
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A symmetric direct ammonia fuel cell using ternary NiCuFe alloy embedded in a carbon network as electrodes†
Mengfei
Zhang
 a,
Jie
Zhang
b,
Georgina
Jeerh
a,
Peimiao
Zou
a,
Boyao
Sun
a,
Marc
Walker
c,
Kui
Xie
b and
Shanwen
Tao
*ad
a,
Jie
Zhang
b,
Georgina
Jeerh
a,
Peimiao
Zou
a,
Boyao
Sun
a,
Marc
Walker
c,
Kui
Xie
b and
Shanwen
Tao
*ad
aSchool of Engineering, University of Warwick, Coventry, CV4 7AL, UK. E-mail: s.tao.1@warwick.ac.uk
bKey Laboratory of Design and Assembly of Functional Nanostructures, Fujian Institute of Research on the Structure of Matter, Chinese Academy of Sciences, Fuzhou, Fujian 350002, China
cDepartment of Physics, University of Warwick, Coventry CV4 7AL, UK
dDepartment of Chemical Engineering, Monash University, Clayton, Victoria 3800, Australia
First published on 22nd August 2022
Abstract
Recently, due to the high energy density of ammonia, low source-to-tank energy cost and carbon free fuel, low-temperature direct ammonia fuel cells (DAFCs) have attracted extensive attention and play an important role in ammonia economy. In DAFCs, Pt-based materials have been the most studied electrocatalysts for the anodic ammonia oxidation reaction (AOR) and cathodic oxygen reduction reaction (ORR) over the past decade. However, the high cost of precious metal has markedly inhibited the large-scale application of DAFCs. Herein, a bifunctional material of ternary Ni4Cu5Fex (x = 0, 0.5, 1, 3, 5) alloy embedded in a carbon network was prepared by a simple solvothermal approach, and showed superior activity and durability towards both AOR and ORR. The excellent bifunctional catalytic activity of the Ni4Cu5Fe1/C sample was analyzed by experimental and calculational methods. Utilizing the bifunctional activities of this alloy, a symmetric DAFC was assembled with Ni4Cu5Fe1/C as both the anode and cathode, with a commercial anion exchange membrane (AEM) as electrolyte. The symmetric DAFC-Ni4Cu5Fe1/C showed a maximum current density of 67 mA cm−2 at 80 °C. To the best of our knowledge, this is the first report on a symmetric DAFC. This work not only reports a new AOR/ORR bifunctional catalyst, but also moves towards the development of low-cost DAFCs with simple structure.
1. Introduction
Fossil fuels contribute approximately 80 percent of the global mainstream energy demand, with this energy system being the source of approximately two thirds of global CO2 emissions.1–3 Due to the greenhouse effect caused by extensive use of traditional fossil energy, many countries have formulated carbon neutral plans to reduce carbon emissions over the next few decades.4 On one hand, the use of carbon-based fuels must be gradually reduced to overcome CO2 emissions; on the other hand, energy demand per capita is projected to continuously increase.5,6 Consequently, to address such demands while diminishing environmental impacts, it is imperative to develop sustainable and environmentally benign carbon-free renewable fuels.7,8 In this regard, hydrogen is often regarded as a promising energy vector, but its storage and transportation remain big challenges and restrict its large-scale applications.9 These problems can be solved if ammonia is used as an energy vector.5,10 Ammonia can be dissolved in water to create a solution with a solubility limit of around 35 wt% that can be kept in a glass container without the need for any extra equipment.2,11 In addition, it also can be readily liquified by increasing pressure to ∼10 bar at room temperature or by cooling to −33 °C at atmospheric pressure, which is much easier than that of hydrogen.5 Except for liquid storage, ammonia is found to be stored as a solid in metal–amine complexes. For example, hydrammine is a non-pressurized ammonia storage material and has an energy density similar to that of liquid ammonia.12 Benefiting from the liquid and solid storage, the technology of shipping and pipeline transfer of ammonia is well established in the current market. Around 175 million tonnes per year are produced worldwide.5,10Due to its carbon-free and easy storage and transportation properties, some pathways have been developed in order to take advantage of ammonia fuel, such as internal combustion engine and catalytic thermal decomposition.10,13–15 Recently, direct utilization of ammonia in fuel cells to generate electricity with high efficiency has been proposed and extensively studied.2,16–22 The direct ammonia fuel cell (DAFC) is an excellent alternative as an ammonia-fueled power source, especially for small-scale and domestic applications.23–25 In DAFCs, the ammonia oxidation reaction (AOR) occurs at the anode and oxygen reduction reaction (ORR) at the cathode. When an anion exchange membrane (AEM) acts as the electrolyte, the reactions for DAFC are shown below:25,26
Anode reaction:
| 2NH3 + 6OH− → N2 + 6H2O + 6e−E0 = −0.77 V | (1) |
Cathode reaction:
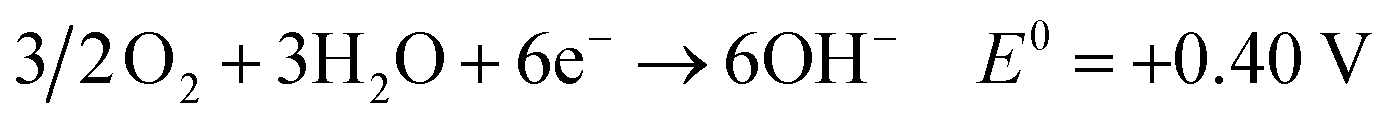 | (2) |
Overall reaction:
 | (3) |
Ammonia is directly oxidized to N2 at the anode side, whilst O2 reduction to OH− occurs at the cathode side. Generally, low temperature DAFCs operate at a temperature range from room temperature to 100 °C.2,26–28 Thus, DAFCs have received a great deal of attention as high-efficient power generation technologies.
While there is no doubt that the low temperature DAFC is a very appealing technology, some of the reasons that commercial DAFC device is at an early stage due to the expensive catalysts and sluggish catalytic reactions at the anode, i.e., electro-catalysts for AOR. Robust alkaline membrane with minimum ammonia cross-over is also desired to commercialize low temperature direct ammonia fuel cells.29,30 Pt-based materials are the most commonly studied electrocatalysts and researchers have systematically screened a range of Pt based bimetallic catalysts for ammonia electro-oxidation over the past two decades.31–34 Both experimental studies and mathematical modeling investigations have indicated that PtIr alloy has the highest AOR activity and lowest ammonia oxidation potential, owing to the catalyst ability to activate bonds and to make bonds.17,32,35–38 Furthermore, Pt-based catalysts have also shown great potential towards ORR, allowing them to act as cathodes in DAFCs. This is due to the straightforward dissociation process of oxygen adsorption on the surface, followed by electron transfer and protonation to create H2O.39 However, high cost, limited resources and poor stability have markedly inhibited this precious-based catalyst's large scale commercial and residential applications. At present, precious metals remain the primary catalysts, and opportunities therefore exist to develop lower-cost materials with equivalent or better activity.40
In the past five years, a wide variety of materials based on non-precious 3d transition metals, such as Ni, Cu, Co, Fe, have gained rapid recognition over Pt as an attempt to increase the practicality of AOR catalysts by lowering the cost.11,41–47 Amongst these metals, experimental studies have reported that Ni-based materials are the most promising catalysts with respect to AOR activity and high selectivity towards N2 in low to intermediate temperature ranges.45,48,49 However, compared to Pt, the d band center of the Ni metal is relatively wide, which means that Ni binds less strongly to the reactants and does not adsorb the reactants as efficiently as Pt.50 So, strengthening the bonding strength of Ni to ammonia is a strategical way to improve the catalytic activity of this catalyst. Previous work reported that the NiCu alloy, which embeds Cu in Ni, performed better due to the synergistic effect between Ni and Cu, leading to a stronger bond between Ni and N atoms.46,47 Furthermore, bimetallic alloys can carefully alter the d-band electron filling, Fermi level, and interatomic spacing, resulting in improved AOR electrocatalytic activity.51 It was found that bimetals would transform to hydroxides after the oxidation process, which were ultimately the active materials towards AOR.46,47,52 Zhu et al. recently reported that further introducing a lower electronegative Fe3+ cation can substantially improve the AOR activity of Ni1−xCuxOOH.42 DFT results showed that the Fe3+ was able to polarize the electrode cloud to a larger extent, decrease the thermodynamic energy and lower the kinetic energy. Additionally, the synergistic effect of the metal atoms in Ni–Cu–Fe–OOH also leads to the improvement in AOR. In this study, ternary Ni4Cu5Fex alloys were prepared by a simple solvothermal method. By optimizing the composition, the ternary Ni4Cu5Fe1 catalyst exhibited the highest AOR activity, which exceeded that of the Ni4Cu5 bimetallic counterpart. Moreover, NiCu bimetallic nanoparticles confined in a porous carbon network were also found to be good ORR catalysts.53 Interestingly, our study demonstrates that introducing Fe into the NiCu alloy further improves ORR performance remarkably. Furthermore, the ORR activity is not affected in the presence of low concentration of ammonia. As this catalyst is found to be active towards both AOR and ORR, a symmetric DAFC based on Ni4Cu5Fe1/C (DAFC-Ni4Cu5Fe1/C) was assembled and tested. Symmetric structures have been used in various electrochemical devices, such as electrolytic cells, supercapacitors and fuel cells.44,54–56 As the same material was used as both the anode and cathode, the symmetric fuel cell not only simplifies the integral device configuration, but also reduces the fabrication cost of a fuel cell.57,58 To the best of our knowledge, this is the first report on a symmetric DAFC. This work sheds light on using non-precious metal catalysts for DAFCs, particularly symmetric DAFCs.
2. Experimental details
2.1 Chemicals
The chemicals used in this experiment are nickel(II) chloride hexahydrate (NiCl2·6H2O, 99.3%, Alfa Aesar), copper(II) chloride dihydrate (CuCl2·2H2O, 99%, Alfa Aesar), iron(II) chloride (FeCl2, 99.5%, Alfa Aesar), sodium hydroxide (NaOH, 98%, Alfa Aesar), potassium hydroxide (KOH, 85%, Alfa Aesar), ammonium chloride (NH4Cl, 99.5%, Alfa Aesar), carbon black (Cabot Vulcan XC-72R), hydrazine monohydrate (N2H4·H2O, 98+%, Alfa Aesar), ammonia solution (NH3·H2O, 35%, 0.88 g mL−1, Fisher Chemical), Nafion solution (5% in lower aliphatic alcohols and water, Sigma-Aldrich), ethylene glycol ((CH2OH)2, Alfa Aesar), ethanol (C2H6O, ≥99.8%, Fisher Chemical), isopropanol (C3H8O, ≥99.8%, Sigma-Aldrich) and deionized water (H2O). All reagents were used directly without further refinement.2.2 Materials synthesis
The Ni4Cu5Fex/C catalysts (x = 0, 0.5, 1, 3, 5) were synthesized by a solvothermal method. In a typical procedure for the Ni4Cu5Fe1/C catalyst, 0.1 g carbon black was firstly added into 50 mL ethylene glycol and ultrasonicated for 30 min to form an ink. Subsequently, 8 × 10−4 mol NiCl2·6H2O, 1 × 10−3 mol CuCl2·2H2O, 2 × 10−4 mol FeCl2 were dissolved in the ink and continuously stirred for another 15 min. Then, 1 g NaOH and 5 mL N2H4·H2O were added into the mixture solution. After being stirred for 1 h, the black mixture was transferred to a Teflon-line stainless autoclave and reacted at 200 °C for 10 h. The sample was separated from the solution by centrifuging at 7000 rpm min−1 for 6 min. The product was then washed three times with deionized water and isopropanol until the solution became transparent. After being dried at 60 °C in an oven for 12 h, the Ni4Cu5Fe1/C powders were obtained. For the preparation of the Ni4Cu5/C catalyst, a similar process was conducted where no FeCl2 was added into the solution.2.3 Electrode preparation
The catalyst ink for AOR was prepared by a similar method reported in our previous work.44 Carbon cloth (Fuel Cell Store, product code: 7302007, wet proofing: 1–5%, thickness: 0.36 mm, plane electrical resistivity < 5 mΩ cm−2) was used as the conductive substrate in this research. Before electrodes were made, carbon cloth (2.5 × 2.5 cm2) was washed and sonicated in dilute hydrochloric acid, deionized water, and isopropanol. 0.1 g catalyst was mixed with 500 μL deionized water, 500 μL isopropanol, and 40 μL Nafion solution. The slurry was ultrasonicated for 30 min and then continuously stirred at room temperature overnight. Subsequently, the as-prepared catalyst ink was brushed onto the washed carbon cloth and the carbon cloth was dried overnight in a fume cupboard. The loading of catalyst was about 2.5 mg cm−2 for both anode and cathode. For comparison, Pt/C electrode (commercial 20 wt% platinum on carbon black, Alfa Aesar) was prepared in the same way and the loading of Pt/C on carbon cloth was 3.5 mg cm−2 (loading for pure Pt was 0.7 mg cm−2).The catalyst ink for ORR was prepared by blending catalyst powder (10 mg) with 100 μL Nafion solution and 1000 μL ethanol in an ultrasonic bath. 5 μL of catalyst ink was then pipetted onto the glassy carbon disk. The working area of the electrode was ∼0.196 cm−2, leading to a catalyst loading of 0.232 mg cm−2 for all samples for rotating electrode tests. The electrode was dried at 80 °C before electrochemical tests.
2.4 Materials characterizations
X-ray diffraction (XRD) patterns of the samples were recorded with a 3rd generation Malvern Panalytical Empyrean equipped with a Cu Kα1/2 radiation (0.15419 nm). Scans were made in the range 20–80° with a step size of 0.01313° and a counting time of ∼34.4 s per step. Scanning electron microscopy (SEM) images and energy dispersive X-ray spectroscopy (EDS) were obtained through a field emission SEM (FE-SEM, Zeiss Supra 55-VP). High resolution transmission electron microscope (HRTEM) and high-angle annular dark-field transmission electron microscope (HAAD-TEM) were conducted by a JEOL ARM 200F equipped with an energy-dispersive X-ray spectrometer that allowed elemental composition analysis. Raman spectra were performed with the 532 nm wavelength excitation (LabRAM HR800, HORIBA Jobin Yvon, Villeneuve-d'Ascq, France). X-ray photoelectron spectroscopy (XPS) measurements were conducted using a monochromated Al Kα X-ray source on a Kratos Axis Ultra DLD spectrometer (Kratos Analytical, Manchester, UK). Data were taken at a take-off angle of 90° with respect to the surface plane and analyzed using the Casa XPS package, using mixed Gaussian–Lorentzian lineshapes and Shirley backgrounds. In order to prevent the surface becoming positively charged during the test, charge neutralization was employed and the spectra was subsequently referenced to the C–C/C–H peak at 285.0 eV during analysis. XRF spectra were acquired using a Rigaku Primus IV wavelength dispersive X-ray fluorescence spectrometer equipped with a 4 kW X-ray tube. Samples were scanned for approximately 5 min to obtain sufficient counts for quantitative analysis to be carried out. The stability of AEM in ammonia fuel was conducted by Fourier transform infrared spectroscopy (FTIR) spectra. One AEM was immersed in 3 M KOH solution and the other was in 3 M KOH + 7 M NH3·H2O. Before FTIR test, both AEMs were left for 24 h and then washed by deionized waster. All FTIR data were collected on a Thermo Fisher iS50R FTIR spectrometer equipped with a SpecAc Golden Gate ATR, using a KBr Beam splitter and a DTGS room temperature detector. FTIR spectra were recorded in the wavenumber range of 550 cm−1 and 4000 cm−1 with a resolution of 4 cm−1. Automatic atmospheric suppression was performed on all data, with a fresh background for each sample.2.5 Electrochemical measurements
Electrochemical AOR and ORR performance tests were both conducted in alkaline solution and room temperature using a three-electrode system. The prepared electrode, Ag/AgCl (sat. KCl) electrode and platinum foil were used as the working, reference, and counter electrode respectively.For the AOR test, the electrochemical characterization was determined by cyclic voltammetry (CV), linear sweep voltammetry (LSV), and chronoamperometry techniques conducted on the Solartron 1287A Electrochemical Station. Before testing, all electrodes were electrochemically activated by potential cycling between 0 V to 0.7 V vs. Ag/AgCl (100 cycles) at scanning rate of 50 mV s−1 to obtain stable results. Then, the CV and LSV measurements were recorded from 0 V to 0.7 V vs. Ag/AgCl with a scan rate of 5 mV s−1 and 2 mV s−1 respectively. Chronopotentiometry tests for the Ni4Cu5/C, Ni4Cu5Fe1/C and Pt/C electrodes were recorded in 0.5 M KOH with 55 mM NH4Cl at a fixed potential of 0.5 V vs. Ag/AgCl. EIS for AOR was conducted using the Solartron 1260A Electrochemical Station at a frequency range of 1 M Hz to 0.01 Hz and fixed potential of 10 mV bias.
For the ORR test, the electrochemical characterization was determined by CV, LSV, and chronopotentiometry techniques conducted on the Solartron 1470E multichannel cell test system. 0.1 M KOH solution was used as the electrolyte. Before electrochemical measurements, the solution was deaerated by continuous purging with high purity Ar/O2 for 30 min. The electrode was activated by running CV scans between 0.1 V and −0.5 V vs. Ag/AgCl at a scan rate of 10 mV s−1 for three cycles. Then CV and LSV curves for ORR were measured at a scan rate of 10 mV s−1. Chronopotentiometry tests for the Ni4Cu5/C and Ni4Cu5Fe1/C electrodes were recorded in 0.1 M KOH at fixed potential of −0.3 V vs. Ag/AgCl.
The rotating disk electrode (RDE, PINE, AFMSRCE 3013) tests were performed at various rotation speeds ranging from 100 to 1600 rpm with a scan rate of 10 mV s−1. The electron transfer number (n) of oxygen molecule was calculated from the Koutecky–Levich plots using following equation:59–61
 | (4) |
| B = 0.62nFC0D02/3V−1/6 | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1), and V is the kinematic viscosity of the electrolyte (0.01 cm2 s−1).
485 C mol−1), C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1), and V is the kinematic viscosity of the electrolyte (0.01 cm2 s−1).
2.6 Density functional theory (DFT) calculations
First-principles and the density functional theory (DFT) calculation method were performed using the Vienna Ab Initio Simulation Package (VASP).62,63 The projected-augmented-wave (PAW) method was used. The generalized gradient approximation (GGA) and Perdew–Burke–Ernzerhof (PBE) functional are chosen to describe the exchange-related interactions.64The plane wave cut-off energy was set to 400 eV, the Ni4Cu5 and Ni4Cu5Fe1 alloys were constructed according to experimental results. 10−4 eV and 0.01 eV Å−1 were used to converge the energies and residual forces respectively. For the Ni4Cu5Fe1 alloy, a 2 × 2 × 2 supercell was calculated on a 4 × 4 × 4 k-point grid (Fig. S14†). The superstructure was employed with five layers and a vacuum gap of 22 Å was established to prevent lower layers interacting with the periodic image. The bottom two layers were fixed and top three layers were relaxed. A 3 × 3 × 1 k-point grid was constructed to sample the Brillouin zone. The slab model of Ni4Cu5 was treated similar to the Ni4Cu5Fe1 superstructure.
For the Ni4Cu5Fe1 alloy, the energy Eads of each adsorbed intermediate was calculated by:
| Eads = Etotal − Esub − Eadatom | (6) |
| ΔG = Eads − ΔZPE − TΔS + ΔGU + ΔGpH | (7) |
ln10 × pH.
The free energy changes of the four processes for ORR could be described as:
| ΔG1 = ΔGOOH* − 4.92 ΔG2 = ΔGO* − ΔGOOH* ΔG3 = ΔGOH* − ΔGO* ΔG4 = −ΔGOH* | (8) |
The overpotential (η) used to evaluate the catalytic performance could be calculated using the following:65
| ηORR = max〈ΔG1, ΔG2, ΔG3, ΔG4〉 + 1.23 eV | (9) |
2.7 Fuel cell testing
An anion exchange membrane (AEM, Fumasep FAA-3-50, product code: 5041622) was employed as the electrolyte. In order to activate the membrane, the AEM was immersed in 1 M KOH solution for 2 h and then washed with deionized water. The symmetric fuel cell was successfully assembled using the same Ni4Cu5Fe1/C as both the anode and cathode. The effective area of the fuel cell was 1 × 1 cm2. A solution of 3 M KOH + 7 M NH3·H2O was pumped as the anodic flow field at 2 mL min−1, whilst 150 mL min−1 humidified air (CO2-free) was flowed as the cathodic flow field. Back pressures of 1.4 barg were applied to the anode and cathode. The polarization curves and power density curves were tested and obtained at different temperatures using the Solartron 1287A Electrochemical Station. EIS of the fuel cell was conducted by the Solartron 1260A Electrochemical Station at a frequency range of 1 M Hz to 0.01 Hz and fixed potential of 10 mV bias.3. Results and discussion
3.1 Structure and morphology
The strategy for synthesizing the Ni4Cu5Fex/C catalyst is illustrated Fig. 1a. Firstly, the precursor ions and carbon black were uniformly dispersed in the ethylene glycol solvent by continuous stirring. Then, an appropriate amount of reducing agent was added to allow for the reduction of all transition metals. After a solvothermal reaction at 200 °C for 10 h, a composite of the ternary Ni4Cu5Fex nanoparticles and carbon black was successfully obtained. Finally, the catalyst was separated and collected by washing and centrifuging. The XRD patterns of Ni4Cu5/C and different amounts of Fe embedded Ni4Cu5Fex/C (x = 0.5, 1, 3, 5) are shown in Fig. 1b. In Ni4Cu5/C, diffraction peaks appeared at 43.3°, 50.5° and 74.2° corresponding to the (111), (200) and (220) planes of the bimetal respectively, which conforms with the face-entered cubic NiCu (JCPDS no. 65-9047). Compared to the control sample of Ni4Cu5/C, similar peaks were observed for Ni4Cu5Fex/C. This is also consistent with a previous report on Ni–Cu–Fe electrodes synthesized by electrodeposition.42,66 It is worth noting that the Ni4Cu5Fe5/C sample was a mixture of NiCuFe, carbon and Fe3O4 (JCPDS no. 89-0691), implying a second phase is formed when x ≥ 5. Therefore, only Ni4Cu5Fex/C samples with 0 ≤ x ≤ 3 were investigated in the following experiment. To clearly determine the precise composition in alloys, the elemental ratio in Ni4Cu5Fex was characterized by XRF. The results showed that the ture compositions of the samples coincide with the designed structures (Table S1†). For example, the elemental ratio in Ni4Cu5Fe1 sample was Ni:Cu:Fe = 4:5.63:0.82. There are some deviation in the final composition from the initial molar ratio which is common when the samples were synthesised by a hydrothermal synthesis process.
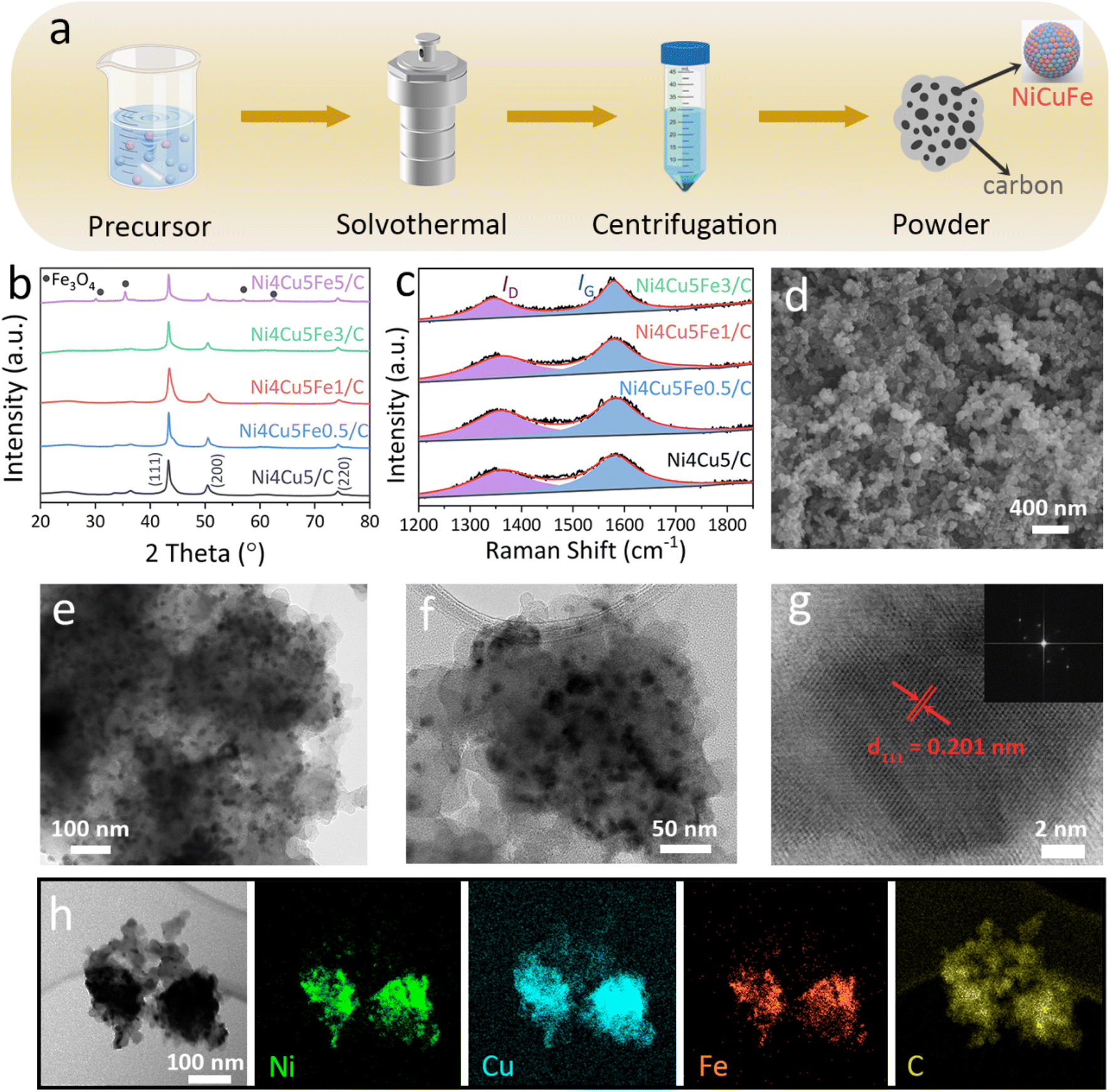 | ||
| Fig. 1 Synthesis and characterization. (a) Schematic illustration of the preparation process of ternary NiCuFe alloy by solvothermal synthesis. (b) XRD patterns of Ni4Cu5/C, Ni4Cu5Fe0.5/C, Ni4Cu5Fe1/C, Ni4Cu5Fe3/C and Ni4Cu5Fe5/C. (c) Raman spectra of Ni4Cu5/C, Ni4Cu5Fe0.5/C, Ni4Cu5Fe1/C and Ni4Cu5Fe3/C. (d) SEM image, (e and f) TEM images and (g) HRTEM image (inset: FFT pattern of the corresponding nanoparticle) of Ni4Cu5Fe1/C sample. (h) HAAD-STEM image and corresponding EDS elemental mappings of Ni, Cu, Fe and C. | ||
Compared to the characteristic high peak intensities of the alloy component, the XRD diffraction peaks of carbon are hardly visible. Fortunately, the existence of the carbon matrix can be confirmed by Raman spectroscopy. Fig. 2c shows the Raman spectra of all of the above four samples, in which the peaks positioned at ∼1357 and ∼1583 cm−1 represent the disorder carbon (D-hand) and graphitic carbon (G-band) respectively.53 Commonly, the graphitization degree and defect density of carbon-based materials can be roughly estimated by calculating the intensity ratio ID/IG of the D and G bands.67 The calculated ID/IG values were found to be 0.81, 0.88, 0.91 and 0.78 for the Ni4Cu5/C, Ni4Cu5Fe0.5/C, Ni4Cu5Fe1/C and Ni4Cu5Fe3/C samples respectively. The comparatively higher ID/IG value of the Ni4Cu5Fe1/C electrocatalyst compared to other samples points to the presence of higher number of defect sites in the Ni4Cu5Fe1/C. Such defect sites are known to be beneficial towards enhancing various activity, such as ORR, OER and HER.48,53,68
 | ||
| Fig. 2 AOR activity. CV curves of Ni4Cu5/C and Ni4Cu5Fe1/C in (a) 0.5 M KOH, (b) 0.5 M KOH + 55 mM NH4Cl at a scan rate of 5 mV s−1. (c) CV curves of Ni4Cu5Fe1/C in 0.2 M/0.5 M/1 M KOH + 55 mM NH4Cl. (d) Nyquist plots of EIS spectra measured for the Ni4Cu5Fe1/C in 55 mM NH4Cl with different KOH concentration. (e) CV curves of Ni4Cu5Fe1/C initially, after 50 cycles and 100 cycles in 0.5 M KOH with 55 mM NH4Cl at a scan rate of 50 mV s−1. (f) Chronoamperogram of Ni4Cu5/C and Ni4Cu5Fe1/C electrodes in 0.5 M KOH with 55 mM NH4Cl at fixed potential of 0.5 V vs. Ag/AgCl. | ||
The morphology of the as-prepared Ni4Cu5Fe1/C electrocatalyst was characterized by FESEM. The sample was composed of nanoparticles and no large particles were observed (Fig. 1d). Nanoparticle spatial distributions may have a large significance on catalyst stability.69 According to the EDS mapping images and EDS spectrum of the Ni4Cu5Fe1/C powders (Fig. S1†), elements Ni, Cu, Fe can be clearly observed and the distribution of these elements is homogeneous. However, it is hard to observe the distribution of C in the powders due to the influence of the conductive tape. In order to reveal the intermetallic microstructure, TEM measurements on Ni4Cu5Fe1/C were carried out. From Fig. 1e and f, small alloy particles embedding carbon nanoparticles can be seen. The ultra-small particle sizes and large surface areas of Ni4Cu5Fe1/C lead to more exposed active sites for the catalytic reaction to take place. It is worth noting that the carbon networks in the catalyst enable fast electron transport, which is a key factor for the catalytic process. The HRTEM image (Fig. 1g) and Fast-Fourier-Transform (FFT) pattern (inset in Fig. 1g) display the interplanar spacing of 0.201 nm corresponding to d111 of fcc-alloy. Ni, Cu, Fe atoms were uniformly distributed over the alloy particles at the same locations, further proving the formation of the Ni–Cu–Fe alloy (Fig. 1h).
3.2 Bifunctional catalytic activities
The AOR activities of the Ni4Cu5Fex/C catalysts were measured in alkaline media. Firstly, CV measurements of binary Ni4Cu5/C and ternary Ni4Cu5Fe1/C were performed in Fig. 2a. Both samples showed a pair of similar redox peaks in 0.5 M KOH media, which are attributed to the transformation between the Ni2+ and Ni3+ species.42,47 In addition, compared to the binary Ni4Cu5/C catalyst, the modified ternary alloy showed a higher current density. When 55 mM NH4Cl was added into the electrolyte, a remarkable increase of anodic current density was observed in both samples, with the ternary alloy presenting a higher AOR activity (Fig. 2b). The LSV curves also demonstrated the positive effects of Fe in the Ni–Cu–Fe catalyst towards the electro-oxidation of ammonia (Fig. S2†), which is consistent with previous results.42 To gain insight into the effect of Fe on the ammonia oxidation performances of Ni4Cu5Fex alloys, different amounts of Fe embedded alloys were synthesised and tested for their catalytic activity. From Fig. S3,† it can be deduced that the optimal x amount of Fe in Ni4Cu5Fex is 1. In order to understand the AOR process, CV curves of Ni4Cu5Fe1/C obtained under different scan rates were acquired (Fig. S4a†). There was a distinct linear trend against scan rate (2–25 mV s−1). A linear relationship (R2 = 0.998) between the square root of the scan rates and the values of current density was obtained, indicating a diffusion-controlled AOR process of Ni4Cu5Fe1 catalyst (Fig. S4b†).70,71The working conditions, such as the alkalinity and ammonia concentration are crucial to the ammonia oxidation performance. The effect of pH value on the AOR activity is shown in Fig. 2c, which reveals that a higher alkaline concentration led to higher current density. When an ammonia solution containing 0.2 M KOH was used as electrolyte, the AOR activity of Ni4Cu5Fe1/C was not good. However, as the OH− concentration increased from 0.5 M to 1 M, the current density enhanced from 31 to 71 mA cm−2 at a potential of 0.55 V vs. Ag/AgCl. The LSV curves also confirmed the positive effect of alkaline conditions (Fig. S5†). By comparing a series of Nyquist plot semicircles, it was clearly revealed that charge transfer resistance was relatively smaller in high KOH concentration (Fig. 2d), indicating the rapid mass transfer and charge transport in the presence of higher concentration of KOH solution.50 When the ammonia content was raised from 20 to 100 mM at a potential of 0.55 V vs. Ag/AgCl, the current density increased from 17 to 31 mA cm−2 (Fig. S6†). These findings show that by increasing the pH value and ammonia concentration, the AOR activity of the Ni4Cu5Fe1/C catalyst improves. This is also compatible with eqn (1), since the reaction shifts to the right as reactant concentration increases.
Besides activity, long-term stability is another essential criterion for the applicability of catalysts. The CV curves of the Ni4Cu5Fe1/C electrode in 0.5 M KOH + 55 mM NH4Cl solution were measured over 100 cycles at a scan rate of 50 mV s−1 (Fig. 2e). In comparison with the initial CV curve, a slightly increase in current density at 0.5 V vs. Ag/AgCl was observed in the last CV curve, which was attributed to the activation of NiCuFe by possible formation of metal hydroxides and oxyhydroxides.47,72 Chronoamperogram testing was further studied to evaluate the stability of the ternary alloy electrode using a potential fixed at 0.5 V vs. Ag/AgCl in 0.5 M KOH + 55 mM NH4Cl (Fig. 2f). Compared to the continuous declining current of un-modified Ni4Cu5/C, the Ni4Cu5Fe1/C catalyst was able to hold excellent AOR activity based on its high current density. It is worth noting that the current density of Ni4Cu5Fe1/C gradually increased during the first 4 h, which is consistent with the observation on CV cycling test. All the above results demonstrate the excellent AOR activity and stability of the ternary Ni4Cu5Fe1/C catalyst.
Considering the potential bifunctional activity, the ORR activity of Ni4Cu5Fe1/C alloy was also evaluated via CV, LSV and chronoamperometric techniques in alkaline solution at room temperature. There was a strong ORR cathodic peak at −0.14 V vs. Ag/AgCl in the CV curves of the Ni4Cu5Fe1/C catalyst recorded in O2-saturated 0.1 M KOH solution. However, there was no obvious reduction peak recorded in Ar-saturated solution (Fig. 3a). Remarkably, Ni4Cu5Fe1/C displayed a more positive oxygen reduction peak and a higher CV peak current density than those of Ni4Cu5/C (Fig. S7†), implying a strong electronic interaction between Fe and Ni/Cu. These results preliminarily confirmed the excellent ORR activity of Ni4Cu5Fe1/C. The ORR polarization curves were recorded at a rotation speed of 1600 rpm and proved that Ni4Cu5Fe1/C demonstrated a much higher onset potential, half-wave potential and larger current density (Fig. 3b). To gain further insight in the ORR kinetics, Tafel plots of these two catalysts were fitted (Fig. 3c). The Tafel slope of Ni4Cu5Fe1/C (103 mV dec−1) was smaller than that of Ni4Cu5/C (111 mV dec−1), indicating that the incorporation of Fe into NiCu induces faster electrokinetic processes of water oxidation on the surfaces of ternary alloy.
 | ||
| Fig. 3 ORR activity. (a) CV curves of Ni4Cu5Fe1/C in Ar or O2-saturated 0.1 M KOH. (b) LSV curves and (c) Tafel plots of Ni4Cu5/C and Ni4Cu5Fe1/C in O2-saturated 0.1 M KOH at 1600 rpm. (d) LSV curves of Ni4Cu5Fe1/C in O2-saturated 0.1 M KOH at different rotation speeds. (e) LSV curves of Ni4Cu5Fe1/C in O2-saturated 0.1 M KOH or 0.1 M KOH + 10 mM NH4Cl. (f) Chronoamperogram of Ni4Cu5/C and Ni4Cu5Fe1/C electrodes in 0.1 M KOH at fixed potential of −0.3 V vs. Ag/AgCl. | ||
Furthermore, RDE measurements were performed at different rotating speeds between 100 to 1600 rpm to obtain the Koutecky–Levich (K–L) plots of the Ni4Cu5Fe1/C catalyst. The limiting current density of this catalyst increased with increasing rotating speed due to the gradually shortened diffusion distance (Fig. 3d).73 The calculated average transfer number of Ni4Cu5Fe1/C varied from 3.8 to 4 (Fig. S8†), illustrating the ORR process of the catalyst is a 4e− dominant process. This result suggests that the Ni4Cu5Fe1/C catalyst could directly reduce oxygen to water and the migration of adsorbed oxygen intermediates is likely to be the rate-determining step for the ternary alloy catalyst.59 The effect of scan rate on the cathodic peak currents at −0.3 V vs. Ag/AgCl is shown in Fig. S9a.† The linear relationship between the square root of the scan rate and current density indicates a diffusion-controlled process for Ni4Cu5Fe1/C (Fig. S9b†).74
For the practical applications of DAFC, tolerance to ammonia crossover is a challenging issue for ORR catalysts since this results in a drop in cell power and efficiency.18 Compared to common precious-metal-based catalysts where ORR activity would significantly decrease due to surface poisoning,75 the polarization curve of the ternary alloy showed negligible change in the presence of NH4Cl. This indicates that ammonia does not noticeably affect ORR activity of the catalyst (Fig. 3e). The carbon embedded structure may play an important role in the ORR catalyst in terms of ammonia tolerance since a similar phenomenon has been confirmed in other methanol-tolerant ORR electrocatalysts.76 In addition, Ni4Cu5Fe1/C showed good electrochemical stability in alkaline solution, maintaining high current applied after 9000 s, surpassing the Ni4Cu5/C stability under the same experimental conditions (Fig. 3f). Overall, the high ORR activity, tolerance to ammonia and robust stability of Ni4Cu5Fe1/C indicates that this catalyst could also be a good cathode in DAFCs. With the AOR/ORR bifunctional activity of Ni4Cu5Fe1/C, it becomes possible to apply it as both anode and cathode in a symmetric direct ammonia fuel cell.
3.3 Bifunctional catalytic mechanism of Ni4Cu5Fe1/C
In the DAFC, the catalytic reaction at the anode side is AOR and at the cathode side is ORR. The Ni4Cu5Fe1/C electrode after AOR is hereby referred to as Ni4Cu5Fe1/C anode and the Ni4Cu5Fe1/C electrode after ORR as Ni4Cu5Fe1/C cathode. SEM images of the Ni4Cu5Fe1/C electrode before and after the catalytic reaction are shown in Fig. 4a–c. Ni4Cu5Fe1/C nanoparticles before the test were uniformly deposited on the carbon cloth, although some agglomeration of the particles was observed (Fig. 4a). According to the EDS mapping images and EDS spectra (Fig. S11†), elements Ni, Cu, and Fe can be clearly observed. The SEM images of the Ni4Cu5Fe1/C anode and cathode showed no observable change in composition and distribution of the Ni4Cu5Fe1 nanoparticles (Fig. 4b, c and S11, S12†), indicating robust structural stability of the carbon capsulated ternary alloy. The EDS analysis confirms that the nanoparticles were still composed of the elements Ni, Cu, and Fe (Fig. S10–S12†). | ||
| Fig. 4 The reaction mechanism. SEM images of (a) Ni4Cu5Fe1/C electrode before test, (b) Ni4Cu5Fe1/C anode and (c) Ni4Cu5Fe1/C cathode after test. (d) XRD patterns, (e) Raman spectra, XPS spectra of (f) Cu 2p3/2 and (g) Cu–Ni 3p in Ni4Cu5Fe1/C, Ni4Cu5Fe1/C anode and Ni4Cu5Fe1/C cathode. For clarity, a larger version of the fit of the Cu 2p3/2 spectrum for the Ni4Cu5Fe1/C anode is given in Fig. S13.† (h) The Ni, Cu, Fe atomic percentage of Ni4Cu5Fe1/C, Ni4Cu5Fe1/C anode and Ni4Cu5Fe1/C cathode. (i) Gibbs free energies for the ORR pathways for Ni4Cu5 and Ni4Cu5Fe1. | ||
To gain further insight on the structural changes, XRD analysis of the Ni4Cu5Fe1/C electrodes before and after tests was conducted (Fig. 5d). Unsurprisingly, before the catalytic reaction, the Ni4Cu5Fe1/C electrode only consisted of the ternary alloy and carbon cloth substrate. After the ammonia oxidation process, two new phases of metal hydroxide and CuO appeared on the surface of the anode. The metal hydroxide belonged to the Ni–Cu–Fe–(OH)2 phase and can be supported by the transformation of NiCuFe → Ni–Cu–Fe–(OH)2 → Ni–Cu–Fe–OOH which has been proven in other Ni-based AOR catalysts.42,47 As mentioned above, nickel hydroxides and/or oxyhydroxides are the real active centers during ammonia oxidation. The formation of CuO at the surface of AOR catalyst has a positive effect to AOR. On one hand, CuO itself is an active catalyst to AOR, which has been proved in previous reports.48,77,78 For example, through Raman and CO-temperature-programmed reduction (TPR) measurements, Han et al. proved that superoxo species (O2−) as key active sites produced on CuO surface have led ammonia molecules to be stepwise oxidized.78 On the other hand, the presence of CuO can improve the performance of the catalysts. Khan et al. reported that CuO dispersed on the surface of the catalyst generate synergy and coupling effects for ammonia adsorption.79 In addition, active oxygen is essential for the AOR and CuO exhibits excellent oxygen storage and activation abilities.48,78 Therefore CuO dispersed on the surface of the catalyst is positive to ammonia oxidation process. The evolution of the composition during the AOR process leads to an improvement of catalytic activity as can be seen in CV cycling and chronoamperogram tests (Fig. 2e and f). For the Ni4Cu5Fe1/C cathode, only a new metal hydroxide phase is observed. A synergic effect between the metal and metal hydroxide accelerates high eletrocatalytic activity and fast electron transfer.80
 | ||
| Fig. 5 Fuel cell performance. (a) The schematic diagram of fuel cell. (b) Polarization curves and power density curves of symmetric DAFC-Ni4Cu5Fe1/C at different temperatures. (c) Nyquist plots of EIS spectra measured for symmetric DAFC-Ni4Cu5Fe1/C. (d) Comparison of OCVs and power densities from this work and those of other direct ammonia fuel cells based on non-precious-metal catalysts. | ||
The structural changes before and after catalytic reactions were also confirmed by Raman spectra (Fig. 4e). Compared to the original ternary alloy catalyst, the electrodes after AOR and ORR both presented two obvious Raman bands, which are located at 230–310 cm−1 and 460–600 cm−1 respectively. According to literature, these two broad Raman bands both correlate to Ni(OH)2, and the broad sharp bands imply that the introduction of metal hydroxides at the surface may be rich in defects.81–83 It is worth noting that the Raman band of 230–310 cm−1 in Ni4Cu5Fe1/C anode was higher and broader than that in the Ni4Cu5Fe1/C cathode. In order to understand the structure better, this Raman peak can be fitted with three peaks at positions of 272, 280 and 323 cm−1. The former fitting peak belonged to Ni(OH)2. The peak observed at 280 and 323 cm−1 are attributed to the Ag mode and Bg of CuO respectively.84–86 Furthermore, the Raman observation is consistent with the XRD results.
The phenomenon of surface reconstruction was also confirmed by XPS analysis. Fig. 4f showed the Cu 2p core level spectra of Ni4Cu5Fe1/C before and after catalytic reaction. All the spectra were corrected using the C 1s signal located at 285.0 eV. The XPS spectra of Ni4Cu5Fe1/C and Ni4Cu5Fe1/C cathode both showed five characteristic peaks located at 933.3 eV (Cu(0)/Cu2O), 935.2 eV (Cu(OH)2), along with components at 939.9 eV, 942.7 eV and 945.0 eV corresponding to Cu(OH)2 shake-up features. It is worth noting that Cu(0) and Cu2O is essentially impossible to distinguish by this deconvolution due to the chemical shift being lower than the resolution of the analyser.87 Conventionally, one would attempt to resolve this using the Cu LMM Auger emission, but the intensity was too low to facilitate an adequate spectrum. As fitting the curves of the Ni4Cu5Fe1/C anode are complex, an enlarged figure is placed in Fig. S13.† Beside the five peaks listed above, the Ni4Cu5Fe1/C anode exhibited five additional components at binding energies of 933.6 eV, 935.0 eV, 941.0 eV, 942.3 eV, 944.2 eV, which could be attributed to CuO.88 The phenomenon of a new CuO phase after AOR is consistent with the observation in XRD. The XPS data from the Cu–Ni 3p regions in Fig. 4g showed that all samples only contained Ni2+ (Ni(OH)2). Unfortunately due to the overlap between Fe LMM Auger emission and the Ni 2p photoemission, it was not possible to extract meaningful conclusions from the Ni 2p energy level. We also noted that the ratios of surface Ni/Cu/Fe varied significantly after catalytic reactions. The atomic percentages of the three different metals before and after catalytic reaction were calculated and presented in Fig. 4h. In the original Ni4Cu5Fe1/C sample, all three metal elements were observed, and the content of Cu was the highest and the content of Fe was the lowest. This coincided with the preparation of the catalyst. After catalytic reactions, more Ni appeared on the external surface of the anode and cathode, indicating the formation of the hydroxide. This once again confirms the important role of the nickel hydroxide in AOR and ORR. More interestingly, the atomic percentages of Fe also increased to 5% and 4% at anode and cathode side respectively. This means more Fe was exposed on the surface during the ammonia oxidation and oxygen reduction process, and is benefical for these two catalytic reactions.
The positive effect of Fe on NiCu-based catalysts for the ammonia oxidation process has been previously demonstrated using DFT simulation.42 It was found that Fe atom preferred to stay close to the Cu atom rather than the Ni atom to form a cluster for the sake of achieving lower formation energy. During the AOR process, the adsorption of ammonia on the catalyst surface was considered to be the limiting step due to the high energy barrier.42,44 When Fe was doped into NiCu, the energy barrier to ammonia adsorption decreased sharply, which was attributed to the decline of thermodynamic energy and the lower of kinetic energy. This calculation is consistent with the result of our experiment, which also confirmed excellent AOR performance of Ni4Cu5Fe1 alloy. In order to further clarify the effect of Fe in the Ni4Cu5Fe1 catalyst for the ORR, the Gibbs free energies for appropriate reaction steps on two models were calculated. A similar calculation was performed for Ni4Cu5. The crystal structure models of these two alloys were built (Fig. S14†). Overpotential is an important parameter to evaluate the catalytic performance of the ORR. Fig. 4i shows the Gibbs free energy diagram of the initial steps in ORR. Ni4Cu5Fe1 exhibits a lower ηORR value (0.86 eV) than that of Ni4Cu5 (1.19 eV), which clearly illustrates the positive effect of Fe on ORR. This suggests that the activity for ORR can be very different at different level of Fe doping.
3.4 Performance of symmetric direct ammonia fuel cell
A schematic illustration of a typical symmetric DAFC is depicted in Fig. 5a. The membrane electrode assembly (MEA) was composed of the Ni4Cu5Fe1/C electrodes and an AEM. Oxygen consumes electrons and generates hydroxyl ions at the cathode side. Hydroxyl ions migrate from cathode to anode side through the AEM. At the anode side, ammonia molecules react electrochemically with hydroxyl ions and are oxidized to provide electrons. Different from other reported low temperature DAFC which consists of two kinds of catalysts, the anode and cathode used in this work are identical, both being based on Ni4Cu5Fe1/C catalyst (loading: 2.5 mg cm−2). The design of a symmetric DAFC not only simplifies the integral device configuration, but also saves time and effort by only preparing a single catalyst. Additionally, in order to be close to practical use, the cathode side is fed with compressed air (CO2-free) instead of pure oxygen in our experiment.According to previous work, cell operating conditions, such as hydroxide concentration and operating temperature, have an important effect on catalytic kinetics, and consequently, fuel cell performance.18 Ideal hydroxide concentration for DAFCs has been reported to be 3 M KOH, since this can minimize the AOR onset potential and obtain a maximum peak current density.17,18 Therefore, in the following experiments, the anode feedstock is fixed to 3 M KOH + 7 M NH3·H2O. Firstly, the stability of AEM under ammonia solution was checked. From the Fig. S15† we can see the structure of AEM did not change when it immersed in 3 M KOH + 7 M NH3·H2O for 24 h. It indicated the good stability of AEM in ammonia solution for a long time. Then, the symmetric DAFC-Ni4Cu5Fe1/C was tested at different operating temperatures and the results are shown in Fig. 5b. When operating at a low temperature (40 °C), the cell OCV and power density were both poor, 0.35 V and 0.29 mW cm−1 respectively. This is because the overpotential of catalytic reactions is high and the DAFC has not been fully activated. The cell performance increased as the operating temperature was raised from 40 °C to 80 °C (Fig. 5b). A significant improvement in AOR was observed at higher temperature, which is due to the enhancement of onset potential and larger peak current density of the AOR.18 To further study the effect of temperature on the fuel cell performance, the EIS spectra for the symmetric DAFC at different operating temperatures were tested. From Fig. 5c, it can be seen that the cell resistance was very high at a low temperature (40 °C). A significant decrease was observed when the temperature increased from 40 °C to 60 °C following by a sluggish increase from 60 °C to 80 °C. The measured area-specific resistance (ASR) was 0.8 Ω cm2 at 80 °C. At 80 °C, a maximum OCV and power density of 0.62 V and 8.9 mW cm−2 were obtained respectively. A comparison of the symmetric DAFC with other reported low temperature ammonia fuel cells based on non-precious metal-based catalysts are noted (Fig. 5d and Table S2†).16,25,89–91 The results demonstrate the excellent bifunctional catalytic activity of ternary Ni4Cu5Fe1 which can be used in a symmetric DAFC. It is worth noting that the stability of the symmetric DAFC is not good at high temperature due to the limitation of the commercial AEM with an operating temperature up to 60 °C and for a very short time at 80 °C. The power density of the symmetric DAFC may be further improved by increasing the operating temperature if a more stable alkaline membrane is used.29
4. Conclusions
In summary, we have successfully constructed a bifunctional ternary Ni4Cu5Fex/C alloy catalyst via a simple solvothermal method for DAFC applications. TEM images showed that alloy particles embedded in a carbon network formed nanoparticle catalysts. The optimal NiCuFe catalyst at a nominal molar ratio of 4:5:1 showed excellent AOR activity and stability as compared to the Ni4Cu5/C electrode in alkaline solution. The kinetic activity of AOR is strongly dependent on the alkaline concentration and ammonia concentration, which resulted in higher pH values and ammonia content facilitating AOR activity of the Ni4Cu5Fe1/C catalyst. In addition, the introduction of Fe into NiCu alloy also improved ORR activity. RDE measurements indicated that the ORR process of Ni4Cu5Fe1/C catalyst is a 4e− dominant process. Negligible change in ORR in the presence of NH4Cl and good electrochemical stability indicated that this ternary alloy catalyst could be a good cathode in DAFC. Experimental results and DFT calculations demonstrated that the excellent bifunctional catalytic activities of Ni4Cu5Fe1/C mainly arise from surface reconstruction during the catalytic reactions and the positive effects of Fe. When this bifunctional catalyst electrode was assembled with a commercial AEM into a single cell, the symmetric DAFC demonstrated an OCV and power density of 0.62 V and 8.9 mW cm−2 at 80 °C respectively. This work not only highlights the importance of optimizing the composition of alloys as a simple strategy to provoke bifunctional activity, but also would provide inspiration on the design and development of symmetric DAFCs.
Author contributions
M. F. Zhang: methodology, investigation, data curation, formal analysis, writing – original draft. J. Zhang: software, DFT calculation. G. Jeerh: writing – editing draft. P. M. Zou: data curation. B. Y. Sun: data curation. M. Walker: XPS measurement and analysis. K. Xie: software, DFT calculation. S. W. Tao: supervision, conceptualization, funding acquisition, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the EPSRC (Grant No. EP/G030995/1) and the Innovate UK (Grant No. 104010 and 133714). We thank Dr Ben Breeze for performing Raman and FTIR spectroscopy, Dr Christopher Waldron for measuring XRF, Dr Yisong Han for TEM observation and Prof. Andre van Veen for access of an oven and a HPLC pump.References
- P. Nejat, F. Jomehzadeh, M. M. Taheri, M. Gohari and M. Z. A. Majid, Renewable Sustainable Energy Rev., 2015, 43, 843–862 CrossRef CAS.
- G. Jeerh, M. Zhang and S. W. Tao, J. Mater. Chem. A, 2021, 9, 727–752 RSC.
- R. Ortega-Lugo, J. A. Fabián-Anguiano, O. Ovalle-Encinia, C. Gómez-Yánez, B. H. Zeifert and J. Ortiz-Landeros, J. Adv. Ceram., 2020, 9, 94–106 CrossRef CAS.
- S. Zhang, K. Wang, W. Xu, U. Iyer-Raniga, A. Athienitis, H. Ge, D. w. Cho, W. Feng, M. Okumiya, G. Yoon, E. Mazria and Y. Lyu, Energy Policy, 2021, 159, 112661 CrossRef.
- D. R. MacFarlane, P. V. Cherepanov, J. Choi, B. H. R. Suryanto, R. Y. Hodgetts, J. M. Bakker, F. M. Ferrero Vallana and A. N. Simonov, Joule, 2020, 4, 1186–1205 CrossRef CAS.
- M. Zhang, G. Jeerh, P. Zou, R. Lan, M. Wang, H. Wang and S. W. Tao, Mater. Today, 2021, 49, 351–377 CrossRef CAS.
- S. J. Davis, N. S. Lewis, M. Shaner, S. Aggarwal, D. Arent, I. L. Azevedo, S. M. Benson, T. Bradley, J. Brouwer, Y.-M. M. Chiang, C. T. M. Clack, A. Cohen, S. Doig, J. Edmonds, P. Fennell, C. B. Field, B. Hannegan, B.-M. M. Hodge, M. I. Hoffert, E. Ingersoll, P. Jaramillo, K. S. Lackner, K. J. Mach, M. Mastrandrea, J. Ogden, P. F. Peterson, D. L. Sanchez, D. Sperling, J. Stagner, J. E. Trancik, C.-J. Yang and K. Caldeira, Science, 2018, 360, eaas9793 CrossRef PubMed.
- Y. Wu, Y. Li, J. Gao and Q. Zhang, SusMat, 2021, 1, 66–87 CrossRef.
- K. Mazloomi and C. Gomes, Renewable Sustainable Energy Rev., 2012, 16, 3024–3033 CrossRef CAS.
- R. Lan, J. T. S. Irvine and S. W. Tao, Int. J. Hydrogen Energy, 2012, 37, 1482–1494 CrossRef CAS.
- M. Zhang, P. Zou, G. Jeerh, B. Sun, M. Walker and S. W. Tao, Adv. Funct. Mater., 2022, 2204881 CrossRef.
- D. Chakraborty, H. N. Petersen, C. Elkjær, A. Cagulada and T. Johannessen, Fuel Cells Bulletin, 2009, 2009, 12–15 CrossRef.
- D.-K. Lim, A. B. Plymill, H. Paik, X. Qian, S. Zecevic, C. R. I. Chisholm and S. M. Haile, Joule, 2020, 4, 2338–2347 CrossRef CAS.
- S. Chatterjee, R. K. Parsapur and K.-W. Huang, ACS Energy Lett., 2021, 6, 4390–4394 CrossRef CAS.
- S. Giddey, S. P. S. Badwal, C. Munnings and M. Dolan, ACS Sustainable Chem. Eng., 2017, 5, 10231–10239 CrossRef CAS.
- M. Zhang, P. Zou, G. Jeerh, S. Chen, J. Shields, H. Wang and S. W. Tao, ACS Sustainable Chem. Eng., 2020, 8, 12817–12824 CrossRef CAS.
- Y. Li, H. S. Pillai, T. Wang, S. Hwang, Y. Zhao, Z. Qiao, Q. Mu, S. Karakalos, M. Chen, J. Yang, D. Su, H. Xin, Y. Yan and G. Wu, Energy Environ. Sci., 2021, 14, 1449–1460 RSC.
- Y. Zhao, B. P. Setzler, J. Wang, J. Nash, T. Wang, B. Xu and Y. Yan, Joule, 2019, 3, 2472–2484 CrossRef CAS.
- R. Lan and S. W. Tao, Frontiers in Energy, 2014, 2, 1–4 Search PubMed.
- I. Dincer and O. Siddiqui, Ammonia Fuel Cells, Elsevier, 2020 Search PubMed.
- Y. Zhao, T. Wang, B. P. Setzler, R. Abbasi, J. Wang and Y. Yan, ACS Energy Lett., 2021, 6, 1996–2002 CrossRef CAS.
- B. Lin, M. Hu, J. Ma, Y. Jiang, S. Tao and G. Meng, J. Power Sources, 2008, 183, 479–484 CrossRef CAS.
- O. Siddiqui and I. Dincer, Therm. Sci. Eng. Prog., 2018, 5, 568–578 CrossRef.
- A. Afif, N. Radenahmad, Q. Cheok, S. Shams, J. H. Kim and A. K. Azad, Renewable Sustainable Energy Rev., 2016, 60, 822–835 CrossRef CAS.
- R. Lan and S. W. Tao, Electrochem. Solid-State Lett., 2010, 13, B83–B86 CrossRef CAS.
- S.W. Tao and R. Lan, WO2010133854A1, 2010.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 1255–1260 RSC.
- S. W. Tao and R. Lan, WO2012007727A1, 2012.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- T. Wang, Y. Zhao, B. P. Setzler, R. Abbasi, S. Gottesfeld and Y. Yan, Cell Rep. Phys. Sci., 2022, 3, 100829 CrossRef CAS.
- Y. Li, X. Li, H. S. Pillai, J. Lattimer, N. Mohd Adli, S. Karakalos, M. Chen, L. Guo, H. Xu, J. Yang, D. Su, H. Xin and G. Wu, ACS Catal., 2020, 10, 3945–3957 CrossRef CAS.
- B. K. Boggs and G. G. Botte, Electrochim. Acta, 2010, 55, 5287–5293 CrossRef CAS.
- F. J. Vidal-Iglesias, J. Solla-Gullón, V. Montiel, J. M. Feliu and A. Aldaz, J. Power Sources, 2007, 171, 448–456 CrossRef CAS.
- J. Liu, Z. Liu, H. Wang, B. Liu, N. Zhao, C. Zhong and W. Hu, Adv. Funct. Mater., 2021, 32, 2110702 CrossRef.
- J. A. Herron, P. Ferrin and M. Mavrikakis, J. Phys. Chem. C, 2015, 119, 14692–14701 CrossRef CAS.
- T. L. Lomocso and E. A. Baranova, Electrochim. Acta, 2011, 56, 8551–8558 CrossRef CAS.
- K. Endo, Y. Katayama and T. Miura, Electrochim. Acta, 2004, 49, 1635–1638 CrossRef CAS.
- X. Yang, C. Priest, Y. Hou and G. Wu, SusMat, 2022 DOI:10.1002/sus2.69.
- X. Ren, Y. Wang, A. Liu, Z. Zhang, Q. Lv and B. Liu, J. Mater. Chem. A, 2020, 8, 24284–24306 RSC.
- B. P. Setzler, Z. Zhuang, J. A. Wittkopf and Y. Yan, Nat. Nanotechnol., 2016, 11, 1020–1025 CrossRef CAS PubMed.
- N. M. Adli, H. Zhang, S. Mukherjee and G. Wu, J. Electrochem. Soc., 2018, 165, J3130–J3147 CrossRef CAS.
- M. Zhu, Y. Yang, S. Xi, C. Diao, Z. Yu, W. S. V. Lee and J. Xue, Small, 2021, 17, 2005616 CrossRef CAS PubMed.
- M. D. Zott, P. Garrido-Barros and J. C. Peters, ACS Catal., 2019, 9, 10101–10108 CrossRef CAS.
- M. Zhang, H. Li, X. Duan, P. Zou, G. Jeerh, B. Sun, S. Chen, J. Humphreys, M. Walker, K. Xie and S. W. Tao, Adv. Sci., 2021, 8, 2101299 CrossRef CAS PubMed.
- G. Jeerh, P. Zou, M. Zhang, S. Chen, J. Humphreys and S. Tao, Sep. Purif. Technol., 2022, 297, 121451 CrossRef CAS.
- W. Xu, D. Du, R. Lan, J. Humphreys, D. N. Miller, M. Walker, Z. Wu, J. T. S. Irvine and S. W. Tao, Appl. Catal., B, 2018, 237, 1101–1109 CrossRef CAS.
- W. Xu, R. Lan, D. Du, J. Humphreys, M. Walker, Z. Wu, H. Wang and S. W. Tao, Appl. Catal., B, 2017, 218, 470–479 CrossRef CAS.
- S. Song and S. Jiang, Appl. Catal., B, 2012, 117–118, 346–350 CrossRef CAS.
- L. Gang, B. G. Anderson, J. van Grondelle and R. A. van Santen, Catal. Today, 2000, 61, 179–185 CrossRef CAS.
- R. Wang, H. Liu, K. Zhang, G. Zhang, H. Lan and J. Qu, Chem. Eng. J., 2021, 404, 126795 CrossRef CAS.
- R. Chen, S. Zheng, Y. Yao, Z. Lin, W. Ouyang, L. Zhuo and Z. Wang, Int. J. Hydrogen Energy, 2021, 46, 27749–27757 CrossRef CAS.
- X. Jiang, D. Ying, X. Liu, M. Liu, S. Zhou, C. Guo, G. Zhao, Y. Wang and J. Jia, Electrochim. Acta, 2020, 345, 136157 CrossRef CAS.
- M. A. Ahsan, A. R. Puente Santiago, Y. Hong, N. Zhang, M. Cano, E. Rodriguez-Castellon, L. Echegoyen, S. T. Sreenivasan and J. C. Noveron, J. Am. Chem. Soc., 2020, 142, 14688–14701 CrossRef CAS PubMed.
- B. Chen and W.-Y. Wong, J. Mater. Chem. A, 2022, 10, 7968–7977 RSC.
- W. Li, Z. Yao, Y. Zhong, C.-a. Zhou, X. Wang, X. Xia, D. Xie, J. Wu, C. Gu and J. Tu, J. Mater. Chem. A, 2019, 7, 10231–10238 RSC.
- Q. Xue, Y. Zhao, J.-Y. Zhu, Y. Ding, T. Wang, H.-Y. Sun, F. Li, P. Chen, P. Jin, S. Yin and Y. Chen, J. Mater. Chem. A, 2021, 9, 8444–8451 RSC.
- D. M. Bastidas, S. W. Tao and J. T. S. Irvine, J. Mater. Chem., 2006, 16, 1603–1605 RSC.
- P. Li, W. Yang, C. Tian, W. Zhao, Z. Lü, Z. Xie and C.-A. Wang, J. Adv. Ceram., 2021, 10, 328–337 CrossRef CAS.
- Z. Sun, Y. Wang, L. Zhang, H. Wu, Y. Jin, Y. Li, Y. Shi, T. Zhu, H. Mao, J. Liu, C. Xiao and S. Ding, Adv. Funct. Mater., 2020, 30, 1910482 CrossRef CAS.
- X. Hai, X. Zhao, N. Guo, C. Yao, C. Chen, W. Liu, Y. Du, H. Yan, J. Li, Z. Chen, X. Li, Z. Li, H. Xu, P. Lyu, J. Zhang, M. Lin, C. Su, S. J. Pennycook, C. Zhang, S. Xi and J. Lu, ACS Catal., 2020, 10, 5862–5870 CrossRef CAS.
- M. Zheyang, Y. Weiyi, G. Shuang, S. Jian Ku, D. Yajun, S. Wuzhu and L. Qi, J. Adv. Ceram., 2021, 10, 714–728 CrossRef.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- T. He, S. K. Matta, G. Will and A. Du, Small Methods, 2019, 3, 1800419 CrossRef.
- P. Zhang, L. Li, D. Nordlund, H. Chen, L. Fan, B. Zhang, X. Sheng, Q. Daniel and L. Sun, Nat. Commun., 2018, 9, 381 CrossRef PubMed.
- Z. Chen, R. Wu, Y. Liu, Y. Ha, Y. Guo, D. Sun, M. Liu and F. Fang, Adv. Mater., 2018, 30, e1802011 CrossRef PubMed.
- J. Zhang, Y. Sun, J. Zhu, Z. Gao, S. Li, S. Mu and Y. Huang, Adv. Sci., 2018, 5, 1801375 CrossRef PubMed.
- X.-T. Wang, T. Ouyang, L. Wang, J.-H. Zhong and Z.-Q. Liu, Angew. Chem., Int. Ed., 2020, 132, 6554–6561 CrossRef.
- A. Yang, J. Wang, K. Su, W. Lei, X. Qiu and Y. Tang, Chem.–Eur. J., 2021, 27, 4869–4875 CrossRef CAS PubMed.
- L. Zhou and Y. F. Cheng, Int. J. Hydrogen Energy, 2008, 33, 5897–5904 CrossRef CAS.
- W. Xu, D. Du, R. Lan, J. Humphreys, Z. Wu and S. W. Tao, New J. Chem., 2017, 41, 4190–4196 RSC.
- T. Zhan, X. Liu, S. Lu and W. Hou, Appl. Catal., B, 2017, 205, 551–558 CrossRef CAS.
- M. G. Hosseini, F. Hosseinzadeh, P. Zardari and M. Darbandi, Int. J. Hydrogen Energy, 2021, 46, 28513–28526 CrossRef CAS.
- Y. Guo, Z. Pan and L. An, J. Power Sources, 2020, 476, 228454 CrossRef CAS.
- J. Xiao, Y. Xu, Y. Xia, J. Xi and S. Wang, Nano Energy, 2016, 24, 121–129 CrossRef CAS.
- T. Curtin, F. O' Regan, C. Deconinck, N. Knüttle and B. K. Hodnett, Catal. Today, 2000, 55, 189–195 CrossRef CAS.
- F. Han, M. Yuan, S. Mine, H. Sun, H. Chen, T. Toyao, M. Matsuoka, K. Zhu, J. Zhang, W. Wang and T. Xue, ACS Catal., 2019, 9, 10398–10408 CrossRef CAS.
- S. Khan, S. S. Shah, M. A. R. Anjum, M. R. Khan and N. K. Janjua, Coatings, 2021, 11, 313 CrossRef CAS.
- P. Kanagavalli, R. Sudha, S. Boopathi and S. S. Kumar, Electrochem. Commun., 2017, 82, 61–65 CrossRef CAS.
- W. Lai, L. Ge, H. Li, Y. Deng, B. Xu, B. Ouyang and E. Kan, Int. J. Hydrogen Energy, 2021, 46, 26861–26872 CrossRef CAS.
- J. Huang, Y. Li, Y. Zhang, G. Rao, C. Wu, Y. Hu, X. Wang, R. Lu, Y. Li and J. Xiong, Angew. Chem., Int. Ed., 2019, 58, 17458–17464 CrossRef CAS PubMed.
- A. Y. Faid, A. O. Barnett, F. Seland and S. Sunde, Electrochim. Acta, 2020, 361, 137040 CrossRef CAS.
- J. F. Xu, W. Ji, Z. X. Shen, W. S. Li, S. H. Tang, X. R. Ye, D. Z. Jia and X. Q. Xin, J. Raman Spectrosc., 1999, 30, 413–415 CrossRef CAS.
- H. Hagemann, H. Bill, W. sadowski, E. Walker and M. François, Solid State Commun., 1990, 73, 447–451 CrossRef CAS.
- A. Chaudhary and H. C. Barshilia, J. Phys. Chem. C, 2011, 115, 18213–18220 CrossRef CAS.
- T. Ghodselahi, M. A. Vesaghi, A. Shafiekhani, A. Baghizadeh and M. Lameii, Appl. Surf. Sci., 2008, 255, 2730–2734 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2010, 257, 887–898 CrossRef CAS.
- P. Zou, S. Chen, R. Lan and S. W. Tao, ChemSusChem, 2019, 12, 2788–2794 CrossRef CAS PubMed.
- P. Zou, S. Chen, R. Lan, J. Humphreys, G. Jeerh and S. W. Tao, Int. J. Hydrogen Energy, 2019, 44, 26554–26564 CrossRef CAS.
- R. Lan and S. W. Tao, ECS Electrochem. Lett., 2013, 2, F37–F40 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta04129d |
| This journal is © The Royal Society of Chemistry 2022 |