5.30 mA cm−2, E1/2 = 0.835 V), and other documented catalysts (Table S1†). Compared with two carbon-based frameworks of NDC and NPDC materials, the promoted ORR activity of Fe-Co2P@NPDC suggests that the metal phosphide component acts as an active site during the oxygen reduction process.63 The worse catalytic efficiency of NDC than NPDC (E1/2: 0.817 vs. 0.803 V) confirms that the introduction of the P-dopant into the NDC matrix can optimize its electronic structure, thus having a certain positive effect on the improvement of ORR performance.64 As expected, the Co2P@NPDC delivers superior electrocatalytic activity to that of NPDC, benefiting from the construction of phosphide active groups in an optimized carbon-based skeleton. However, due to the lack of reasonable charge rearrangement induced by Fe-doping, its intrinsic activity is inferior to that of the doped samples. Compared with Fe-Co2P@NPDC-1 (Co/Fe = 5.2:1) and Fe-Co2P@NPDC-2 (Co/Fe = 3.7:1) samples, the first-rank ORR performance of Fe-Co2P@NPDC (Co/Fe = 4.4:1) demonstrates that rational iron doping can achieve an optimized distribution of local charges and in turn modulates the electronic interactions of the catalyst with key oxygen species (*OOH, *O, and *OH), thus ultimately accelerating the kinetic process of the catalytic reaction.65 Furthermore, the most advantageous M–N content in the Fe-Co2P@NPDC sample is also an important factor for its optimal ORR activity (Table S2†).5
 | ||
| Fig. 3 ORR performances: (a) CV curves (solid line: O2-saturated 0.1 M KOH solution; dotted line: N2-saturated 0.1 M KOH solution), (b) LSV (1600 rpm), and (c) Tafel plots; (d) H2O2 yield with the Fe-Co2P@NPDC catalyst (inset: electron transfer number of n). OER performances: (e) LSV, (f) Tafel, and (g) Nyquist plots; (h) long-term stability testing with the Fe-Co2P@NPDC catalyst at a current density of 10 mA cm−2. (i) ORR/OER LSV curves of as-developed catalysts and the coupling of Pt/C and RuO2. Notes: (red) Fe-Co2P@NPDC, (pink-purple) Fe-Co2P@NPDC-1, (light green) Fe-Co2P@NPDC-2, (sky blue) Co2P@NPDC, (dark purple) NPDC, (orange) NDC, (black) Pt/C, and (navy blue) RuO2. | ||
In general, the Tafel slope represents the charge-transfer ability of the electrocatalyst during the electrochemical processes.66 A smaller Tafel slope indicates a lower overpotential required to maintain a certain current density, thus leading to better electrocatalytic activity.67 As displayed in Fig. 3c, the as-obtained Fe-Co2P@NPDC yields the lowest Tafel slope of 91 mV dec−1 among all the catalysts and the commercial Pt/C sample, thus suggesting its most favorable reaction kinetics to activate an efficient ORR process. Based on the measured LSV plots with different rotation speeds (Fig. S12†), the electron transfer number (n) of the Fe-Co2P@NPDC product is calculated to be ∼4 by using eqn (1) (Koutecky–Levich equation) and eqn (2) (Fig. 3d and the ESI Section†), demonstrating a typical four-electron reaction mechanism towards the ORR.68,69 Expectedly, this champion catalyst also affords a decent value of jL and the current measured from the Pt ring corresponding to the contribution of hydrogen peroxide production is nearly ignorable according to eqn (3) (see the ESI Section†), thereby indicating that the major product in the ORR process is OH− species. The tolerance of the Fe-Co2P@NPDC catalyst to the poisoning effect of fuel molecules was also studied.70 When methanol was added to the electrolyte, the Fe-Co2P@NPDC electrode remained largely immune to the effect of methanol oxidation with only a weak current decay (Fig. S13a†). In contrast, the Pt/C catalyst exhibits high sensitivity to methanol, manifesting a dramatic current decrease. To satisfy the standards for industrial utilization, the stability of the Fe-Co2P@NPDC catalyst was measured by a chronoamperometric method. Impressively, this well-designed product can maintain its excellent ORR activity during 20 h of continuous operation with 5.3% decay of the original current density, much better than that of the Pt/C electrode (37.7% of decrease), as demonstrated in Fig. S13b.† Fig. S13c† illustrates the LSV plots of this sample before and after the long-term durability test.71 The weak difference between these two polarization curves further confirms the excellent stability of the Fe-Co2P@NPDC catalyst. After the long-term test, it can be easily observed that the microscopic morphology, valence state (Fe, Co and P) and chemical composition of the catalyst do not change significantly (Fig. S13d–g†), which may be attributable to the reductive environment in the ORR process and the robust protective carbon matrix.5,37 However, the Raman spectrum shows that the catalyst surface is partially oxidized after the ORR stability test, probably related to the strong alkaline corrosive electrolyte (Fig. S13h†).
OER performance of the Fe-Co2P@NPDC catalyst
The Fe-Co2P@NPDC catalyst also affords remarkable OER activity, thus providing an essential pairing reaction for the subsequent construction of a reversible ZAB device. The LSV plots in Fig. 3e suggest that the well-managed phosphide-based nanohybrid displays a decent overpotential of 320 mV at 10 mA cm−2, superior to that of Fe-Co2P@NPDC-1 (350 mV), Fe-Co2P@NPDC-2 (334 mV), Co2P@NPDC (419 mV), NPDC (negligible), NDC (negligible), and other reported catalysts (Table S1†). Although the overpotential of the Fe-Co2P@NPDC product at a current density of 10 mA cm−2 is slightly higher than that of RuO2, the resulting current density increases rapidly as the operating voltage continues to rise, thus quickly showing an obvious advantage over the comparative noble-metal catalyst. Unsurprisingly, two samples with the worst OER activity are NDC and NPDC, indicating that the active sites of the OER process are contributed by the metal phosphide moiety. Similar to the ORR situation, the electronic structure modulation of the Co2P component by Fe-doping and the optimal regulation of the microenvironment of the carbon matrix by the P doping synergistically enhance the OER activity of the Fe-Co2P@NPDC catalyst. As displayed in Fig. 3f, the corresponding Tafel slope demonstrates that the Fe-Co2P@NPDC sample exhibits the lowest Tafel slope (61 mV dec−1), suggesting its highest charge-transfer rate during the OER process.72–74 Moreover, electrochemical impedance spectra depicted in Fig. 3g further reveal that the Fe-Co2P@NPDC sample delivers the smallest charge-transfer resistance than those of Fe-Co2P@NPDC-2, Fe-Co2P@NPDC-1, Co2P@NPDC, NPDC, and NDC, which demonstrates that the porous carbon nanocage with the embedded Fe-Co2P component is beneficial for charge-transfer during the OER process.73The turnover frequency (TOF) test is an important descriptor for measuring the intrinsic activity of catalysts.75 The relevant measurement methods are described in ESI.† Obviously, this Fe-Co2P@NPDC product yields the topmost TOF value (47.58 s−1) with an operating voltage of 1.68 V (Fig. S14†), surpassing those of Fe-Co2P@NPDC-1 (8.73 s−1), Fe-Co2P@NPDC-2 (22.28 s−1), Co2P@NPDC (1.01 s−1), NPDC (0.08 s−1), and NDC (0.005 s−1), coinciding with the trend of their electrocatalytic activity. Besides, the double layer capacitance (Cdl), which is proportional to the electrochemical active surface area (ECSA), is calculated by measuring the cyclic voltammetry (CV) plots at different scan speeds in the non-faradaic potential window.76 Compared with five reference samples, the resulting Fe-Co2P@NPDC delivers the highest Cdl value. Therefore, the above findings both prove that this champion catalyst possesses abundant active sites with high-quality to catalyze the oxygen-related catalytic reactions (Fig. S15†). Interestingly, Fe-Co2P@NPDC exhibits a larger specific surface area (637.47 m2 g−1) than Fe-Co2P@NPDC-2 (587.56 m2 g−1), Fe-Co2P@NPDC-1 (566.38 m2 g−1), and Co2P@NPDC (595.26 m2 g−1), but it is much lower than that of NPDC (863.86 m2 g−1) and NDC (927.36 m2 g−1), as demonstrated in Fig. S16.† Therefore, for the four metal-based catalysts, the enhanced specific surface area contributes to efficient electrocatalytic reactions. But for the two metal-free samples (NPDC and NDC), the lack of the necessary high-quality catalytic centers makes them inert to the ORR and OER processes. As displayed in Fig. S17,† the theoretical and experimental amounts of oxygen produced are in good agreement with each other, implying that no side reaction occurs during the OER process. Chronoamperometry was conducted to investigate the durability of the catalyst.77Fig. 3h shows that 95.9% of the initial applied voltage at a current density of 10 mA cm−2 is retained after 24 h in the presence of the Fe-Co2P@NPDC catalyst. The SEM image of this sample after the OER stability test demonstrates that its morphology changes significantly from the original dodecahedron to the final irregular particle (Fig. S18a†). More importantly, the surface of the catalyst is severely oxidized, as demonstrated in Fig. S18b–e.† Specifically, the content of oxides or hydroxides on the catalyst surface is greatly increased after the OER reaction, while the species corresponding to metal phosphides decrease significantly, which may be attributed to the characteristics of the strong alkaline medium and strong oxidizing environment during the OER process.33 Lastly, to visually assess the reversible oxygen catalytic activity of the catalysts, the potential gap ΔE (between Ej=10 and E1/2) of the OER and ORR over all the samples was measured. As revealed in Fig. 3i, the as-obtained Fe-Co2P@NPDC unsurprisingly exhibits the smallest ΔE of 0.655 V, especially far superior to those of the mixed noble-metal catalysts (RuO2 and Pt/C) and other bi-functional electrocatalysts documented in the literature (Table S1†), obviously benefiting from its best ORR and OER activities.
ZAB performance of the Fe-Co2P@NPDC catalyst
To explore the practical utilization of the as-designed Fe-Co2P@NPDC catalyst, a homemade primary ZAB device was first assembled based on the model depicted in Fig. 4a and S19,† affording an elevated peak power density of 340 mW cm−2 at 390 mA cm−2, outperforming that of the Pt/C benchmark (165 mW cm−2 at 310 mA cm−2) using the equal cell configuration (Fig. 4b) and other documented catalysts (Table S3†). Beyond that, the calculated specific capacity of this Fe-Co2P@NPDC-based battery reaches 762 mA h gZn−1, as evidenced in Fig. 4c. In contrast, the performance of the Pt/C-based cell is much inferior, with a moderate specific capacity of 651 mA h gZn−1. Galvanostatic discharge measurements at 10 mA cm−2 show that the Fe-Co2P@NPDC electrode can afford a decent initial voltage of 1.27 V (Fig. 4d). After continuous discharge for 83 h, the terminal discharge voltage can be maintained at 1.13 V. As for the Pt/C-based battery, both discharge voltage (Einitial = 1.04 V) and lifetime (55 h) are far inferior to those of our designed electrocatalyst. | ||
| Fig. 4 (a) Schematic illustration, (b) polarization and power density, (c) specific capacity, and (d) discharge curves of the primary ZAB device. Notes: Fe-Co2P@NPDC (red) and Pt/C (black). (e) Schematic illustration, (f) discharge/charge polarization, (g) discharge/charge cycling curves, (h) the open-circuit voltage of the rechargeable ZAB device, (i) the LED viewing screen (1 V) and (j) the LED light (1.5 V) powered by the rechargeable ZAB device. Notes: Fe-Co2P@NPDC (red) and Pt/C + RuO2 (black). | ||
Based on the outstanding bi-functional oxygen activity of the Fe-Co2P@NPDC product, the proof-of-concept application was conducted to confirm its feasibility in rechargeable ZABs. Note that the rechargeable battery was fabricated similarly to the primary counterpart, except that the electrolyte was replaced by 6.0 M KOH and 0.2 M Zn(Ac)2 solutions (Fig. 4e). As stated in Fig. 4f, the Fe-Co2P@NPDC electrode exhibits more satisfactory galvanodynamic charge–discharge performance than the Pt/C/RuO2 reference. In particular, this advantage is more pronounced at higher current densities. At 1 mA cm−2, this metal phosphide-based electrode enables the assembled ZAB device to continuously charge and discharge for 83 hours with a small voltage difference (Fig. 4g), thus demonstrating its promoted long-term durability compared to the benchmark catalyst of Pt/C-RuO2 and most reported bi-functional catalysts (Table S4†). Moreover, Fig. 4h reveals that the ZAB using the Fe-Co2P@NPDC catalyst as the air electrode exhibits a competitive open-circuit potential of 1.438 V. As an impressive exemplification, this battery can power the LED viewing screen and an LED (Fig. 4i and j). Thus, the above results indicate that the bi-functional Fe-Co2P@NPDC catalyst can endow the assembled battery with remarkable cell efficiency and long cycling life, suggesting its wide adaptability and feasibility in future ZAB applications.
Mechanism analysis of the elevated bi-functional oxygen activity
According to previous findings, *OH desorption at cobalt sites in the Co2P microstructure is the rate-determining step (RDS) of the ORR process.78 Recent studies have shown that the electronic structure of Co2P can be optimized by doping with transition metals, which in turn modulates the adsorption/desorption energy barriers between the active sites and reaction intermediates, and ultimately promotes its ORR performance.79 With this in mind, to confirm the expected electronic-structure regulation of the catalysts, ultraviolet photoemission spectroscopy was performed to measure their electron-donating abilities.80,81 As depicted in Fig. S20a–d,† the calculated working function (φ) of Fe-Co2P@NPDC is found to be 4.26 eV, smaller than those of Co2P@NPDC (4.79 eV), NPDC (6.62 eV) and NDC (6.70 eV). It is noted that P-doping enables the φ of the carbon matrix to be reduced, thus producing a lower φ value of NPDC than that of the NDC counterpart (6.62 vs. 6.70 eV). By comparing the two samples, Fe-Co2P@NPDC and Co2P@NPDC, it can also be found that iron doping has a similar function. Therefore, benefitting from the synergistic effect of multi-doping, this Fe-Co2P@NPDC catalyst is endowed with the smallest φ value and highest Fermi level, thus suggesting that it possesses the most suitable d-band center. The reduction of the φ value means that the binding effect of the catalyst on electrons becomes weaker.82 As a result, more electrons will escape from the surface of the electrocatalyst to participate in the related electrocatalytic reactions. Not surprisingly, the valence band maximum value of Fe-Co2P@NPDC blue-shifts to the vacuum level, in comparison to the other three reference samples (Fig. S20e–h).† It is generally believed that the valence electron near the Fermi level contributes to the d-band state.83 Therefore, the modulation of work function and the expected shift of this valence band demonstrate the effective regulation of the d-band center and easier electron transfer, thus supporting this Fe-Co2P@NPDC catalyst with increased concentrations of active sites through regulating the coordination valence states and chemical environment of the electrocatalyst, as well as a suitable binding force between the catalytic centers and the adsorbed intermediates.84,85 All of these help the Fe-Co2P@NPDC catalyst product to overcome the sluggish *OH desorption step and enhance its ORR activity, which is consistent with previous research reports (Fig. 5).79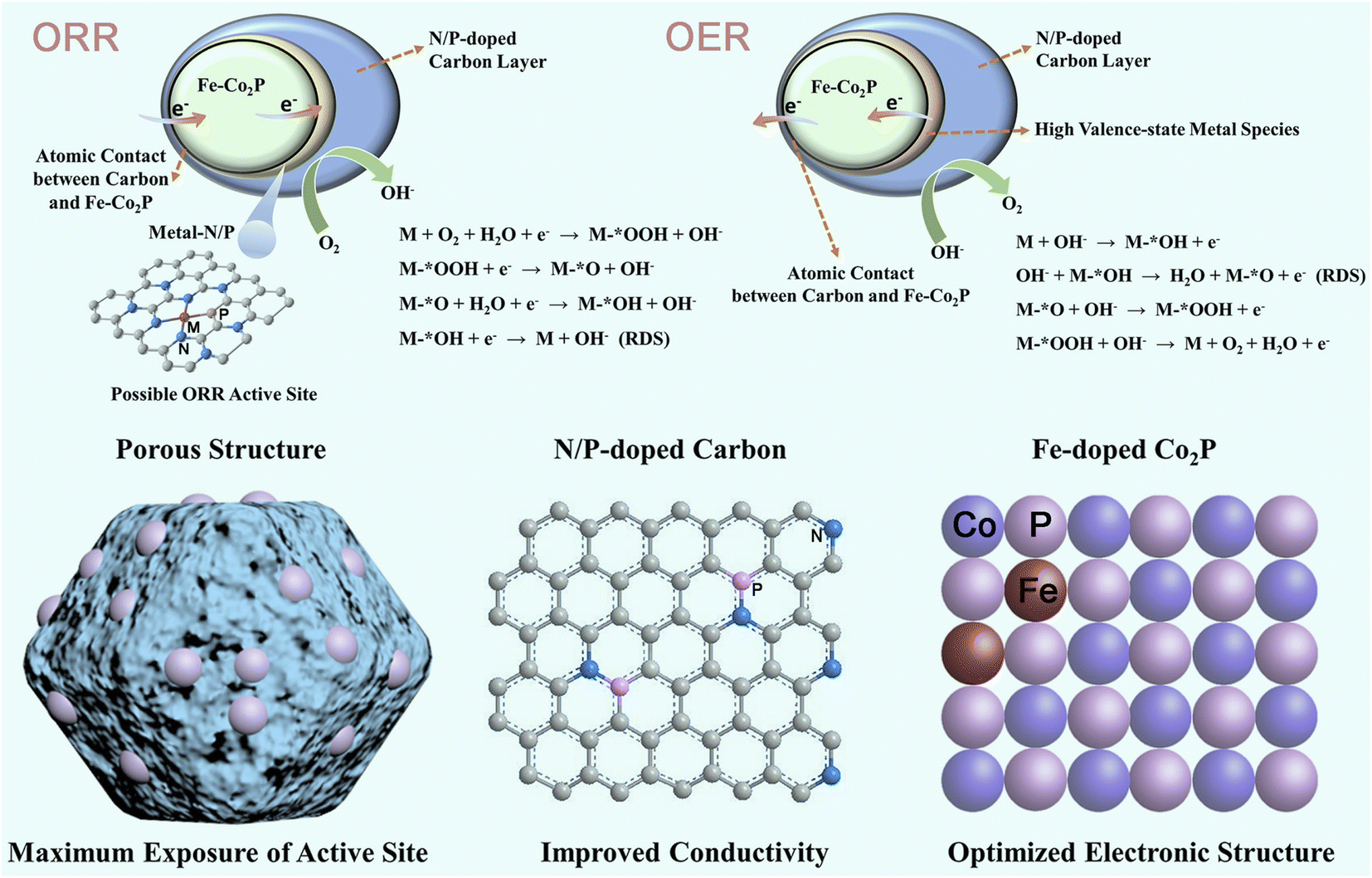 | ||
| Fig. 5 Schematic diagram of the possible activity-enhancing mechanism. | ||
As discussed above (Fig. 1 and S7†), the expected partial charge-transfer from the cobalt moiety to the P domain is facilitated when a secondary iron species is introduced into the Co2P phase, thus giving rise to the formation of high-valence-state metal species with stronger oxidizing power. A previous report has demonstrated that the OER rate-limiting step for many mixed metal phosphide precatalysts including Fe–Co–P may be a chemical step after the first electron transfer step, described as follows (Krasil'shchikov's Path 2): M–*OH + OH− → M–*O + H2O + e−.86 In our case, the formation of M–*OH and the chemical adsorption process of OH− groups may be promoted in the presence of high-valence-state cobalt species because of the optimized local electrostatic interaction between cobalt ions and OH− species, thereby contributing to fast OER kinetics and excellent catalytic efficiency (Fig. 5).87
In addition, because phosphorus (P) has a weaker electronegativity than N, incorporating the secondary heteroatom P would lead to the formation of an unsymmetrical N/P coordination structure, which could undergo a symmetry-breaking charge transfer process and trigger the modulation of the electronic structure of active metal sites.88 Due to its highest P–C content (Table S5†), the optimized electronic structure of the Fe-Co2P@NPDC catalyst enables the metal–N/P active sites to exhibit suitable adsorption/desorption energy barriers for various reaction intermediates (*OOH, *O, and *OH), thus resulting in the promoted electrocatalytic ORR/OER activities (Fig. 5).39 Lastly, the porous structure and highly graphitized carbon matrix can not only accelerate the mass and charge transfer during the electrocatalytic reactions, but also provide effective protection for the embedded phosphide active sites, giving satisfactory activity necessary for the remarkable performance of the ZAB device (Fig. 5).89 This activity enhancement results from the synergistic effect of microenvironment optimization brought about by the multi-doping engineering of Co2P components and the carbon framework, thus collectively contributing to the optimized elementary reactions in the involved ORR/OER process and finally exhibiting significantly enhanced reversible oxygen catalytic activity.
Conclusions
In this study, we explored a delicate rhombic dodecahedron nanoreactor featuring the intercalation of Fe-Co2P active sites into the N/P co-doped carbon framework through the sequential etching–coordination–phosphorization process. Benefiting from the efficient electronic structure regulation by multi-doping engineering and the fast charge/species transport endowed by the porous conductive carbon support, the expected Fe-Co2P@NPDC sample enabled remarkable ORR and OER bi-functional electrochemical activities and stabilities, with a promoted half-wave potential of 0.895 V for the ORR and a low overpotential of 320 mV at 10 mA cm−2 for the OER both in alkaline media. As a proof of concept, a rechargeable Fe-Co2P@NPDC-based ZAB device delivered an outstanding peak power density of 340 mW cm−2, a notable specific capacity of 762 mA h gZn−1, and a good round-trip efficiency, thus making this nanohybrid useful for utilization to replace commercial precious metal catalysts. This work provides a model for efficient and stable electrocatalyst design in sustainable energy storage and conversion devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Anhui Provincial Natural Science Foundation (2208085Y06), the National Natural Science Foundation of China (No. 21975001), the University Synergy Innovation Program of Anhui Province (GXXT-2021-012), the Support Program of Excellent Young Talents in Anhui Provincial Colleges and Universities (gxyqZD2022034), the National Natural Science Foundation of China-Yunnan Joint Fund (U2002213), and the Double Tops Joint Fund of the Yunnan Science and Technology Bureau and Yunnan University (2019FY003025). Thomas Wågberg acknowledges the support from Vetenskapsrådet (2017-04862, and 2021-04629), Energimyndigheten (45419-1), and SSF-Agenda 2030-PUSH.References
- T. T. Nguyen, J. Balamurugan, D. H. Kim, N. H. Kim and J. H. Lee, Small, 2020, 16, 2004661 CrossRef CAS.
- N. K. Wagh, D. H. Kim, S. H. Kim, S. S. Shinde and J. H. Lee, ACS Nano, 2021, 15, 14683–14696 CrossRef CAS PubMed.
- H. L. Guo, Q. C. Feng, J. X. Zhu, J. S. Xu, Q. Q. Li, S. L. Liu, K. W. Xu, C. Zhang and T. X. Liu, J. Mater. Chem. A, 2019, 7, 3664–3672 RSC.
- D. C. Nguyen, D. T. Tran, T. L. L. Doan, D. H. Kim, N. H. Kim and J. H. Lee, Adv. Energy Mater., 2020, 10, 1903289 CrossRef CAS.
- L. Zhang, Y. X. Zhu, Z. C. Nie, Z. Y. Li, Y. Ye, L. L. Li, J. Hong, Z. H. Bi, Y. T. Zhou and G. Z. Hu, ACS Nano, 2021, 15, 13399–13414 CrossRef CAS PubMed.
- A. I. Douka, Y. Y. Xu, H. Yang, S. Zaman, Y. Yan, H. F. Liu, M. A. Salam and B. Y. Xia, Adv. Mater., 2020, 32, 2002170 CrossRef CAS PubMed.
- M. A. Ahsan, A. R. Puente Santiago, Y. Hong, N. Zhang, M. Cano, E. Rodriguez-Castellon, L. Echegoyen, S. T. Sreenivasan and J. C. Noveron, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- S. L. Liu, R. D. Wan, Z. S. Lin, Z. Liu, Y. G. Liu, Y. Tian, D. D. Qin and Z. H. Tang, J. Mater. Chem. A, 2022, 10, 5244–5254 RSC.
- T. L. L. Doan, D. T. Tran, D. C. Nguyen, D. H. Kim, N. H. Kim and J. H. Lee, Adv. Funct. Mater., 2021, 31, 2007822 CrossRef CAS.
- Y. L. Zhang, Y. K. Dai, B. Liu, X. F. Gong, L. Zhao, F. Cheng, J. J. Cai, Q. Y. Zhou, B. Liu and Z. B. Wang, J. Mater. Chem. A, 2022, 10, 3112–3121 RSC.
- H. Begum, M. S. Ahmed and S. Jung, J. Mater. Chem. A, 2021, 9, 9644–9654 RSC.
- G. Zhang, G. C. Wang, Y. Liu, H. J. Liu, J. H. Qu and J. H. Li, J. Am. Chem. Soc., 2016, 138, 14686–14693 CrossRef CAS PubMed.
- S. Huang, E. Huixiang, Y. Yang, Y. F. Zhang, M. Ye and C. C. Li, J. Mater. Chem. A, 2021, 9, 7458–7480 RSC.
- Z. M. Zheng, H. H. Wu, H. D. Liu, Q. B. Zhang, X. He, S. C. Yu, V. Petrova, J. Feng, R. Kostecki, P. Liu, D. L. Peng, M. L. Liu and M. S. Wang, ACS Nano, 2020, 14, 9545–9561 CrossRef CAS PubMed.
- J. Q. Chi, X. Y. Zhang, X. Ma, B. Dong, J. Q. Zhang, B. Y. Guo, M. Yang, L. Wang, Y. M. Chai and C. Liu, ACS Sustainable Chem. Eng., 2019, 7, 17714–17722 CrossRef CAS.
- N. N. Wang, Z. C. Bai, Z. W. Fang, X. Zhang, X. Xu, Y. Du, L. F. Liu, S. X. Dou and G. H. Yu, ACS Mater. Lett., 2019, 1, 265–271 CrossRef CAS.
- H. T. Liu, J. Y. Guan, S. X. Yang, Y. H. Yu, R. Shao, Z. P. Zhang, M. L. Dou, F. Wang and Q. Xu, Adv. Mater., 2020, 32, 2003649 CrossRef CAS.
- Y. Li, Z. H. Dong and L. F. Jiao, Adv. Energy Mater., 2019, 10, 1902104 CrossRef.
- X. Xiao, L. M. Tao, M. Li, X. W. Lv, D. K. Huang, X. X. Jiang, H. P. Pan, M. K. Wang and Y. Shen, Chem. Sci., 2018, 9, 1970–1975 RSC.
- E. L. Hu, J. Q. Ning, D. Z. Zhao, C. Y. Xu, Y. Y. Lin, Y. J. Zhong, Z. Y. Zhang, Y. J. Wang and Y. Hu, Small, 2018, 14, 1704233 CrossRef.
- N. K. Wagh, S. S. Shinde, C. H. Lee, J. Y. Jung, D. H. Kim, S. H. Kim, C. Lin, S. U. Lee and J. H. Lee, Appl. Catal., B, 2020, 268, 118746 CrossRef CAS.
- A. A. Eissa, S. G. Peera, N. H. Kim and J. H. Lee, J. Mater. Chem. A, 2019, 7, 16920–16936 RSC.
- G. L. Li, Z. F. Lu, X. Wang, S. Cao and C. Hao, ACS Sustainable Chem. Eng., 2022, 10, 224–233 CrossRef CAS.
- G. J. Zhang, F. Y. Tang, X. Wang, L. Q. Wang and Y. N. Liu, ACS Catal., 2022, 12, 5786–5794 CrossRef CAS.
- G. Y. Zhou, Y. R. Ma, X. M. Wu, Y. Z. Lin, H. Pang, M. Y. Zhang, L. Xu, Z. Q. Tian and Y. W. Tang, Chem. Eng. J., 2020, 402, 126302 CrossRef CAS.
- H. S. Shang, X. Y. Zhou, J. C. Dong, A. Li, X. Zhao, Q. H. Liu, Y. Lin, J. J. Pei, Z. Li, Z. L. Jiang, D. N. Zhou, L. R. Zheng, Y. Wang, J. Zhou, Z. K. Yang, R. Cao, R. Sarangi, T. T. Sun, X. Yang, X. S. Zheng, W. S. Yan, Z. B. Zhuang, J. Li, W. X. Chen, D. S. Wang, J. T. Zhang and Y. D. Li, Nat. Commun., 2020, 11, 3049 CrossRef CAS PubMed.
- W. D. Xue, Q. X. Zhou, X. Cui, S. R. Jia, J. W. Zhang and Z. Q. Lin, Nano Energy, 2021, 86, 106073 CrossRef CAS.
- C. Chen, Y. Huang, Z. Y. Meng, M. W. Lu, Z. P. Xu, P. B. Liu and T. H. Li, J. Mater. Sci. Technol., 2021, 76, 11–19 CrossRef CAS.
- X. F. Lu, L. Yu, J. T. Zhang and X. W. Lou, Adv. Mater., 2019, 31, 1900699 CrossRef.
- X. F. Lu, L. Yu and X. W. Lou, Sci. Adv., 2019, 5, eaav6009 CrossRef CAS.
- X. X. Wang, Z. L. Na, D. M. Yin, C. L. Wang, Y. M. Wu, G. Huang and L. M. Wang, ACS Nano, 2018, 12, 12238–12246 CrossRef CAS PubMed.
- J. Cheng, N. Liu, L. Q. Hu, Y. N. Li, Y. L. Wang and J. H. Zhou, Chem. Eng. J., 2019, 364, 530–540 CrossRef CAS.
- Y. X. Zhu, L. Zhang, X. Zhang, Z. Y. Li, M. Zha, M. Li and G. Z. Hu, Chem. Eng. J., 2021, 405, 127002 CrossRef CAS.
- Y. M. Kang, Y. I. Kim, M. W. Oh, R. Z. Yin, Y. Lee, D. W. Han, H. S. Kwon, J. H. Kim and G. Ramanath, Energy Environ. Sci., 2011, 4, 4978–4983 RSC.
- X. L. Jiang, X. Q. Yue, Y. X. Li, X. J. Wei, Q. J. Zheng, F. Y. Xie, D. M. Lin and G. X. Qu, Chem. Eng. J., 2021, 426, 130718 CrossRef CAS.
- Y. Qi, Q. X. Zhang, S. X. Meng, D. Li, W. X. Wei, D. L. Jiang and M. Chen, Electrochim. Acta, 2020, 334, 135633 CrossRef CAS.
- X. Zhang, L. Zhang, Y. X. Zhu, Z. Y. Li, Y. Wang, T. Wagberg and G. Z. Hu, ChemSusChem, 2021, 14, 467–478 CrossRef CAS PubMed.
- X. Hu, Y. Min, L. L. Ma, J. Y. Lu, H. C. Li, W. J. Liu, J. J. Chen and H. Q. Yu, Appl. Catal., B, 2020, 268, 118405 CrossRef CAS.
- S. Mukherjee, D. Cullen, S. Karakalos, K. X. Liu, H. Zhang, S. Zhao, H. Xu, K. More, G. F. Wang and G. Wu, Nano Energy, 2018, 48, 217–226 CrossRef CAS.
- X. J. Zheng, J. Wu, X. C. Cao, J. Abbott, C. Jin, H. B. Wang, P. Strasser, R. Z. Yang, X. Chen and G. Wu, Appl. Catal., B, 2019, 241, 442–451 CrossRef CAS.
- C. Tang, R. Zhang, W. B. Lu, L. B. He, X. Jiang, A. M. Asiri and X. P. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef.
- J. Yang, X. Wang, B. Li, L. Ma, L. Shi, Y. X. Xiong and H. X. Xu, Adv. Funct. Mater., 2017, 27, 1606497 CrossRef.
- Y. Pan, K. A. Sun, Y. Lin, X. Cao, Y. C. Cheng, S. J. Liu, L. Y. Zeng, W. C. Cheong, D. Zhao, K. L. Wu, Z. Liu, Y. Q. Liu, D. S. Wang, Q. Peng, C. Chen and Y. D. Li, Nano Energy, 2019, 56, 411–419 CrossRef CAS.
- Y. Y. Guo, P. F. Yuan, J. N. Zhang, H. C. Xia, F. Y. Cheng, M. F. Zhou, J. Li, Y. Y. Qiao, S. C. Mu and Q. Xu, Adv. Funct. Mater., 2018, 28, 1805641 CrossRef.
- Y. Y. Wang, W. P. Kang, X. J. Pu, Y. C. Liang, B. Xu, X. Q. Lu, D. F. Sun and Y. L. Cao, Nano Energy, 2022, 93, 106897 CrossRef CAS.
- L. S. Ni, G. Chen, X. H. Liu, J. Han, X. Xiao, N. Zhang, S. Q. Liang, G. Z. Qiu and R. Z. Ma, ACS Appl. Energy Mater., 2019, 2, 406–412 CrossRef CAS.
- M. J. Wu, G. X. Zhang, J. L. Qiao, N. Chen, W. F. Chen and S. H. Sun, Nano Energy, 2019, 61, 86–95 CrossRef CAS.
- E. Vijayakumar, S. Ramakrishnan, C. Sathiskumar, D. J. Yoo, J. Balamurugan, H. S. Noh, D. Kwon, Y. H. Kim and H. Lee, Chem. Eng. J., 2022, 428, 131115 CrossRef CAS.
- Q. Shi, Q. Liu, Y. Ma, Z. Fang, Z. Liang, G. Shao, B. Tang, W. Y. Yang, L. Qin and X. S. Fang, Adv. Energy Mater., 2020, 10, 1903854 CrossRef CAS.
- J. C. Gao, J. M. Wang, L. J. Zhou, X. Y. Cai, D. Zhan, M. Z. Hou and L. F. Lai, ACS Appl. Mater. Interfaces, 2019, 11, 10364–10372 CrossRef CAS.
- X. W. Lv, W. S. Xu, W. W. Tian, H. Y. Wang and Z. Y. Yuan, Small, 2021, 17, 2101856 CrossRef CAS.
- C. C. Hou, L. L. Zou, Y. Wang and Q. Xu, Angew. Chem., Int. Ed., 2020, 59, 21360–21366 CrossRef CAS PubMed.
- G. Q. Zhao, T. Wang, L. L. Li, Y. Tang, Q. Qin and C. Wu, Carbon, 2021, 183, 291–300 CrossRef CAS.
- K. Yuan, D. Lutzenkirchen-Hecht, L. B. Li, L. Shuai, Y. Z. Li, R. Cao, M. Qiu, X. D. Zhuang, M. K. H. Leung, Y. W. Chen and U. Scherf, J. Am. Chem. Soc., 2020, 142, 2404–2412 CrossRef CAS PubMed.
- T. T. Gebremariam, F. Y. Chen, Y. C. Jin, Q. Wang, J. L. Wang and J. P. Wang, Catal. Sci. Technol., 2019, 9, 2532–2542 RSC.
- B. B. Pan, X. R. Zhu, Y. L. Wu, T. C. Liu, X. X. Bi, K. Feng, N. Han, J. Zhong, J. Lu, Y. F. Li and Y. G. Li, Adv. Sci., 2020, 7, 2001002 CrossRef CAS PubMed.
- H. F. Wang, X. X. Wang, M. L. Li, L. J. Zheng, D. H. Guan, X. L. Huang, J. J. Xu and J. H. Yu, Adv. Mater., 2020, 32, 2002559 CrossRef CAS PubMed.
- H. Y. Gong, X. J. Zheng, K. Zeng, B. Yang, X. Liang, L. Li, Y. Y. Tao and R. Z. Yang, Carbon, 2021, 174, 475–483 CrossRef CAS.
- C. L. Lai, J. Y. Fang, X. P. Liu, M. X. Gong, T. H. Zhao, T. Shen, K. L. Wang, K. Jiang and D. L. Wang, Appl. Catal., B, 2021, 285, 119856 CrossRef CAS.
- H. C. Yang, X. J. Guo, R. R. Chen, Q. Liu, J. Y. Liu, J. Yu, C. G. Lin, J. Wang and M. L. Zhang, Appl. Surf. Sci., 2020, 525, 146611 CrossRef CAS.
- D. C. Nguyen, T. L. Luyen Doan, S. Prabhakaran, D. T. Tran, D. H. Kim, J. H. Lee and N. H. Kim, Nano Energy, 2021, 82, 105750 CrossRef CAS.
- P. B. Li, X. Q. Qi, L. Zhao, J. J. Wang, M. Wang, M. H. Shao, J. S. Chen, R. Wu and Z. D. Wei, J. Mater. Chem. A, 2022, 10, 5925–5929 RSC.
- S. J. Li, R. H. Wang, X. Yang, J. Wu, H. Y. Meng, H. L. Xu and Z. Y. Ren, ACS Sustainable Chem. Eng., 2019, 7, 11872–11884 CrossRef CAS.
- H. Zhang, T. T. Wang, A. Sumboja, W. J. Zang, J. P. Xie, D. Gao, S. J. Pennycook, Z. L. Liu, C. Guan and J. Wang, Adv. Funct. Mater., 2018, 28, 1804846 CrossRef.
- Y. Lin, K. A. Sun, X. M. Chen, C. Chen, Y. Pan, X. Y. Li and J. Zhang, J. Energy Chem., 2021, 55, 92–101 CrossRef.
- H. Zhao, R. J. Jiang, Y. Zhang, B. B. Xie, J. L. Fu, X. N. Yuan, W. X. Yang, Y. Wu and R. J. Zhang, J. Mater. Chem. A, 2021, 9, 5848–5856 RSC.
- J. J. Shi, X. X. Shu, C. C. Xiang, H. Li, Y. Li, W. Du, P. F. An, H. Tian, J. T. Zhang and H. B. Xia, J. Mater. Chem. A, 2021, 9, 6861–6871 RSC.
- O. Y. Bisen, S. Atif, A. Mallya and K. K. Nanda, ACS Appl. Mater. Interfaces, 2022, 14, 5134–5148 CrossRef CAS.
- H. Y. Zou, G. Li, L. L. Duan, Z. K. Kou and J. Wang, Appl. Catal., B, 2019, 259, 118100 CrossRef CAS.
- Z. Ahmed, P. K. Sachdeva, R. Rai, R. Kumar, T. Maruyama, C. Bera and V. Bagchi, ACS Appl. Energy Mater., 2020, 3, 3645–3652 CrossRef CAS.
- Y. Ye, L. Zhang, Z. C. Nie, N. P. Li, S. X. Zhou, H. S. Wang, T. Wågberg and G. Z. Hu, Chem. Eng. J., 2022, 446, 137210 CrossRef CAS.
- Y. X. Zhu, L. Zhang, G. G. Zhu, X. Zhang and S. Y. Lu, J. Colloid Interface Sci., 2020, 562, 42–51 CrossRef CAS PubMed.
- Y. X. Zhu, L. Zhang, G. G. Zhu, X. Zhang and S. Y. Lu, Nanoscale, 2020, 12, 5848–5856 RSC.
- H. Singh, M. Marley-Hines, S. Chakravarty and M. Nath, J. Mater. Chem. A, 2022, 10, 6772–6784 RSC.
- Z. Ahmed, Krishankant, R. Rai, R. Kumar, T. Maruyama, C. Bera and V. Bagchi, ACS Appl. Mater. Interfaces, 2021, 13, 55281–55291 CrossRef CAS.
- M. I. Jamesh and M. Harb, J. Energy Chem., 2021, 56, 299–342 CrossRef.
- F. Zeng, C. Mebrahtu, L. F. Liao, A. K. Beine and R. Palkovits, J. Energy Chem., 2022, 69, 301–329 CrossRef.
- H. Li, Q. Li, P. Wen, T. Williams, S. Adhikari, C. C. Dun, C. Lu, D. Itanze, L. Jiang, D. Carroll, G. Donati, P. Lundin, Y. J. Qiu and S. Geyer, Adv. Mater., 2018, 30, 1705796 CrossRef.
- L. C. Diao, T. Yang, B. Chen, B. Zhang, N. Q. Zhao, C. S. Shi, E. Z. Liu, L. Y. Ma and C. N. He, J. Mater. Chem. A, 2019, 7, 21232–21243 RSC.
- Z. S. Li, S. H. Xu, Y. D. Shi, X. H. Zou, H. Wu and S. Lin, Chem. Eng. J., 2021, 414, 128814 CrossRef CAS.
- L. Zhang, B. Wang, J. S. Hu, X. H. Huang, W. Y. Ma, N. P. Li, T. Wagberg and G. Z. Hu, J. Colloid Interface Sci., 2022, 625, 521–531 CrossRef CAS PubMed.
- S. S. Sun, X. Zhou, B. W. Cong, W. Z. Hong and G. Chen, ACS Catal., 2020, 10, 9086–9097 CrossRef CAS.
- K. Q. Zhong, L. Z. Huang, H. Li, Y. Dai, H. G. Zhang, R. Y. Yang, S. R. Babu Arulmani, X. J. Liu, L. Huang and J. Yan, Carbon, 2021, 183, 62–75 CrossRef CAS.
- J. Sun, L. Du, B. Y. Sun, G. K. Han, Y. L. Ma, J. J. Wang, H. Huo, P. J. Zuo, C. Y. Du and G. P. Yin, J. Energy Chem., 2021, 54, 217–224 CrossRef.
- S. Hao, L. B. Yang, D. N. Liu, R. Kong, G. Du, A. Asiri, Y. C. Yang and X. P. Sun, Chem. Commun., 2017, 53, 5710–5713 RSC.
- R. L. Doyle and M. E. G. Lyons, Photoelectrochemical Solar Fuel Production, Springer, 2016, 41–104, ch. 2 Search PubMed.
- J. Y. Xu, J. J. Li, D. H. Xiong, B. S. Zhang, Y. F. Liu, K. Wu, I. Amorim, W. Li and L. F. Liu, Chem. Sci., 2018, 9, 3470–3476 RSC.
- X. K. Wang, X. K. Zhou, C. Li, H. X. Yao, C. H. Zhang, J. Zhou, R. Xu, L. Chu, H. L. Wang, M. Gu, H. Q. Jiang and M. H. Huang, Adv. Mater., 2022, 34, 2204021 CrossRef CAS.
- L. L. Chen, Y. L. Zhang, L. L. Dong, X. J. Liu, L. Long, S. Y. Wang, C. Y. Liu, S. J. Dong and J. B. Jia, Carbon, 2020, 158, 885–892 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta04482j |
| This journal is © The Royal Society of Chemistry 2022 |