
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDose rate dependent cation & anion radiation enhanced diffusion in hematite†
Kayla H.
Yano
 *a,
Aaron A.
Kohnert
b,
Tiffany C.
Kaspar
c,
Sandra D.
Taylor
c,
Steven R.
Spurgeon
a,
Hyosim
Kim
b,
Yongqiang
Wang
b,
Blas P.
Uberuaga
b and
Daniel K.
Schreiber
a
*a,
Aaron A.
Kohnert
b,
Tiffany C.
Kaspar
c,
Sandra D.
Taylor
c,
Steven R.
Spurgeon
a,
Hyosim
Kim
b,
Yongqiang
Wang
b,
Blas P.
Uberuaga
b and
Daniel K.
Schreiber
a
aEnergy & Environment Directorate, Pacific Northwest National Laboratory, Richland, WA 99354, USA. E-mail: kayla.yano@pnnl.gov
bMaterial Science and Technology Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA
cPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99354, USA
First published on 4th November 2022
Abstract
Irradiation induced non-equilibrium point defect populations influence mass transport in oxides, which in turn affects their stability and performance in hostile environments. In this study, we combine theoretical predictions and experimental observations to examine the dose rate dependence (2 × 10−5 to 2 × 10−3 dpa s−1) of mass transport in irradiated (400 keV Ar+ at 300 and 500 °C) hematite (α-Fe2O3). Both cation (57Fe) and anion (18O) tracers are precisely embedded in an epitaxial single crystal hematite film deposited by molecular beam epitaxy. Atom probe tomography is used to experimentally measure isotope movement to determine diffusion coefficients, and a chemical rate-theory model is developed to interpret radiation enhanced transport. Radiation enhanced diffusivities are similar for both species, and are found to be at least 20 orders of magnitude faster than the Arrhenius extrapolation of high-temperature values. In the recombination dominant regime (300 °C, 2 × 10−4 dpa s−1) the experimental results for the anion species deviate from the model, suggesting that the cation lattice limits the anion diffusivity in this regime. Ultimately, a strong dose rate dependence is observed driven by irradiation induced defect populations.
1. Introduction
Oxidation and corrosion of structural alloys in high-temperature environments (such as energy-generating systems) impact material reliability and performance.1,2 Sustained corrosion, or conversely its inhibition, is oftentimes dictated by atomic transport across oxide films formed naturally on the alloy surface that separate the alloy from the environment. Materials used in nuclear reactors face additional challenges for corrosion resistance because radiation fields create a high density of point defects within the oxide film and underlying alloy. Broadly speaking, radiation effects in structural alloys are well studied and lead to a variety of effects including an increase in hardness and loss of ductility, radiation induced segregation and precipitation, and void swelling.3 However, the impact of irradiation on corrosion is much less studied.4 It is known that materials exposed to aqueous solutions or heavy liquid metals develop a protective duplex oxide structure, while contact with oxygen-free molten salts does not lead to protective surface layers, but in all cases corrosion alters the base metal by depleting one or more alloying elements.4 To understand how irradiation alters alloy corrosion, there is a fundamental and critical need to develop predictive models informed by direct measurement of how radiation changes mass transport mechanisms and kinetics through oxide films.Irradiation of crystalline materials results in the generation of point defects (often Frenkel pairs of a vacancy and interstitial). As such, the density of point defects in a radiation environment is often substantially higher than the population at thermal equilibrium and types of defects that are negligible under thermal conditions can be present in very high concentrations under irradiation; both effects contribute to increased diffusion.5–8 These defects diffuse through the bulk and can recombine, reach sinks, or aggregate with each other to become defect clusters or higher-dimension defects.9 Defect behaviour in oxides is complicated by the presence of multiple lattices and their accompanying charge. Still, the supersaturated population of defects can accelerate atomic diffusion by many orders of magnitude over thermal diffusion, increasing oxide thicknesses under irradiation.10–12
To understand radiation enhanced diffusion, we first need to establish thermal diffusion behaviour of the oxide system. Existing literature studies on mass transport in hematite (α-Fe2O3), the model oxide system studied here, have evaluated cation13,14 or anion15–17 diffusion at elevated temperatures, with one study examining both.18 In these studies,15–17 oxygen self-diffusion coefficients were measured between 897–1066 °C with coefficients varying between 4.9 × 10−20 to 3.2 × 10−15 m2 s−1. In comparison, cation diffusion is slightly slower,13,14 with values between 5.2 × 10−20 to 1.7 × 10−16 m2 s−1 at temperatures ranging between 814–1067 °C.18 Similar studies exist for anion and cation diffusion through chromia (Cr2O3), a more protective oxide than hematite on structural alloys, in both single crystals and polycrystals at elevated temperatures. These include studies of oxygen diffusion in single crystals19,20 and polycrystals,21,22 one study with anion (18O) and cation (54Cr) tracers,23 another with two cation tracers (54Cr and 50Cr),20 and even Fe diffusion into Cr2O3.24,25 Anion diffusivities in Cr2O3 are found to range from 3.2 × 10−22 to 2.2 × 10−16 m2 s−1 between 1100 and 1450 °C, which is a couple of orders of magnitude slower at higher temperatures than oxygen in Fe2O3.19,26 Likewise, Cr cation diffusivities are also slower than Fe, ranging between 1 × 10−22 to 6.3 × 10−21 m2 s−1 between 1200-1450 °C.
We emphasize here that these previous studies on mass transport in oxides were conducted at elevated temperatures (>900 °C) where vacancy-mediated thermal diffusion dominates; nuclear reactors operate at significantly lower temperatures. For example, light-water reactors operate around 280–350 °C and molten salt cooled, next-generation reactor designs call for operating temperatures of <700 °C.27 It is possible that the fundamental mechanism of diffusion could differ in these relatively moderate temperature regimes. In particular, a transition was predicted for Fe2O3 from vacancy to interstitialcy mediated transport at 900 °C.10 Additionally, materials used in existing and next-generation reactor designs could experience a wide range of dose rates. Radiation damage and dose for a material is calculated in units of displacements per atom (dpa), which can be readily simulated for most materials and forms of radiation.28 In commercial reactors, the damage rate could be as high as 6 × 10−7 dpa s−1 (ref. 29) in fusion reactor designs, significantly less in light-water reactors (∼10−7 dpa s−1), and much lower in high-level nuclear waste forms (10−13 dpa s−1).
Mass transport under irradiation is affected by a rate balance for point defect generation and annihilation, which is temperature and dose rate dependent. Prior experimental observations and modelling efforts on the radiation-enhanced transport in hematite10 and chromia30 predict three regimes (Fig. 1). Temperature ranges vary based on the material system and the values used herein are for hematite. At relatively low temperatures (<300 °C), radiation-enhanced transport is strongly temperature dependent and dominated by the recombination behaviour of Frenkel pairs. At moderate temperatures (300–800 °C), transport is weakly temperature dependent and radiation-enhanced transport is dominated by (and thus dependant on) the density of sinks, which are sites for annihilation of radiation-induced point defects. Finally, at high temperatures (>800 °C), thermal transport dominates as the thermal concentration of defects overwhelms the radiation-induced content, and radiation effects are relatively minor. Conceptually, varying the dose rate shifts the temperature-independent diffusivity plateau (and correspondingly the transition temperature between regimes) in the sink dominant regime by increasing or decreasing the defect production rate and thus the steady-state defect population. This regime also covers most of the practical temperatures that structural alloys would experience in reactor service. In the other two regimes, this effect is expected to be relatively minor as the additional defects are immediately annihilated (recombination dominant regime) or too few to matter (thermal dominant regime).
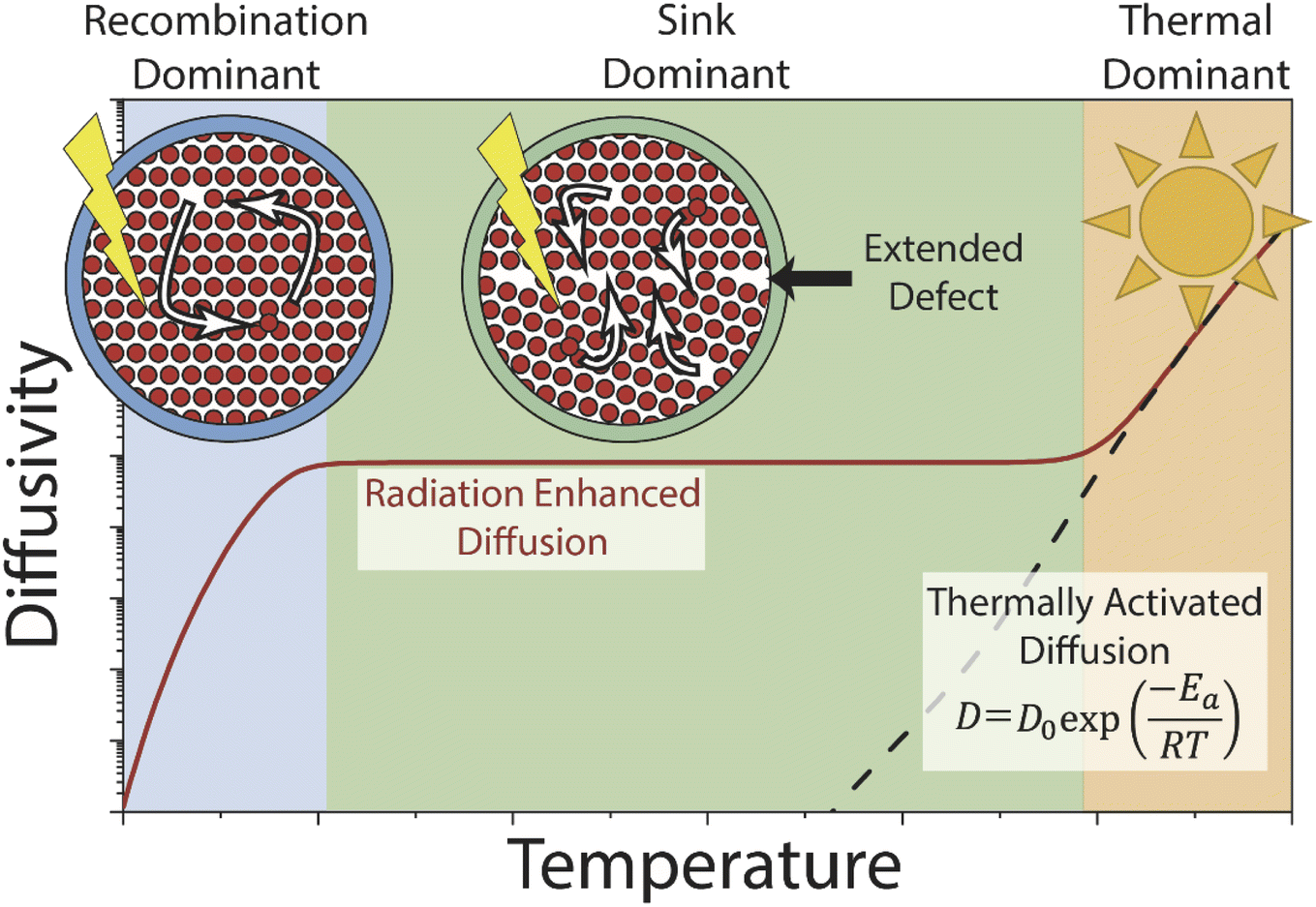 | ||
| Fig. 1 Mass transport regimes under irradiation and thermal anneal. Thermal (orange) dominant regime begins at high temperatures, following an Arrhenius expression. Irradiation driven transport regimes are split into sink dominant at mid-range temperatures (green) and recombination dominant at the lowest temperatures (blue). | ||
Building from our previous work that developed the use of isotopic tracers deposited into epitaxial Fe2O310 and Cr2O330 films for diffusion quantification, the current work combines anion (18O) and cation (57Fe) tracers into a model heterogenous oxide (see schematic in Fig. 2a) to study the effect of irradiation dose rate on mass transport. 3D atom probe tomography (APT) is used to measure nanoscale interdiffusion of the isotopic tracers and the magnitude of radiation-enhanced diffusion within predicted recombination (300 °C) and sink dominated (500 °C) regimes. A chemical rate theory model is used to predict diffusivities under irradiation. Comparison of the model with our experimental results provides insights into the dominant transport regimes in Fe2O3 and Cr2O3 under irradiation.
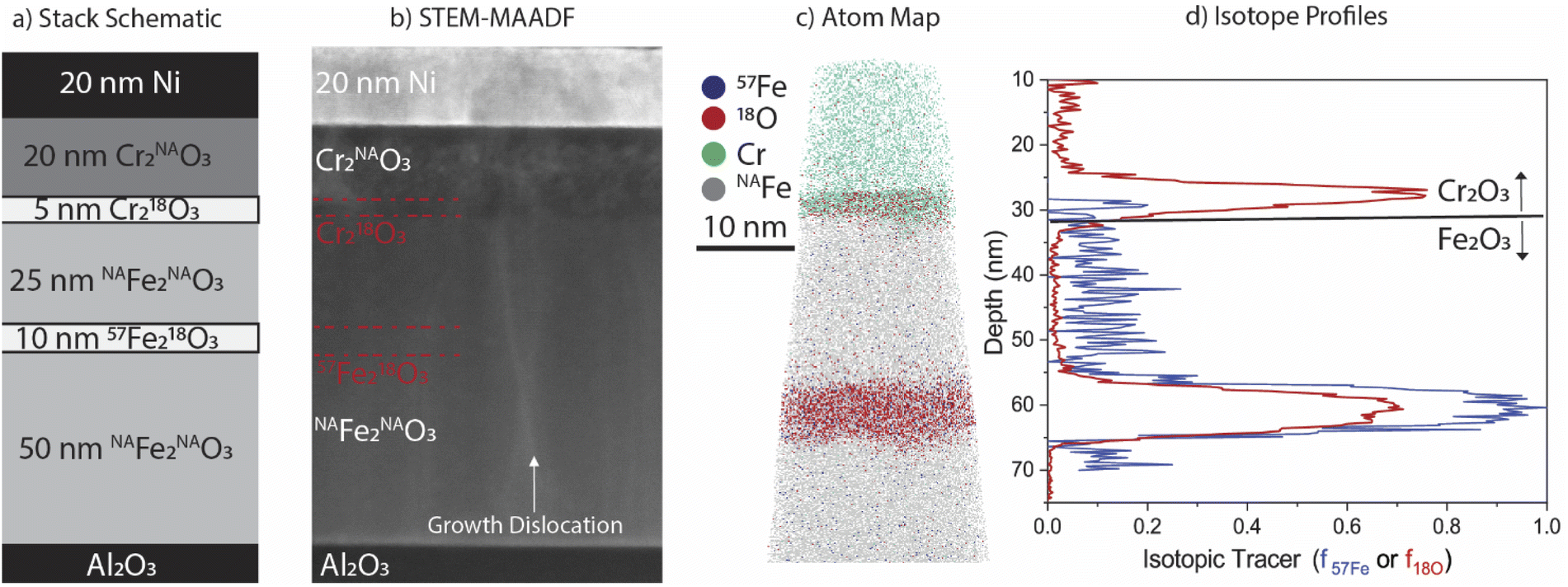 | ||
| Fig. 2 (a) Stack schematic of as-grown epitaxial films with (b) STEM-MAADF image showing uniformity within layers. (c) A representative 3D APT atom map highlighting the isotopic tracer layers in both the Cr2O3 and Fe2O3 layers, and (d) profiles of the isotopic distribution in the APT results for both 57Fe and 18O. | ||
2. Experiment
2.1 Model film design and deposition by molecular beam epitaxy
Growth of a degrading oxide layer is often opportunistic, attacking the alloy at surface imperfections (i.e. grain boundaries, scratches). In such cases, fast diffusion along grain boundaries allows the oxygen to penetrate the alloy.31–33 By embedding our tracer layer in a defect-free single-crystalline oxide, we can measure the bulk diffusion coefficients of both species in the absence of enhanced diffusion along structural imperfections. A model multilayer film stack (Fig. 2a) was specifically designed with embedded isotopic tracers to enable both heavy ion irradiation and subsequent characterization by APT. The entire film stack is ∼130 nm thick, deposited onto a 10 × 10 mm2 single crystal sapphire, α-Al2O3(0001), substrate. Nanoscale layers of chromia (Cr2O3) and hematite (α-Fe2O3) with natural abundance (NA) O (99.8% 16O) and Fe (91.7% 56Fe) and isotopically enriched O (99% 18O) and Fe (95% 57Fe) were controllably deposited using molecular beam epitaxy (MBE). Films were deposited at 730 °C at a rate of 0.1 Å s−1; layers containing 57Fe were deposited at 0.04 Å s−1. The final 20 nm Ni layer was deposited ex situ by magnetron sputtering at room temperature. This is a sacrificial capping layer to protect the underlying tracers from direct vacuum exposure during irradiation, and also helps with subsequent sample preparation for APT and scanning transmission electron microscopy (STEM). Likewise, the Cr2O3 layer provides protection to the hematite film during the vacuum irradiation and in this study, radiation enhanced diffusion was only measured in the Fe2O3. The 10 × 10 mm2 sample was cleaved into four pieces for subsequent irradiation. For thermal annealing studies, a second film was grown on a 10 × 10 mm2 α-Al2O3(0001) substrate, omitting 18O from the Cr2O3 capping layer and the Ni cap altogether. The full stack design is included in the ESI.†2.2 Heavy ion irradiation and thermal annealing
Heavy ion irradiation using 400 kV Ar2+ was accomplished via a Danfysik implanter. Separate areas of the film surface were irradiated at dose rates of 2 × 10−3, 2 × 10−4, and 2 × 10−5 dpa s−1, to a total fluence of 1.15 × 1015 ions per cm2 in all cases. This corresponds to a dose of 0.62 dpa at the 57Fe218O3 tracer position, as calculated by SRIM.34 The irradiation times were 4 min, 45 min, and 390 min (6.5 h), respectively. Irradiations at dose rates of 10−3 and 10−5 dpa s−1 were conducted at 500 °C, while the irradiation at 10−4 dpa s−1 was conducted at 300 °C, to span different transport regimes as illustrated in Fig. 1. Final irradiation parameters and the SRIM profiles used to calculate final dose and Ar implantation peaks are included in the ESI.† To provide baseline data on thermal transport in Fe2O3 at a temperature lower than reported previously, the second, unirradiated film was annealed in air in a Thermo Scientific Thermolyne Model # FB1415M furnace for 1000 h at 650 °C. Air was chosen to minimize reduction of the sample to Fe3O4.2.3 Scanning transmission electron microscopy
STEM characterization was conducted using both a JEOL JEM-ARM 200CF at 200 kV and a JEOL GrandARM-300F at 300 kV. Mid-angle annular dark field (MAADF) images were collected with a probe size of 1.06 Å, a convergence angle of 27.5 mrad, and collection angles of 30–120 mrad. High-angle annular dark field images (HAADF) consisted of collection angles between 68–280 mrad. Dislocation imaging was conducted off the [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 20] zone axis with tilts of 5–10° towards the [0001] direction with a probe size of 1.7 Å, a convergence angle of 6.9 mrad, and collection angles of 6.8–28 mrad to maximize dislocation contrast and minimize bend contours.35 TEM through-focus imaging was conducted to capture void population throughout the film. Defocuses of ±1 μm were used.
20] zone axis with tilts of 5–10° towards the [0001] direction with a probe size of 1.7 Å, a convergence angle of 6.9 mrad, and collection angles of 6.8–28 mrad to maximize dislocation contrast and minimize bend contours.35 TEM through-focus imaging was conducted to capture void population throughout the film. Defocuses of ±1 μm were used.
2.4 Atom probe tomography
A CAMECA local electrode atom probe (LEAP) 4000X HR was used to analyse APT samples. Data were collected at a base temperature of 40 K and pressures <2.7 × 10−9 Pa with laser-assisted field evaporation (λ = 355 nm) at pulse rates of 200 and 250 kHz and a pulse energy of 60 pJ. The detection rate was maintained at 0.003 detected ions per pulse by varying the applied voltage. Using the Integrated Visualization and Analysis Software (IVAS 3.8.5a45), the data were reconstructed in 3D. Reconstructions were scaled using the O bilayer spacing of hematite (2.3 Å) within the APT data (ESI Fig. S9 and S10†).36 This was best achieved using the voltage reconstruction method (see ESI† for details). Representative mass spectra (Fig. S6†) and the comparison between 18O enriched and NA hematite layers (Fig. S7†) are provided in the ESI.† The isotopic tracer distribution was quantified using an isotopic fraction (f18O or f57Fe) according to eqn (1) and (2):10,30| f18O = N181+O/(N181+O + N161+O) | (1) |
| f57Fe = N57Fe2+/(N572+Fe + N562+Fe) | (2) |
N values are the counts for each respective peak at each data point in the corresponding concentration profiles. For the oxygen isotope analysis, the peaks used are at 18 and 16 Da in the mass-to-charge state ratio spectra (16,18O1+). For the iron isotopic calculation, the 2+ peaks are used at 28 and 28.5 Da (56,57Fe2+). Isotope profiles are extracted using a 1D concentration profile (0.2 nm step size) from a cylindrical region of interest with a diameter of 20 nm. The long axis of the cylindrical region is parallel to the [0001] pole. Some regions of the irradiated samples experienced phase transformation from Fe2O3 to Fe3O4, as indicated in the APT reconstructions by both a change in the Fe![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O ratio and a change in the lattice spacing. These cases were not included in the quantitative analysis.
:O ratio and a change in the lattice spacing. These cases were not included in the quantitative analysis.
2.5 APT and STEM sample preparation
Samples were prepared for STEM and APT using dual-beam focused ion beam (FIB)/scanning electron microscope (SEM) systems. Samples were lifted out using an FEI Quanta 3D-FEG, and final thinning (for STEM) or sharpening (for APT) was conducted on a FEI Helios NanoLab. According to established procedures, samples were thinned for TEM37 using progressive steps at 30 kV, 5 kV, and 2 kV Ga+. Samples for APT38 were annularly milled at 30 kV with Ga+ to diameters of ∼150 nm, then polished with 2 kV Ga+ to a final diameter of ∼100 nm.2.6 Chemical rate-theory model
Irradiation has a profound impact on elemental diffusivity. To predict its impact, it is essential to identify the production, recombination, and annihilation of point defects in the irradiated material. A chemical rate-theory model is often used to balance these various contributions to determine the non-equilibrium point defect concentration. In this model, two equations are used to define the rates of change in concentration of interstitial defects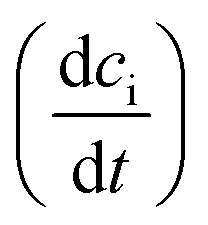 and vacancies
and vacancies 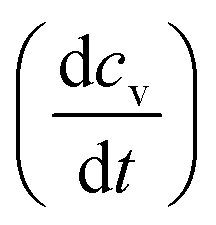 .39
.39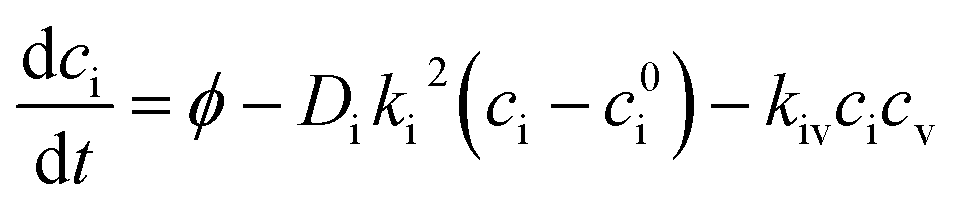 | (3) |
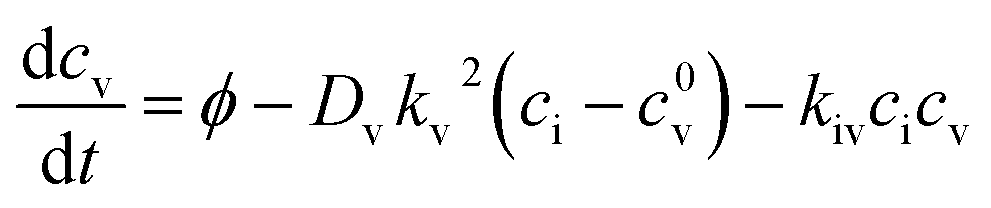 | (4) |
These are evaluated at steady state, with ϕ the defect production rate, Di,v the defect diffusivities, k2 the sink strength, and kiv the recombination coefficient for vacancies, v, and self-interstitials, i. From these equations the irradiation generated point defect concentration can be related to the elemental diffusivity. Self-diffusion included both vacancy and self-interstitial contributions,
| D = Dvcv + Dici | (5) |
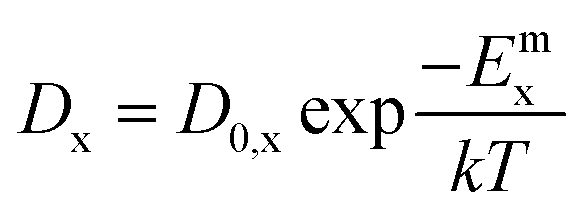 | (6) |
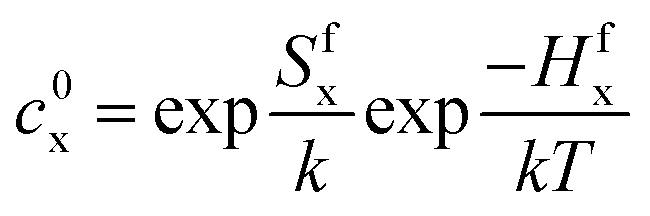 | (7) |
3. Results
3.1 High-resolution characterisation of as-deposited films
Characterisation of as-deposited films via STEM and APT are presented in Fig. 2, along with the nominal stack schematic of the film used for irradiation (Fig. 2a). STEM-MAADF imaging (Fig. 2b) shows Z-contrast differences between layers of the film and phase contrast around vertical growth dislocations. Note that dislocation loops and voids are absent in this micrograph of the as-grown film, and further confirmed in larger field of view micrograph in Fig S2.† Approximate locations of isotopic tracer layers are marked in the image but are not visible by STEM imaging contrast. Corresponding 3D APT atom maps clearly reveal the well-defined Cr218O3 and 57Fe218O3 isotopic tracer layers within the film (Fig. 2c). The abrupt and planar interfaces between the isotopically enriched 57Fe218O3 and surrounding Fe2O3, as well as the Cr218O3/Fe2O3 interface, confirm that little if any intermixing occurred during the film deposition.APT quantification of the as-deposited isotopic tracer distribution for a representative dataset is presented in Fig. 2d. The isotopic tracer layer in the Fe2O3 has a full width at half maximum (FWHM) of 6.4 nm in the 18O and 7.5 nm in the 57Fe, with average isotopic fractions of f18O = 0.61 and f57Fe = 0.91, respectively. The Cr218O3 FWHM is 3.8 nm, with f18O = 0.74. The difference in 18O enrichment maxima arises from differences in the rate of anion exchange between 18O and 16O at the film growth front42 and exchange in the underlayers43 when depositing Cr218O3 at 0.1 Å s−1 compared to 57Fe218O3 at 0.04 Å s−1; both the lower mobility of oxygen in Cr2O3 and the reduced time required for deposition of this layer likely contribute to a higher f18O than in 57Fe218O3. To confirm reproducibility of the APT analysis, two as-grown samples were measured, and profiles for each are included in the ESI (Fig. S8†).
3.2 Irradiated film microstructure
(S)TEM imaging on the high dose rate and low dose rate specimens (500 °C) establishes the defects introduced into the oxide microstructure upon combined irradiation and elevated temperature annealing. Fig. 3 presents data from three imaging modes for each sample: STEM-HAADF to provide a microstructural overview, STEM-BF to reveal dislocations, and overfocus TEM images (Fresnel contrast) that highlight nanoscale voids and dislocations. The HAADF images of both samples (Fig. 3a and b) show localized regions adjacent to the Cr2O3 overlayer where Fe2O3 has reduced to Fe3O4, as outlined by dashed white lines. The transformation is more extensive at the lower dose rate, likely because of the longer time at temperature to reach the desired dose. Moiré contrast, common to Fe2O3 in these films,10,42 does not extend to the transformed Fe3O4 regions, providing additional contrast of the phase boundary. A high-resolution STEM-HAADF image of the interface between phases in the low dose rate sample is also presented in Fig. 3b, illustrating the local change in crystal structure and confirming that an epitaxial relationship between Fe2O3 and Fe3O4 is maintained. Off-zone STEM-BF imaging of dislocations and over-focus TEM images of voids are presented in Fig. 3b and c, respectively, for the high dose rate specimen, and Fig. 3e and f, respectively for the low dose rate specimen. Dislocation loops formed under irradiation in both samples align with the crystal basal plane. These loops appear predominantly as black spot damage in the STEM-BF view because the features have very small diameters (2–5 nm) and have the highest density within the Cr2O3 layer (Fig. 3b). In separate regions of the film, a single larger loop (20 nm diameter) can be seen in the TEM overfocus images in both Fig. 3c and f in the Fe2O3. Dislocation lines, likely persisting from the initial population of vertical growth defects, are present in each film. Small voids (1–2 nm diameter) are present throughout the film, with a smaller population of larger voids (5 nm diameter) grouped near the interface between the Cr2O3 and Fe2O3. Voids were not apparent within the Cr2O3 film, suggesting their formation may be associated with Fe-oxide reduction. In the TEM overfocus image of the low dose rate sample (Fig. 3f), a region of Fe3O4 and associated voids at the Fe2O3/Cr2O3 interface is captured on the left side of the image. Complementary darkfield imaging and the accompanying diffraction pattern of this island of Fe3O4 are included in ESI Fig. S13.† The Ni/Cr2O3 interface is uniform, with no evidence for ballistic mixing between the layers due to irradiation when compared to the as-grown interface. | ||
| Fig. 3 STEM characterization of high (a–c) and low (d–f) dose rate films. (a and c) HAADF images show lighter contrast regions where Fe2O3 reduced to Fe3O4 and include inset high resolution atomic column imaging of the interface between phases. Dislocation imaging of each film (b and e) show loops primarily in the basal plane with the small loops (∼5 nm diameter) in the Cr2O3 and (c and f) large loops (∼10 nm) in the Fe2O3. Vertical dislocations remain in the film after irradiation. Voids (2–3 nm) are present throughout the irradiated films, with larger voids (5 nm) near the Cr2O3/Fe2O3 interface. | ||
3.3 Isotopic redistribution due to radiation-enhanced diffusion
APT measurements were performed to quantify the evolution of the embedded isotopic tracers upon irradiation. Fractional isotopic profiles of the irradiated samples are compared against the as-deposited isotope profiles in Fig. 4 for 18O and 57Fe tracers (top and bottom, respectively) at three irradiation conditions (left to right). The solid black lines in the f18O profiles denote the approximate position of the Cr2O3/Fe2O3 interface. Qualitatively, some 18O movement can be observed from the Cr2O3 tracer layer at the low dose rate sample, likely as a result of its longer irradiation and annealing time to reach the same dose, while no significant mobility is noticed at the other dose rates. We therefore do not further quantify the diffusion from the Cr2O3 tracer in the current study and focus the remainder of our analyses on the Fe2O3 dual-tracer layer.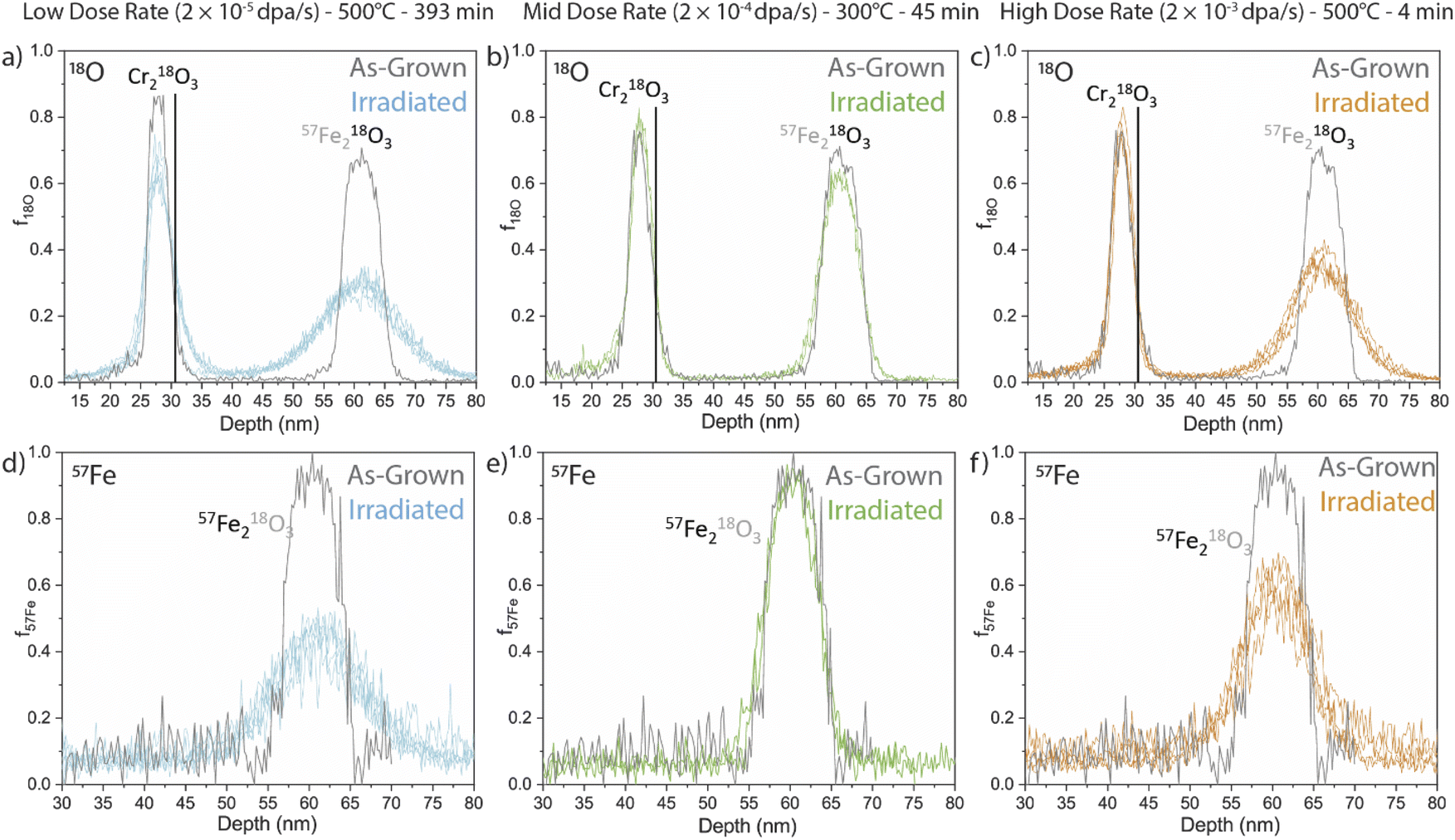 | ||
| Fig. 4 Fractional isotopic profiles from irradiated samples for 18O (upper row) and 57Fe (low row) split by dose rate, low (left), middle (centre), and high (right). Solid black lines in the 18O plots delineate the Cr2O3/Fe2O3 interface. As-grown profiles (gray) are included for comparison. | ||
Qualitatively, each tracer profile from the Fe2O3 film shows clear isotopic redistribution relative to the as-deposited profiles. We note here that some APT samples intersected regions where the Fe2O3 had reduced to Fe3O4. In these cases, diffusion coefficients are not quantified or tabulated as they would represent some unknown combination of transport in both Fe2O3 and Fe3O4. No APT samples from the mid dose rate at the lower temperature (300 °C) showed reduction, possibly due to the small analysis volume of the APT specimens. The transformation to Fe3O4 had a significant effect on the cation tracer diffusion, which was dramatically faster in the samples where the transformation directly encountered the tracer layer. Interestingly, when the reduction did not fully reach the tracer layer the adjacent reduction had no discernible effect on the anion or cation profiles. This phenomenon is not further discussed in this work, though the fractional isotopic profiles are included in the ESI (Fig. S15†).
Both low and high dose rate samples displayed qualitatively similar 18O diffusion into the surrounding NAFe2O3 regions. As a reminder, all specimens were irradiated to the same dose at different dose rates. Considering the significantly longer irradiation time at elevated temperature for the low-dose rate specimen, this implies a faster anion diffusion coefficient for the higher dose rate (shorter time duration) specimen. Conversely, the 57Fe fractional profiles for the low dose and high dose look qualitatively different. The largest spread appears in the low dose rate sample (longest time), whereas the high dose rate profile (shortest time) shows somewhat less spread. In contrast, the irradiation at 300 °C shows different behaviour with very little diffusion.
To determine the rate of thermal diffusion expected at these moderate temperatures, an unirradiated specimen was air annealed at 650 °C for 1000 h. Comparing the as-grown f18O and f57Fe profiles presented in Fig. 5 reveals relatively modest diffusion. It is visually apparent that the 18O tracer has spread, while the changes in the 57Fe profile are far more subtle.
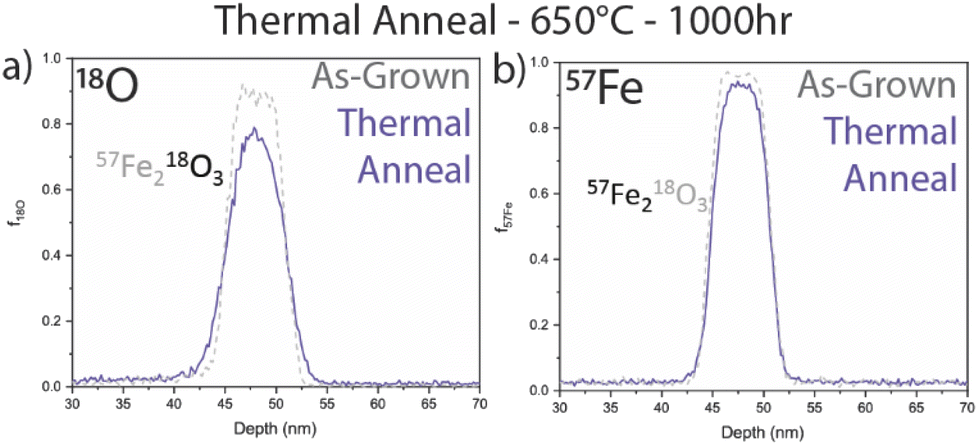 | ||
| Fig. 5 (a) 18O and (b) 57Fe isotopic fraction profiles for a thermally annealed sample at 650 °C, 1000 h compared to the as-grown profile. | ||
Diffusion coefficients were quantified for each profile by numerically aging the as-grown profiles (Fig. 2d) via Fick's Law and minimizing the difference between the numerically aged and experimentally measured profiles. This approach has significant advantages since the imperfections of the as-deposited isotope profiles can be exactly leveraged, rather than approximated by available analytical solutions. Further details on this methodology can be found in prior work10,30 and in the ESI.† Values were averaged over two or three separate APT runs for each irradiation and annealing condition (with profiles that experienced Fe3O4 transformation removed). Table 1 summarizes the resulting diffusion coefficients, as well as results from our preceding study of anion diffusion under proton irradiation at a significantly lower dose rate using the same methods described here.10 The indicated error interval (Dmin and Dmax range) from the fitting procedure is provided, and describes the combined effect of two sources of uncertainty. The first is the standard deviation of the mean of diffusivity collected from each averaged set of samples. The second is the confidence interval of the numerical aging, bounded by the diffusivity values where the square residual increases over the best fit by a factor of two. Finally, an estimate of the APT's diffusivity detection limit (Dlimit) is reported in the last column. As reported elsewhere,30 this estimate derives from an assumption that the APT concentration profiles are sensitive to a characteristic diffusion distance, 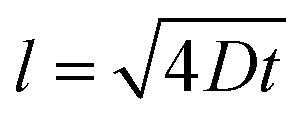 , of l = 0.5 nm. We can solve for the diffusion coefficient, D, assuming the time (t) is the irradiation time. Any extracted or predicted diffusivities below this detection limit should be dismissed as below APT's reliable detection threshold. As shown in Table 1, in all cases the calculated diffusion coefficient is at least an order of magnitude larger than Dlimit, indicating that the isotope diffusion can be detected by APT. A more detailed summary of the APT resolution and diffusion detectability assumptions can be found in the ESI (Fig. S9–S12†).
, of l = 0.5 nm. We can solve for the diffusion coefficient, D, assuming the time (t) is the irradiation time. Any extracted or predicted diffusivities below this detection limit should be dismissed as below APT's reliable detection threshold. As shown in Table 1, in all cases the calculated diffusion coefficient is at least an order of magnitude larger than Dlimit, indicating that the isotope diffusion can be detected by APT. A more detailed summary of the APT resolution and diffusion detectability assumptions can be found in the ESI (Fig. S9–S12†).
| Dose rate (dpa s−1) | Temp. (°C) | Time (min) | D (10−22 m2 s−1) | D min (10−22 m2 s−1) | D max (10−22 m2 s−1) | APT Dlimit (10−22 m2 s−1) |
|---|---|---|---|---|---|---|
| 2 × 10−5 | 500 | 393 | 6.66 | 5.36 | 8.36 | 0.027 |
| 2 × 10−4 | 300 | 45 | 3.98 | 5.00 | 3.08 | 0.23 |
| 2 × 10−3 | 500 | 4 | 299 | 376 | 239 | 2.47 |
| N/A | 650 | 60000 |
3.2 × 10−3 | 2.3 × 10−3 | 4.4 × 10−3 | 1.7 × 10−4 |
| 2 × 10−5 | 500 | 393 | 9.85 | 5.43 | 20.0 | 0.027 |
| 2 × 10−4 | 300 | 45 | 3.67 | 2.87 | 4.6 | 0.23 |
| 2 × 10−3 | 500 | 4 | 251 | 157 | 418 | 2.47 |
| N/A | 650 | 60000 |
5.4 × 10−4 | 2.8 × 10−4 | 8.4 × 10−4 | 1.7 × 10−4 |
| 1 × 10−6 | 450 | 1440 | 0.185 | 0.12 | 0.26 | 7.2 × 10−3 |
The tabulated values of both anion and cation diffusion coefficients vary by two orders of magnitude between the three irradiation conditions, with the previously reported anion diffusion at lower dose rates exhibiting an additional order of magnitude lower diffusivity. From the current set of measurements, the mass transport in the high dose rate is fastest by 1–2 orders of magnitude for both anions and cations, while the mid-dose rate (lower temperature) and low-dose rate (higher temperature) coincidentally display similar diffusion coefficients. By contrast, the extracted diffusion coefficients of the unirradiated air anneal sample were 3 to 4 orders of magnitude lower than the smallest value measured in the irradiated samples, despite its higher annealing temperature.
3.4 Model results
Predictions from the chemical rate-theory model of diffusion coefficients for both anions and cations through Fe2O3 are shown in Fig. 6. In the temperature range shown, the thermal contribution to diffusivity is negligible. In the model, the sink strength describes the mean square distance a point defect must diffuse prior to absorption at an interface, free surface, dislocation, or other such extended defect. A value of 5 × 1015 m−2 was assumed for these calculations, intended to reflect the thickness of the sample and the spacing of voids evident in Fig. 3. This microstructure is generally on the higher end of sink densities observed in irradiated materials. The model includes the elimination of point defects through either recombination or absorption at sinks (the first two regimes described in Fig. 1). At lower temperatures, where point defects are less mobile and accumulate to higher densities in the lattice, the defect mean free path is limited by recombination. As temperature increases, the defect supersaturations are lower as defects reach sinks prior to recombining. In this model, the sink density is assumed to be independent of temperature, leading to a plateau in radiation-enhanced diffusivity. These regimes are highlighted in Fig. 6 for both species. With increasing dose rate, more defects are generated resulting in faster diffusivities, which is reflected in the shifting sink dominant plateaus.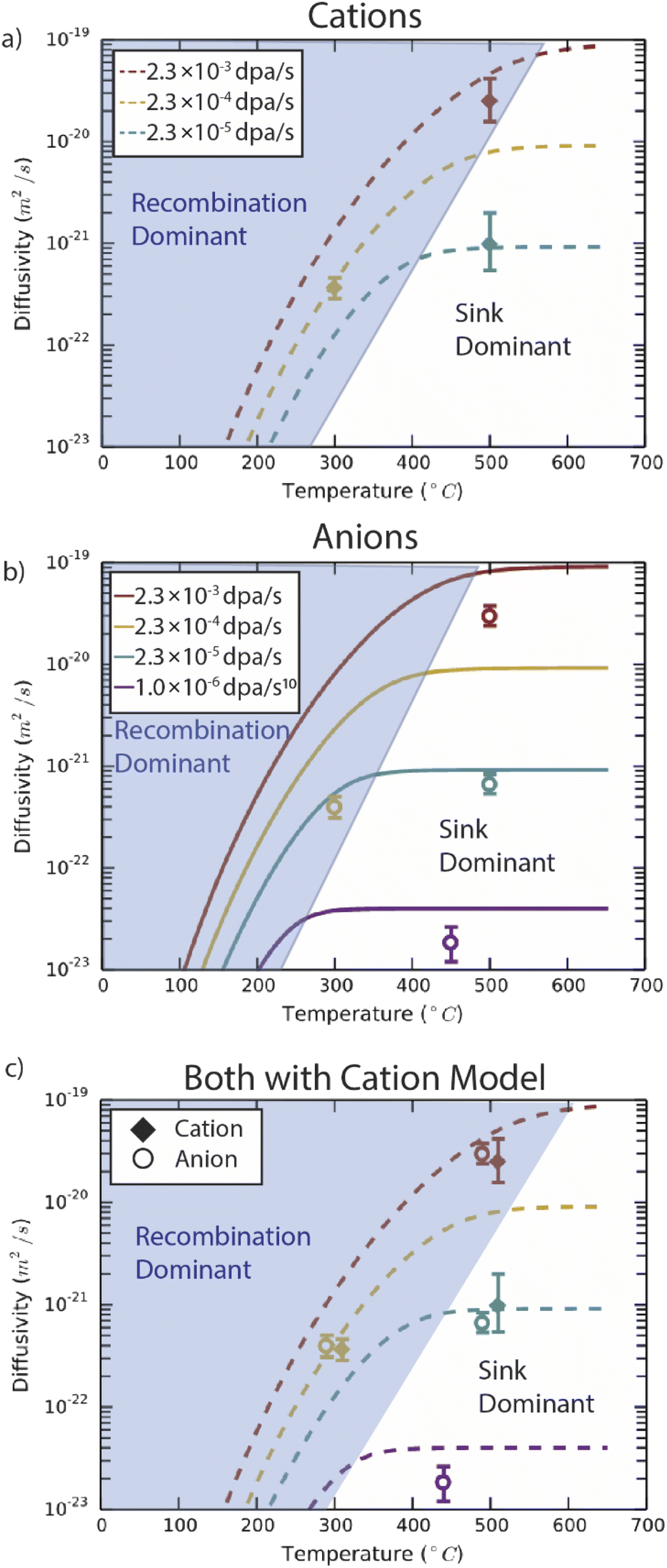 | ||
| Fig. 6 Chemical rate theory model results for (a) anions, (b) cations and (c) both species in Fe2O3, compared with experimental data. The model examines the dose-rate dependence of species transport under non-equilibrium point defect population generated through irradiation. Experimental data points are the average values from each irradiation condition. Included in the (b) and (c) panels is data from prior work on proton irradiated Fe2O3 with a dose rate of 10−6 dpa s−1.10 Panels (a) and (b) compare results from the model with experiment for anions and cations, respectively. The final panel (c) compares experimental results for both anion and cation species with the model results for cations, emphasizing the seemingly coupled behaviour of anions and cations in the experiments. | ||
In Fe2O3 at these temperatures, mobility is predicted to be lower for cation vacancies than anion vacancies. This leads to a slight shift to higher temperatures before sink strength dominates transport for cation defects. For cation transport, this model slightly overestimates the diffusivity at the high dose rate (2 × 10−3 dpa s−1) compared to the experimental values, but closely follows the experimentally observed behaviour as temperature and dose rate change. However, in the anion case, the model predicts a transition between recombination and sink dominance which occurs at lower temperatures than for cations. The model somewhat overestimates the experimental data points at the higher temperatures (450 °C and 500 °C). However, the model significantly overestimates the anion diffusivity at 300 °C compared to the experimental measurement. Because the experimental diffusivity measurements are nearly identical for the two sublattices, both closely follow the cation model, as illustrated in Fig. 6c.
4. Discussion
4.1 Dose rate dependence
The chemical rate theory model predicts differences in diffusivity depending on the dose rate. For the high and low dose rates at 500 °C, the experimental results agree very well with the model for both cation and anion diffusivity. However, at the 300 °C, 2 × 10−4 dpa s−1 irradiation conditions, the model predicts a difference between anion and cation behaviour that is not observed experimentally. Within the model, the lower vacancy mobility in the cation lattice extends the recombination dominant regime to higher temperatures (400–500 °C, Fig. 6b) as compared to the anion model (300–400 °C, Fig. 6a). Yet, the experimental results for anions closely follow the cation diffusivity (Fig. 6c). This suggests that, in the recombination dominant regime, the defects on the cation lattice are rate-limiting for both cation and anion diffusion. The defects on the cation lattice may be attracting anion defects, slowing their migration down, such that the anion recombination regime should also extend to higher temperatures.As an additional check of model performance, a prior experimental result (proton irradiation at 450 °C, 1 × 10−6 dpa s−1) is included in Fig. 6b and c.10 This result is in the sink dominant regime and appears to match reasonably with the model curve at this lower dose rate. The inclusion of this data point highlights the applicability of the model at lower dose rates. Reactor conditions, and in particular light water reactor conditions, are at much lower dose rates (10−8 to 10−7 dpa s−1)44,45 than studied here. When heavy ion irradiations are used as neutron irradiation substitutes the dose rate must be considered.46 Yet even in these conditions, radiation enhanced diffusion would be expected to proceed at least six orders of magnitude faster than thermal diffusion under this model. For reference, the dose rates expected in cladding,44 reactor pressure vessels,47 and glass waste forms48 are 10−7, 10−11, and 10−13 dpa s−1, respectively.
These results confirm that any prediction of performance in systems with corrosion and irradiation must consider the dose rate dependence of cation and anion diffusion. While in engineering materials, grain boundaries and multiphase composite microstructures would play an important role in diffusivity, these results highlight a distinct change in diffusivity due to irradiation through the matrix material that cannot be disregarded. Further experimental work using isotopic tracers and APT to quantify transport along fast pathways and across the phase boundaries could help develop more sophisticated models to account for these real-world features.
4.2 Radiation enhanced diffusivity
Extracting the true amplification of mass transport in Fe2O3 from irradiation is challenging, given the disparate temperature regimes for our irradiations compared to previous thermal transport data. However, as a starting point, we present a comparison of our experimentally measured diffusion coefficients against previous thermal diffusion measurements in Fig. 7. Arrhenius extrapolation of these data to our experimental temperatures results in a predicted diffusion coefficient for oxygen in Fe2O3 of 10−45 m2 s−1 at 300 °C, assuming the activation enthalpy is consistent.18 In comparison, experimental results at 300 °C for oxygen in Fe2O3 were measured to be 3.98 × 10−22 m2 s−1, an increase of >20 orders of magnitude. For context, anion transport measured between 250-550 °C in Fe3O4, also plotted in Fig. 7, is ∼1.1 × 10−24 m2 s−1 at 300 °C.52 Here, even Fe3O4 anion diffusivity is 2–3 orders of magnitude less than experimental RED measurements in Fe2O3. However, the validity of these Arrhenius expressions for thermal diffusivity when extrapolating to relevant temperatures for reactors (RT – 500 °C) is suspect.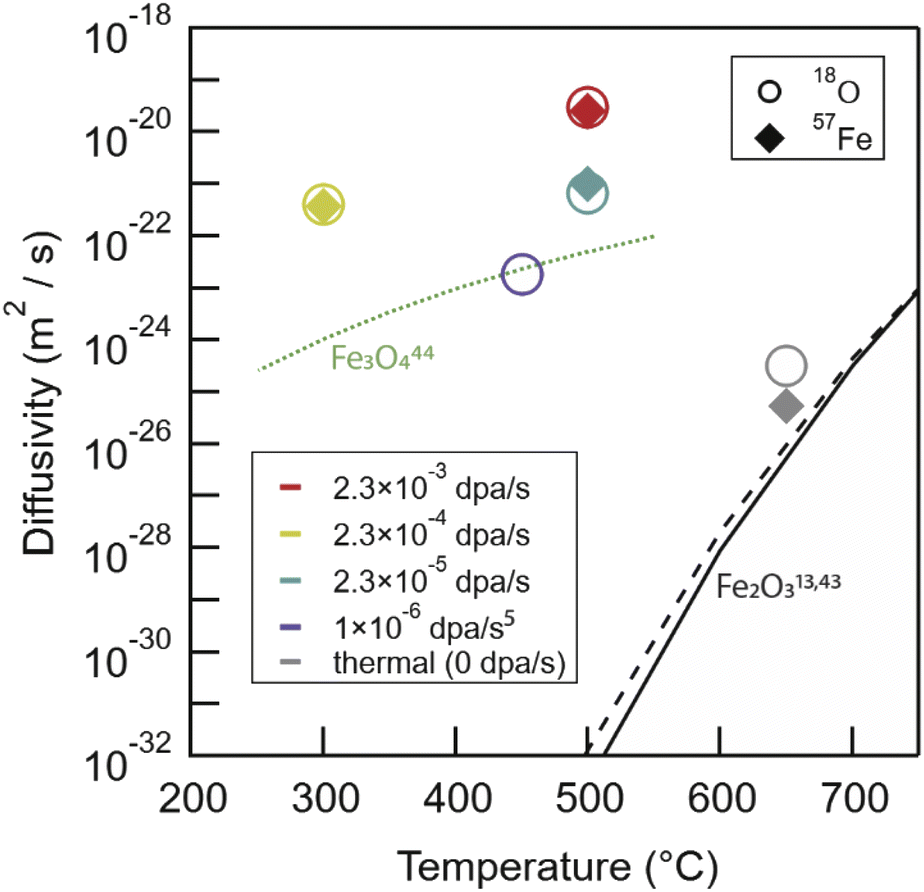 | ||
| Fig. 7 The broad picture of our irradiation and thermal anneal experimental results compared against literature Arrhenius extrapolations for Fe2O3 cation49 and anion18 species, and experimental anion Fe3O4 (ref. 50) transport. Fe3O4 cation transport is much faster with diffusivities between 1 × 10−12 to 1 × 10−16 m2 s−1 between 900 and 1400 °C,51 and as such are not plotted here. RED in Fe2O3 leaves both anion and cation transport tens of orders of magnitude faster than Arrhenius extrapolations would predict and 2–4 orders of magnitude faster than anion transport in Fe3O4. | ||
The thermal anneal (650 °C 1000 h in air) data for both 57Fe and 18O in this study falls near to the vacancy only, simple Arrhenius extrapolation for both species. The chemical rate theory model would expect 2–3 orders of magnitude faster diffusion due to a vacancy and interstitialcy mechanism. There are multiple indications of an as-yet ill-defined deviation from previously described high-temperature Arrhenius relationships, both in our prior work10 and in a study by Atkinson and Taylor49 using 55Fe as an isotopic tracer to measure diffusivities down to 700 °C. In both cases, a change in transport mechanism was noted. For anions, both our data and the chemical rate-theory model suggest a possible change from vacancy to interstitialcy mediated transport around 900 °C. The Arrhenius extrapolation of high-temperature literature 18O data closely follows the vacancy only curve.18 For cations, Atkinson and Taylor attributed a change in activation energy to impurity dominated transport in their specimens. Curiously, this largely coincides with temperatures at which the activation energies reported for anions appear to change as well, as captured by the full model. Atkinson's 55Fe experimental data closely follows this trend, and a possible alternative explanation is that self-diffusion on the two-sublattices is linked, as was clearly seen in the present radiation enhanced diffusion experiments across a wide range of dose rates.
On the other hand, the transition temperature to interstitialcy dominated transport remains uncertain, as discussed in detail in prior work,10 with contributions from entropy and chemical potential which are not yet firmly established. More data are needed for low temperature diffusion behaviour in oxides to distinguish between these possibilities.
4.3 Model and experimental limitations
Both the model and the experiment have some limitations. On the experimental side, the resolution of the APT to a specific diffusion distance is conservatively estimated to be ∼0.5 nm in depth, but could be significantly worse laterally (∼1–3 nm). In the current experiments, we exploit the geometry of the epitaxially deposited isotope tracers to align with the optimal APT resolution direction to enable sensitive quantification of interdiffusion. Further discussion of the APT resolution has been discussed in prior work30 and a summary is included in the ESI.†The practical limitations of experimental measurements do, however, highlight the importance of developing and parameterizing models that can extrapolate beyond our experimental limits. To that end, we note several needs for further model development. Defect charge states are dependent on the Fermi level in the oxide and, in the current model, changes in Fermi level are not taken into account.10 Perhaps more importantly, there is some uncertainty in mapping the formation energies calculated with DFT to a particular location in the phase diagram. Depending on the details of theory used (e.g., the choice of exchange–correlation functional), DFT may incorrectly predict the binding energy of the oxygen molecule or the formation energy of the oxide. While correction schemes have been proposed,53 it is not entirely clear whether the chemical potential of the anion, cation, or both species ought to be modified to correct the oxide phase energy. As such, it remains possible that DFT energetics are offset from actual chemical potentials. This influences the thermal component of self-diffusion.
Under irradiation, interactions other than recombination are likely to play a significant role but are not captured in the current model. For example, the sink strength in this study is kept constant, while previous studies have shown that the sink strength is dependent on temperature and increases with dose.30 These dependences come from the aggregation of point defects into larger clusters, leading to an evolving microstructure during radiation damage. Finally, as discussed above, the defect pathways are not entirely illuminated. The anion defect behaviour does appear to be strongly linked to the cation defects. Interactions between defects on the different sublattices are one possible explanation for this, promoting the clustering of individual defects into larger complexes. These clusters could then either diffuse together as a unit or act as traps and promote recombination, linking self-diffusion behaviour for the two species. These possibilities are being examined with ongoing atomistic simulations.
5. Conclusions
In conclusion, combined experimental and theoretical techniques captured a strong dose-rate dependence in both anion and cation transport through α-Fe2O3. Computational and experimental results agreed well in the sink dominant regime for irradiations at 500 °C. In the recombination regime (300 °C irradiation), cation transport appeared to dominate; experimental anion transport was limited compared to the computational model. In addition, radiation enhanced diffusion was at least 20 orders of magnitude faster than Arrhenius extrapolations from literature.These results emphasize the need to experimentally inform predictive models by considering the impact of irradiation induced non-equilibrium point defect populations on mass transport in oxide films relevant to structural alloys. Irradiation dose-rate can result in 2–3 orders of magnitude difference in transport, leading to significant different macroscale oxide growth. With increasing dose rates in next generation reactor designs, this parameter becomes even more important in designing robust materials.
Author contributions
Using the CRediT (Contributor Roles Taxonomy) the authors contributed as follows: Conceptualization – Kayla H. Yano, Aaron A. Kohnert, Tiffany C. Kaspar, Sandra D. Taylor, Blas P. Uberuaga, and Daniel K. Schreiber. Data curation – Kayla H. Yano, Aaron A. Kohnert. Formal Analysis – Kayla H. Yano, Aaron A. Kohnert. Funding acquisition – Blas P. Uberuaga, Daniel K. Schreiber. Investigation – Kayla H. Yano, Aaron A. Kohnert, Sandra D. Taylor. Methodology – Kayla H. Yano, Aaron A. Kohnert, Tiffany C. Kaspar, Sandra D. Taylor, Blas P. Uberuaga, and Daniel K. Schreiber. Project administration – Sandra D. Taylor, Blas P. Uberuaga, and Daniel K. Schreiber. Resources – Tiffany C. Kaspar, Hyosim Kim, Yongqiang Wang. Software – Aaron A. Kohnert, Blas P. Uberuaga. Supervision – Sandra D. Taylor, Blas P. Uberuaga, and Daniel K. Schreiber. Validation – Kayla H. Yano, Aaron A. Kohnert, Sandra D. Taylor, Blas P. Uberuaga, and Daniel K. Schreiber. Visualization – Kayla H. Yano, Aaron A. Kohnert. Writing – original draft – Kayla H. Yano, Aaron A. Kohnert. Writing – review & editing – Kayla H. Yano, Aaron A. Kohnert, Tiffany C. Kaspar, Sandra D. Taylor, Steven R. Spurgeon, Hyosim Kim, Yongqiang Wang, Blas P. Uberuaga, and Daniel K. Schreiber.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported as part of FUTURE (Fundamental Understanding of Transport Under Reactor Extremes), an Energy Frontier Research Center funded by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences. A portion of the research was performed using EMSL (grid.436923.9), a DOE Office of Science User Facility sponsored by the Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory (PNNL). PNNL is a multiprogram national laboratory operated by Battelle for the U.S. DOE under Contract DE-AC05-79RL01830. This research used resources provided by the Los Alamos National Laboratory (LANL) Institutional Computing Program. Irradiations were conducted at the Ion Beam Materials Laboratory at LANL. LANL, an affirmative action equal opportunity employer, is managed by Triad National Security, LLC for the U.S. DOE's NNSA, under contract 89233218CNA000001.Notes and references
- R. A. Rapp, J. H. Devan, D. L. Douglass, P. C. Nordine, F. S. Pettit and D. P. Whittle, Mater. Sci. Eng., 1981, 50, 1–17 CrossRef CAS.
- J. Stringer, High Temp. Technol., 1985, 3, 119–141 CrossRef CAS.
- J. Stubbins, B. Heuser, P. Hosemann and M. Li, Fundamental Studies of Irradiation- Induced Modifications in Microstructural Evolution and Mechanical Properties of Advanced Alloys, Urbana-Champaign, 2018 Search PubMed.
- F. Schmidt, P. Hosemann, R. O. Scarlat, D. K. Schreiber, J. R. Scully and B. P. Uberuaga, Annu. Rev. Mater. Res., 2021, 51, 293–328 CrossRef CAS.
- E. M. Schulson, J. Nucl. Mater., 1979, 83, 239–264 CrossRef CAS.
- D. J. Bacon and T. Diaz de la Rubia, J. Nucl. Mater., 1994, 216, 275–290 CrossRef CAS.
- B. D. Wirth, X. Hu, A. Kohnert and D. Xu, J. Mater. Res., 2015, 30, 1440–1455 CrossRef CAS.
- C. A. Taylor, D. C. Bufford, B. R. Muntifering, D. Senor, M. Steckbeck, J. Davis, B. Doyle, D. Buller and K. M. Hattar, Materials, 2017, 10(10), 1148 CrossRef PubMed.
- J. O. Stiegler and L. K. Mansur, Annu. Rev. Mater. Sci., 1979, 9, 405–454 CrossRef CAS.
- K. H. Yano, A. A. Kohnert, A. Banerjee, D. J. Edwards, E. F. Holby, T. C. Kaspar, H. Kim, T. G. Lach, S. D. Taylor, Y. Wang, B. P. Uberuaga and D. K. Schreiber, Chem. Mater., 2021, 33, 2307–2318 CrossRef CAS.
- D. Frazer, S. Qvist, S. Parker, D. L. Krumwiede, M. Caro, J. Tesmer, S. A. Maloy, Y. Q. Wang and P. Hosemann, J. Nucl. Mater., 2016, 479, 382–389 CrossRef CAS.
- S. S. Raiman, Irradiation Accelerated Corrosion of 316L Stainless Steel in Simulated Primary Water, University of Michigan, 2016 Search PubMed.
- A. C. S. Sabioni, A. M. Huntz, A. M. J. M. Daniel and W. A. A. MacEdo, Philos. Mag., 2005, 85, 3643–3658 CrossRef CAS.
- K. Hoshino and N. L. Peterson, J. Phys. Chem. Solids, 1985, 46, 1247–1254 CrossRef CAS.
- K. P. R. Reddy and A. R. Cooper, J. Am. Ceram. Soc., 1983, 66, 664–666 CrossRef CAS.
- R. P. Kingery, W. D. Hill and D. C. Nelson, J. Am. Ceram. Soc., 1960, 43, 473–475 CrossRef.
- W. C. Hagel, Trans. AIME, 1966, 236, 179–184 CAS.
- B. Amami, M. Addou, F. Millot, A. Sabioni and C. Monty, Ionics, 1999, 5, 358–370 CrossRef CAS.
- Wi. C. Hagel, J. Am. Ceram. Soc., 1965, 48, 70–75 CrossRef CAS.
- A. C. S. Sabioni, B. Lesage, A. M. Huntz, J. C. Pivin and C. Monty, Philos. Mag. A, 1992, 66, 333–350 CrossRef CAS.
- H. G. Sockel, B. Saal and M. Heilmaier, Surf. Interface Anal., 1988, 12, 531–533 CrossRef.
- W. E. King and J. H. Park, MRS Proc., 1988, 122, 193–198 CrossRef CAS.
- S. C. Tsai, A. M. Huntz and C. Dolin, Mater. Sci. Eng. A, 1996, 212, 6–13 CrossRef.
- R. E. Lobnig, H. P. Schmidt, K. Hennesen and H. J. Grabke, Oxid. Met., 1992, 37, 81–93 CrossRef CAS.
- A. C. S. Sabioni, A. M. Huntz, F. Silva and F. Jomard, Mater. Sci. Eng., A, 2005, 392, 254–261 CrossRef.
- A. C. S. Sabioni, A. M. Huntz, F. Millot and C. Monty, Philos. Mag. A, 1992, 66, 351–360 CrossRef CAS.
- S. J. Zinkle and J. T. Busby, Mater. Today, 2009, 12, 12–19 CrossRef CAS.
- R. E. Stoller, M. B. Toloczko, G. S. Was, A. G. Certain, S. Dwaraknath and F. A. Garner, Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms, 2013, 310, 75–80 CrossRef CAS.
- L. El-Guebaly, L. Mynsberge, A. Davis, C. D'Angelo, A. Rowcliffe and B. Pint, Fusion Sci. Technol., 2017, 72, 17–40 CrossRef.
- K. H. Yano, A. A. Kohnert, T. C. Kaspar, S. D. Taylor, S. R. Spurgeon, H. Kim, Y. Wang, B. P. Uberuaga and D. K. Schreiber, J. Phys. Chem. C, 2021, 125, 27820–27827 CrossRef CAS.
- H. Falk-Windisch, P. Malmberg, M. Sattari, J. E. Svensson and J. Froitzheim, Mater. Charact., 2018, 136, 128–133 CrossRef CAS.
- A. Brückman, R. Emmerich and S. Mrowec, Oxid. Met., 1972, 5, 137–147 CrossRef.
- J. Zurek, D. J. Young, E. Essuman, M. Hänsel, H. J. Penkalla, L. Niewolak and W. J. Quadakkers, Mater. Sci. Eng. A, 2008, 477, 259–270 CrossRef.
- J. F. Ziegler, Stopping Range of Ions in Matter (SRIM), SRIM Company, 2013 Search PubMed.
- Y. Zhu, C. Ophus, M. B. Toloczko and D. J. Edwards, Ultramicroscopy, 2018, 193, 12–23 CrossRef CAS PubMed.
- M. P. Moody, B. Gault, L. T. Stephenson, D. Haley and S. P. Ringer, Ultramicroscopy, 2009, 109, 815–824 CrossRef CAS PubMed.
- L. A. Giannuzzi and F. A. Stevie, Micron, 1999, 30, 197–204 CrossRef.
- K. Thompson, D. Lawrence, D. J. Larson, J. D. Olson, T. F. Kelly and B. Gorman, Ultramicroscopy, 2007, 107, 131–139 CrossRef CAS PubMed.
- G. S. Was, Fundamentals of Radiation Materials Science, Springer, New York, 2007 Search PubMed.
- A. Banerjee, A. A. Kohnert, E. F. Holby and B. P. Uberuaga, Phys. Rev. Mater., 2021, 5, 1–6 Search PubMed.
- A. Banerjee, A. A. Kohnert, E. F. Holby and B. P. Uberuaga, J. Phys. Chem. C, 2020, 124, 23988–24000 CrossRef CAS.
- T. C. Kaspar, S. D. Taylor, K. H. Yano, T. G. Lach, Y. Zhou, Z. Zhu, E. K. Still, P. Hosemann, S. R. Spurgeon and D. K. Schreiber, Adv. Mater. Interfaces, 2021, 8, 2001768 CrossRef CAS.
- T. C. Kaspar, P. Hatton, K. H. Yano, S. D. Taylor, S. R. Spurgeon, B. P. Uberuaga and D. K. Schreiber, Nano Lett., 2022, 22, 4963–4969 CrossRef CAS PubMed.
- T. Allen, J. Busby, M. Meyer and D. Petti, Mater. Today, 2010, 13, 14–23 CrossRef CAS.
- R. L. Klueh and D. R. Harries, High-Chromium Ferritic and Martensitic Steels for Nuclear Applications, 2001 Search PubMed.
- S. J. Zinkle and L. L. Snead, Scr. Mater., 2018, 143, 154–160 CrossRef CAS.
- L. Malerba, in Understanding and Mitigating Ageing in Nuclear Power Plants, 2010, pp. 456–543 Search PubMed.
- W. J. Weber, A Review of Radiation Effects in Solid-Nuclear-Waste Forms, 1981 Search PubMed.
- A. Atkinson and R. I. Taylor, J. Phys. Chem. Solids, 1985, 46, 469–475 CrossRef CAS.
- J. E. Castle and P. L. Surman, J. Phys. Chem., 1969, 73, 632–634 CrossRef CAS.
- R. Dieckmann and H. Schmalzried, Ber. Bunsen Ges., 1977, 81, 344–347 CrossRef CAS.
- J. E. Castle and P. L. Surman, J. Phys. Chem., 1967, 71, 4255–4259 CrossRef CAS.
- A. Jain, G. Hautier, S. P. Ong, C. J. Moore, C. C. Fischer, K. A. Persson and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 1–10 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta03403d |
| This journal is © The Royal Society of Chemistry 2022 |