
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Li trapping in nanolayers of cation ‘disordered’ rock salt cathodes†
Maria
Diaz-Lopez
 *a,
Philip A.
Chater
a,
Olivier
Proux
b,
Yves
Joly
b,
Jean-Louis
Hazemann
b,
Pierre
Bordet
b and
Valerie
Pralong
c
*a,
Philip A.
Chater
a,
Olivier
Proux
b,
Yves
Joly
b,
Jean-Louis
Hazemann
b,
Pierre
Bordet
b and
Valerie
Pralong
c
aDiamond Light Source Ltd, Diamond House, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK. E-mail: maria.diaz-lopez@diamond.ac.uk
bGrenoble Alpes Univ, CNRS, Inst Neel, 38000 Grenoble, France
cNormandie Univ, Ensicaen, Unicaen, CNRS, Crismat, 14000 Caen, France
First published on 22nd August 2022
Abstract
Disordered rock salt (DRS) cathodes have attracted considerable attention because of their high first charge capacities and relatively low cost. Here we investigate operando the structure and charge evolution of the Li1.1Mn0.7Ti0.2O2 Li-excess rock salt cathode with a first charge capacity of 270 mA h g−1. We associate a certain extent of the capacity fade in DRS to the in situ formation of locally ordered layered nanodomains during the electrochemical cycling of the long-range cation disordered rock salt. We quantify the short-range ordering of cations during cycling and evaluate its effect on the lithium-ion diffusion and charge compensation using operando studies based on X-ray total scattering and advanced spectroscopic methods at the Mn K-edge, namely high energy resolution fluorescence detected XANES and emission spectroscopies including main and valence-to-core transitions.
Introduction
There is an intensifying demand for high-performance Li-ion batteries for their application in mobile devices, electric-powered vehicles, and energy storage systems to enable transformational carbon-neutral solutions.1 Within the Li-ion cell, the cathode material currently limits the energy density and dominates the battery cost.2 Improving the capacity and longevity performance of Li-ion cathode materials remains a significant challenge for the battery community. In recent years, there has been a surge of interest in Li-rich cathodes with a cation Disordered Rock Salt (DRS) structure which where once thought to be electrochemically inactive.3 DRS offers a tantalizing promise for high-energy-density Li-ion batteries with capacities beyond 300 mA h g−1, low percolation energies of 3D Li-diffusion paths and absence of phase transformations during cycling.4,5In a recent study we identified an improved electrochemical performance of a nanostructured Li2MnO3 (nano- Li2MnO3) DRS cathode material displaying reversible capacities of 290 mA h g−1 for over 10 cycles;6 whilst its crystalline counterpart, the well-researched Li2MnO3, displays a 50% capacity drop over the first five cycles.7 A joint neutron and X-ray total scattering approach evidenced a phase transformation during a high energy ball milling process, where Li2MnO3 synthesized at 900 °C with a layered structure (C2/m) transformed into a nanostructured and disordered cubic MnO-type rock salt (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). Interestingly, the well-known layered-to-spinel phase transformation of Li and Mn-rich layered cathodes8 could not explain the diffuse scattering reflections in nano-Li2MnO3 that were instead attributed to the formation of a short-range ordered superstructure due to the Li/Mn layering of ∼1 nm into alternating (111) planes in the rock salt structure, as previously reported in LixNi2−xO2.32.9
m). Interestingly, the well-known layered-to-spinel phase transformation of Li and Mn-rich layered cathodes8 could not explain the diffuse scattering reflections in nano-Li2MnO3 that were instead attributed to the formation of a short-range ordered superstructure due to the Li/Mn layering of ∼1 nm into alternating (111) planes in the rock salt structure, as previously reported in LixNi2−xO2.32.9
Other work identified short-range-ordering (SRO) in further DRS compositions which breaks the general assumption that all the cations are randomly distributed in these cathode materials. In Li1.25Nb0.25Mn0.5O2,10 the Li/(Nb, Mn) cations order in an ABAB sequence parallel to both [110] and [−110] of the rock salt sublattice resulting in a tetragonal I41/amd structure displayed by γ-LiFeO2.11 The correlation length of Li/(Nb, Mn) ordering was found to depend on the synthesis conditions, and had negative implications for the electrochemistry of these phases with a decrease of the first charge capacity by 5% and an increase of the capacity fade by 8% when the correlation length of SRO domains was increased from 1 to 13 nm.
DRS compositions containing several transition metal species including electrochemically inactive transition metals with a d0 electronic configuration have been extensively investigated. The presence of d0 transition metals can stabilize disordered structures,12–14 and their ionic character can also stabilise the superoxide species formed during charge.15–19 Within this group of DRS, Li1.2Mn0.4Ti0.4O2 displayed a superior electrochemical performance15 compared to the isoelectronic Zr4+-doped Li1.2Mn0.4Zr0.4O2. This result is counterintuitive as the performance of the Zr-doped rock salt is expected to be better based on its larger lattice parameters, which generally benefits Li diffusion. Here, the different electrochemical performance was reported to be due to different and more pronounced SRO of the Zr-doped rock salt.20
In this work, we investigate operando the evolution of the average structure, SRO, and charge during the electrochemical cycling of a Li-excess Ti4+-doped DRS with composition Li1.1Mn0.7Ti0.2O2 (LMTO). Pristine LMTO did not show significant SRO, as the presence of the Ti4+-dopant aids to stabilize the disorder in the structure. However, during electrochemical cycling, we identify the appearance of diffuse peaks in the Bragg data which matched the SRO previously observed in nano-Li2MnO3.6 During the electrochemical cycling, the cation rearrangements in LMTO form in situ a SRO superstructure with nanodomains of Li:M layering, where M = Ti and Mn, that continue to grow over the first few cycles (up to 15 investigated in this work). Cation SRO, amid other subtle structural changes, has a profound effect on the population and connectivity of Li-migration channels and influence the electrochemical performance of DRS as a result. We have investigated the impact of these layered nanodomains on the material's electrochemical performance through a combination of total scattering and advanced X-ray spectroscopic techniques including High Energy Resolution Fluorescence Detected (HERFD-)XANES and emission spectroscopies including main, and valence-to-core (V2C) transitions performed operando. We identify the trapping of Li in the layered domains of DRS as a significant source of capacity fade alongside contributions from oxygen redox irreversibility. These results indicate the importance of SRO and provide another important handle to tailor the performance of DRS cathode materials. The results of this study provide insight into the relationship between the local chemistry and Li transport properties in DRS, enabling future design of better-performing cathode materials.
Results
Synthesis and electrochemical activity
We synthesized Li1.1Mn0.7Ti0.2O2 using the solid-state method proposed for Li1.2Mn0.4Ti0.4O2.20 We identified TiO2 impurities for Li1+xMn1−3xTi2xO2 with x > 0.1 and the x = 0.1 composition (hereafter referred to as LMTO) was used in the study (see Pawley fit in Fig. 1a). LMTO delivers a large first-cycle capacity of 270 mA h g−1 (0.9 Li+ f.u.) as shown in the galvanostatic voltage profile in Fig. 1b. The sloped galvanostatic voltage profile is consistent with those of other cation DRS materials. LMTO displays an irreversible capacity of 30 mA h g−1 (0.1 Li+ f.u.) after the first charge, after which the capacity is stable at around 240 mA h g−1 (0.8 Li+ f.u.) with a further decrease at a slower rate of 17 mA h g−1 (0.6 Li+ f.u.) for 15 cycles tested.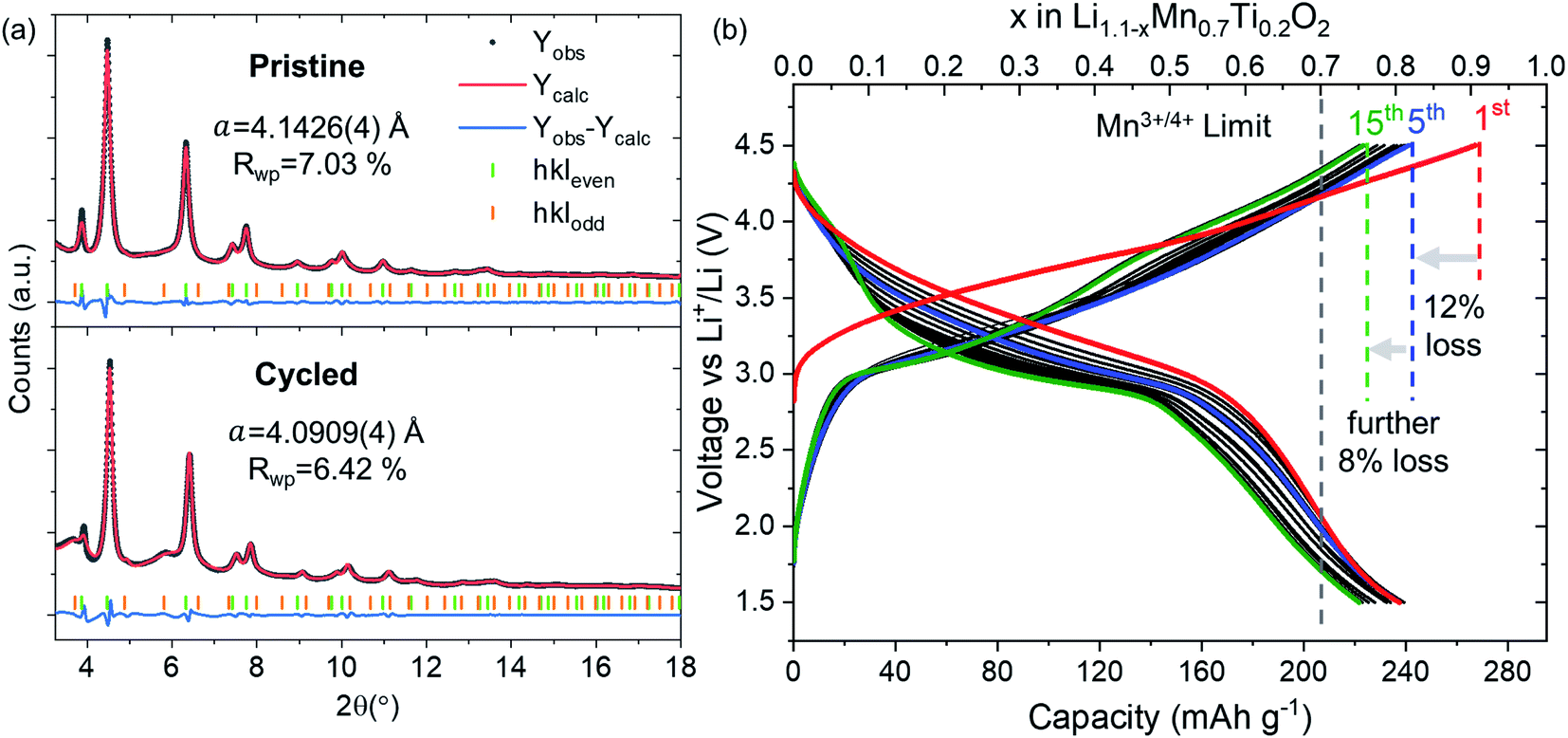 | ||
| Fig. 1 Characterisation of LMTO. (a) Pawley fits of pristine (top) and cycled compositions (bottom) using an hkl-dependent model. These fits correspond to the first (t = 0 h, 2.8 V) and last (t = 37 h, 3.24 V) points of the operando Bragg data in Fig. 2. (b) Galvanostatic cycling at a rate of C/20. | ||
Structural evolution during battery cycling
Previous work by Ji et al.20 reports diffuse scattering patterns surrounding the Bragg reflection spots in electron diffraction patterns of a crystalline Li1.2Mn0.4Ti0.4O2 sample synthesized at high temperatures, suggesting the existence of SRO. In this work, we investigate the ball-milled LMTO electrode, and the extent of structural disorder after the milling is such that the structural characterization by electron diffraction is inhibited. Instead, we investigate the SRO in LMTO by total scattering analysis, combining Bragg and PDF data. The evolution of broad diffuse scattering features in the data could be successfully identified owing to the accurate background subtraction allowed by the DRIX electrochemical cells.21While the Bragg data of pristine LMTO can be well explained with the average DRS model, broad diffuse scattering peaks appear during the first charge, which continued to increase in intensity during the subsequent (dis)charge cycles. Such diffuse scattering peaks have previously been attributed to the formation of a second phase in other work.22 However, these diffuse peaks could be explained by the formation of a SRO superstructure with nanodomains of Li/M layering into alternating (111) planes in the rock salt structure, as previously reported in materials such as LixNi2−xO2 (ref. 9) and nano-Li2MnO3.6 Both sets of broad and sharper reflections, corresponding to the layered (Rm) and DRS (Fmm) sublattices, respectively, were fitted using the rhombohedral model. In the hexagonal unit cell description, reflections derived from the cubic structure have Miller indices with even values of l (hkleven), whereas hexagonal reflections that are forbidden in the cubic unit cell, but arise due to Li and Mn layering, exhibit odd values of l (hklodd).9 The double-Voigt approach23 was used to estimate the structural coherence of the layered and DRS components based on the peak broadening of hklodd and hkleven reflections, respectively. This refinement approach allowed us to estimate the average crystallite size of the nanostructured material to be 4.6(2) nm, and the extent of Li/M layering to be 0.50(7) nm, which approximately equals four alternative layers of Li and M.
Operando Bragg data of LMTO cycled over the potential window of 1.5–4.5 V over the course of two charge–discharge cycles and a subsequent cycle discharged down to 3.2 V are shown in Fig. 2. The evolution of the lattice parameters (Fig. 2b) is in good agreement with the expected breathing behaviour of the cubic M framework of DRS with the lattice parameters contracting from 4.1426(4) Å in the lithiated compound to 4.0358(4) Å upon delithiation.
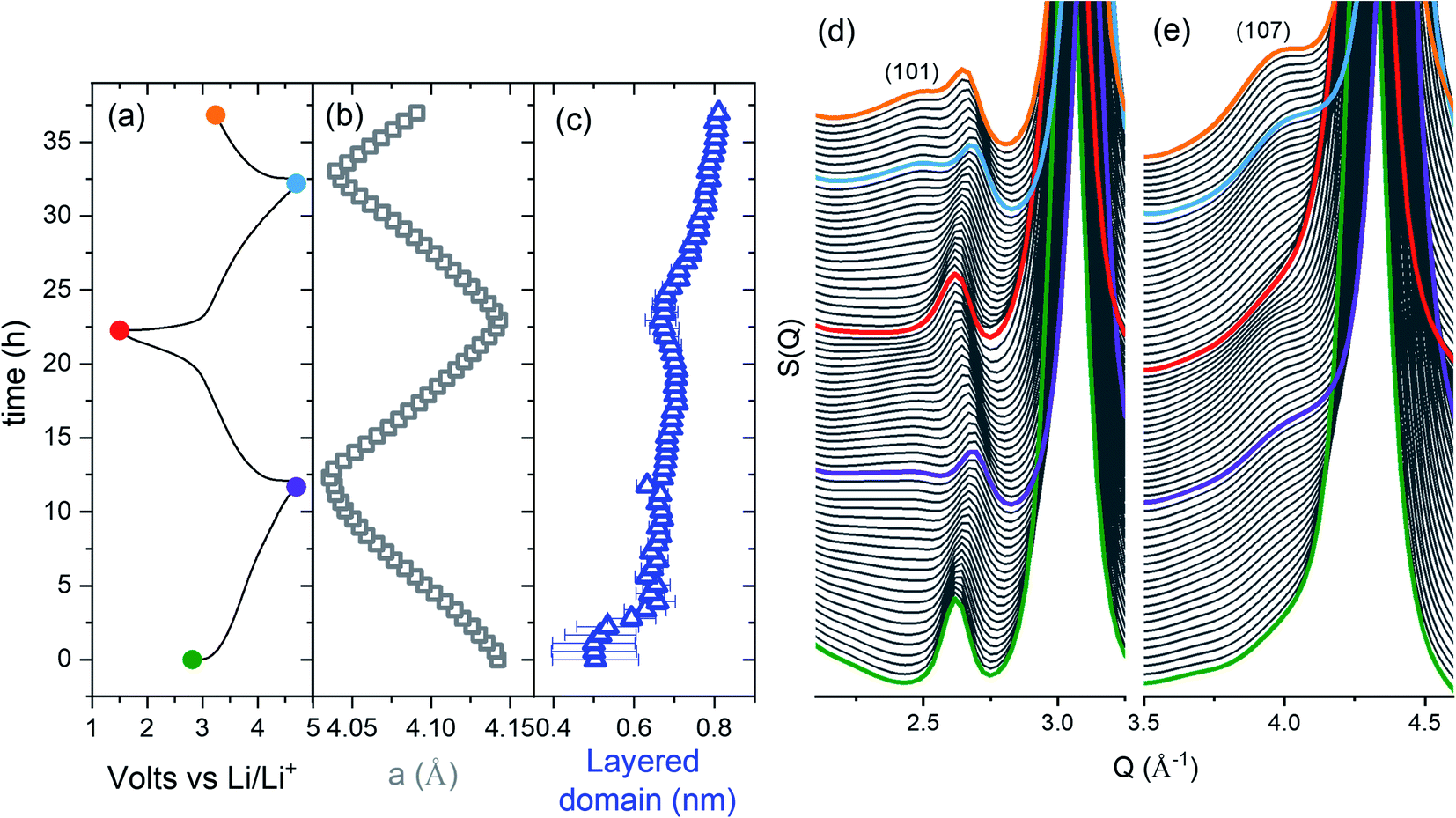 | ||
| Fig. 2 LMTO structural evolution. (a) Electrochemical performance, (b) lattice parameters and (c) structural coherence of the layered sublattice from a sequential refinement. The fits for the first and last datasets are given in Fig. 1. (d and e) Operando Bragg data showing the increased intensity of the (101) and (107) SRO superstructure peaks with cycling. | ||
Diffuse peaks in the pristine LMTO composition increase in intensity during battery cycling, as shown in Fig. 2d and e. We have performed a sequential refinement of the Bragg data to quantify the increase in the structural coherence of the layered nanodomains (see Fig. 2c). The Bragg refinement of operando data shows that during cycling, there is a steady increase of the layered nanodomain size of approximately 17% per half cycle over the course of the first two cycles.
The structural coherence of the layered domains was further investigated ex situ after 7 and 15 cycles (see Fig. S1†). The refined structural coherence of the layered domains of 1.0(2) and 2.25(9) nm after 7 and 15 cycles, respectively corresponds to a growth rate of 21(1)%, congruent with the growth rate observed operando. Thus, the SRO nanolayers at the initial stages of cycling evolve into long-range ordered domains after several cycles.
The refined atomistic models were analysed by the bond valence site energy (BVSE) approach,24 where potentially accessible sites for lithium are identified by the calculation of the mismatch of the bond valence sum with respect to Li+ formal valence. The BVSE approach exploits the analogy between the squared bond valence mismatch and the Morse-type potential to transform valence into energy units.25Fig. 3 shows graphical representations of isosurfaces in which Li+ could diffuse according to this method. The importance of basing BVSE calculations in atomistic models becomes clear when comparing the percolated volume fraction of average structural models of around 5%, which increases to 8.1(5) and 9(2)% for the layered and DRS models, respectively. The BVSE maps in Fig. 3 reveal the expected 3D Li diffusion for the DRS model, and 2D Li diffusion for the layered domains, where for both sublattices Li diffusion occurs along tetrahedral sites. To further analyse the Li transport environments in the two sublattices of the LMTO structure, we have quantified the variation of Li-diffusion energy barriers as a function of the number of M within the vicinity of the tetrahedral sites that participate in the Li diffusion.26Fig. 4 summarizes the occurrence of tetrahedral cluster types as a function of M = Ti and Mn, and Li neighbours (M4, LiM3, Li2M2, Li3M and Li4) in layered and DRS domains. The tetrahedral cluster occurrence follows a standard distribution in the disordered model centred around the Li2M2 clusters. In the layered model, LiM3 and Li3M sites are favoured as expected since these are the only two sites present in stoichiometric LiMO2 (see Fig. S4†). The different distribution of tetrahedral clusters in Fig. 4 indicates that the induced SRO during cycling of LMTO strongly modifies the population of local tetrahedral clusters. Despite the different cluster distribution, the calculated energies of each cluster type are similar in both models, as shown in Fig. 4a, which is indicative of the cluster energy strongly depending on the closest neighbours within the tetrahedra and not on the extended structure. Therefore, we consider occurrence of clusters and their distribution within a percolating network as the primary factors to sustain the migration of Li+ cations throughout LMTO.
 | ||
| Fig. 3 Local structure and Li diffusion of pristine LMTO based on structures derived from PDF refinement with electrostatic potential constraints. Left: Magnified view of the short r-range of the PDF refinements (to see the whole r-range used in the refinements the interested reader is referred to Fig. S5†) Right: Refined average structural models (top) and supercells of atomistic disorder (bottom). The compositions for the refined supercells are Li18Mn11Ti3O32 and Li26Mn17Ti5O48 for the disordered and layered models, respectively. Green, purple, blue and red spheres denote Li, Mn, Ti and O, respectively. The yellow isosurfaces represent the regions with a constant BVSE of −2.2 eV energy threshold. | ||
 | ||
| Fig. 4 Cation clusters energy from the refined models in Fig. 3 (a) and probabilistic occurrence (b) in disordered (blue) and layered (red) LMTO. | ||
We observe that the occurrence of Li4 tetrahedra of 8.5%, which is the most important for good Li transport, is larger in the disordered model (against 5.5% in the layered model). In addition, the migration barrier through the Li4 tetrahedra in the disordered model is on average 170 meV lower than that of the layered model, further aiding the Li4-mediated Li diffusion in the disordered sublattice. On the other hand, the occurrence of Li3M tetrahedra, whose range of energies partially lie below the energy threshold for Li-mobility of −2.2 eV, is higher in the layered (45.7%) than in the disordered model (31.8%). Therefore, to fully address the lithium migration in LMTO, we must also consider the distribution and evolution of the Li4 and Li3M tetrahedra with suitable Li migration barriers in both layered and DRS sublattices.
In other systems, the complete depletion of lithium from layered structures (Rm) is known to increase the repulsive interactions between adjacent layers of MO2, which in turn results in oxygen loss and structural rearrangements where the layered structure tends to transform into a mixture of spinel and rock salt phases.27,28 The decomposition of LMTO into other phases is not observed in this study, where changes in the structure with cycling entail an isotropic breathing of the cubic framework. This could indicate that the complete depletion of lithium between the MO2 slabs in the layered nanodomains is not reached and the growth of these domains with the cycling is thereby acting as a Li trap (a structural feature that irreversibly immobilises Li+-ions), which correlates with the decay of the capacity with cycling. The formation of layered domains results in a higher occurrence of LiM3 clusters where lithium becomes trapped away from the lithium percolation pathway where it was originally connected to.
Charge evolution during battery cycling
The charge evolution and coordination environment of electrochemically active Mn3+ were studied operando over the course of the first charge–discharge cycle.Mn K-edge HERFD-XANES data provide information on the evolution of the local environment around MnO6. The data in Fig. S6† show the expected evolution for Li–Mn–O rock salts in previous work,6 which are (1) fading of the shoulder in the white line transition due to change in composition and (2) increase of intensity of the eg transition attributed to Mn3+ to Mn4+ oxidation. In this new experiment, the improved sensitivity of HERFD-XANES gave better resolved pre-edges, in which we clearly distinguish the quadrupole transitions toward the t2g and eg states, whereas conventional XANES shows a partial overlap of the eg state with the white line.29,30 HERFD-XANES data show an increase in the intensity of the eg transition during the charge that could correspond to the emptying of electrons in the corresponding level because of the oxidation of Mn3+ (d4) to Mn4+ (d3). However, since the position and shape of the absorption edge are strongly influenced by structural effects due to the large overlap between the outer 4p orbitals of Mn and the oxygen ligands, the charge evolution of Mn was further investigated by the combined interpretation of main and V2C emission transitions (see Fig. S7†). Here, the main XES 3p-1s energy transition serves as a better probe of the number of unpaired 3d electrons in Mn, which are indirectly inferred from the magnitude of 3p–3d exchange interactions. In addition, the V2C energy transitions from hybridized Mn–O orbitals were recorded during the same experiment to further aid the identification of changes in the charge density and coordination environment of both Mn and the oxygen ligands.
Fig. 5 shows the comparison of the most intense kβ1,3 and of kβ2,5 of the main and V2C transitions respectively, as a gauge of the Mn oxidation state at different states of charge. The energy transitions of MnO2 and Mn2O3 standards are given in the same figure for reference. In contrast with the high reversibility of HERFD-XANES transitions in the charge and discharge states, both kβ1,3 and Kβ2,5 transitions that were measured operando during the same experiment show some irreversibility on the reduction of Mn4+ to Mn3+ during the discharge. Based on the energy of the kβ1,3 transition, and the linear response of this transition with the change in the oxidations state,30 we estimate that 13% of manganese remains as Mn4+ in the cycled composition, which amounts to approximately 0.09 Mn per f.u.
 | ||
| Fig. 5 (a) kβ1,3 and (b) kβ2,5 XES transitions at t = 0, 16 and 28 h, corresponding to 2.4, 4.4 and 1.5 V respectively. | ||
The irreversible charge evolution of Mn which is not appreciable by XAS, can be detected by XES and could be ascribed to the loss of accessible cation vacancy sites in the layered domains for the reincorporation Li+-ions during discharge.
Discussion
Capacity fade in DRS is generally associated to the lack of reversibility of oxygen redox. Here we use the results of our LMTO study in combination with other reported systems to explore the impact of the SRO evolution during cycling with a drop in capacity. We aim to compare the performance of LMTO to DRS with different extents of oxygen redox participation and SRO.Fig. 6 summarises the theoretical, 1st charge capacity and capacity fade rate of cathode materials cycled in half cells at room temperature. We compare the reported performance of cathode materials with no extra capacities (LiFePO4,31 LiCoO2,31 NMC811 (ref. 32) and NMC311 (ref. 32)) to DRS cathodes with different extents of oxygen participation ranging from 25 to 100% of the extra capacities beyond the theoretical value given by the transition metal redox limit. As shown in Fig. 5, the higher 1st charge capacities of DRS are accompanied by a higher rate of capacity fade. While the capacity fade of cathode materials with no oxygen redox typically ranges from 0.2 up to 0.5%, the typical value of DRS is around one order of magnitude higher.
 | ||
| Fig. 6 Comparison of the theoretical capacities based on TM redox, experimental 1st charge capacities and capacity fade%. The experimental values were obtained in half cells cycled at room temperature,6,10,20,31,32 and the theoretical capacities were calculated using the transition metal redox activity. | ||
We can further compare the performance of two DRS compositions in the LMTO system with different extent of O redox participation; Li1.2Mn0.4Ti0.4O2 (reported in this work) and Li1.1Mn0.7Ti0.2O2.20 To calculate the expected capacity fade in Li1.1Mn0.7Ti0.2O2, we subtract the typical capacity fade value of 0.4% ascribed to other mechanisms than anion redox in Li1.2Mn0.4Ti0.4O2 and assume that the remaining capacity loss is due to oxygen redox. This calculated expected capacity fade has a value of 0.97% in Li1.1Mn0.7Ti0.2O2, which is significantly lower than the observed value of 1.9%. Such higher-than-expected capacity fade could be linked to the formation and growth of layered domains during the initial charge–discharge cycles. The results for the LMTO system agree with previous work on a DRS cathode material with composition Li1.25Nb0.25Mn0.5O2 (LNbMO), where the coherence length of the layered sublattice was successfully controlled by the change of the synthetic conditions. Slowly cooled LNbMO resulted in a DRS structure containing layered domains with a structural coherence of 12.5 nm, whereas the structural coherence of the layered domains in rapidly cooled LNbMO was only 1 nm. This work demonstrated a better capacity retention of LNbMO (−1.9%) vs. LNbMO with SRO (−2.9%). However, the study did not show the structural evolution of the ordered domains with battery cycling. As shown in this work, the structural changes during (de)lithiation can impact the structural coherence of these domains, and their correlation with the capacity should be also investigated during battery cycling. Future work in DRS should not only focus on the stabilization of oxygen redox to improve the capacity fade of DRS, but also to control the structural coherence of SRO domains both during the synthesis and their evolution with cycling.
We recently identified the presence of SRO in the nano-Li2MnO3 cathode material with an average DRS structure and a high first charge capacity of ca. 335 mA h g−1. In nano-Li2MnO3, layered domains of 1 nm were already present in the pristine structure and their structural coherence remained constant during the first charge–discharge cycle.6 In pristine LMTO, cation SRO was inhibited by the presence of electrochemically inactive Ti4+ which stabilised the formation of a disordered structure.12,14 However, during electrochemical cycling the SRO nanolayers were formed, which indicated that unfavourable cation rearrangements can still occur in spite of the presence of Ti4+ acting as a stabiliser of disorder in DRS.
While the growth of layered domains could be associated with a higher capacity fade in Li1.1Mn0.7Ti0.2O2vs. Li1.2Mn0.4Ti0.4O2, we believe there are qualities of the layered materials such as their inherently high capacity and rate capability that could benefit the performance of DRS cathode materials. As a matter of fact, the interplay between DRS and layered sublattices was recently exploited to enhance the performance of a Li1.2Ni0.4Ru0.4O2 cathode with a layered Rm structure and rock salt (Fmm) domains.33 Although containing only 30 mol% of the rock salt domains, Li1.2Ni0.4Ru0.4O2 displays isotropic structural changes resembling those of ‘pure’ DRS, as the interwoven rock salt structure prevents the anisotropic structural change that is typical for layered oxides. The Li1.2Ni0.4Ru0.4O2 cathode displays a preferred extraction of Li+ cations from the rock salt rather than layered phase, whilst the layered structure is believed to facilitate extraction of Li+ from the intergrown rock salt thanks to the 2D Li+ diffusion along the layers. Understanding the interplay between the layered and disordered sublattices, how they can be controlled during synthesis and their evolution with cycling is critical to harness the favoured figures of merit from each layered and disordered component and design better cathode materials that meet industrial demands for performance.
Strategies for improvement
We believe future work in DRS could be aimed at controlling the coherence length of layered domains during cycling either through the control of the particle size or the structural coherence of the layered domains. If the layered domains continue to grow during cycling, we will ultimately have a phase transition into a layered structure, which is not experimentally observed. Thus, there must be a limit value the layered domains will grow up to. Further work is needed to find the effect of particle size and the limit of growth of layered domains in each DRS system to inform future synthesis of DRS with optimal properties.Alternatively, future efforts on DRS optimization could be aimed at tuning a favourable SRO through the choice of different co-dopants so that small, SRO- domains can be maintained through cycling. In Ni rich layered oxides, the introduction of electrochemically inactive Mg2+ (ref. 34 and 35) and Al3+ (ref. 36) cations acting as pillars can prevent the undesirable migration of atoms mitigating structural degradation. A similar doping strategy could potentially enhance the performance of DRS, although here the migration of transition metals into the vacancies generated from Li+ extraction is partly responsible for the high capacities observed (see Fig. S8†), and doping strategies aimed at preventing the transition metal diffusion should be exercised with care. An alternative doping strategy that seems not to interfere with the migration of transition metals into lithium vacancies has been demonstrated in a metastable LiMnO2 nanostructured DRS integrating phosphorus ions located at a tetrahedral site.37
Conclusion
The combined use of potential constraints, PDF data and BVS mismatch mapping revealed that Li trapping in LMTO originating from SRO can perturb the percolating Li-diffusion network in DRS. The lithium and cation vacancies in the layered domain formed during cycling become less accessible in the subsequent charge cycles, as evidenced by total scattering and XES. The comparison between LMTO and other DRS in the literature suggests that the trapping of Li in the layered domains could be associated with the capacity fade of DRS and could be a significant source of capacity fade alongside contributions from oxygen redox irreversibility.The formation of SRO in DRS cathodes, either formed during the composite preparation by ball milling or the evolution of these domains with electrochemical cycling, should be further investigated in future materials. Preventing the formation of layered domains during cycling or controlling their size could be key to reduce capacity losses of DRS. This could be potentially achieved by nanostructuration or introducing electrochemically inactive dopants acting as pillars to mitigate the migration of the TM.
Although the growth of layered domains could effectively act as a trap for lithium, layered cathode materials have an inherently high capacity and rate capability. Thus, the interplay between the layered and DRS sublattices needs to be further investigated and the impact of SRO should be optimised. This work highlights the importance of local structures in cyclability of battery materials and provides insight into the design of better DRS cathodes. Successfully controlling the coexistence of layered and DRS sublattices exemplifies a new route to electrode design that opens a new path to develop high-performance cathode materials.
Experimental
Synthesis and sample preparation
We synthesized LMTO by a solid-state method from stoichiometric amounts of high purity Li2CO3, Mn2O3 and TiO2. A small excess (∼5 wt%) of Li2CO3 was added to compensate for lithium evaporation at high temperatures. We obtained phase pure LMTO after annealing the sample at 1100 °C for several hours under Ar flow. The phase purity was confirmed by X-ray diffraction analyses.The electrode material was prepared by combining LMTO with amorphous super P carbon in a 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 weight ratio. The homogeneity of the electrode mixture was aided by a short cycle of mechanical milling performed in a Fritsch Planetary Micro Mill PULVERISETTE 7 Premium Line. The electrode mixture was milled at 250 rpm for a set of two cycles of 30 min milling followed by a 20 min pause.
:3 weight ratio. The homogeneity of the electrode mixture was aided by a short cycle of mechanical milling performed in a Fritsch Planetary Micro Mill PULVERISETTE 7 Premium Line. The electrode mixture was milled at 250 rpm for a set of two cycles of 30 min milling followed by a 20 min pause.
Electrochemistry
The electrochemical performance was tested with the as-prepared composite electrodes without the addition of binder. The composite electrode mixture was pressed to give a good electronic contact between the grains to minimize the cell polarization and tested in a Swagelok half-cell using lithium metal as a counter electrode and a cycling rate of C/10. The galvanostatic voltage profile of LMTO cycled in a voltage window of 1.5–4.5 V is consistent with previous reports by Yabuuchi et al.38 and Ji et al.20Total scattering
In operando total scattering measurements were performed at the I15-1 XPDF beamline of the Diamond light source (UK) using high energy X-rays (76.6 keV) with a wavelength of 0.161669 Å. The measurements were performed in DRIX in situ electrochemical cells with a radial geometry and minimal background for optimal PDF extraction, where the only background contributions considered were the empty cell and acetylene black conductive additive present in the cathode mixture in 30 wt%. The real and reciprocal space data are given in Fig. S9.†Samples for the pristine compositions and LMTO:C composite electrodes at cycles 7 and 15 were prepared in borosilicate glass capillaries of 1 mm internal diameter and measured ex situ.
The data collection was carried out using two PerkinElmer detectors at ∼20 cm from the sample for XPDF data collection and at 80 cm for Bragg data collection. ∼5 minute data collection gave excellent data quality for XPDF data analysis. In the operando study, four duplicates of these batteries were measured that gave identical results.
The relative geometry of the batteries and the detectors was calibrated with Si standards and the collected 2D data were processed into 1D diffraction patterns using the DAWN software. Total scattering data were background corrected and normalized with the GudrunX software using a Qmax of 25 Å−1.
The Pawley fits were performed using the TOPAS software.39 The scale factor, lattice parameters, and isotropic thermal parameters (Biso) were refined. The backgrounds were fitted with Chebyshev polynomials and the peak profile shapes were described using a modified Thompson–Cox–Hastings pseudo-Voigt function,40 where instrumental profiles were determined by the refinement of standards. The scale factors for the scattering signal from the carbon black and cell were included in the refinement as shown in Fig. S10 and S11.† Including these backgrounds in the refinement was key to improve the fit, given the large overlap of these signals with the diffuse scattering peaks of LMTO.
PDF refinements
The PDF data of pristine LMTO measured ex situ were fitted using electrostatic potentials implemented in TOPAS as the General Real Space (GRS) interaction41 with the Lennard-Jones potential as the repulsion term. The supercells for these refinements were produced from the averaged structural models refined by a Pawley fit. DRS supercells were built by doubling the unit cell in every direction, and layered supercells by doubling the unit cell in the ab plane.Li+ diffusion in LMTO
BVSE pathways also known as energy-scaled bond-valence mismatch landscapes were derived from the proposed structural models by Pawley refinements, and the bond distances extracted from PDF refinements. BVSE maps were constructed by summing the BVSE contributions to a hypothetical Li+ ion at each point with a 7 × 10−4 Å3 volume up to 8 Å. SoftBV parameters42 were used to account for the influence of higher coordination shells and avoid cut-off effects at the boundaries of coordination shells.The volume fraction of a percolating pathway with a 3 eV threshold, and the percolation threshold energy values along each axes were calculated from Monte-Carlo simulations using the BondStr software embedded in Fullprof-Suite.43 The figures illustrating the local environments and diffusion pathways were produced with the VESTA software.44
In operando absorption and emission spectroscopies
Commercial manganese oxide standards were diluted in BN (5 wt%), thoroughly mixed and uniaxially pressed into pellets that were mounted on a sample holder equipped with Kapton windows that allow the entry of X-rays. A C/LMTO cathode mixture was investigated operando and prepared in an in situ cell45 equipped with a 200 μm thick window made of beryllium; more details on the preparation of the cell are given in ref. 30.Operando HERFD-XANES and XES experiments were carried out at I20 beamline of the Diamond Light source46 using angle dispersive analysis of the fluorescence X-ray signal emitted from the sample. The spectrometer was equipped with five spherically bent Si(440) crystal analysers focusing the diffracted beams onto a silicon drift detector for the recording of the Mn Kβ lines. The energy and experimental setup were calibrated by the recording a Mn foil spectrum. The energy resolution of the spectrometer, including contributions from both the monochromator and the CAS, was estimated from the recorded width of a pseudo-elastic peak.
For operando data acquisitions, the spectrometer was positioned at the maximum of the Mn Kβ emission line prior to the recording of the XANES spectra. HERFD-XANES spectra were then collected at the selected emission, which allowed discrimination of the detected photons with an energy resolution of 0.8 eV, smaller than the core-hole lifetime of the fluorescence line. This allows obtaining spectra with almost a core-hole lifetime-free broadening, that is, providing optimal energy resolution. XES and V2C data were averaged and background corrected using the PyMCA software.47
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge Justine Jean at CRISMAT for technical assistance, Dr Matteo Aramini for support during beamtime at the I20 beamline of the Diamond Light Source and Diamond Light Source for beamtime under proposals CY24582 and SP2958.References
- The European Commission, A Clean Planet for All A European Strategic Long-Term Vision for a Prosperous, Modern, Competitive and Climate Neutral Economy, 2018 Search PubMed.
- S. G. Booth, N. N. Anthonisamy, R. Boston, H. Bronstein, S. J. Clarke, E. J. Cussen, N. A. Fleck, H. S. Geddes, N. Gollapally, J. M. Griffin, A. R. Haworth, M. A. Hayward, S. Hull, S. Price and D. O. Scanlon, APL Mater., 2021, 9, 109201 CrossRef CAS.
- M. N. Obrovac, O. Mao and J. R. Dahn, Solid State Ionics, 1998, 112, 9 CrossRef CAS.
- R. J. Clement, Z. Lun and G. Ceder, Energy Environ. Sci., 2020, 13, 345 RSC.
- N. Yabuuchi, Curr. Opin. Electrochem., 2022, 34, 100978 CrossRef CAS.
- M. Diaz-lopez, P. A. Chater, Y. Joly, O. Proux, J. Hazemann and V. Pralong, J. Mater. Chem. A, 2020, 2, 10998 RSC.
- M. Oishi, K. Yamanaka, I. Watanabe, K. Shimoda, T. Matsunaga, H. Arai, Y. Ukyo, Y. Uchimoto, Z. Ogumi and T. Ohta, J. Mater. Chem. A, 2016, 4, 9293 RSC.
- K. Shimoda, M. Oishi, T. Matsunaga, M. Murakami, K. Yamanaka, H. Arai, Y. Ukyo, Y. Uchimoto, T. Ohta, E. Matsubara and Z. Ogumi, J. Mater. Chem. A, 2017, 5, 6695 RSC.
- P. T. Barton, Y. D. Premchand, P. A. Chater, R. Seshadri and M. J. Rosseinsky, Chem.–Eur. J., 2013, 14521 CrossRef CAS PubMed.
- M. A. Jones, P. J. Reeves, I. D. Seymour, M. J. Cliffe, S. E. Dutton and C. P. Grey, Chem. Commun., 2019, 55, 9027 RSC.
- G. C. Mather, C. Dussarrat, J. Etourneau and A. R. West, J. Mater. Chem., 2000, 10, 2219 RSC.
- A. Urban, A. Abdellahi, S. Dacek, N. Artrith and G. Ceder, Phys. Rev. Lett., 2017, 176402, 1 Search PubMed.
- R. Chen, R. Witte, R. Heinzmann, S. Ren, S. Mangold, H. Hahn, R. Hempelmann and S. Indris, Phys. Chem. Chem. Phys., 2016, 7695 RSC.
- L. Xu, W. Liu, X. Li, S. Rashid, C. Shen and Y. Wen, RSC Adv., 2015, 5, 12248 RSC.
- D. Chen, J. Wu, J. K. Papp, B. D. Mccloskey, W. Yang and G. Chen, Small, 2020, 16, 2000656 CrossRef CAS PubMed.
- N. Yabuuchi, M. Takeuchi, M. Nakayama, H. Shiiba, M. Ogawa, K. Nakayama, T. Ohta, D. Endo, T. Ozaki, T. Inamasu, K. Sato and S. Komaba, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 7650 CrossRef CAS PubMed.
- J. Lee, D.-H. Seo, M. Balasubramanian, N. Twu, X. Li and G. Ceder, Energy Environ. Sci., 2015, 3255 RSC.
- Y. Li, X. Zhao, Q. Bao, M. Cui, W. Qiu and J. Liu, Energy Storage Mater., 2020, 32, 253 CrossRef.
- Y. Kobayashi, M. Sawamura, S. Kondo, M. Harada, Y. Noda, M. Nakayama, S. Kobayakawa, W. Zhao, A. Nakao, A. Yasui, H. B. Rajendra, K. Yamanaka, T. Ohta and N. Yabuuchi, Mater. Today, 2020, 37, 43 CrossRef CAS.
- H. Ji, A. Urban, D. A. Kitchaev, D. Kwon, N. Artrith, C. Ophus, W. Huang, Z. Cai, T. Shi, J. C. Kim, H. Kim and G. Ceder, Nat. Commun., 2019, 10, 592 CrossRef CAS PubMed.
- M. Diaz-lopez, G. L. Cutts, P. K. Allan, D. S. Keeble, A. Ross, V. Pralong and P. A. Chater, J. Synchrotron Radiat., 2020, 27, 1 CrossRef PubMed.
- K. Zhou, S. Zheng, H. Liu, C. Zhang, H. Gao, M. Luo, N. Xu, Y. Xiang, X. Liu, G. Zhong and Y. Yang, ACS Appl. Mater. Interfaces, 2019, 11, 45674 CrossRef CAS PubMed.
- D. Balzar, N. Audebrand, M. R. Daymond, A. Fitch, A. Hewat, J. I. Langford, A. Le Bail, D. Louer, O. Masson, C. N. McCowan, N. C. Popa, P. W. Stephens and B. H. Toby, J. Appl. Crystallogr., 2004, 37, 911 CrossRef CAS.
- S. Adams, Solid State Ionics, 2006, 177, 1625 CrossRef CAS.
- S. Adams and P. Rao, Struct. Bond., 2006, 119, 193 CrossRef.
- A. Van der Ven and G. Ceder, J. Power Sources, 2001, 97–98, 529 CrossRef CAS.
- N. Tran, L. Croguennec, M. Ménétrier, F. Weill, P. Biensan, C. Jordy and C. Delmas, Chem. Mater., 2008, 20, 4815 CrossRef CAS.
- S. Jung, H. Gwon, J. Hong, K. Park and D. Seo, Adv. Energy Mater., 2014, 4, 1300787 CrossRef.
- M. Diaz-lopez, P. Bordet, C. V Colin, M. Freire and V. Pralong, Chem. Mater., 2017, 30, 8598 Search PubMed.
- M. Diaz-lopez, Y. Joly, M. Freire, C. Colin, O. Proux, V. Pralong and P. Bordet, J. Phys. Chem. C, 2018, 122, 29586 CrossRef CAS.
- M. Diaz-lopez, P. A. Chater, P. Bordet, M. Freire, C. Jordy, O. I. Lebedev and V. Pralong, Adv. Energy Mater., 2020, 1902788, 1 Search PubMed.
- H. Noh, S. Youn, C. Seung and Y. Sun, J. Power Sources, 2013, 233, 2 CrossRef.
- N. Li, M. Sun, W. H. Kan, Z. Zhuo, S. Hwang, S. E. Renfrew, M. Avdeev, B. D. Mccloskey, D. Su, W. Yang and W. Tong, Nat. Commun., 2021, 2348 CrossRef CAS PubMed.
- Q. Xie, W. Li and A. Manthiram, Chem. Mater., 2019, 31, 938–946 CrossRef CAS.
- H. Li, P. Zhou, F. Liu, H. Li, F. Cheng and J. Chen, Chem. Sci., 2019, 10, 1374 RSC.
- M. Guilmard, L. Croguennec, D. Denux and C. Delmas, Chem. Mater., 2003, 2, 4476 CrossRef.
- M. Sawamura, S. Kobayakawa, J. Kikkawa, N. Sharma, D. Goonetilleke, A. Rawal, N. Shimada, K. Yamamoto, R. Yamamoto, Y. Zhou, Y. Uchimoto, K. Nakanishi, K. Mitsuhara, K. Ohara, J. Park, H. R. Byon, H. Koga, M. Okoshi, T. Ohta and N. Yabuuchi, ACS Cent. Sci., 2020, 6, 2326 CrossRef CAS PubMed.
- N. Yabuuchi, M. Nakayama, M. Takeuchi, S. Komaba, Y. Hashimoto, T. Mukai, H. Shiiba, K. Sato, Y. Kobayashi, A. Nakao, M. Yonemura, K. Yamanaka, K. Mitsuhara and T. Ohta, Nat. Commun., 2016, 7, 13814 CrossRef CAS PubMed.
- A. A. Coelho, Topas-Academic, Version 6, 2016 Search PubMed.
- P. Thompson, D. E. Cox and J. B. Hastings, J. Appl. Crystallogr., 1987, 20, 79 CrossRef CAS.
- A. A. Coelho and R. W. Cheary, Comput. Phys. Commun., 1997, 4655, 15 CrossRef.
- S. Adams, softBVS Web Page, 2003 Search PubMed.
- J. Rodríguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272 CrossRef CAS.
- J. B. Leriche, S. Hamelet, J. Shu, M. Morcrette, C. Masquelier, G. Ouvrard, M. Zerrouki, P. Soudan, S. Belin, E. Elkaïm and F. Baudelet, J. Electrochem. Soc., 2010, 157, A606 CrossRef CAS.
- S. Diaz-Moreno, S. Hayama, M. Amboage, A. Freeman, J. Sutter and G. Duller, J. Phys.: Conf. Ser., 2009, 190, 012038 CrossRef.
- V. A. Solé, E. Papillon, M. Cotte, P. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62, 63 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta04262b |
| This journal is © The Royal Society of Chemistry 2022 |