
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThiol-decorated covalent organic frameworks as multifunctional materials for high-performance supercapacitors and heterogeneous catalysis†
Dawid
Pakulski
 *ab,
Verónica
Montes-García
c,
Adam
Gorczyński
d,
Włodzimierz
Czepa
abd,
Tomasz
Chudziak
bd,
Paolo
Samorì
*c and
Artur
Ciesielski
*bc
*ab,
Verónica
Montes-García
c,
Adam
Gorczyński
d,
Włodzimierz
Czepa
abd,
Tomasz
Chudziak
bd,
Paolo
Samorì
*c and
Artur
Ciesielski
*bc
aAdam Mickiewicz University Foundation, Poznań Science and Technology Park, Rubież 46, 61-612 Poznań, Poland. E-mail: dawid.pakulski@amu.edu.pl
bCentre for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, 61-614 Poznań, Poland
cUniversité de Strasbourg, CNRS, ISIS 8 allée Gaspard Monge, 67000 Strasbourg, France. E-mail: ciesielski@unistra.fr; samori@unistra.fr
dFaculty of Chemistry, Adam Mickiewicz University, Uniwersytetu Poznańskiego 8, 61-614 Poznań, Poland
First published on 13th July 2022
Abstract
Tunable physicochemical properties combined with the high chemical and thermal stabilities of covalent organic frameworks (COFs) make them ideal candidates for the next generation of energy storage systems. The integration of redox-active moieties (e.g., thiols) in COFs imparts them a pseudocapacitive characteristic and represents an efficient strategy to boost their performance as electrochemical supercapacitors (SCs). We report the synthesis of two thiol-decorated COFs (SH-COF-1 and SH-COF-2) via the condensation between 2,5-diaminobenzene-1,4-dithiol (DABDT) and benzene-1,3,5-tricarboxaldehyde (TBA), or 1,2,4,5-tetrakis-(4-formylphenyl)benzene (TFPB), respectively. SH-COF-1, which possesses a higher number of thiol groups per structural repeat unit compared to SH-COF-2, exhibits a higher surface area (227 m2 g−1) and enhanced electrochemical performance (areal capacitance of 118 mF cm−2 and a capacitance retention >95% after 1000 cycles), being superior to previously reported COFs missing redox-active units in their scaffolds. Moreover, to demonstrate the multifunctionality resulting from the presence of thiol groups, AuNPs were in situ grown using SH-COFs as templates. By taking advantage of the strength of the bonding between the AuNPs and the SH-COFs, Au-SH-COF hybrids were used as heterogeneous catalysts for the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP), showing an excellent catalytic activity kobs, of 1.01 min−1 and 0.71 min−1 for Au-SH-COF-1 and Au-SH-COF-2, respectively, and long-term performance (4-NP to 4-AP conversion above 95% after 10 catalytic cycles). This work highlights the importance of COFs' molecular design towards the development of highly efficient (multi)functional materials.
Introduction
Supercapacitors (SCs) are energy storage devices that exhibit cyclabilities and charge–discharge rates exceeding those of market-dominating technologies, i.e., batteries and conventional capacitors.1 These features make them effective in applications where short power bursts are needed, e.g., in transportation, consumer electronics or memory backup systems.2 However, SCs suffer from a major drawback: their typical energy densities are lower compared to those of batteries (>100 W h kg−1). This limitation can be overcome by developing novel nanostructured electrode materials, including oxides, sulphides, metal organic frameworks and/or covalent organic frameworks (COFs).3–9 The latter ones constitute an emerging and rapidly growing class of crystalline organic polymeric materials,10 which combine numerous unique features including the tuneability of their well-defined chemical structures, extraordinary thermal and chemical stabilities, high porosity, and large internal surface areas. COFs have been successfully used in gas storage and separation,11 catalysis,12 sensors,13 and drug delivery.14 Moreover, COFs display low solubility, minimal volumetric changes during charge–discharge, and can be electrochemically tuned, rendering them ideal candidates when integrated in energy storage systems (ESSs).15 COFs (i.e., β-keto-enamine-linked COFs) were first used in 2013 as a supercapacitor electrode, with a specific capacitance of 48 F g−1 (at 1 A g−1 current density). Since then, numerous attempts to improve the electrochemical performance of COFs in SCs have been reported.15In general, COFs are pseudocapacitive materials capable of storing energy through two well-known mechanisms: (i) an electrical double layer (EDL) mechanism via ion adsorption and swapping of co-ions for counter-ions at the electrode/electrolyte interface, and (ii) through faradaic electron charge transfer with redox reactions, also known as a pseudocapacitive mechanism. While the EDL is promoted by long-range ordered surfaces that facilitate ionic mobility at the electrolyte/electrode interface, chemical functionalization of the active materials incorporating numerous redox-active groups can be employed to introduce pseudocapacitance, which can substantially improve the overall capacitance and energy densities of ESSs.16 Therefore, COFs featuring redox-active groups hold enormous potential for the construction of next-generation high-performance SCs.17 Preliminary studies have shown that the integration of different redox-active organic moieties such as benzoquinone,18 anthraquinone,19–21 orthoquinone,22 or N-atom rich groups (porphyrins, porphyrinoids,23 hexaazaphenylenes (HATs),24 and triazines25) in COF skeletons leads to an increased capacitance when compared to those of non-redox-active COFs. The deliberate incorporation of heteroatoms (e.g., N, B, P, S, and O) into the COF skeleton is also an effective method to positively influence the EDL and pseudocapacitive mechanism. For instance, Li et al., reported the synthesis of a benzobisthiazole-linked COF involving strong intramolecular hydrogen bonds, which in addition to thiazole moieties stabilize the ordered porous structure of the COF. Its application as an electrode material for SCs showed an astonishing capacitance value of 724 F g−1 and an energy density of 69 W h kg−1.26 Among the heteroatom dopants, sulfur exhibits a high electronegativity (2.58) compared to that of carbon (2.55), and as a result, the addition of sulfur atoms to a carbon-based material's lattice improves not only its electrical conductivity but also the specific capacitance due to the redox activity of sulfur-based functional groups.27 For instance, a S-doped graphene aerogel reported by Yu et al. delivered a high specific capacitance of 445.6 F g−1 in 1 M H2SO4, which is four-fold greater than that of graphene aerogel without S-doping.28 There are very few examples involving the post-synthetic grafting of sulfur atoms as pendant groups in COF backbones.29,30 In these examples, the introduction of sulfur moieties increased the electrochemical performance of COFs used as cathode materials in Li–S batteries.29,30 Redox moieties can be post synthetically grafted in the COFs' structure, or simply, they can be structurally embedded in their molecular building blocks. Although COFs based on thioether-functionalized monomers have been extensively used for efficient removal of Hg2+ from water,13,31,32 to the best of our knowledge, their application as electrodes in SCs remains unexplored.
A suitable incorporation of chemically programmed building blocks opens new perspectives toward the formation of novel 2D or 3D crystalline (multi)functional architectures. For example, thiol-decorated COFs have been successfully used as supports of catalysts such as metal nanoparticles (NPs) (e.g., gold,33 palladium34 or platinum34 NPs) with their incomparable advantages: (i) the strong interaction of metal NPs with thiol groups minimizes the migration/coalescence of the AuNPs in the catalytic process, guaranteeing a high recyclability, (ii) the narrow size distribution of metal NPs, whose growth is pore-size driven, guarantees a high catalytic activity.
Herein, we report the synthesis of two redox-active COFs via solvothermal condensation of 2,5-diaminobenzene-1,4-dithiol (DABDT) with benzene-1,3,5-tricarboxaldehyde (TBA), (SH-COF-1) or with 1,2,4,5-tetrakis-(4-formylphenyl)benzene (TFPB) (SH-COF-2), respectively. Two different aldehydes were selected as monomers for the generation of two distinct COF skeletons, i.e., hexagonal SH-COF-1 and rhomboidal SH-COF-2. The application of SH-COFs as SC electrodes was explored in three and two-electrode systems using 1 M tetraethylammonium tetrafluoroborate in acetonitrile as an electrolyte. Both SH-COFs exhibited an excellent chemical stability as well as a high areal capacitance (118 mF cm−2 for SH-COF-1 and 74 mF cm−2 for SH-COF-2) at current 0.5 mA cm−2, which are the highest values ever achieved in pristine COF based SCs and comparable to those of other porous carbon-based electrode materials (see Table S2†). Moreover, to demonstrate the multifunctionality of thiol-COFs, we have performed the in situ growth of gold nanoparticles (AuNPs) in the presence of SH-COF templates (Au-SH-COF-1 and Au-SH-COF-2) and studied their catalytic activities in the reduction of 4-nitrophenol. AuNPs with narrow size distribution (10.6 ± 2.8 nm and 13.7 ± 3.2 nm for Au-SH-COF-1 and Au-SH-COF-2, respectively), high density and homogeneous spatial distribution have been successfully prepared, and both Au-SH-COFs showed excellent catalytic activity and recyclability.
Results and discussion
Synthesis and structural characterization
Two different aldehydes are selected as precursors in this study to construct COFs with different skeleton structures and to evaluate the effect of appended thiol groups on multifunctionality of the obtained systems. Specifically, the overall SH-group density per amount of the aldehyde carbon atoms will be the crucial variable observed herein responsible for radical differences in the observed performance. The synthesis of thiol-decorated SH-COF-1 and SH-COF-2 is accomplished by the conventional solvothermal strategy26 illustrated in Fig. 1 (see the Experimental section for details). A condensation between 2,5-diaminobenzene-1,4-dithiol (DABDT) with benzene-1,3,5-tricarboxaldehyde (TBA), or 1,2,4,5-tetrakis-(4-formylphenyl)benzene (TFPB) is performed in 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (DABDT:TBA) and 2:1 (DABDT:TFBP) molar ratios to account for the exhaustive imine formation. Specifically, the system is heated at 120 °C for 3 days in the n-butyl alcohol/1,2-dichlorobenzene/6 M AcOH solvent mixture (5:5:1 volume ratio) to ensure that the cyclization of the thiol group to form benzothiazole rings does not occur.26 Acetic acid is responsible for the pH control and is routinely used for the imine condensation reactions.35 Attempts to perform the reactions using other known solvent mixtures such as DMF/AcOH or mesitylene/dioxane36 are unsuccessful i.e. non-uniform mixtures of products with different colors and poor crystallinity are obtained (see Fig. S1†).
:2 (DABDT:TBA) and 2:1 (DABDT:TFBP) molar ratios to account for the exhaustive imine formation. Specifically, the system is heated at 120 °C for 3 days in the n-butyl alcohol/1,2-dichlorobenzene/6 M AcOH solvent mixture (5:5:1 volume ratio) to ensure that the cyclization of the thiol group to form benzothiazole rings does not occur.26 Acetic acid is responsible for the pH control and is routinely used for the imine condensation reactions.35 Attempts to perform the reactions using other known solvent mixtures such as DMF/AcOH or mesitylene/dioxane36 are unsuccessful i.e. non-uniform mixtures of products with different colors and poor crystallinity are obtained (see Fig. S1†).
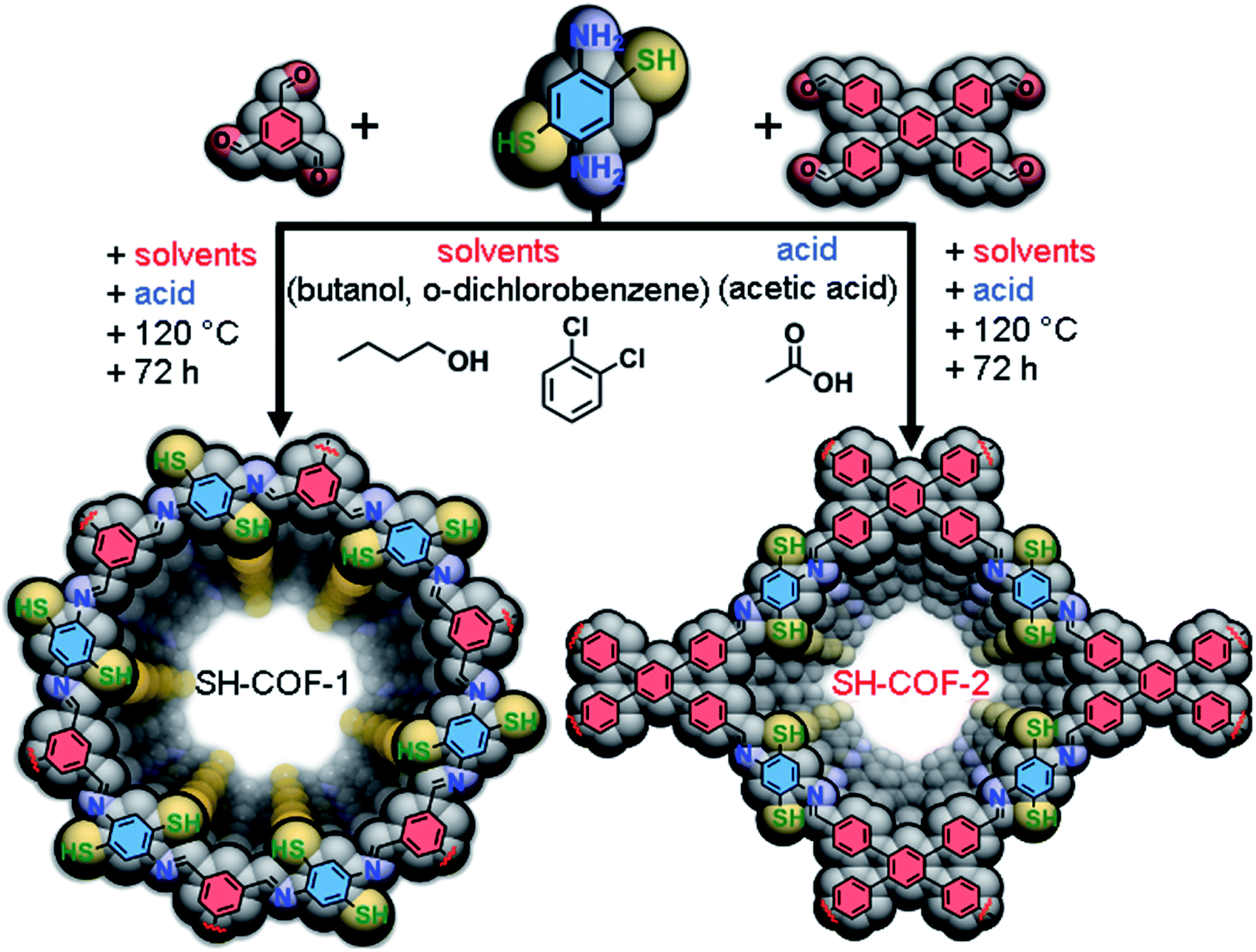 | ||
| Fig. 1 Synthetic pathway of SH-COF-1 and SH-COF-2. | ||
The solid-state nuclear magnetic resonance (NMR) analysis of SH-COFs is performed on the basis of classical 13C cross-polarization magic angle spinning (CP-MAS) experiments. In the 13C CP-MAS NMR spectra of SH-COF-1 and -2 (Fig. 2), the chemical shift at ∼150 ppm is assigned to the carbon atoms in imine groups (–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N),37 and the chemical shift at ∼170 ppm is assigned to the carbon atoms functionalized with thiol groups (C-SH).31 Other peaks present in both SH-COFs (chemical shifts 115–145 ppm) are related to the aromatic structure of each SH-COF.37 Importantly, the signals of the CHO subunits (chemical shift 185 nm) from the starting monomers are absent, which confirm the quantitative and successful generation of COF structures.38
N),37 and the chemical shift at ∼170 ppm is assigned to the carbon atoms functionalized with thiol groups (C-SH).31 Other peaks present in both SH-COFs (chemical shifts 115–145 ppm) are related to the aromatic structure of each SH-COF.37 Importantly, the signals of the CHO subunits (chemical shift 185 nm) from the starting monomers are absent, which confirm the quantitative and successful generation of COF structures.38
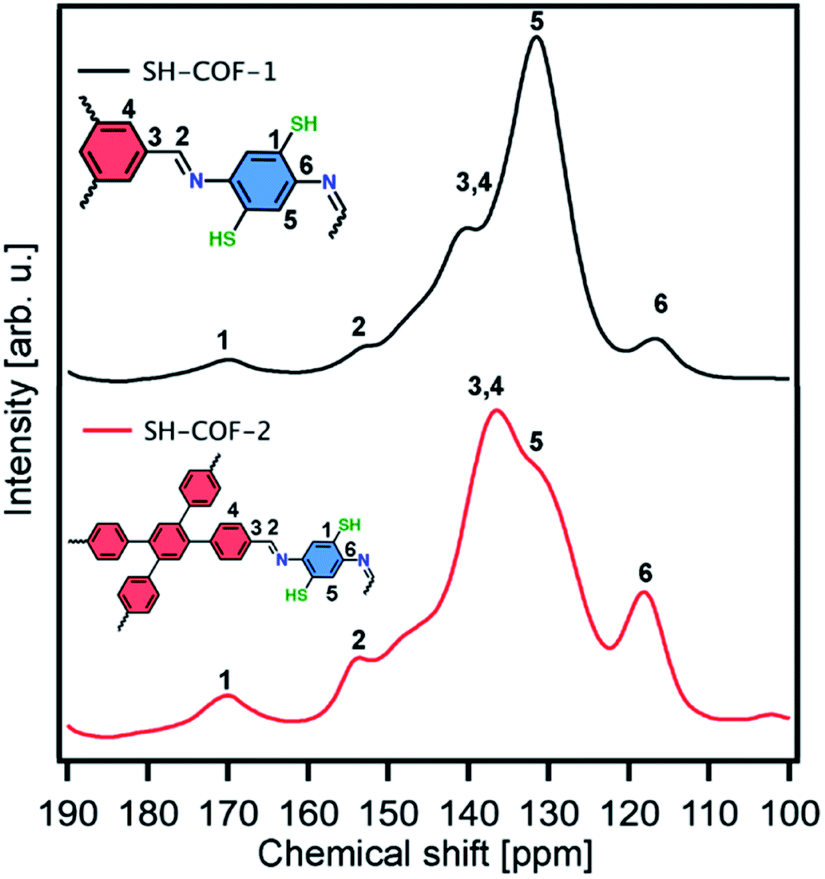 | ||
| Fig. 2 13C solid-state NMR spectra of SH-COF-1 (black curve) and SH-COF-2 (red curve). | ||
The Fourier transform infrared (FT-IR) spectra of SH-COF-1 and SH-COF-2 (Fig. S2†) reveal signals at ∼1600 and ∼1240 cm−1, corresponding to CC stretching vibrations and C–N stretch moieties, respectively, which provides the evidence for the successful COF formation.31,39,40 The characteristic CN bond stretching vibration bands at around ∼1620 cm−1 are also observed.41 Noteworthily, the band at ∼2570 cm−1 observed in the FT-IR spectra of COF-SH-1 and SH-COF-2 indicates the presence of sulfhydryl groups, which further confirms the successful synthesis of SH-COFs.42 Moreover, the FT-IR spectra of SH-COF-1 and SH-COF-2 show the absence of the –CHO stretching band at ∼1690 cm−1, which indicates the complete consumption of the starting materials (Fig. S3†).
To gain insight into the chemical composition of SH-COFs, X-ray photoelectron spectroscopy (XPS) analysis is performed (Fig. S4 and S5†). The analysis of the C 1s (Fig. S5a and b†), N 1s (Fig. S5c and d†) and S 2p (Fig. S5e and f†) spectra of SH-COF-1 (left panels) and SH-COF-2 (right panels) shows similar results for both SH-COFs. The analysis of C 1s spectra reveals the presence of three peaks at 285, 285.9 and 287.6, eV corresponding to the chemical bonds CC, C–S and C–N, respectively.43 On the other hand, N 1s spectra display a peak at ∼400 eV corresponding to the chemical bond CN respectively, thereby indicating the formation of imine bonds.44
The elemental analysis from energy-dispersive X-ray (EDX) spectroscopy (Fig. S6, S7 and Table S1†) indicates that the ratios of C/S are 7 and 19 for SH-COF-1 and SH-COF-2, respectively. These ratios are in full agreement with the ratio of C/S of each COF unit for SH-COF-1 and SH-COF-2 and with the results obtained from XPS analysis. Importantly, the C, N and S elements are homogeneously distributed on both SH-COFs (Fig. S6 and S7†).
The crystalline structures of both SH-COFs are characterized by powder X-ray diffraction (PXRD) (Fig. S8†). SH-COF-1 and SH-COF-2 exhibit a broad band in the region from 3–15° due to the thiol group modification,31 as well as an intense diffraction peak at 2θ = 28°, which originates from the spacing between the layers (100 plane), as previously reported for other COF-SHs.31 This suggests that both SH-COFs have a layered and polycrystalline nature. The d values corresponding to the (100) and (001) planes of SH-COF-1 and SH-COF-2 are calculated to be 3.2 Å and 5.9 Å, respectively.
The thermal stability of SH-COFs is investigated using thermogravimetric analysis (TGA) (Fig. S9†). SH-COF-1 and SH-COF-2 show an extraordinary thermal stability, with values Td10 (thermal decomposition of 10% weight) of approximately 413 and 407 °C, respectively. Noteworthily, the thermal decomposition of SH-COF-1 and SH-COF-2 is observed from 600 °C, which is related to the rupture of the imine bonds and the decomposition of COFs into oligomers and combustion products.45
The morphology and microstructure of the as-prepared SH-COF materials are evaluated by means of scanning electron microscopy (SEM) (Fig. 3a and b). The SEM images of SH-COF-1 (Fig. 3a) and SH-COF-2 (Fig. 3b) show the presence of spherical particles with a diameter of ∼200 nm. The porosity of SH-COF-1 and SH-COF-2 is evaluated by acquiring N2 adsorption–desorption isotherms at 77 K (Fig. 3c) in their completely activated state, i.e., after treating the samples under vacuum (<1 torr pressure) and heating at 100 °C for 12 h. The adsorption isotherms of the two SH-COFs exhibit Type-I sorption isotherms, with steep increases appearing at low relative pressure and also Type-IV sorption features with adsorption/desorption hysteresis at higher pressure. The calculated Brunauer–Emmett–Teller (BET) surface areas of SH-COF-1 and SH-COF-2 amount to 227 m2 g−1 and 93 m2 g−1, respectively. The pore size distribution (Fig. 3d) shows the dominance of micropores with typical sizes of 1.9 nm and 2.2 nm for SH-COF-1 and SH-COF-2, respectively.
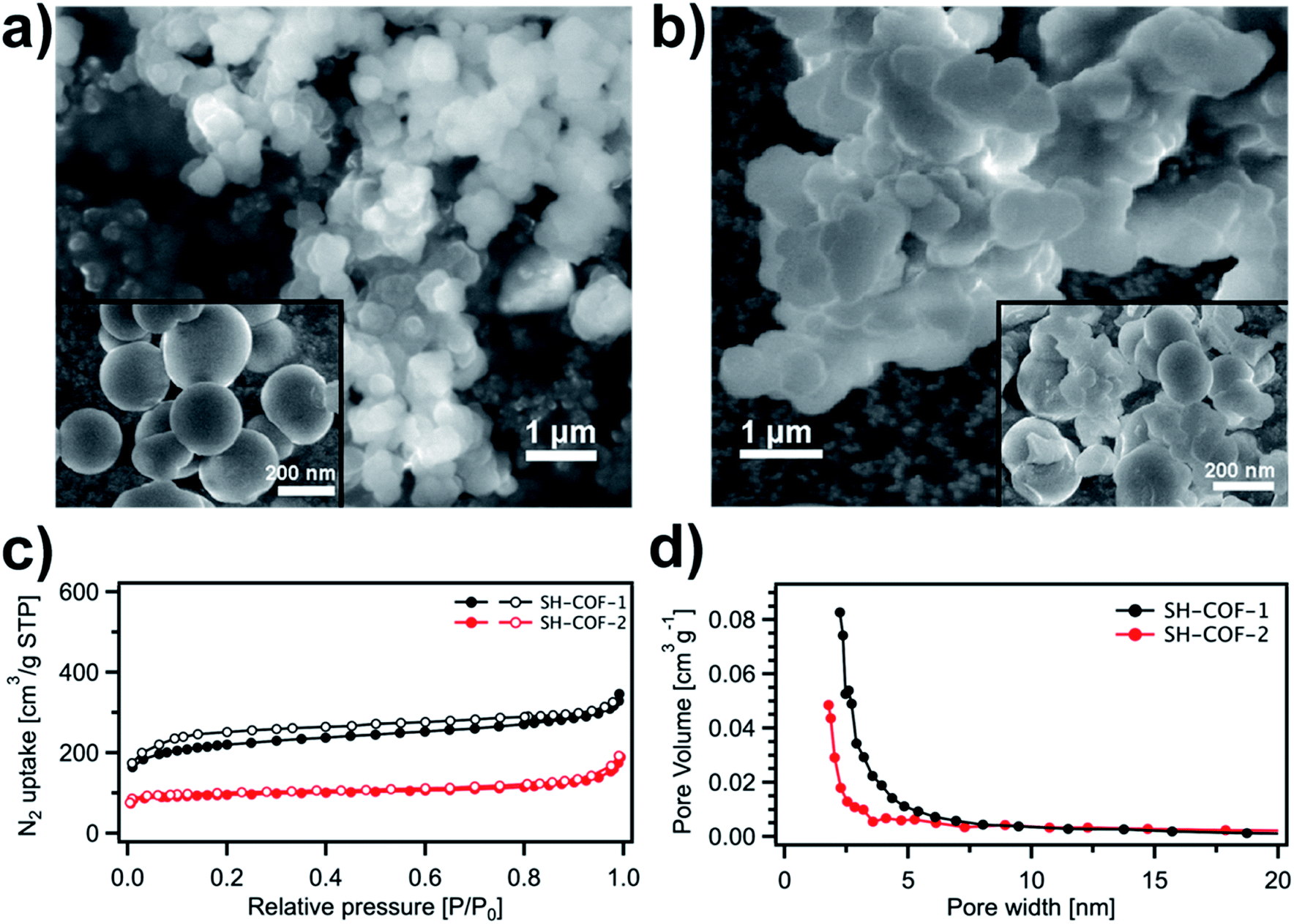 | ||
| Fig. 3 (a and b) SEM images of (a) SH-COF-1 and (b) SH-COF-2; (c) BET surface area of SH-COF-1 (black curve) and SH-COF-2 (red curve). (d) Pore size distribution of SH-COF-1 (black curve) and SH-COF-2 (red curve). | ||
Electrochemical analysis
Both SH-COFs are then used for the preparation of working electrodes, and their electrochemical performance is investigated using a three-electrode configuration (Fig. 4). This system includes SH-COF-1 or SH-COF-2 as the working electrode, a platinum wire as the counter electrode, and a reference electrode (Hg/Hg2Cl2 electrode) in tetraethylammonium tetrafluoroborate in acetonitrile electrolyte. Fig. 4a and b display the cyclic voltammograms of (a) SH-COF-1 and (b) SH-COF-2 between −1.8 and 2.2 V at scan rates from 1 to 1000 mV s−1. A comparison of the CV profiles of SH-COF-1 and SH-COF-2 reveals a greater contribution of the pseudocapacitive mechanism in the case of SH-COF-1 as evidenced by the appearance of multiple redox peaks. On the other hand, the quasi-rectangular shape suggests that the EDL-type energy storage mechanism is dominant for SH-COF-2, whose sulfur content is lower when compared to SH-COF-1 (see Fig. S4 and Table S1†). The rate capability is also an important feature for supercapacitors. The cyclic voltammograms maintain box-like shapes with a little deviation at lower potentials, even when the scan rate increases to 1000 mV s−1, implying a quick charge propagation capability of both the EDL and pseudocapacitance mechanism and hence a good rate capability in both cases.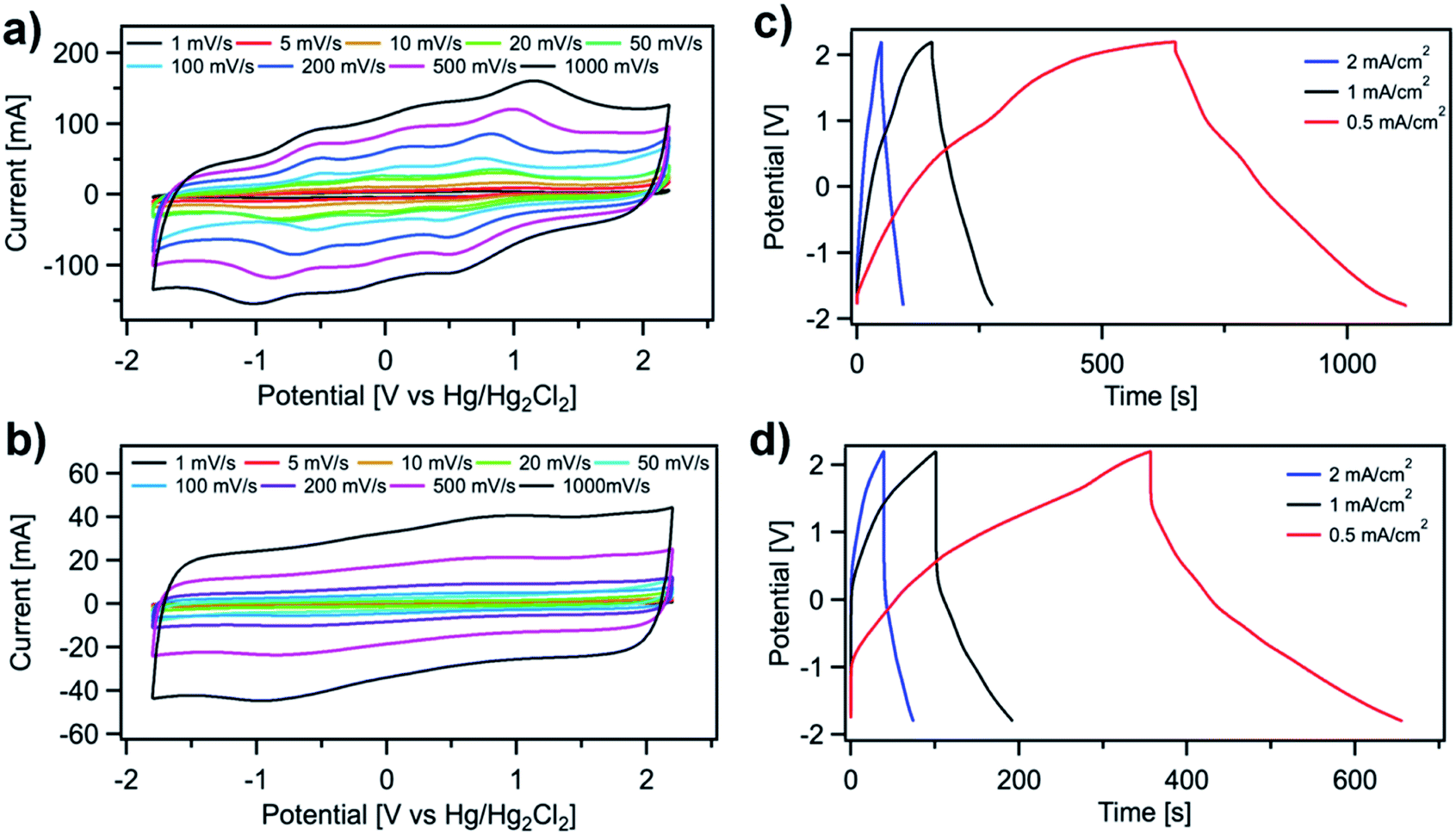 | ||
| Fig. 4 Electrochemical characterization of SH-COFs in a three-electrode configuration. CV curves at different scan rates for (a) SH-COF-1 and (b) SH-COF-2. GCD at different current densities for (c) SH-COF-1 and (d) SH-COF-2. | ||
To prove the key role played by the thiol groups in the electrochemical behaviour of COFs, we perform a control experiment where COFs without thiol units (i.e., the 2,5-diaminobenzene-1,4-dithiol monomer is replaced by 1,4-phenylenediamine), COF-1 and COF-2, are synthesized (see the Experimental section and Fig. S10–S12†). The electrochemical performance of COF-1 and COF-2 is assessed using a three-electrode configuration (see Fig. S12†) in the same manner as SH-COFs. The CV test results of COF-1 and -2 do not exhibit obvious oxidation and reduction peaks in the same potential range of SH-COF-1 and SH-COF-2, indicating the absence of redox activity in COF-1 and COF-2. Besides, the maximum areal capacitances obtained are 59 and 31 mF cm−2 for COF-1 and COF-2, respectively, which represent much lower values than the ones obtained for SH-COF-1 (118 mF cm−2) and SH-COF-2 (74 mF cm−2) calculated from GCD (Fig. 4c and d). This is unequivocal proof that the presence of redox-active groups effectively boosts the electrochemical performance of COFs.
The type of heteroatoms, their amount and their accessibility should be considered as the main factors influencing the COF electrochemical performance.10,15 Although imine bonds do not actively participate in redox reactions, their presence enhances the electrochemical performance by providing a conjugated structure.46–48 On the other hand, the redox-active heteroatom-containing groups, i.e., thiols, can be easily oxidized to the sulfonic form during electrochemical processes providing the pseudocapacitive behaviour.49 Considering the stereochemistry of the two SH-COFs, the thiol groups in the case of SH-COF-1 seem to be more accessible for electrolyte ions, while functional groups of SH-COF-2 are more hindered by side aromatic chains. Moreover, SH-COF-1 exhibits a much greater specific surface area, a larger amount of thiol units per cavity, and an optimal pore size for electrolyte penetration, which suggests that SH-COF-1 as an ideal indicator for energy storage device applications.50 Finally, to investigate the electrochemical stability of SH-COFs, galvanostatic charge–discharge cycling is performed at a current density of 100 mA g−1 (Fig. S13†). After 5000 cycles, the capacitance decay is 5% for SH-COF-1 and 12% for SH-COF-2, indicating that both SH-COFs have high cycle durability.
To further investigate the electrochemical performance of the SH-COF electrodes for practical application in supercapacitors, a symmetric supercapacitor with a single electrode mass loading of 2 mg is assembled by using tetraethylammonium tetrafluoroborate in acetonitrile as the electrolyte. Fig. 5a and b display the CVs of (a) SH-COF-1 and (b) SH-COF-2 between 0 and 2.5 V and 0 and 2 V, respectively, at scan rates from 10 to 1000 mV s−1. In analogy to the measurements carried out using a three-electrode configuration, the CV curves of SH-COFs show a typical rectangular shape together with the presence of redox peaks. This confirms the coexistence of an EDL and battery-type mechanism due to the pseudocapacitive behavior of SH-COFs. The electrochemical kinetics of both materials have been investigated by exploiting the procedure proposed by Wu et al., (see Fig. S14†).51 The relation of the peak current (I) and scan rate (ν) from CV complies with the equation i = aνb. The value of b gives a quantitative determination of the diffusion-controlled contribution and capacitive contribution in the charge storage mechanism, being 0.5 for pure battery-type materials and 1 for pure electrical double layer capacitor (EDLC) materials.52 The average value of b for SH-COF-2 is 0.94, indicating a predominantly capacitive controlled contribution and partial diffusion contribution (see Fig. S14c†). On the other hand, the average value of b for SH-COF-1 is 0.79, which indicates a higher weight of the diffusion contribution in agreement with the CV profiles (see Fig. S14d†). At the same time, the galvanostatic charge/discharge curves of the SH-COF symmetric supercapacitors (Fig. 5c and d) show a distorted triangular shape due to the redox behavior of the thiol functional groups. The absence of IR drop at the current densities from 0.5 to 3 mA cm2 indicates fast kinetics for charge-storage processes. Moreover, the SH-COF symmetric supercapacitors reach high areal capacitances of 40 mF cm−2 and 16 mF cm−2 at a current density of 0.5 mF cm−2. The areal capacitance of our symmetric supercapacitor is higher than those of other reported COFs with no redox-active moieties in their structure (see Table S2†).
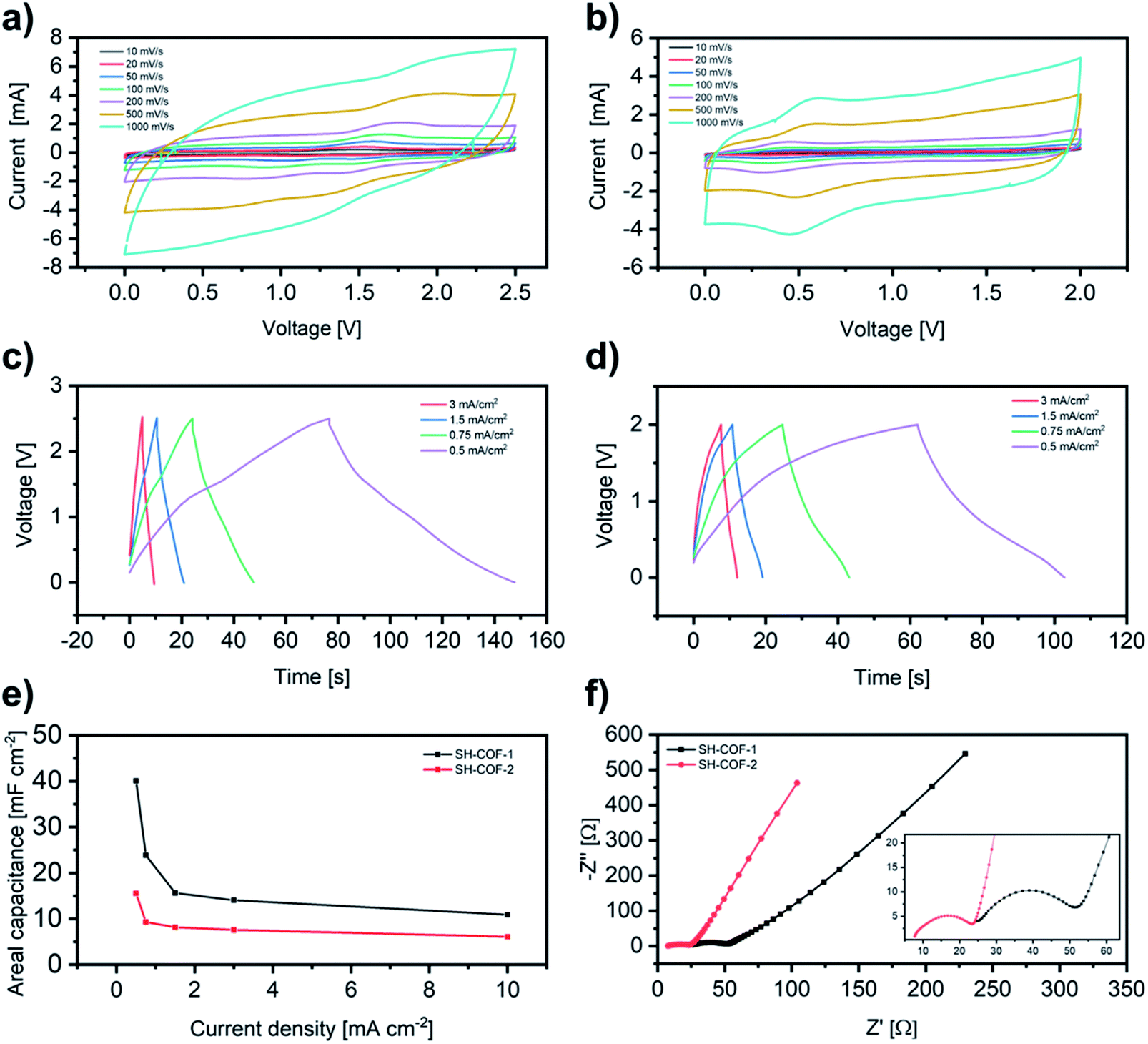 | ||
| Fig. 5 Electrochemical characterization of SH-COFs in a two-electrode configuration. CV curves at different scan rates and GCD curves at different current densities for (a and c) SH-COF-1 and (b and d) SH-COF-2. (e) Areal capacitance as a function of current density and (f) EIS spectra of SH-COF-1 (black) and SH-COF-2 (red). | ||
Furthermore, the electrochemical impedance spectroscopy data are analyzed using Nyquist plots (Fig. 5f, S15 and Tables S3, S4†) and Bode plots (Fig. S16†). As depicted in Fig. 4f, a semicircle is observed within the high-frequency region representing the foremost supercapacitor's resistive nature, which consists of an electrode/electrolyte/current collector. The experimental results are well fitted with the two indicated circuits (Fig. S15†). Rs is the combined internal resistance, including the interfacial contact resistance of the material with the current collector, the ohmic resistance of the electrolyte and the intrinsic resistance of the current collector, which can be obtained from the high-frequency region of the Nyquist plot by intersection on the real axis. Rct is the interfacial charge transfer resistance, corresponding to the diameter of the semicircle, which represents the resistance of electrochemical reactions at the electrode surface. Furthermore, the semicircle can also reflect a constant phase element (CPE) due to the double-layer behavior. In the middle frequency region, the sloping transmission line corresponds to the Warburg element ZW, describing the transfer and diffusion of the electrons and electrolyte ions in the pores of the electrode materials. The nearly vertical line in the low-frequency region reflects an excellent capacitive behavior of the electrodes. The fitting parameters are reported in Tables S3 and S4.† The magnitudes of the Rs and Rct for both SH-COFs are as low as ∼2–25 Ω. The low internal resistance is consistent with the fact that at a scanning rate as high as 1 V s−1, the CV curve still shows a rectangular shape with small distortion, indicating that the SH-COF samples possess a good rate capability. Moreover, a low charge transfer resistance is consistent with the fact that SH-COFs exhibit a good conductivity due to the thiol functional groups. The small charge transfer resistance of SH-COF-2 is correlated with the higher conjugation of its chemical structure resulting in a less resistive COF. However, the presence of a higher amount of redox-active thiol groups in SH-COF-1 allows it to outperform SH-COF-2.
Heterogeneous catalysis
In addition to improving the electrochemical properties of COFs, the presence of thiol groups in the scaffold imparts other properties to the material, thereby widening the functional complexity of COFs and making them suitable for other applications. Due to the high affinity for metal surfaces, including Au and Ag, thiol-derived molecules have been widely employed to chemically stabilize metal NPs for their use in different applications.53 Gold is well-known for its chemical inertness and for its high catalytic activity.54 Although the stability of pristine AuNPs is usually compromised during the catalytic processes, this weakness can be overcome when AuNPs are effectively anchored on a solid support.55,56 Inspired by a recent study by Zhang et al.33 using a thiol-decorated COF as a support for gold nanoparticle (AuNP) growth, and considering that our SH-COFs possess a much higher surface area, we have predicted a higher catalytic activity of the hybrid comprising Au and our SH-COFs.AuNPs are grown in situ in the presence of SH-COF templates by following a reported procedure.33
The transmission electron microscopy (TEM) images of Au-SH-COF-1 and Au-SH-COF-2 (Fig. S17†) portray Au spheres with a narrow size distribution of 10.6 ± 2.8 nm and 13.7 ± 3.2 nm for Au-SH-COF-1 and Au-SH-COF-2, respectively (see histograms in Fig. S18†). High-resolution TEM (HR-TEM) imaging of Au-SH-COFs reveals that the AuNPs are anchored on the COF structure with an interplanar spacing of 0.20 nm, corresponding to the plane of a typical Au face-centered cubic (fcc) structure (Fig. S17†). Moreover, the corresponding selected area electron diffraction (SAED) pattern displays the polycrystalline nature of AuNPs (Fig. S17f†).
The PXRD peaks in Fig. S19† located at 2θ of 38.3°, 44.2, 64.7° and 77.7° are ascribed to the 111, 200, 220 and 311 crystallographic planes of the face-centered cubic gold crystals, respectively, which agrees with the reported values.57 The (111) peak has the highest intensity, indicating that (111) is the preferred orientation.
XPS measurements (Fig. S20 and S21†) show that the Au/C ratio is higher in Au-SH-COF-1, meaning that Au content is higher. This result is in accordance with the higher surface area of SH-COF-1. Fig. S12† displays the high-resolution spectrum of the Au 4f core level, characterized by a pair of peaks due to Au 4f7/2 and Au 4f5/2 spin–orbit coupling (binding energies of 84 and 87.68 eV), which is related to elemental gold (Au0).58 XPS measurements of S 2p in Au-SH-COFs exhibit an energy red shift compared with S 2p in SH-COF, which could be ascribed to the metal–ligand interactions.33
The porosity of Au-SH-COF-1 and Au-SH-COF-2 is evaluated by acquiring N2 adsorption–desorption isotherms at 77 K (Fig. S22†) in their completely activated state, i.e., after treating the samples under vacuum (<1 torr pressure) and heating at 100 °C for 12 h. Analogously to the SH-COFs, the adsorption isotherms of the two Au-SH-COFs exhibit Type-I sorption isotherms, with steep increases appearing at low relative pressure and also Type-IV sorption features with adsorption/desorption hysteresis at higher pressure. The calculated BET surface areas of Au-SH-COF-1 and Au-SH-COF-2 (Fig. S22a†) decrease remarkably to 3.29 ± 0.02 m2 g−1 and 8.65 ± 0.04 m2 g−1, respectively, as compared to those of SH-COFs. This huge decrease in the surface area is expected as AuNPs are formed both on the surface and inside the SH-COF pores. The pore size distribution (Fig. S22b†) shows the dominance of micropores and mesopores.
The nitrophenol reduction to aminophenol is presently considered as the benchmark reaction to evaluate the catalytic activity of metal-nanoparticles.59,60 The catalytic activities of Au-SH-COF-1 and Au-SH-COF-2 are evaluated by the reduction of 4-nitrophenol (4-NP) in water in the presence of NaBH4 by ultraviolet-visible (UV-vis) spectroscopy. The typical UV-vis spectrum of 4-NP shows a broad band centered at 400 nm (Fig. 6a and b), whose intensity is proportional to the 4-NP concentration. When the reduction to 4-aminophenol (4-AP) takes place, the band at 400 nm decreases, and a new band at 300 nm appears, corresponding to the formation of 4-AP (Fig. 6a and b).60 When no catalyst is added, i.e., only NaBH4 (Fig. S23a†) or a mixture of NaBH4 with SH-COF-1 (Fig. S23b†) or SH-COF-2 (Fig. S23c†), the absorption band of 4-NP remains stable, even after 150 min. This indicates the lack of catalytic activity of pristine SH-COFs. However, when the heterogeneous catalyst Au-SH-COF-1 or Au-SH-COF-2 is added to the mixture of 4-NP and NaBH4 in water, the reduction is initiated immediately (Fig. 6a and b). As can be seen in Fig. 6c, the 4-NP band completely disappears after 4 and 5 min for the catalysts Au-SH-COF-1 and Au-SH-COF-2, respectively. The kinetics of the catalytic process (Fig. S24†) are fitted to a pseudo-first-order reaction, and the calculated observed pseudo-first-order rate constant, kobs, amounts to 1.01 min−1 for Au-SH-COF-1 and 0.71 min−1 for Au-SH-COF-2. Compared with other heterogeneous catalytic systems based on metal NPs, Au-SH-COFs exhibit higher catalytic activity (Table S5†). The only example that exhibits better catalytic activity than our Au-SH-COFs is based on Ag NPs,61 whose tendency to oxidation hampers their wider applicability, and hence, the higher chemical stability of AuNPs is preferred. Besides, the higher catalytic activity of Au-SH-COF-1 is related to the higher content of SH groups in the SH-COF structures, as demonstrated by XPS.
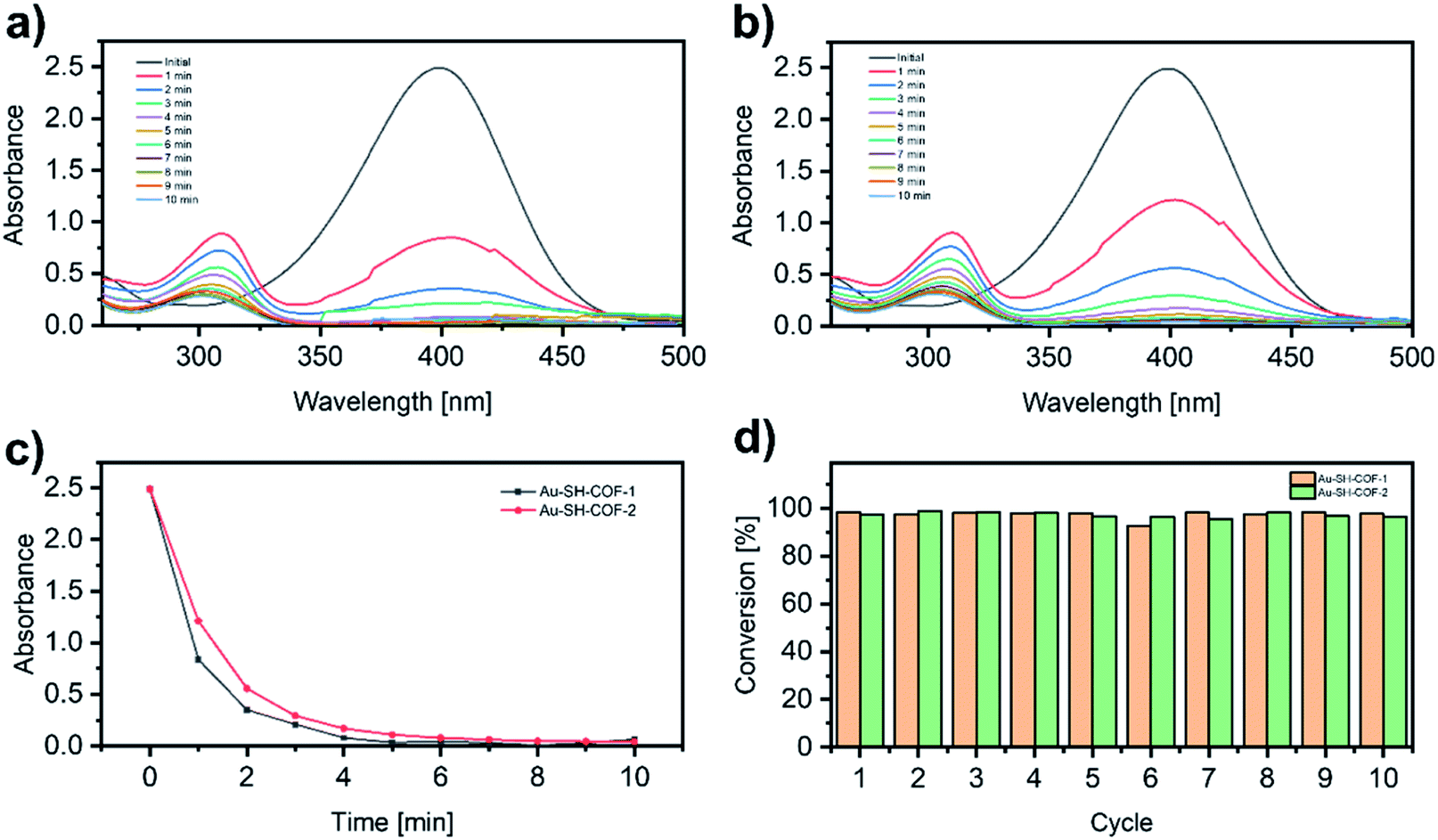 | ||
| Fig. 6 UV-vis spectra of the reduction of 4-nitrophenol in the presence of (a) Au-SH-COF-1 and (b) Au-SH-COF-2; (c) kinetic trace of the absorbance at 400 nm (4-NTP) for Au-SH-COF-1 (black) and Au-SH-COF-2 (red); (d) recyclability of Au-SH-COFs after ten catalytic cycles. | ||
Apart from the catalytic activity, recyclability is another key characteristic that heterogeneous catalysts should possess for practical applications. To test the cyclability of Au-SH-COFs, after the first catalytic cycle, the second addition of 4-NP and NaBH4 is performed, and the reduction reaction is monitored again by UV-vis spectroscopy. This protocol was repeated ten times, and as can be seen in Fig. 6d, the conversion of 4-NP to 4-AP still exceeds 95%, after ten catalytic cycles for both SH-COFs, indicating the excellent stability of Au-SH-COFs under the reaction conditions. Moreover, the STEM analysis after the ten cycles (Fig. S25†) confirms the high stability of the AuNPs strongly anchored on the SH-COF supports, as no morphological changes are observed after the catalytic process.
Conclusions
In summary, we have synthesized two unique COFs decorated with redox-active thiol moieties to boost the electrochemical performance of pristine COFs. Two different aldehydes were used to explore and correlate their chemical structure with electrochemical performance. Both SH-COFs were successfully synthesized based on the Schiff-base reactions by a solvothermal solution-suspension method. We explored their application as supercapacitors in a three-electrode configuration obtaining a maximum areal capacitance of 118 and 74mF cm−2 for SH-COF-1 and SH-COF-2, respectively, at a current of 0/5 A cm−2. Interestingly, we demonstrated the pseudocapacitive nature of both SH-COFs by cyclic voltammetry. We obtained a good rate capability up to 1000 mV s−1 scan rate and excellent stability (capacitance retention 95% and 88% after 5000 cycles for SH-COF-1 and SH-COF-2, respectively). In a symmetric supercapacitor configuration, SH-COF-1 and SH-COF-2 exhibited high areal capacitances of 40 and 16 mF cm−2 at a current density of 0.5 mF cm−2, largely exceeding the analogous performance indicators of COFs lacking redox groups in their structures. Our findings provide evidence for the exceptional potential of designing efficient redox-active COFs to be used as supercapacitors in energy storage applications. Importantly, the post-modification of SH-COFs with AuNPs showcases the multifunctional COFs which can be used as catalyst supports. Au-SH-COFs exhibited a high catalytic activity (kobs, of 1.01 min−1 and 0.71 min−1 for Au-SH-COF-1 and Au-SH-COF-2, respectively) and high recyclability after ten catalytic cycles (conversion of 4-NP to 4-AP above 95%). Overall, this work highlights the importance of rational molecular design of COFs to render them a multifunctional nature and to maximize their catalytic and electrochemical performances.Experimental section
Materials
All reagents including organic compounds and solvents were purchased from Merck Chemical Company and used without further purification. Tetraethylammonium tetrafluoroborate, acetonitrile (electronic grade), Whatman® glass microfiber filters as separators, the binder poly(tetrafluoroethylene) (PTFE) and 1-methyl-2-pyrrolidinone were purchased from Sigma Aldrich. Conductive Carbon Black Super P (H30253) was acquired from Alfa Aesar, and the carbon AvCarb P75 substrate was purchased from FuelCellStore. Gold chloride trihydrate (HAuCl4·3H2O) and sodium borohydride (NaBH4) were purchased from Sigma Aldrich.Characterization
Electrode preparation and electrochemical measurements in the three-electrode configuration
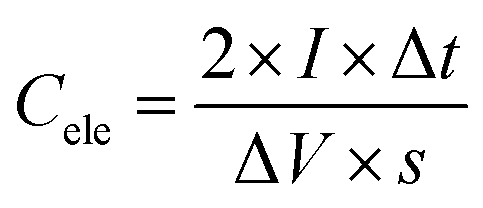 | (1) |
Electrode preparation and electrochemical measurements in the two-electrode system
The preparation procedure of the electrodes was as follows. First, a paste was prepared by fully mixing 80 wt% of the sample, 10 wt% of carbon P, 10 wt% of the polytetrafluoroethylene (PTFE) binder and a certain amount of N-methyl-2-pyrrolidone (NMP) using an agate mortar and pestle. The paste was coated over the carbon electrode, which was then subjected to quick drying at 80 °C. The electrodes were completely dried at 80 °C in a vacuum oven for 16 h. The mass loading of the SH-COF was ∼2 mg in each electrode.
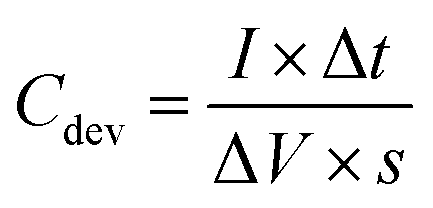 | (2) |
Synthesis of SH-COF-1 and SH-COF-2
COFs were obtained using two types of precursor (aldehydes) through a one-pot hydrothermal process with the Schiff-base reaction. 0.1 mmol (17.2 mg) of 2,5-diaminobenzene-1,4-dithiol and 0.66 mmol (10.7 mg) of benzene-1,3,5-tricarboxaldehyde (for SH-COF-1) or 0.05 mmol (24.7 mg) of 1,2,4,5-tetrakis-(4-formylphenyl)benzene (for SH-COF-2) were precisely weighed and moved into a Pyrex tube. Next, the solvents (1 mL of dichlorobenzene, 1 mL of butyl alcohol and 0.2 mL of 6 M acetic acid) were added directly to the Pyrex tube. The mixture was ultrasonicated for 30 min to get well dispersed homogeneous suspensions. Then, the tube was flash frozen at 77 K in liquid nitrogen, degassed by 3 freeze–pump–thaw cycles and sealed. The reaction mixture was heated at 120 °C in an oven for 72 h to obtain yellow (SH-COF-1) and yellow–brown (SH-COF-2) precipitates. After the reaction, the products were washed with anhydrous THF, DMF and MeOH several times to remove unreacted substrates. Subsequently, the material was activated by Soxhlet extraction in MeOH for 48 h. Finally, the products referred to as SH-COF-1 and SH-COF-2 were isolated by filtration and dried in an oven at 150 °C for 12 h.Synthesis of COF-1 and COF-2
The synthesis of control COFs (COF-1 and COF-2) was performed by following a reported protocol with slight modification.62,63 Briefly, trialdehyde(1,3,5-triformylbenzene 48 mg, 0.3 mmol) or tetraldehyde(1,2,4,5-tetrakis-(4-formylphenyl)benzene, 0.1 mmol 49.4 mg) and 1,4 phenylenediamine (0.45 mmol (48.6 mg) for trialdehyde and 0.2 mmol (21.6 mg) for tetraldehyde) were weighed into a vial and were dissolved in 1,4-dioxane (3 mL); then the mixture was transferred into a glass ampoule. To the mixture, 0.6 mL of 3 M acetic acid was added. Then the ampoule was flash frozen in a liquid nitrogen bath, evacuated and flame sealed. The ampoules were placed in an oven at 120 °C for 72 hours, yielding a yellow solid along the tube. The solids were isolated by centrifugation and washed several times with DMF, THF and MeOH and finally dried at 80 °C under vacuum for 12 hours to yield control COFs (COF-1 and COF-2) as a yellow powder with 85–87% yield.Synthesis of Au-SH-COFs
The synthesis of Au-SH-COFs was performed by following a reported protocol.33 Briefly, 4 mg of SH-COF-1 or SH-COF-2 were well dispersed in 3 mL of methanol and sonicated for 30 min. Then 2 mL of HAuCl4 (1.3 × 10−2 mM) were added under stirring. The mixture was stirred for 48 h and then evaporated to dryness at 80 °C under vacuum overnight. Then, 2 mL of NaBH4 methanol solution (0.25 M) was added dropwise to the above solid, and the mixture was stirred for 48 h. Finally, the product was collected by centrifugation, washed with water and methanol three times and dried at 80 °C under vacuum overnight.Reduction of 4-nitrophenol catalyzed by Au-SH-COFs
In a quartz cuvette, 1 mL of water, 0.3 mL of 4-nitrophenol aqueous solution (1 × 10−3 M) and 1 mL NaBH4 aqueous solution (0.5 M) were added. A well dispersed suspension of Au-SH-COF-1 or -2 in water (0.5 mg mL−1, 100 μL) was added to the cuvette. The reaction was monitored by UV-vis spectroscopy for 10 min. For the control experiments, the concentrations of 4-NP and NaBH4 were the same, and for SH-COF-1 or -2, 100 μL of a 0.5 mg mL−1 stock solution was used. The reaction was monitored by UV-vis spectroscopy for 150 min.Recyclability of Au-SH-COFs
After the first reaction was performed by using the protocol described above, 18.5 mg of NaBH4 and 0.03 mL of 1 × 10−2 M 4-nitrophenol aqueous solution were added to the same reaction system. The reaction was monitored by UV-vis spectroscopy for 10 min. This procedure was repeated another 8 times. The catalytic efficiency is the ratio of the amount of change in ultraviolet absorption intensity at 400 nm within 10 min to the initial ultraviolet absorption intensity. After ten cycles, the Au-SH-COFs were collected by centrifugation, washed with ethanol three times, and dried under vacuum for further characterization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Centre, Poland (grant no. 2020/36/C/ST5/00247 and grant no. 2019/35/B/ST5/01568), the Agence Nationale de la Recherche through the Interdisciplinary Thematic Institute SysChem via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d'Avenir, the International Center for Frontier Research in Chemistry (icFRC), and the Institut Universitaire de France (IUF). The authors thank Dr Michal Bielejewski from the Institute of Molecular Physics Polish Academy of Sciences for the high-resolution solid-state NMR data and Dr Wojciech Kukułka for the BET analysis of Au-SH-COFs.References
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- W. Raza, F. Ali, N. Raza, Y. Luo, K.-H. Kim, J. Yang, S. Kumar, A. Mehmood and E. E. Kwon, Nano Energy, 2018, 52, 441–473 CrossRef CAS.
- Y. Ren, T. Zhu, Y. Liu, Q. Liu and Q. Yan, Small, 2021, 17, 2008047 CrossRef CAS PubMed.
- T. Zhu, Z. He, Y. Ren, W. Zeng, J. Mao and L. Zhu, Sol. RRL, 2021, 5, 2100021 CrossRef CAS.
- J. Ren, M. Shen, Z. Li, C. Yang, Y. Liang, H.-E. Wang, J. Li, N. Li and D. Qian, J. Power Sources, 2021, 501, 230003 CrossRef CAS.
- C. Miao, C. Zhou, H.-E. Wang, K. Zhu, K. Ye, Q. Wang, J. Yan, D. Cao, N. Li and G. Wang, J. Power Sources, 2021, 490, 229532 CrossRef CAS.
- Y.-F. Ren, Z.-L. He, H.-Z. Zhao and T. Zhu, Rare Met., 2022, 41, 830–835 CrossRef CAS.
- B. Xu, H. Zhang, H. Mei and D. Sun, Coord. Chem. Rev., 2020, 420, 213438 CrossRef CAS.
- S. Wang, Y.-z. Guo, F.-x. Wang, S.-h. Zhou, T.-y. Zeng and Y.-b. Dong, New Carbon Mater., 2022, 37, 109–135 CrossRef.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- Y. Zeng, R. Zou and Y. Zhao, Adv. Mater., 2016, 28, 2855–2873 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang, O. M. Yaghi and C. J. Chang, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- S.-Y. Ding, M. Dong, Y.-W. Wang, Y.-T. Chen, H.-Z. Wang, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2016, 138, 3031–3037 CrossRef CAS PubMed.
- Q. Fang, J. Wang, S. Gu, R. B. Kaspar, Z. Zhuang, J. Zheng, H. Guo, S. Qiu and Y. Yan, J. Am. Chem. Soc., 2015, 137, 8352–8355 CrossRef CAS PubMed.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- I. E. Rauda, V. Augustyn, B. Dunn and S. H. Tolbert, Acc. Chem. Res., 2013, 46, 1113–1124 CrossRef CAS PubMed.
- M. Li, J. Liu, T. Zhang, X. Song, W. Chen and L. Chen, Small, 2021, 17, 2005073 CrossRef CAS PubMed.
- S. Chandra, D. Roy Chowdhury, M. Addicoat, T. Heine, A. Paul and R. Banerjee, Chem. Mater., 2017, 29, 2074–2080 CrossRef CAS.
- C. R. DeBlase, K. E. Silberstein, T.-T. Truong, H. D. Abruña and W. R. Dichtel, J. Am. Chem. Soc., 2013, 135, 16821–16824 CrossRef CAS PubMed.
- C. R. DeBlase, K. Hernández-Burgos, K. E. Silberstein, G. G. Rodríguez-Calero, R. P. Bisbey, H. D. Abruña and W. R. Dichtel, ACS Nano, 2015, 9, 3178–3183 CrossRef CAS PubMed.
- A. Halder, M. Ghosh, A. Khayum M, S. Bera, M. Addicoat, H. S. Sasmal, S. Karak, S. Kurungot and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 10941–10945 CrossRef CAS PubMed.
- M. Li, J. Liu, Y. L. Li, G. Xing, X. Yu, C. Peng and L. Chen, CCS Chem., 2020, 2, 696–706 Search PubMed.
- M. Chen, H. Li, C. Liu, J. Liu, Y. Feng, A. G. H. Wee and B. Zhang, Coord. Chem. Rev., 2021, 435, 213778 CrossRef CAS.
- X. Wang, Z. Zhou, X. Lin, Z. Pei, D. Liu and S. Zhao, Chem. Eng. J., 2022, 427, 130995 CrossRef CAS.
- S. Haldar, R. Kushwaha, R. Maity and R. Vaidhyanathan, ACS Mater. Lett., 2019, 1, 490–497 CrossRef CAS.
- T. Li, X. Yan, Y. Liu, W.-D. Zhang, Q.-T. Fu, H. Zhu, Z. Li and Z.-G. Gu, Polym. Chem., 2020, 11, 47–52 RSC.
- S. Shaheen Shah, S. M. Abu Nayem, N. Sultana, A. J. Saleh Ahammad and M. Abdul Aziz, ChemSusChem, 2022, 15, e202101282 CrossRef CAS PubMed.
- X. Yu, S. K. Park, S.-H. Yeon and H. S. Park, J. Power Sources, 2015, 278, 484–489 CrossRef CAS.
- D.-G. Wang, N. Li, Y. Hu, S. Wan, M. Song, G. Yu, Y. Jin, W. Wei, K. Han, G.-C. Kuang and W. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 42233–42240 CrossRef CAS PubMed.
- S. Wang, Y. Liang, T. Dai, Y. Liu, Z. Sui, X. Tian and Q. Chen, J. Colloid Interface Sci., 2021, 591, 264–272 CrossRef CAS PubMed.
- Z. Ma, F. Liu, N. Liu, W. Liu and M. Tong, J. Hazard. Mater., 2021, 405, 124190 CrossRef CAS PubMed.
- Z. Yang, Y. Gu, B. Yuan, Y. Tian, J. Shang, D. C. W. Tsang, M. Liu, L. Gan, S. Mao and L. Li, J. Hazard. Mater., 2021, 403, 123702 CrossRef CAS PubMed.
- Q.-P. Zhang, Y.-l. Sun, G. Cheng, Z. Wang, H. Ma, S.-Y. Ding, B. Tan, J.-h. Bu and C. Zhang, Chem. Eng. J., 2020, 391, 123471 CrossRef CAS.
- S. Lu, Y. Hu, S. Wan, R. McCaffrey, Y. Jin, H. Gu and W. Zhang, J. Am. Chem. Soc., 2017, 139, 17082–17088 CrossRef CAS PubMed.
- R. W. Layer, Chem. Rev., 1963, 63, 489–510 CrossRef CAS.
- J. Xiao, J. Chen, J. Liu, H. Ihara and H. Qiu, Green Energy Environ., 2022 DOI:10.1016/j.gee.2022.05.003.
- S.-Y. Ding, X.-H. Cui, J. Feng, G. Lu and W. Wang, Chem. Commun., 2017, 53, 11956–11959 RSC.
- D.-H. Yang, Z.-Q. Yao, D. Wu, Y.-H. Zhang, Z. Zhou and X.-H. Bu, J. Mater. Chem. A, 2016, 4, 18621–18627 RSC.
- S. Karak, S. Kandambeth, B. P. Biswal, H. S. Sasmal, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 1856–1862 CrossRef CAS PubMed.
- L. Huang, R. Shen, R. Liu and Q. Shuai, J. Hazard. Mater., 2020, 392, 122320 CrossRef CAS PubMed.
- A. F. M. El-Mahdy, M. G. Mohamed, T. H. Mansoure, H.-H. Yu, T. Chen and S.-W. Kuo, Chem. Commun., 2019, 55, 14890–14893 RSC.
- Q. Sun, B. Aguila, J. Perman, L. D. Earl, C. W. Abney, Y. Cheng, H. Wei, N. Nguyen, L. Wojtas and S. Ma, J. Am. Chem. Soc., 2017, 139, 2786–2793 CrossRef CAS PubMed.
- D. Chen, C. Gan, X. Fan, L. Zhang, W. Li, M. Zhu and X. Quan, Materials, 2019, 12, 2800 CrossRef CAS PubMed.
- A. M. Khattak, Z. A. Ghazi, B. Liang, N. A. Khan, A. Iqbal, L. Li and Z. Tang, J. Mater. Chem. A, 2016, 4, 16312–16317 RSC.
- M. Afshari and M. Dinari, J. Hazard. Mater., 2020, 385, 121514 CrossRef CAS PubMed.
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- E. Troschke, M. Oschatz and I. K. Ilic, Exploration, 2021, 1, 20210128 CrossRef.
- G. A. M. Ali, E. Y. Lih Teo, E. A. A. Aboelazm, H. Sadegh, A. O. H. Memar, R. Shahryari-Ghoshekandi and K. F. Chong, Mater. Chem. Phys., 2017, 197, 100–104 CrossRef CAS.
- R. Heimböckel, F. Hoffmann and M. Fröba, Phys. Chem. Chem. Phys., 2019, 21, 3122–3133 RSC.
- X. Wu, J. J. Hong, W. Shin, L. Ma, T. Liu, X. Bi, Y. Yuan, Y. Qi, T. W. Surta, W. Huang, J. Neuefeind, T. Wu, P. A. Greaney, J. Lu and X. Ji, Nat. Energy, 2019, 4, 123–130 CrossRef CAS.
- Y. Jiang and J. Liu, Energy Environ. Mater., 2019, 2, 30–37 CrossRef.
- V. Montes-García, M. A. Squillaci, M. Diez-Castellnou, Q. K. Ong, F. Stellacci and P. Samorì, Chem. Soc. Rev., 2021, 50, 1269–1304 RSC.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852–7872 CrossRef CAS PubMed.
- A. Corma and H. Garcia, Chem. Soc. Rev., 2008, 37, 2096–2126 RSC.
- M. N. Owaid, M. A. Rabeea, A. Abdul Aziz, M. S. Jameel and M. A. Dheyab, Environ. Nanotechnol., Monit. Manage., 2019, 12, 100270 Search PubMed.
- V. Montes-García, R. F. de Oliveira, Y. Wang, A. Berezin, P. Fanjul-Bolado, M. B. González García, T. M. Hermans, D. Bonifazi, S. Casalini and P. Samorì, Adv. Funct. Mater., 2021, 31, 2008554 CrossRef.
- S. Rodal-Cedeira, V. Montes-García, L. Polavarapu, D. M. Solís, H. Heidari, A. La Porta, M. Angiola, A. Martucci, J. M. Taboada, F. Obelleiro, S. Bals, J. Pérez-Juste and I. Pastoriza-Santos, Chem. Mater., 2016, 28, 9169–9180 CrossRef CAS.
- T. Aditya, A. Pal and T. Pal, Chem. Commun., 2015, 51, 9410–9431 RSC.
- R.-L. Wang, D.-P. Li, L.-J. Wang, X. Zhang, Z.-Y. Zhou, J.-L. Mu and Z.-M. Su, Dalton Trans., 2019, 48, 1051–1059 RSC.
- S.-Y. Ding, J. Gao, Q. Wang, Y. Zhang, W.-G. Song, C.-Y. Su and W. Wang, J. Am. Chem. Soc., 2011, 133, 19816–19822 CrossRef CAS PubMed.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31, 1901640 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta03867f |
| This journal is © The Royal Society of Chemistry 2022 |