
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improving poly(arylene piperidinium) anion exchange membranes by monomer design†
Dong
Pan
 ,
Pegah Mansouri
Bakvand
,
Thanh Huong
Pham
and
Patric
Jannasch
*
,
Pegah Mansouri
Bakvand
,
Thanh Huong
Pham
and
Patric
Jannasch
*
Polymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden. E-mail: patric.jannasch@chem.lu.se
First published on 5th July 2022
Abstract
Energy conversion devices such as alkaline membrane fuel cells and water electrolyzers rely critically on durable anion exchange membranes (AEMs) with high hydroxide conductivity. In this context, poly(arylene piperidinium)s have emerged as one of the top candidate materials. Here, we report on the preparation and properties of poly(arylene alkylene piperidinium)s (PAAPs) with significantly higher alkaline stability than current state-of-the-art poly(arylene piperidinium)s derived from piperidones. A new piperidine trifluoromethyl ketone monomer (TFPip) was designed, synthesized and employed in superacid-mediated polyhydroxyalkylations with p- and m-terphenyl, biphenyl and fluorene, respectively. The pendant piperidine rings of the resulting polymers were then quaternized and cycloquaternized to form N,N-dimethylpiperidinium (DMP) and 6-azonia-spiro[5.5]undecane (ASU) cations, respectively. Polymers based on p- and m-terphenyl were cast into mechanically strong AEMs which reached OH− conductivities close to 80 and 180 mS cm−1 at 20 and 80 °C, respectively. The AEMs also displayed an excellent resistance against OH− attack. For example, AEMs carrying DMP cations showed a mere 14% ionic loss after storage in 5 M aq. NaOH at 90 °C during 20 days. In comparison, a corresponding benchmark poly(arylene piperidinium) AEM lost three times as many DMP cations (42%) under the same conditions. The results of the study demonstrate that the overall properties and alkaline stability of AEMs can be considerably improved by rational monomer design.
Introduction
A broad world-wide implementation of fuel cell powered vehicles and devices calls for the development of a more sustainable and cost-effective alternative to the proton exchange membrane fuel cell (PEMFC). The most viable option is the anion exchange membrane fuel cell (AEMFC), which has the potential to operate without the use of platinum-group metal catalysts and perfluorinated membranes.1–4 In parallel, the anion exchange membrane water electrolyzer (AEMWE) is developed to efficiently produce green hydrogen under alkaline conditions in a low-cost and sustainable way.5–7 Consequently, considerable research efforts are currently devoted to the development of durable and functional anion exchange membranes (AEMs) for AEMFCs and AEMWEs.8–13 AEMs should possess high ion conductivity at a limited ionic content to avoid excessive water uptake and swelling that will compromise the mechanical strength. The long-term structural integrity and properties of the AEMs are however greatly challenged by the severe alkaline operating conditions in these devices. Both the polymer backbone and the tethered cations are prone to degrade via hydroxide attack, resulting in loss of mechanical integrity and ion conductivity, respectively.14To avoid backbone cleavage, polymers devoid of vulnerable ether bonds have been employed in AEMs, including polyethylenes,15,16 polystyrenes,17 polybenzimidazoles,18 polyphenylenes19 and poly(arylene alkylene)s.20–22 In addition, the alkaline stability of various organic cations, including imidazolium,23 quaternary ammonium (QA)24–27 and organometallic cations,28,29 have been studied as model compounds to identify suitable candidates.30 In this context, different N-heterocyclic QA cations have emerged as synthetically accessible and generally highly alkali-stable ion-exchange groups. Especially the N,N-dimethylpiperidinium (DMP) and 6-azonia-spiro[5.5]undecane (ASU) cations have shown especially high alkaline stability,24 which has prompted a large number of studies by us31–33 and others.34–36
Poly(arylene piperidinium)s (PAPs) have piperidinium rings directly attached to the backbone structure and constitute a rather new class of polymers which has drawn increasing attention as membrane materials, primarily because of their stiff aromatic backbone, high alkaline stability, tunable structure, and relative ease of synthesis and processing (Scheme 1). Commercially available 4-piperidone and N-methyl-4-piperidone monomers and different arenes enable straightforward synthesis of high-molecular weight PAPs by superacid-mediated polyhydroxyalkylations, followed by a suitable quaternization to introduce the desired piperidinium-type cation. In general, AEMs based on PAPs with moderate ion exchange capacities (IECs) show a well-controlled water uptake, high hydroxide ion conductivity and chemical stability. For example, an AEM based on poly(p-terphenyl dimethylpiperidinium) displayed no structural change after storage in 2 M aq. NaOH at 60 °C for 15 days.37 Subsequently, the ex-situ stability of PAP-based AEMs has been verified to exceed 1500 h in 1 M aq. NaOH at 80 °C.38,39 A number of different successful approaches have been reported to further improve membrane durability and performance, including the introduction of flexible spacers,40 arrangements with multiple cations,41,42 crosslinking,43–45 branching46 and reinforcement.47 For example, we recently reported on a series of cationically modified poly(biphenyl piperidine)s which retained >92% of the cations after storage in 2 M aq. NaOH at 90 °C for 30 days.48 This was achieved by leaving the piperidine groups in the backbone non-quaternized and hence far less sensible to hydroxide attack and degradation.
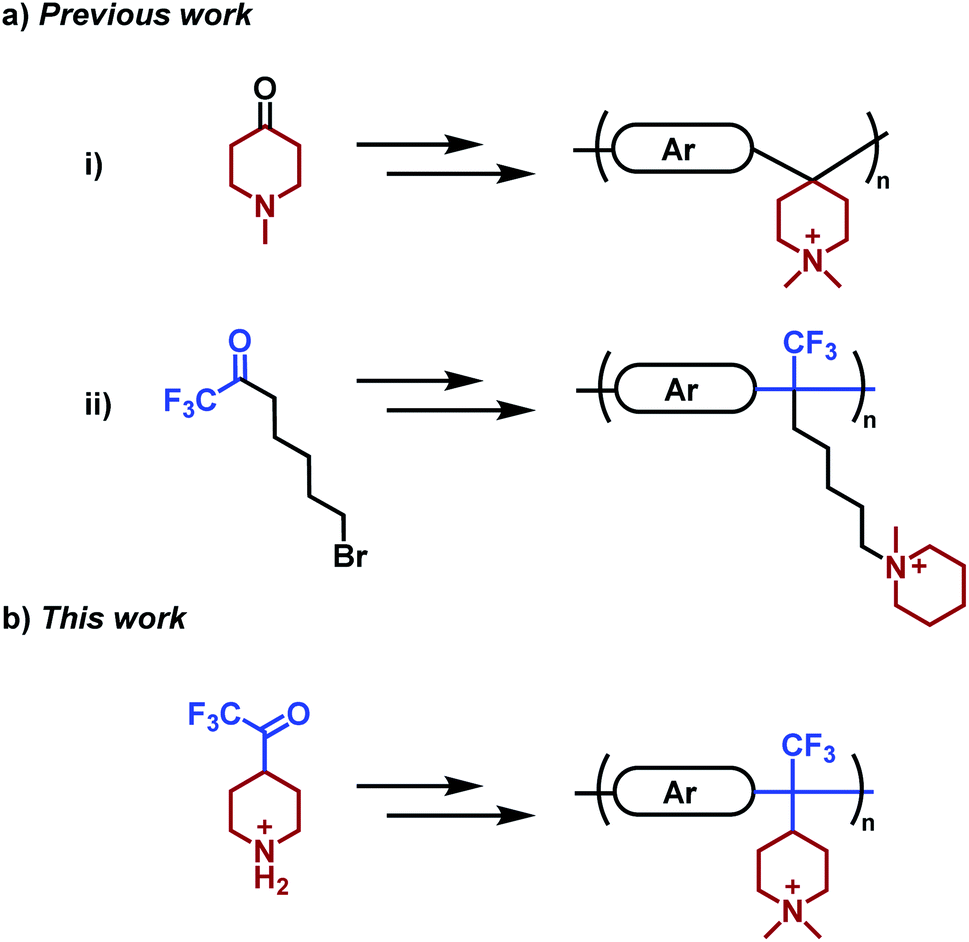 | ||
| Scheme 1 Monomers and corresponding polymers prepared in two steps by polyhydroxyalkylation and subsequent quaternization to produce AEMs. Key: (a) previous work based on (i) poly(arylene piperidinium)s [e.g., poly(p-terphenyl piperidinium)] and (ii) poly(arylene alkylene)s [e.g., poly(biphenyl alkylene)] prepared from prepared from piperidone37 and 7-bromo-1,1,1-trifluoro-2-heptanone,53 respectively. (b) The present work is based on poly(arylene alkylene piperidinium)s derived from the TFPip monomer. Ar = arene. | ||
Although quite alkali-stable, PAP-based AEMs are still susceptible to ionic loss via, e.g., ring-opening Hofmann elimination and substitution in harsh alkaline environments. A high ionic loss relative to model piperidinium cations has been partly attributed to the conformational distortion of the piperidinium ring directly attached to the rigid PAP backbone.37 Furthermore, the ionic loss has been observed to accelerate when the possibilities for ring relaxation was further restricted by the spirocyclic arrangement of the ASU cation.49 Hence, the alkaline stability of these cations is highly dependent on precisely how they are attached to the polymer backbone,9,50 and in many cases on the rigidity of the backbone itself.17,51 Providing a high local mobility of the cations by tethering via flexible spacers has shown to increase their resistance against hydroxide attack. In order to reduce ionic loss by β-elimination at the spacer chain, it is advantageous to attach the piperidinium rings via the 4-position instead of the 1(N)-position. In this way, all the β-hydrogens are placed in the better protected ring structure, which produces substantially more stable AEMs.52
With the aim to further enhance membrane properties and stability beyond that of AEMs based on PAP, we have in the present work designed and synthesized a new piperidine-functional trifluoromethylketone monomer, and then prepared and studied a series of poly(arylene alkylene piperidinium)s (PAAPs) tethered with DMP and ASU cations (Scheme 1). The monomer was designed to place the cations pendent to the polymer backbone in order to lessen the conformational distortion of the rings by the stiff backbone and increase the ring strain relaxation. The PAAPs were prepared in superacid-mediated polycondensations with different arene monomers, followed by quaternization reactions to produce the cations. Solution-cast AEMs were characterized with regard to water uptake, hydroxide conductivity, morphology, thermal and alkaline stability. A special emphasis was placed on alkaline stability and ion-loss mechanisms, and the results were benchmarked against PAP reference AEMs.
Results and discussion
Monomer synthesis and characterization
Piperidone and various trifluoromethyl ketones have been widely employed in Friedel–Crafts type polycondensations with electron-rich aromatic compounds to prepare linear, high-molecular-weight polymers functionalized with different cationic groups (Scheme 1).20,37 In the present work, the monomer 2,2,2-trifluoro-1-(4-piperidine)ethanone (TFPip) was designed to improve the alkaline stability of AEMs, and was subsequently synthesized as a hydrochloride salt in two steps, starting from commercially available 4-piperidinecarboxylic acid (Fig. 1). In the first step, the carboxylic moiety was converted to a trifluoroacetyl group by a reaction with trifluoroacetic anhydride (TFAA), in the presence of pyridine, followed by hydrolysis/decarboxylation in water. In parallel, the piperidine N-H functionality underwent N-trifluoroacetylation. The majority of the piperidine groups remained in the protected form after the hydrolysis, and a mixture of the intermediate and the final TFPip product (salt-free) was detected in the crude product (Fig. S1†). Still, this did not interfere with the subsequent synthesis and purification. In the second step, a complete deprotection of the mixed intermediate product was carried out in concentrated HCl at 100 °C overnight. Finally, the pure TFPip hydrochloride salt was obtained after recrystallization in acetonitrile. The synthesis was based on 26 g 4-piperidinecarboxylic acid which was divided in two separate reactions conducted in parallel before combining the batches in the second step.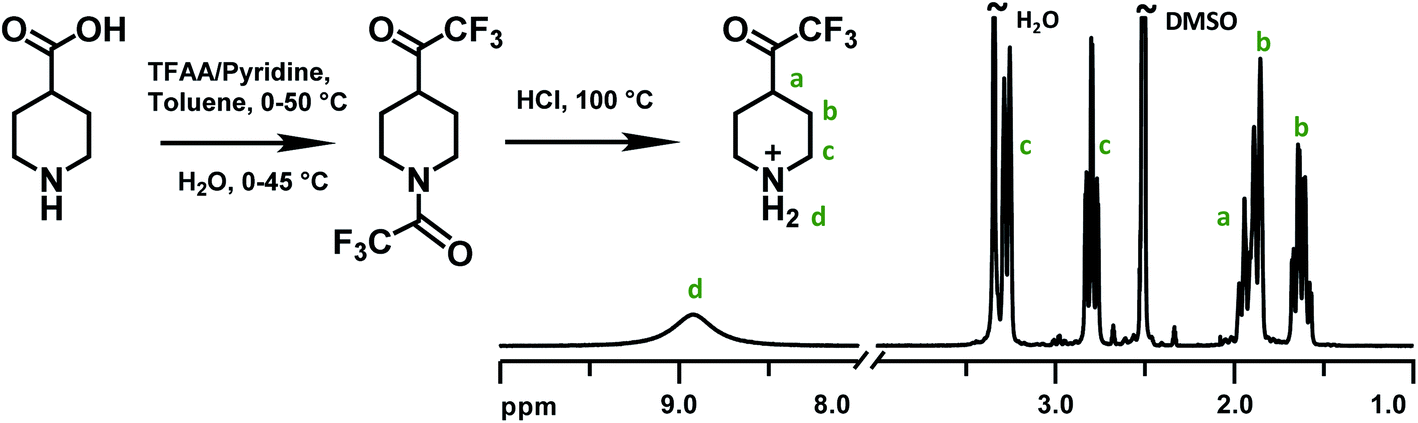 | ||
| Fig. 1 Synthetic route to monomer TFPip and 1H NMR spectrum of an isolated sample in the hydrochloride salt form, recorded in DMSO-d6. | ||
The successful synthesis and purification of TFPip was confirmed by 1H NMR spectroscopy (Fig. 1). The α (c) and β (b) protons of the piperidine ring were observed as split signals located between 2.8–3.3 ppm and 1.6–1.9 ppm, respectively. The methine proton (a) emerged at around 1.9 ppm and was partly overlapped with one of the β (b) proton signals. A broad singlet at 8.9 ppm was assigned to the protonated secondary piperidine ring (d). The molar mass of TFPip was also confirmed by high-resolution mass spectroscopy (Table S1†).
Synthesis and characterization of poly(arylene alkylene piperidinium)s
Superacid-mediated polyhydroxyalkylations involving TFPip and different electron-rich arene monomers were carried out as shown in Scheme 2. The electron-withdrawing trifluoromethyl substituent gave the TFPip monomer sufficient electrophilicity to react with common arenes in the superacid medium.54 During the polycondensations, the ketone moiety of TFPip is protonated to generate a dication to react with the arene monomer. In general, TFPip showed moderate reactivity, as evidenced by the long polymerization periods required to reach high molecular weights. Consequently, we used a high molar excess of TFPip (up to 40%) in order to accelerate the polymerization rate. The polycondensations were carried out in dichloromethane (DCM) at 20 °C and the resulting polymers containing p-terphenyl (pT), m-terphenyl (mT), biphenyl (B) and 9,9-dimethyl-9H-fluorene (dF) units were denoted PpTPip, PmTPip, PBPip and PdFPip, respectively. Their structures were subsequently verified by 1H NMR spectroscopy (Fig. 2 and S2†) after addition of trifluoroacetic acid (TFA) to the DMSO-d6 solutions. The signals located between 1.5 and 3.9 ppm corresponded to piperidine ring protons (a)–(c) and were found in all the polymer spectra. Moreover, the signal of the protonated secondary N+–H2 groups (d) appeared between 8.0 and 8.5 ppm as distinctly split singlets. In polyhydroxyalkylations involving piperidone, signals originating from vinylic groups formed in elimination reactions are frequently observed at 6.2 and 4.0 ppm.37 These side reactions limit the molecular weight, and thus the AEM properties. In the present case, no traces of elimination products were detected in any of the polycondensation experiments, which shows that the TFPip monomer is less sensitive to elimination than piperidone during polyhydroxyalkylations. In the protonated form, the polymers were readily soluble in common organic solvents, such as dimethyl sulfoxide (DMSO), N-methyl-2-pyrrolidone (NMP) and acetone, but were non-soluble in DCM, chloroform and tetrahydrofuran (THF).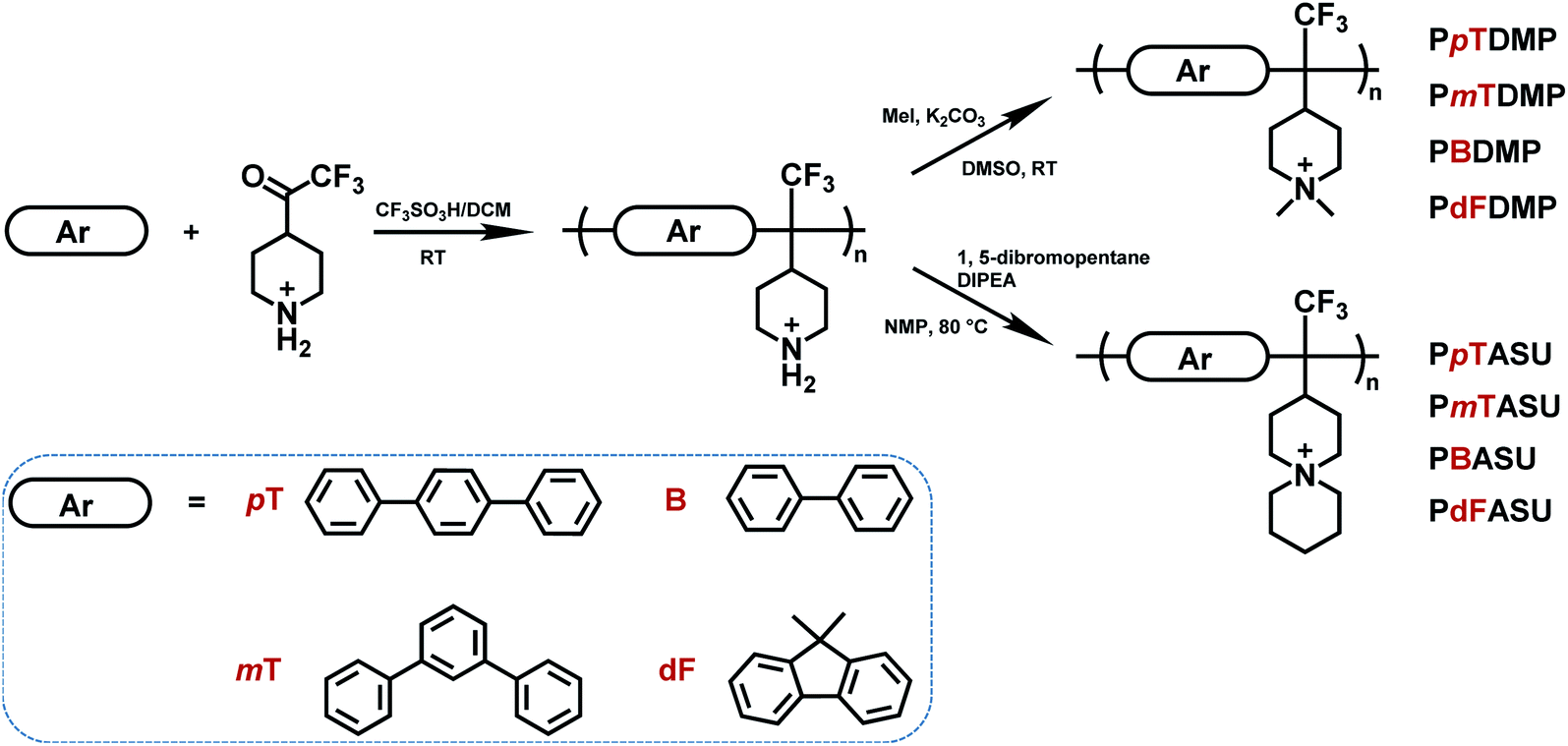 | ||
| Scheme 2 Synthesis and designations of the cationic polymers prepared via superacid-mediated polyhydroxyalkylations and quaternizations. | ||
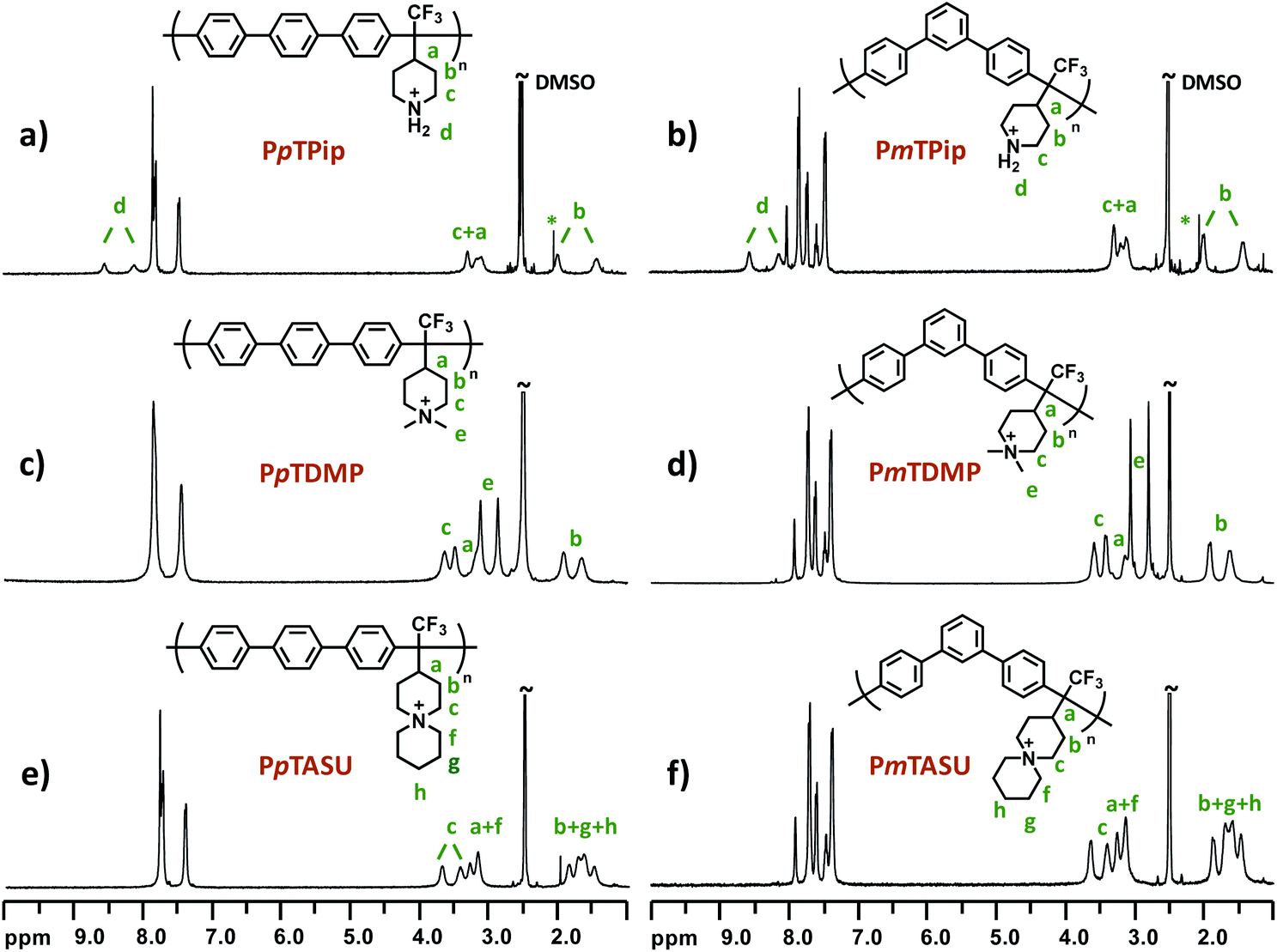 | ||
| Fig. 2 1H NMR spectra of sample (a) PpTPip, (b) PmTPip, (c) PpTDMP, (d) PmTDMP, (e) PpTASU and (f) PmTASU, recorded in DMSO-d6 with 5 vol% TFA. | ||
PAAPs were prepared by quaternizations of the precursor polymers (PpTPip, PmTPip, PBPip and PdFPip) using methyl iodide and 1,5-dibromopentane, respectively, which resulted in the formation of DMP and ASU cations, respectively. Because of the limited solubility in solvents commonly used in size exclusion chromatography (e.g., chloroform and THF), as well as a lack of suitable reference samples, the molecular weight of the polymers carrying the DMP cation was estimated by intrinsic viscosity ([η]) measurements in 0.1 M LiBr in DMSO at 25 °C. The results (Table 1) showed that high molecular weights were achieved in polycondensations involving the terphenyls (pT and mT), which was further supported by the good film-forming properties of these samples. However, it was not possible to reliably measure the intrinsic viscosities of PBDMP and PdFDMP, suggesting low molecular weights when the polymerizations involved biphenyl and dimethylfluorene, respectively. This may be attributed to the comparatively low reactivity of the latter arenes in the polycondensations. Although free-standing membranes were cast from DMSO solutions of PBDMP and PBASU, they were rather brittle in the dry state, and extremely soft when hydrated and thus difficult to handle. This was most probably a consequence of the high IEC, the rather high backbone flexibility, combined with limited molecular weights. Therefore, cationic polymers based on pT and mT were excellent candidates for use as AEMs, whereas those based on biphenyl and dimethylfluorene may be suitable as high-IEC ionomers in the electrode layers. Because of the high reactivity of pT, we chose to further optimize this polycondensation. The results showed values of [η] between 0.52 and 1.37 dL g−1 depending on the reaction conditions. Details of the different polymerizations and the products are described and summarized in the experimental section (Table 2). The PpTPip sample reaching the highest intrinsic viscosity (Table 2, entry 4) was used in the preparation of the AEMs further described below.
| AEM | IEC (mequiv. g−1) | WUb (%) (20 °C) | WUb (%) (80 °C) | σ (mS cm−1) (20 °C) | σ (mS cm−1) (80 °C) | λ (80 °C) | T d,95 (°C) | [η]c (dL g−1) | |
|---|---|---|---|---|---|---|---|---|---|
| Theoreticala | Titrated | ||||||||
| a Calculated from the polymer structures in the Br− form (OH− form within parentheses). b Measured in the OH− form. c Measured in DMSO at 25 °C. | |||||||||
| PpTDMP | 1.99 (2.27) | 2.03 (2.32) | 78 | 104 | 79 | 178 | 25 | 452 | 1.37 |
| PpTASU | 1.84 (2.08) | 1.85 (2.09) | 43 | 81 | 70 | 157 | 22 | 389 | — |
| PmTDMP | 1.99 (2.27) | 2.02 (2.31) | 144 | 446 | 78 | 169 | 107 | 428 | 0.25 |
| PmTASU | 1.84 (2.08) | 1.83 (2.07) | 105 | 341 | 67 | 147 | 92 | 394 | — |
| Entry | TFPip:pT | TFSA:TFPip | TFSA:DCM | Polymerization time (h) | [η]a (dL g−1) | WUb (%) (20 °C) | WUb (%) (80 °C) | σ (mS cm−1) (20 °C) | σ (mS cm−1) (80 °C) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in DMSO at 25 °C. b Measured in the OH− form. | |||||||||
| 1 | 1.2 | 10 | 2 | 288 | 0.52 | 102 | 174 | 75 | 167 |
| 2 | 1.3 | 18 | 2 | 168 | 0.83 | 93 | 128 | 67 | 155 |
| 3 | 1.4 | 13 | 2 | 144 | 0.78 | 88 | 121 | 68 | 156 |
| 4 | 1.4 | 18 | 2 | 576 | 1.37 | 78 | 104 | 79 | 178 |
Membrane preparation and morphology
Transparent membranes with excellent robustness and flexibility were prepared from PpTDMP, PpTASU, PmTDMP and PmTASU solutions in NMP at 80 °C. The AEMs were ion-exchanged to the Br− form and subsequently stored in deionized water prior to characterization. The ion exchange capacity (IEC) was determined by Mohr titrations. As shown in Table 1, the results were in excellent accordance with the theoretical values expected from the polymer structures, which indicated the completeness of the quaternizations. The morphology of the AEMs was studied by small-angle X-ray scattering (SAXS) and the results are shown in Fig. 3. As indicated by the arrows, ionomer peaks indicated by scattering maxima were observed for PpTDMP and PpTASU. Specifically, the ionomer peak of PpTDMP were found at q = 1.6 nm−1, which corresponded to a characteristic separation distance (d) of 3.9 nm. PpTASU displayed a similar scattering profile with the ionomer peak at q = 1.3 nm−1 and a corresponding d-spacing of 4.8 nm. In addition, a second scattering maximum was observed for these two AEMs at q = 5.6 nm−1. This corresponded to d = 1.1 nm, which approximately equals the length of a terphenylene unit. In comparison, benchmark poly(p-terphenyl piperidinium) (PTPipQ100) and poly(biphenyl piperidinium) (PBPipQ100), both derived from piperidone (Scheme 1), displayed single ionomer peaks at q = 1.6 and 2.5 nm−1, respectively, and showed no scattering maxima around q = 5.6 nm−1. Furthermore, AEMs based on mT showed a distinctly different SAXS profile than their pT counterparts. A broad ionomer peak centered around q = 3.1 nm−1 was detected for PmTDMP. This can be explained by the higher flexibility of the m-terphenylene conformation which potentially facilitated ionic aggregation. PBDMP and PBASU exhibited an ionomer peak around 2.3 and 2.2 nm−1, respectively, indicating their distinct phase-separated morphology.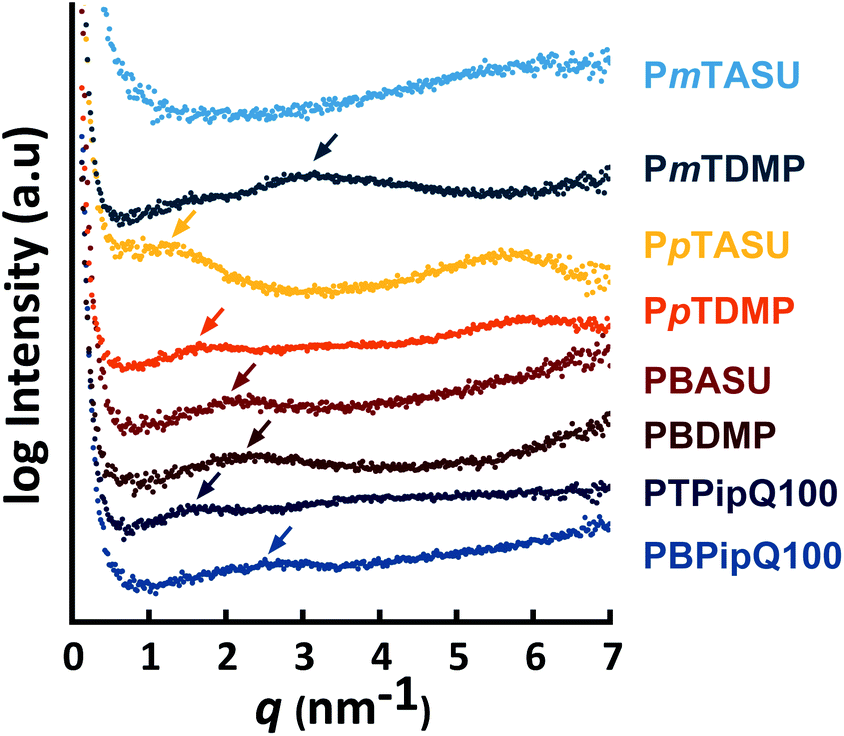 | ||
| Fig. 3 SAXS profiles of the PAAP AEMs. For comparison, scattering data of PTPipQ100 and PBPipQ100 are also shown. | ||
Thermal stability
The thermal stability of the precursor polymers and the AEM samples were studied by thermogravimetrical analysis (TGA) (Fig. 4) by recording the thermal decomposition temperature at 95% weight loss (Td,95) (Table 1). The precursor polymers PmTPip and PpTPip both degraded in single steps at Td,95 = 376 and 385 °C, respectively, attributed to their stiff and aromatic backbone structures. After the quaternizations, the AEMs surprisingly reached an even higher thermal stability. The PmTDMP and PpTDMP samples displayed significantly higher Td,95 values at 428 and 452 °C, respectively, while PpTASU and PmTASU had lower values, i.e., Td,95 = 389 and 394 °C, respectively. As frequently reported, QA functionalized AEMs normally decompose at lower temperatures than the non-functionalized precursors. For example, the Td,95 values of poly(arylene piperidinium)s37,43 and poly(arylene alkylene)s20,55 have previously been reported to be lower than their corresponding precursors polymers. In the present study, the opposite trend can be attributed to the nature of the precursor polymers, where the piperidine groups remained protonated and carried TFSA− counter ions. We speculated that the presence of the triflate ions weakened the piperidine unit and therefore impaired its thermal stability. The thermal stability of samples based on the two different backbone configurations, i.e., pT and mT, showed little difference. TGA traces of samples containing biphenyl and dimethylfluorene units indicated a similar degradation behavior as those based on pT and mT (Fig. S3†).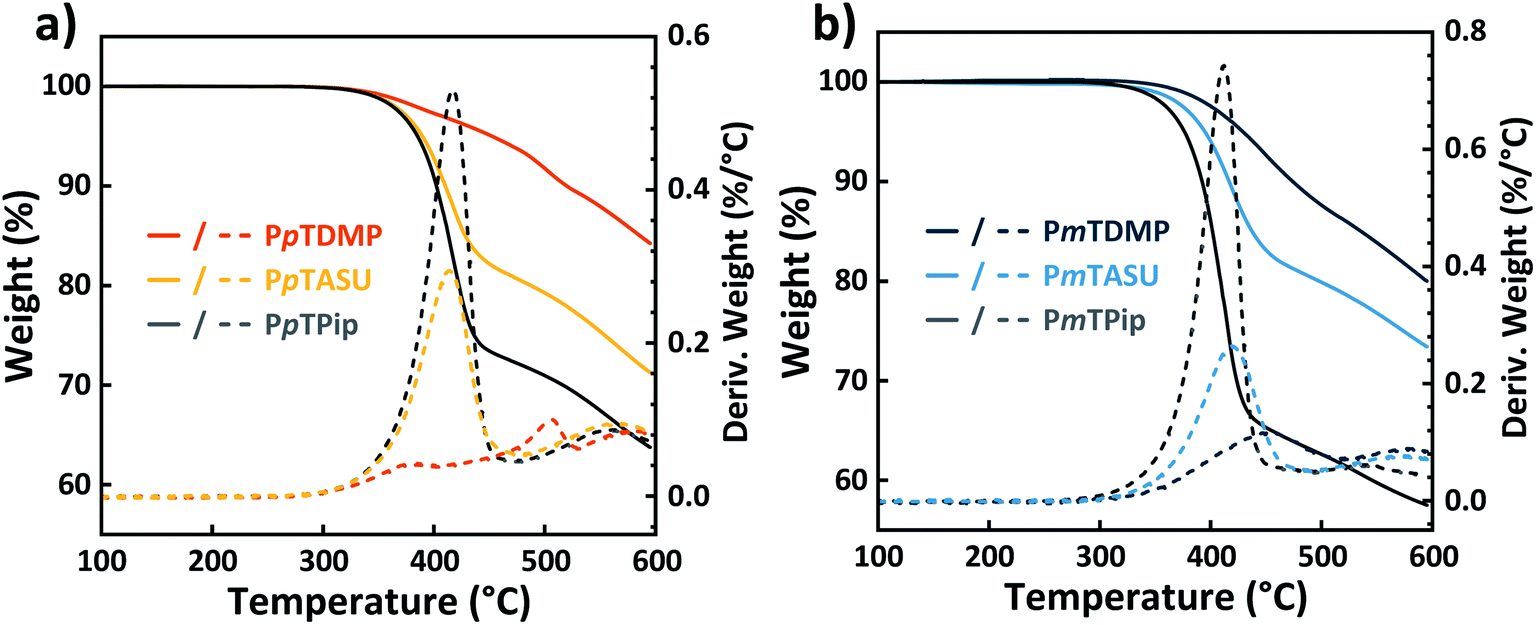 | ||
| Fig. 4 TGA traces of the protonated precursor polymers in the TFSA− form and the AEMs in the Br− form recorded under N2 atmosphere at a heating rate of 10 °C min−1: samples based on (a) pT and (b) mT, respectively. | ||
Water uptake and swelling ratio
The presence of water is crucial for the function of an AEM. An ideal uptake of water facilitates efficient ion transport in the membrane, while an excessive water uptake leads to swelling and loss of mechanical robustness and even to disintegration. The water uptake and swelling behavior of the present AEMs in the OH− form were gravimetrically determined (Fig. 5a–d and Table 1). As expected, the water uptake of the AEMs increased with temperature and IEC. The PBDMP and PBASU membranes had a high water uptake because of their high IECs and backbone flexibility. The fully hydrated samples were difficult to handle and hence the water uptake and swelling were not measured. When the m-terphenyl backbone was employed, the water uptake was considerably more restricted. For example, PmTASU showed a water uptake of 105 and 341% at 20 and 80 °C, respectively. At a higher IEC, the water uptake of PmTDMP reached 144% and over 400% at 20 and 80 °C, respectively. Notably, the AEM samples, although soft, remained intact with such high water contents. As expected, when the stiffer p-terphenyl was employed, the water uptake of the AEMs became even more constrained. PpTDMP showed a water uptake between 78 and 104% between 20 and 80 °C. With the bulkier ASU cation and slightly lower IEC, PpTASU demonstrated a lower water uptake, i.e., 43–81% in the same temperature range. These AEMs also displayed moderate in- and through-plane swelling, around 20% even at 80 °C.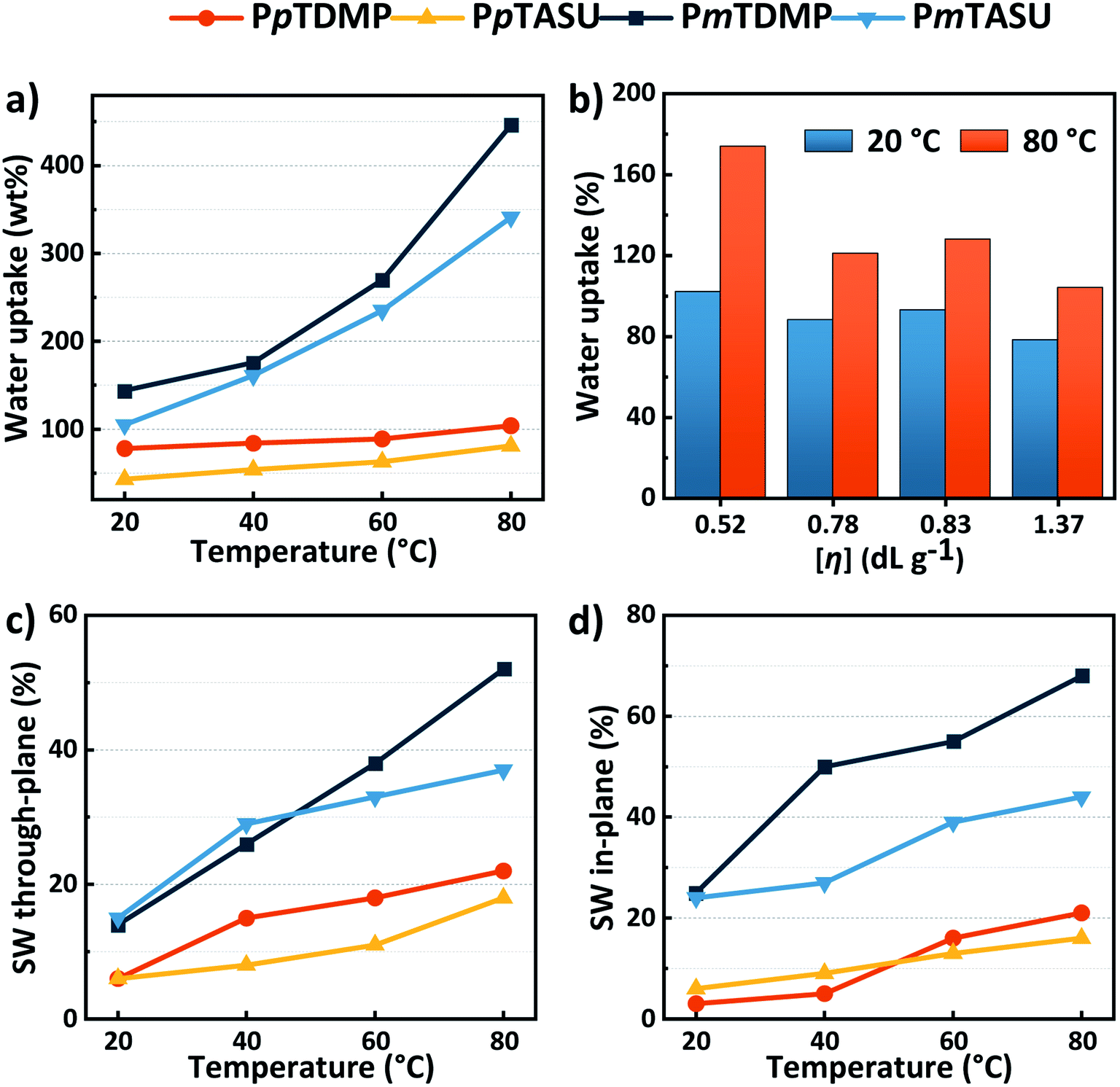 | ||
| Fig. 5 Water uptake of AEMs based on pT and mT (a), and of AEMs based on PpTDMP samples with different [η] values (b), as well as through- (c) and in-plane (d) swelling ratios of AEMs based on pT and mT. Data were measured between 20 and 80 °C on fully hydrated (immersed) samples in the OH− form. | ||
The restricted water uptake of the PpT AEMs may primarily be explained by the higher rigidity of the p-terphenylene backbone compared with the m-terphenylene counterpart. However, the higher molecular weight of the former, as indicated by the [η] values, also limited the water uptake and swelling by improving the viscoelastic properties. This relationship was corroborated by data obtained with AEMs prepared from PpTDMP samples with different [η] values, which showed that the water uptake decreased from 174 to 104% at 80 °C as [η] increased from 0.52 to 1.37 dL g−1 (Fig. 5b). The large dependence on the molecular weight emphasized the necessity to optimize the polymerization to obtain high-performance AEMs.
Ion conductivity
High OH− conductivity is a key property for AEMs which is dependent on several factors, including the IEC, water uptake, morphology and temperature. In the present study, the OH− conductivity of PAAP AEMs in the fully hydrated (immersed) state was measured by electrochemical impedance spectroscopy (EIS) between 20 and 80 °C. As shown in Fig. 6a–c, the conductivities of the PAAP AEMs increased with temperature and water uptake as anticipated. Overall, high conductivities were reached for all the AEMs (Table 1) and an Arrhenius behavior was shown (Fig. 6a and b). The most conductive PAAP AEM was PpTDMP, reaching a OH− conductivity of 79 and 178 mS cm−1 at 20 and 80 °C, respectively. Similarly, PmTDMP had OH− conductivities of 78–169 mS cm−1 at the same IEC. Furthermore, AEMs carrying the bulky ASU cations (PpTASU and PmTASU) also displayed high conductivities, between 67 and 157 mS cm−1, despite the lower IEC. The relationship between the OH− conductivity and the hydration number (λ) of the AEMs is given in Fig. 6c. When AEMs with different backbone structures were compared, it was obvious that PpT AEMs were more effective ion conductors than the PmT AEMs, as seen by the much steeper conductivity increase with λ compared to the latter AEMs. A dilution effect by which excessive water uptake decreased the charge carrier concentration, resulting in less efficient ion transport, was clearly observed for the PmT AEMs.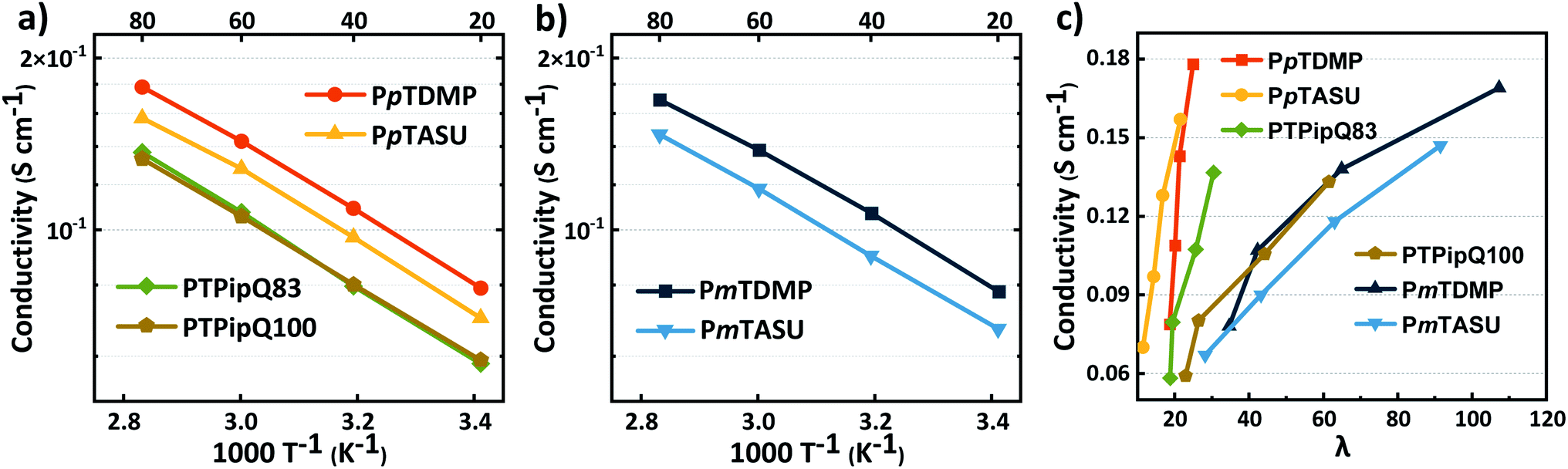 | ||
| Fig. 6 OH− conductivity of the AEMs in the fully hydrated (immersed) state as a function of (a) and (b) T−1 and (c) the hydration number (λ). | ||
The OH− conductivity of PpTDMP was compared with benchmark PAP AEMs. Here, the homopolymer PTPipQ100 (ref. 56) was selected because it has the same terphenyl backbone and piperidinium cation (Scheme 3) as PpTDMP. The copolymer PTPipQ83 was included because of a similar IEC and water uptake in comparison with PpTDMP. As shown in Fig. 6a, PpTDMP reached higher conductivity values than the two benchmarks based on PAP. This could be attributed to the combination of the high molecular weight, reasonably high IEC, and the cationic arrangement of PpTDMP, which led to a more distinct phase separation of the AEM (Fig. 3). Moreover, the conductivity of PTPipQ100 increased only slowly with increasing water content (λ), most probably due to the high IEC and overall water uptake, leading to a dilution effect (Fig. 6c).
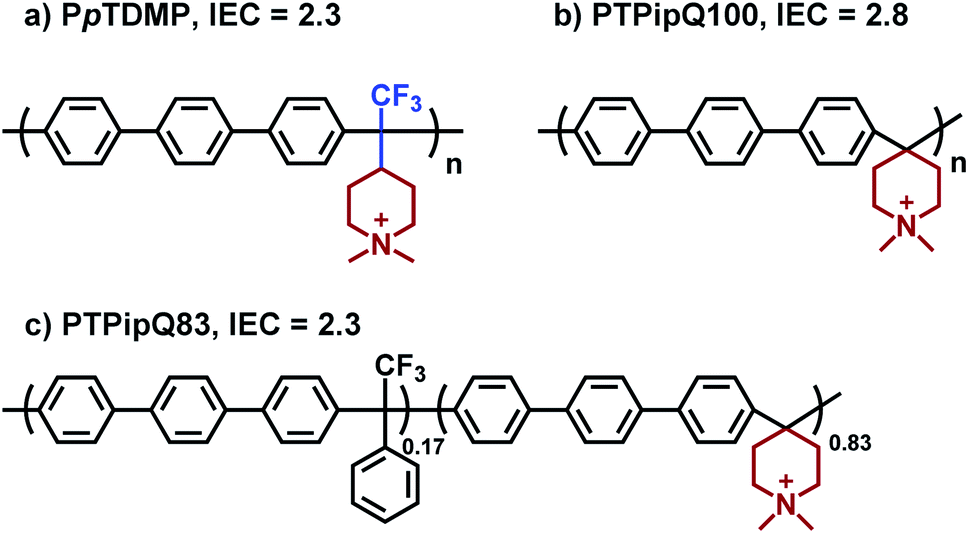 | ||
| Scheme 3 Molecular structures of a representative PAAP, PpTDMP prepared from TFPip (a), and two corresponding benchmark PAPs for reference: PTPipQ100 (b) and PTPipQ83 (c), both derived from piperidone (counter ions omitted). | ||
Alkaline stability
One critical factor that hampers the development of durable AEMFCs is the lack of sufficient long-time chemical stability of currently available AEMs under alkaline environment. The rational design of monomer TFPip was expected to provide polymers with a cationic arrangement which enhances the resistance against OH− attack, especially in relation to PAP AEMs based on piperidone (Scheme 1). In order to investigate this, the two benchmark AEMs based on PAPs prepared from piperidone, PTPipQ83 and PTPipQ100, were included in the alkaline stability study (Scheme 3). The copolymer PTPipQ83 had a similar water uptake as PpTDMP, which presumably gave a comparable effect of the hydration level on the stability.57 Here, the alkaline stability and degradation were investigated by 1H NMR spectroscopy in order to probe the active degradation mechanisms and assess the ex situ lifetime of the AEMs. Samples were first immersed in alkaline media, initially 2 M aq. NaOH at 90 °C during 20–100 days. Subsequently, the AEM samples were also studied after storage in 5 M aq. NaOH at the same temperature for 10 and 20 days. All the samples retained their mechanical integrity and robustness after these severe testing conditions. 1H NMR spectra of the AEM polymers before and after storage in alkali were recorded after dissolution in DMSO-d6 with addition of TFA (Fig. 7, 8 and S4–S10†).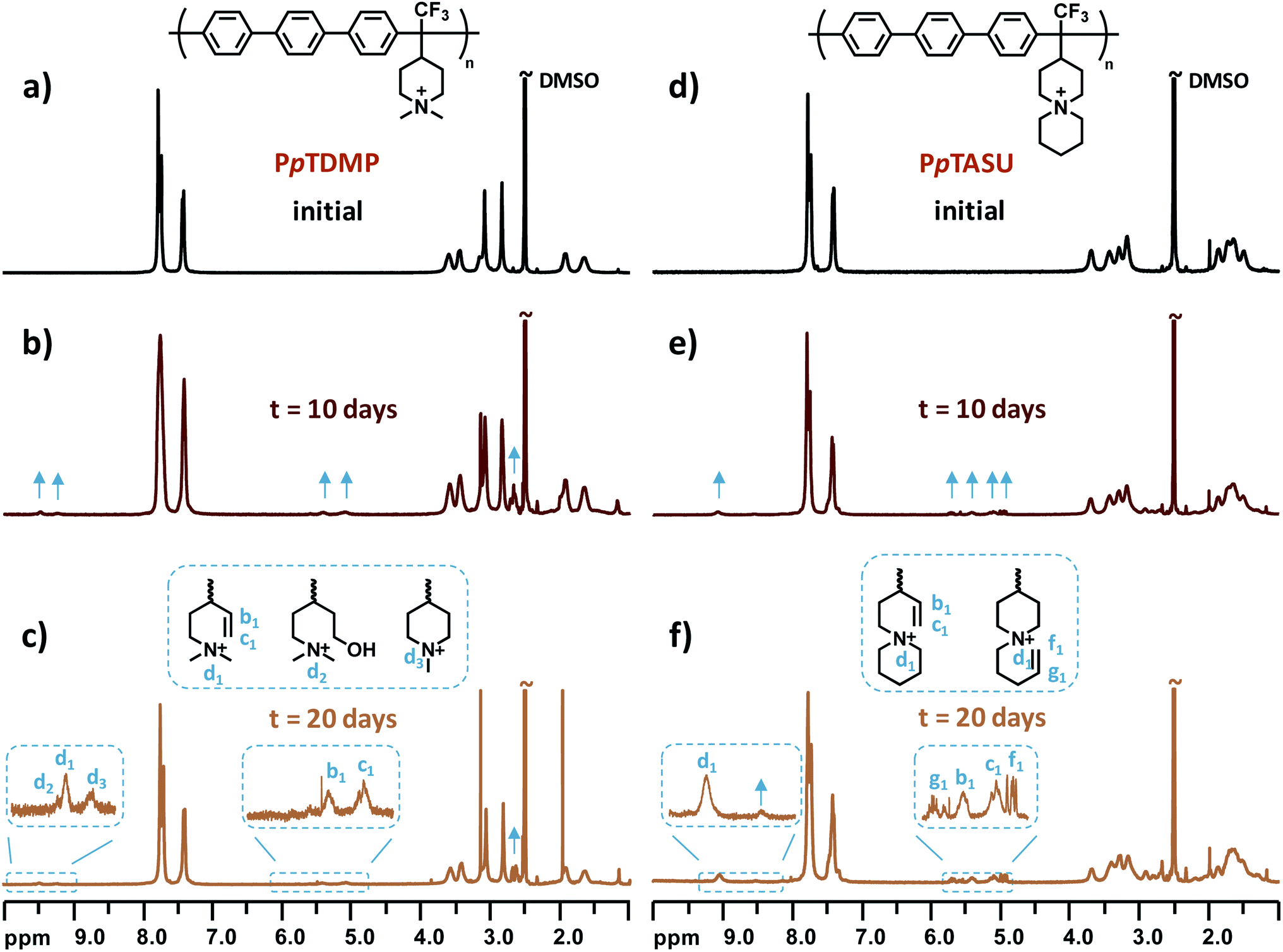 | ||
| Fig. 7 1H NMR spectra of (a)–(c) PpTDMP and (d)–(f) PpTASU before and after storage in 5 M aq. NaOH at 90 °C for 10 and 20 days, respectively. | ||
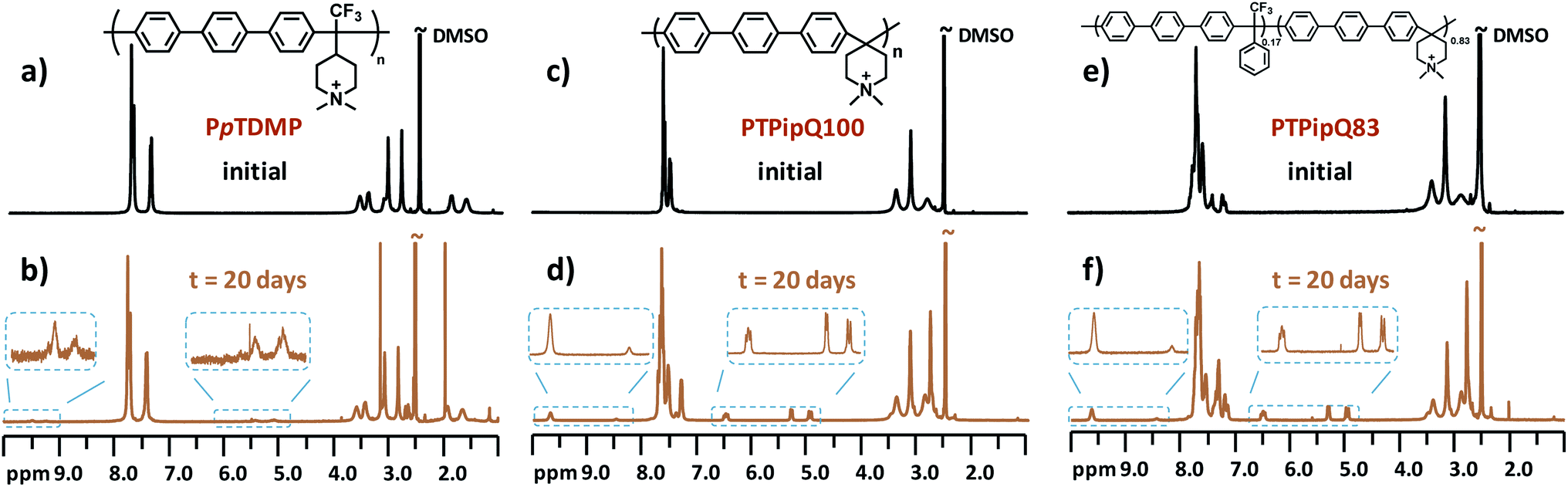 | ||
| Fig. 8 1H NMR spectra of (a) and (b) PpTDMP and the benchmark AEMs (c) and (d) PTPipQ100 and (e) and (f) PTPipQ83 before and after storage in 5 M aq. NaOH at 90 °C for 20 days. | ||
The degradation mechanisms of the DMP and ASU cations have already been widely studied and reported.37,48,49 Typically, in a severe alkaline environment, both cations undergo Hofmann elimination at β-hydrogens, or ring-opening nucleophilic substitution at α-carbons. Methyl substitution occurs exclusively for the DMP cations. Several studies on QA model compounds (salts) have revealed the high alkaline stability of DMP and ASU because of the constraints imposed by the ring structure.58–60 For instance, Hofmann elimination is depressed for the DMP cation due to the high energy transition state, which instead cause degradation via methyl substitution to dominate. This may in turn be circumvented by replacing the methyl groups with a second 6-membered ring to form a spiro cation, i.e., the ASU cation. This cation successfully demonstrated a longer half-life (110 h for ASU versus 87.3 h for DMP at 160 °C in 6 M aq. NaOH).24 However, the reverse situation has often been observed when these N-alicyclic cations are incorporated into various polymer structures, such as PAPs.50,52 Hofmann elimination is dominant in most cases,37 which has been attributed to a distortion of the ring conformation imposed by the rigid arylene backbone. In fact, ionic loss through elimination has been found to increase with the rigidity of the backbones.51 In AEMs the ASU cation has been found to be more prone to degrade compared with the DMP cation, which is in contrast to the results on the corresponding model compounds.
In the present study, structural changes of the AEM samples were detected by 1H NMR spectroscopy after storage in 2 M aq. NaOH at 90 °C for 20 days, even though the new signals from the degradation products were very small, and the ionic loss was only quantifiable after 60 day's storage (Fig. S5–S8, d–g†). Consequently, the AEMs were treated under harsher conditions to accelerate the ionic loss, and 1H NMR spectra were recorded after immersion in 5 M aq. NaOH at 90 °C for 10 and 20 days. Fig. 7 shows the spectra of PpTDMP and PpTASU before and after the aforementioned treatment, and the spectra of the other AEM samples are provided in Fig. S7–S10.† As shown in Fig. 7c, the signals at 5.1 (c1) and 5.4 (b1) ppm were ascribed to the vinyl groups of the Hofmann elimination products, along with the tertiary proton signal at 9.48 ppm (d1). As expected, the three signals had intensity ratios of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1. Traces of methyl and ring-opening substitution were also detected, as indicated by two small additional signals (d2 and d3) from protonated tertiary piperidine between 9.0 and 9.5 ppm. When it comes to PpTASU, another set of vinylic signals was detected (Fig. 7f), suggesting Hofmann elimination in both ASU rings (inner and outer). In addition, the alkaline stability of PpTDMP was compared with the benchmark PAP AEMs, as shown in Fig. 8. After alkaline storage, the PAP reference materials showed distinct signals from vinylic protons between 4.8 and 6.8 ppm, originating from Hofmann elimination.37 Lastly, the 1H NMR spectra of PmTDMP and PmTASU, presented in Fig. S7 and S8,† showed that AEMs based on PmT degraded in a similar manner as the PpT-based AEMs, despite the different backbone configurations.
:1:1. Traces of methyl and ring-opening substitution were also detected, as indicated by two small additional signals (d2 and d3) from protonated tertiary piperidine between 9.0 and 9.5 ppm. When it comes to PpTASU, another set of vinylic signals was detected (Fig. 7f), suggesting Hofmann elimination in both ASU rings (inner and outer). In addition, the alkaline stability of PpTDMP was compared with the benchmark PAP AEMs, as shown in Fig. 8. After alkaline storage, the PAP reference materials showed distinct signals from vinylic protons between 4.8 and 6.8 ppm, originating from Hofmann elimination.37 Lastly, the 1H NMR spectra of PmTDMP and PmTASU, presented in Fig. S7 and S8,† showed that AEMs based on PmT degraded in a similar manner as the PpT-based AEMs, despite the different backbone configurations.
The ionic loss of the AEMs was calculated by comparing the integral of the (protonated) amine signals around 9 ppm, originating from the degradation of piperidinium, with the intensity of all the aromatic signals (7–8 ppm), which remained unaffected by the alkaline treatment. The results are summarized in Table 3 and revealed that PpTDMP was the most stable AEM in the present study, with a mere 14% ionic loss, after storage in 5 M NaOH at 90 °C for 20 days. Under the same conditions, the reference samples PTPipQ83 and PTPipQ100 showed an ionic loss of 52 and 42%, respectively, which was significantly higher than that of PpTDMP. Moreover, in PpTDMP half of the degradation occurred via Hofmann elimination, accounting for 7% ionic loss. In comparison, Hofmann elimination was the primary pathway in the benchmark AEM, corresponding to 42% ionic loss for PTPipQ83 (out of a total 52% ionic loss). The most possible reason for the superior alkaline stability of the present AEMs lies in the more favorable cationic arrangement. First of all, the piperidinium rings are attached in the 4-position. This greatly enhances the alkaline stability of the cation,52 in comparison to corresponding piperidinium rings tethered at the 1(N)-position.55 Furthermore, the ring strain imposed by the rigid polymer chain are alleviated in, e.g., PpTDMP, as opposed to the benchmark PTPipQ100 where the cations are directly incorporated in the backbone. In PpTDMP, the piperidinium rings are separated from the backbone, which significantly facilitates the ring relaxation to impede ionic loss. This was supported by the reduced ionic loss via Hofmann elimination, in comparison to the PAP benchmark materials. Electron-withdrawing substituents located in the vicinity of cations usually decrease their alkaline stability.59 However, in the present case the quite strongly electron-withdrawing trifluoromethyl group seemed to be distant enough from the piperidinium ring in the polymer structure not to deteriorate its alkaline stability.
| AEM | Ionic loss (%) | |
|---|---|---|
| Total | By Hofmann elimination | |
| PpTDMP | 14 | 7 |
| PpTASU | 41 | 12 + 17 (outer + inner ring) |
| PmTDMP | 18 | 10 |
| PmTASU | 39 | 14 + 21 (outer + inner ring) |
| PTPipQ83 | 52 | 42 |
| PTPipQ100 | 42 | 35 |
Experimental
Materials
The following chemicals were purchased from Sigma-Aldrich: p-terphenyl (99%), 1,1,1-trifluoroacetic acid (TFA, 99%), N,N-diisopropylethylamine (DIPEA, ≥99%), NaNO3 (99%), AgNO3 (99%), 1,5-dibromopentane (97%), chloroform-d (99.8 at% D), dimethyl sulfoxide (DMSO, reagent grade), DMSO-d6 (99.96 at% D), K2CO3 (99%), N-methyl-2-pyrrolidone (NMP, reagent grade), triflic acid (TFSA, 99%), pyridine (≥99%, reagent grade), trifluoroacetic anhydride (TFAA, ≥99%), methyl iodide (MeI, 98%, stabilized), isonipecotic acid (97%), ethyl acetate (EtOAc, ≥99.5%). Diethyl ether (Et2O, reagent grade), isopropanol (IPA, reagent grade), NaOH (99%, pellets), toluene (99.5%, reagent grade), Na2CO3 (99%) and NaBr (99%) were obtained from VWR. Biphenyl (Acros, 99%), HCl (ThermoFischer, 37%), m-terphenyl (Alfa Aesar), acetonitrile (HPLC grade, Fisher scientific), 9,9-dimethyl-9H-fluorene (Ambeed) and all the chemicals listed above were used as received. Dichloromethane (DCM) used in the polymerizations was collected from MBraun dry solvent dispenser system MB-SPS 800.Polymer synthesis
:heptane (0:100 to 55:45) as eluent. The still impure product was added to a 100 mL round bottom flask filled with concentrated HCl (50 mL) and deionized water (150 mL) and heated to 100 °C and left stirring overnight. The solution was cooled to room temperature, and the solvent was evaporated to obtain a dark solid, which was recrystallized from acetonitrile to gain the final product as a light tan crystal. This gave 25.2 g product at a total isolated yield of 58%.
The precursor polymers were also functionalized with pendant N-spirocyclic cations via cyclo-quaternization using 1,5-dibromopentane in the presence of DIPEA as the base. Here, the preparation of PpTASU will serve as an example. A solution of PpTPip (0.6 g, 1.1 mmol, 1 equiv.) in NMP (10 mL) was heated to 80 °C. Meanwhile, a solution of 1,5-dibromopentane (0.164 mL, 1.2 mmol, 1.1 equiv.) and DIPEA (1.04 mL, 5.52 mmol, 5 equiv.) in NMP (14 mL) was vigorously stirred in a 50 mL round bottom flask at 80 °C. Subsequently, the polymer solution was added slowly dropwise to the reagent flask. The reaction solution was stirred at 80 °C for 48 h. Next, the homogeneous dark-red solution was cooled to room temperature and the product was precipitated in a diethyl ether/IPA mixture, further washed with fresh diethyl ether, 1 M aq. NaBr and then with deionized water. Finally, the product was dried under vacuum at room temperature to give an orange product.
Description of membrane preparation and characterization methods can be found as ESI.†
Conclusions
A series of PAAPs were prepared and studied with the aim to further enhance the alkaline stability of N-alicyclic QA cations attached to rigid polymer backbones. To that end, the TFPip monomer was first designed with a reactive trifluoromethyl ketone group and a piperidine functionality, and then synthesized using a straightforward procedure. PAAPs based on TFPip were successfully prepared in superacid-mediated polycondensations and subsequent quaternizations to obtain DMP and ASU cations, respectively. Polymers based on biphenyl and dimethylfluorene took up too much water, and may instead be suitable ionomers for catalyst layers. In contrast, the corresponding p- and m-terphenyl polymers formed AEMs with excellent mechanical robustness and high OH− conductivity. Optimization of the polymerization conditions was important in order to reach high molecular weights, which resulted in suppressed water uptake and swelling of the corresponding AEMs without compromising their high OH− conductivity. Moreover, benefiting from the cationic arrangement, poly(terphenyl alkylene piperidinium)s showed significantly enhanced the alkaline stability compared to corresponding benchmark poly(terphenyl piperidinium)s prepared from piperidone where the cyclic cations are directly connected to the rigid polymer backbone. Incorporating the cations into the polymer structure in such a way that the backbone and the cyclic cation do not share any atoms, as in the present case, most probably significantly facilitated the ring relaxation which impeded ionic loss. This work provides valuable insights into the molecular design of high performance alkali-stable AEMs. Future work will focus on evaluations of the new PAAP materials in AEMFC and AEMWE.Author contributions
Dong Pan: methodology, investigation, validation, writing - original draft, writing - review & editing. Pegah Mansouri Bakvand: investigation, methodology, validation, writing – original draft. Thanh Huong Pham: conceptualization, methodology, investigation. Patric Jannasch: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Swedish Energy Agency (grants 50519-1, 45057-1, and 37806-3), the Swedish Research Council (grants 45397-1 and 2015-04820), the Swedish Foundation for Strategic Research, SSF (grants EM16-0060 and ARC19-0026), and the Royal Physiographic Society of Lund for financial support. We are also grateful to Peter Holmqvist for the support with the SAXS experiments and data treatment, and Sofia Essén for the help with HRMS measurements and data analysis.References
- J. R. Varcoe and R. C. T. Slade, Fuel Cells, 2005, 5, 187–200 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- J. R. Varcoe, P. Atanassov, D. R. Dekel, A. M. Herring, M. A. Hickner, P. A. Kohl, A. R. Kucernak, W. E. Mustain, K. Nijmeijer, K. Scott, T. W. Xu and L. Zhuang, Energy Environ. Sci., 2014, 7, 3135–3191 RSC.
- M. Hren, M. Božič, D. Fakin, K. S. Kleinschek and S. Gorgieva, Sustainable Energy Fuels, 2021, 5, 604–637 RSC.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernäcker, T. Weißgärber, L. Röntzsch and J. Meier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- C. Santoro, A. Lavacchi, P. Mustarelli, V. Di Noto, L. Elbaz, D. R. Dekel and F. Jaouen, ChemSusChem, 2022, 15, e202200027 CrossRef CAS PubMed.
- C. Li and J.-B. Baek, Nano Energy, 2021, 87, 106162 CrossRef CAS.
- N. J. Chen and Y. M. Lee, Trends Chem., 2022, 4, 236–249 CrossRef CAS.
- J. Xue, J. Zhang, X. Liu, T. Huang, H. Jiang, Y. Yin, Y. Qin and M. D. Guiver, Electrochem. Energy Rev., 2022, 5, 348–400 CrossRef CAS.
- W. You, K. J. T. Noonan and G. W. Coates, Prog. Polym. Sci., 2020, 100, 101177 CrossRef CAS.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 15456–15477 RSC.
- Y. S. Kim, ACS Appl. Polym. Mater., 2021, 3, 1250–1270 CrossRef CAS.
- M. Mandal, ChemElectroChem, 2020, 8, 36–45 CrossRef.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y.-K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- W. You, J. M. Ganley, B. G. Ernst, C. R. Peltier, H. Y. Ko, R. A. DiStasio, R. R. Knowles and G. W. Coates, Chem. Sci., 2021, 12, 3898–3910 RSC.
- K. J. Noonan, K. M. Hugar, H. A. t. Kostalik, E. B. Lobkovsky, H. D. Abruna and G. W. Coates, J. Am. Chem. Soc., 2012, 134, 18161–18164 CrossRef CAS PubMed.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2020, 53, 4722–4732 CrossRef CAS PubMed.
- A. G. Wright, J. Fan, B. Britton, T. Weissbach, H.-F. Lee, E. A. Kitching, T. J. Peckham and S. Holdcroft, Energy Environ. Sci., 2016, 9, 2130–2142 RSC.
- A. G. Wright, T. Weissbach and S. Holdcroft, Angew. Chem., Int. Ed. Engl., 2016, 55, 4818–4821 CrossRef CAS PubMed.
- W.-H. Lee, Y. S. Kim and C. Bae, ACS Macro Lett., 2015, 4, 814–818 CrossRef CAS PubMed.
- R. Ren, S. M. Zhang, H. A. Miller, F. Vizza, J. R. Varcoe and Q. G. He, ACS Appl. Energy Mater., 2019, 2, 4576–4581 CrossRef CAS.
- T. Jiang, C. Wu, Y. Zhou, S. Cheng, S. Yang, H. Wei, Y. Ding and Y. Wu, J. Membr. Sci., 2022, 647, 120342 CrossRef CAS.
- K. M. Hugar, H. A. t. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- L. Gu, H. Dong, Z. Sun, Y. Li and F. Yan, RSC Adv., 2016, 6, 94387–94398 RSC.
- S. A. Nuñez, C. Capparelli and M. A. Hickner, Chem. Mater., 2016, 28, 2589–2598 CrossRef.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 RSC.
- T. Zhu, S. Xu, A. Rahman, E. Dogdibegovic, P. Yang, P. Pageni, M. P. Kabir, X. D. Zhou and C. Tang, Angew. Chem., Int. Ed. Engl., 2018, 57, 2388–2392 CrossRef CAS PubMed.
- X. Liu, N. Xie, J. Xue, M. Li, C. Zheng, J. Zhang, Y. Qin, Y. Yin, D. R. Dekel and M. D. Guiver, Nat. Energy, 2022, 7, 329–339 CrossRef CAS.
- W. You, K. M. Hugar, R. C. Selhorst, M. Treichel, C. R. Peltier, K. J. T. Noonan and G. W. Coates, J. Org. Chem., 2021, 86, 254–263 CrossRef CAS PubMed.
- T. H. Pham and P. Jannasch, ACS Macro Lett., 2015, 4, 1370–1375 CrossRef CAS PubMed.
- J. S. Olsson, T. H. Pham and P. Jannasch, Macromolecules, 2017, 50, 2784–2793 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- X. Chu, Y. Shi, L. Liu, Y. Huang and N. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- X. M. Chu, L. Liu, Y. D. Huang, M. D. Guiver and N. W. Li, J. Membr. Sci., 2019, 578, 239–250 CrossRef CAS.
- N. Chen, C. Hu, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, J. Y. Bae, J. H. Park and Y. M. Lee, Angew. Chem., Int. Ed. Engl., 2021, 60, 7710–7718 CrossRef CAS PubMed.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. Ben Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- N. Chen, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, C. Hu, J. Y. Bae, E. S. Sim, Y. C. Chung, J. H. Jang, S. J. Yoo, Y. Zhuang and Y. M. Lee, Nat. Commun., 2021, 12, 2367 CrossRef CAS PubMed.
- F. Wang, Y. Li, C. Li and H. Zhu, J. Membr. Sci., 2021, 620, 118919 CrossRef CAS.
- W. W. Gou, W. T. Gao, X. L. Gao, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2022, 645, 120200 CrossRef CAS.
- C. Lu, C. Long, Y. Li, Z. Li and H. Zhu, J. Membr. Sci., 2020, 598, 117797 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, J. Membr. Sci., 2019, 578, 183–195 CrossRef CAS.
- N. J. Chen, J. H. Park, C. Hu, H. H. Wang, H. M. Kim, N. Y. Kang and Y. M. Lee, J. Mater. Chem. A, 2022, 10, 3678–3687 RSC.
- X. Wang and R. G. Lammertink, J. Mater. Chem. A, 2022, 10, 8401–8412 RSC.
- X. Wu, N. Chen, H. A. Klok, Y. M. Lee and X. Hu, Angew. Chem., Int. Ed. Engl., 2022, 61, e202114892 CAS.
- H. H. Wang, C. Hu, J. H. Park, H. M. Kim, N. Y. Kang, J. Y. Bae, W. H. Lee, N. Chen and Y. M. Lee, J. Membr. Sci., 2022, 644, 120160 CrossRef CAS.
- D. Pan, T. H. Pham and P. Jannasch, ACS Appl. Energy Mater., 2021, 4, 11652–11665 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6, 16537–16547 RSC.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 15895–15906 RSC.
- D. Pan, J. S. Olsson and P. Jannasch, ACS Appl. Energy Mater., 2022, 5, 981–991 CrossRef CAS.
- T. H. Pham, A. Allushi, J. S. Olsson and P. Jannasch, Polym. Chem., 2020, 11, 6953–6963 RSC.
- D. Tian, T. L. Gu, S. N. Yellamilli and C. Bae, Energies, 2020, 13, 1924 CrossRef CAS.
- M. Zolotukhin, S. Fomine, R. Salcedo and L. Khalilov, Chem. Commun., 2004, 1030–1031 RSC.
- A. Allushi, T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2019, 7, 27164–27174 RSC.
- T. Novalin, D. Pan, G. Lindbergh, C. Lagergren, P. Jannasch and R. W. Lindström, J. Power Sources, 2021, 507, 230287 CrossRef CAS.
- D. R. Dekel, S. Willdorf, U. Ash, M. Amar, S. Pusara, S. Dhara, S. Srebnik and C. E. Diesendruck, J. Power Sources, 2018, 375, 351–360 CrossRef CAS.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- N. Chen, Y. Jin, H. Liu, C. Hu, B. Wu, S. Xu, H. Li, J. Fan and Y. M. Lee, Angew. Chem., Int. Ed., 2021, 60, 19272–19280 CrossRef CAS PubMed.
- W. You, K. M. Hugar, R. C. Selhorst, M. Treichel, C. R. Peltier, K. J. Noonan and G. W. Coates, J. Org. Chem., 2020, 86, 254–263 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta03862e |
| This journal is © The Royal Society of Chemistry 2022 |