
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModel structures of molten salt-promoted MgO to probe the mechanism of MgCO3 formation during CO2 capture at a solid–liquid interface†
Alexander. H.
Bork
 a,
Norbert
Ackerl
ab,
Joakim
Reuteler
c,
Sachin
Jog
a,
David
Gut
a,
Robert
Zboray
d and
Christoph. R.
Müller
*a
a,
Norbert
Ackerl
ab,
Joakim
Reuteler
c,
Sachin
Jog
a,
David
Gut
a,
Robert
Zboray
d and
Christoph. R.
Müller
*a
aLaboratory of Energy Science and Engineering, Department of Mechanical and Process Engineering, ETH Zurich, CH-8092 Zürich, Switzerland. E-mail: muelchri@ethz.ch
bNAPho – Norbert Ackerl Photonics, CH-8049 Zürich, Switzerland
cScientific Center for Optical and Electron Microscopy, ETH Zurich, CH-8093 Zurich, Switzerland
dCenter for X-ray Analytics, Empa, Swiss Federal Laboratories for Materials Science and Technology, CH-8600 Dübendorf, Switzerland
First published on 8th July 2022
Abstract
MgO is a promising solid oxide-based sorbent to capture anthropogenic CO2 emissions due to its high theoretical gravimetric CO2 uptake and its abundance. When MgO is coated with alkali metal salts such as LiNO3, NaNO3, KNO3, or their mixtures, the kinetics of the CO2 uptake reaction is significantly faster resulting in a 15 times higher CO2 uptake compared to bare MgO. However, the underlying mechanism that leads to this dramatic increase in the carbonation rate is still unclear. This study aims to determine the most favourable location for the nucleation and growth of MgCO3 and more specifically, whether the carbonation occurs preferentially at the buried interface, the triple phase boundary (TPB), and/or inside the molten salt of the NaNO3–MgO system. For this purpose, a model system consisting of a MgO single crystal that is structured by ultra-short pulse laser ablation and coated with NaNO3 as the promoter is used. To identify the location of nucleation and growth of MgCO3, micro X-ray computed tomography, scanning electron microscopy, Raman microspectroscopy and optical profilometry were applied. We found that MgCO3 forms at the NaNO3/MgO interface and not inside the melt. Moreover, there was no preferential nucleation of MgCO3 at the TPB when compared to the buried interface. Furthermore, it is found that there is no observable CO2 diffusion limitation in the nucleation step. However, it was observed that CO2 diffusion limits MgCO3 crystal growth, i.e. the growth rate of MgCO3 is approximately an order of magnitude faster in shallow grooves compared to that in deep grooves.
Introduction
CO2 absorption from large point sources such as power plants and its subsequent storage (carbon dioxide capture and storage, CCS) is a cost-effective approach to significantly reduce anthropogenic CO2 emissions. Alkaline earth metal oxide-based sorbents that reversibly ab- and desorb CO2 have attracted attention in the framework of CCS due to favourable cost estimates for CO2 capture compared to amine scrubbing.1–3Magnesium oxide-based sorbents that capture CO2 reversibly through the reaction MgO + CO2 ↔ MgCO3 at intermediate temperatures (300–500 °C) have shown great potential for CCS owing to its high theoretical gravimetric uptake (1.09 gCO2 gMgO−1) and low energy requirement for sorbent regeneration (2.68 kJ g−1 CO2) when compared to other metal oxide sorbents (e.g. 4.04 kJ g−1 CO2 for CaO).4,5 However, bulk MgO exhibits sluggish carbonation kinetics resulting in low experimentally obtained uptakes of only 0.02 gCO2 gMgO−1 after 1 hour of exposure to CO2.4,5 To accelerate the kinetics of CO2 uptake of MgO-based sorbents, different approaches, such as the increase of surface area, have been reported.4,6,7 However, the addition of alkali metal salts e.g. NaNO3, KNO3, LiNO3 or mixtures thereof have resulted in the largest increases in the CO2 uptake kinetics.8–11 It is important to notice, that an enhancement in the CO2 uptake only occurs when the salt is in the molten state and for an optimized system containing 20 wt% NaNO3 an CO2 uptake of 0.31 gCO2 gMgO−1 (after 60 min of carbonation) has been reported.9,10,12 Although there is consensus on the promoting effect of alkali metal salts, there is an ongoing debate on the mechanism(s) through which alkali metal salt promotes carbonate formation with accelerated rates.1,9,10,13–16 One hypothesis considers alkali metal salts (AMS) as phase transfer catalysts that dissolve MgO yielding solvated ionic [Mg2+…O2−] pairs that have weaker bonds compared to the strong ionic bond of bulk MgO. According to density functional theory calculations by Zhang et al. the dissociation of MgO into a [Mg2+…O2−] pair requires 7.1 eV (in vacuum) while only 5.3 eV is required in NaNO3 due to solvation effects.9 In addition AMS have the ability to dissolve CO2 promoting further its reaction with solvated [Mg2+…O2−] pairs.9,17 Besides the mechanism itself, currently also the location of MgCO3 growth and the rate limiting step is under debate, i.e. the dissolution of MgO or the dissolution of CO2.1,9,10
Kinetic analysis of CO2 absorption on AMS promoted MgO using thermogravimetric analysis (TGA) has revealed that the formation of MgCO3 is characterized by a nucleation and growth process.10 In such a process the nucleation and growth of magnesium carbonate would rely on the dissolution of MgO and CO2 in the AMS leading to a supersaturated solution of a magnesium carbonate precursor (e.g. [Mg2+…CO32−]) yielding ultimately crystalline MgCO3. A recent study into the growth process of MgCO3 has shown that MgCO3 grows in the form of 2D crystals with a sector plate morphology at the interface of NaNO3/MgO, providing further evidence for a nucleation and growth process in which MgCO3 precipitates as crystals from a supersaturated solution.15 While there is a general agreement on the nucleation and growth process for MgCO3 formation,14,16 the location at which MgCO3 precipitates most favorably is still unclear. Conceptually, MgCO3 can form within the molten salt, at the interface of the salt and MgO (NaNO3/MgO), often referred to as the “buried interface” or at the triple phase boundary (TPB), i.e. the interface of NaNO3/MgO that is exposed to the CO2 atmosphere. Answering this question, will provide important sorbent design guidelines as to whether an effective sorbent shall have a high TPB length or a large NaNO3/MgO interface area.
Previous work aiming to address this question have applied in situ TEM analysis on AMS promoted MgO nanoparticles and observed that MgCO3 precipitates at the TPB.14,18 Somewhat contradictory, a study using a MgO(100) single crystal coated completely with NaNO3 revealed the formation of MgCO3 in the absence of a TPB.16 In another example, in situ X-ray diffraction on a sample of MgO(100) promoted with NaNO3 that is dispersed on the surface, evidenced the formation of crystalline MgCO3 at the NaNO3/MgO interface.15 Moreover, ex situ microscopy analysis of the formed MgCO3 indicate a favorable growth of MgCO3 at the buried interface.15 The indication (importantly not a direct proof) of growth at the buried interface was based on the observation of a 2D hemispherical zone of etched dissolution pyramidal pits at the surface of MgO(100) under and around the 2D MgCO3 crystals. Since both NaNO3 and CO2 are required for both the formation of such etching pits in MgO and the subsequent growth of MgCO3, the growth of a crystal ought to occur at the buried interface as it is unlikely that a dynamically expanding TPB exists at the perimeter of the (growing) crystal.15 However, besides some structural advantages of the NaNO3–MgO(100) model system it has a key disadvantage in that the dispersed molten AMS is mobile on the surface under operating conditions making the determination of the location of the AMS/MgCO3 interface very difficult, if not impossible. Therefore, a clear answer to the question whether MgCO3 forms favorably at the TPB or the buried interface is still missing. To provide an unequivocal answer to this question, a model system of MgO that confines the (molten) AMS to a fixed location is required. To this end we use laser micro processing to manufacture model geometries in MgO.19,20 An ultra-short pulsed (USP) laser system with pulse durations below a picosecond was used to fabricate grooves of controlled (yet varying) depth ensuring that the molten AMS remains at a fixed location during operation while providing both a TPB and a buried interface. Such well-defined model structures are subsequently characterized by micro X-ray computed tomography (μCT), scanning electron microscopy (SEM), Raman spectroscopy and optical profilometry to identify the location and quantify the amount of MgCO3 grown. Our experimental results show clearly that MgCO3 grows equally favorably at the buried interface and TPB. Furthermore, by varying the depth of the grooves in MgO allows for an assessment of the effect of the CO2 diffusion path length from the gas atmosphere to the reaction interface on the growth rate, showing a faster growth of MgCO3 in the shallow grooves i.e. for shorter CO2 diffusion path lengths.
Results and discussion
Fabrication of model NaNO3–MgO structures
The fabrication of the NaNO3–MgO model structure is illustrated in Fig. 1. We used ultra-short pulse (USP) laser ablation to microstructure MgO(100) single crystals with grooves of fixed width and varying depths, which are subsequently filled with NaNO3, as shown in Fig. 1a (see also ESI Fig. S1† for illustration of the USP laser setup and ablation process). Fig. 1 shows grooves of different depths, dgroove, and arrows point to the location of the (buried) NaNO3/MgO interface and the triple phase boundary (CO2/NaNO3/MgO) line in such model sorbent structures. Two sets of MgO model grooves were fabricated and are referred here to as MgO_A and MgO_B. The sample MgO_A, shown in Fig. 1, was designed to contain groves with a large difference between the most shallow (dgroove = 5 μm) and the deepest groove (dgroove = 238 μm) to explore also the effect of the diffusion length of CO2 on the formation of MgCO3. Optical profilometry measurements rely on the reflection of incoming light. However, the steep walls of the groove in MgO_A limited reflection and consequently prevented the measurement of this sample by optical profilometry. Therefore, an additional sample referred to as MgO_B, with groove of depths between dgroove = 6–180 μm was designed. The grooves had a wide and flat bottom to enable (top view) characterization of MgCO3 formation by SEM and optical profilometry measurements (Fig. S2 and S3†). The shape and roughness of the microstructured grooves with and without NaNO3 are visualized by SEM and are given in Fig. 1b–g). A top view of six grooves in MgO_A at different magnifications is shown in Fig. 1b and e, while cross-sections (with and without NaNO3) are imaged in Fig. 1c–g. For MgO_A, all the grooves have the same length (and width of 3000 μm and 100 μm respectively, while the groove depths vary between dgroove = 5–238 μm. In sample MgO_B all grooves have an identical length (3000 μm) and the depths varied between dgroove = 6–180 μm. In addition, in sample MgO_B the width of the grooves was wider at the top (200 μm) and the grooves had a smaller taper angle than the ones in sample MgO_A allowing for a wide flat bottom to enable optical profilometry measurements as described earlier, Table S1.† The average roughness of the most shallow groove was ca. 0.41 μm, and increased to ca. 1.1 μm for the deepest groove (ESI Fig. S3c†). Here, the average roughness is calculated as the standard deviation from the mean value along the length of the groove measured over a 600 μm wide window in the y-direction. A cross-sectional view of the alkali salt coated sample, i.e. NaNO3–MgO_A, shows an intimate contact between the surface of MgO and the promoter NaNO3 confirming an excellent wetting of the surface of MgO by NaNO3 (Fig. 1g).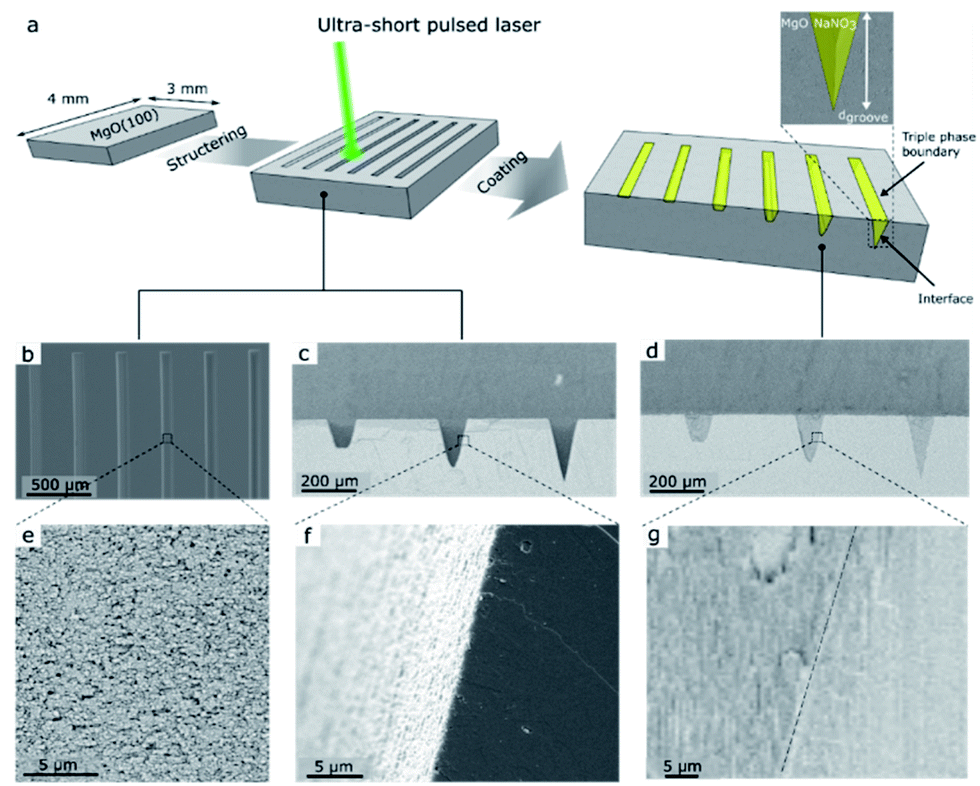 | ||
| Fig. 1 Fabrication and SEM characterization of the NaNO3–MgO_A model structure to determine where MgCO3 nucleates and grows most favorably during CO2 capture. (a) Schematic of the fabrication process of the NaNO3–MgO_A model system using USP laser ablation to microstructure grooves in a MgO(100) single crystal of depth, dgroove and top width wgroove, which are subsequently filled with NaNO3. The black arrows point to the triple phase boundary (TPB) line (CO2/NaNO3/MgO) and the (buried) NaNO3/MgO interface. (b–g) SEM images of grooves without and with NaNO3. (b) Top view of six laser ablated grooves in a MgO(100) single crystal. (c) Mechanically cleaved sample exposing cross-sections of three grooves without a NaNO3 promoter and (d) shows the same three grooves with the NaNO3 promoter. (e–g) high magnification images of the respective structures shown in (b–d). | ||
Carbonation and characterization of the NaNO3–MgO model system
The micro-structured, model NaNO3–MgO samples were exposed to typical CO2 capture conditions of molten-salt promoted MgO.9,10,21 To this end, the as-prepared NaNO3–MgO_A and NaNO3–MgO_B samples were placed in a thermogravimetric analyser (TGA) and heated to 330 °C (heating rate of 10 °C min−1) in a flow of N2 (80 ml min−1), followed by exposure to a flow of CO2 (80 ml min−1) for a given time (0.5 hour to 24 hours, e.g. NaNO3–MgO_A24hours-CO2). For the duration of the experiment a purge flow of N2 (25 ml min−1) was used in the TGA. For subsequent ex situ analyses the sample is cooled down in CO2 to 30 °C (cooling rate of 10 °C min−1) to avoid the decomposition of MgCO3. To confirm the presence of salt (NaNO3) inside the grooves after the sample has been exposed to reaction conditions 3D X-ray computed tomography was applied.3D visualization of NaNO3 in the model system after CO2 capture
To visualize and quantify the volume of NaNO3 in the grooves of the model samples after CO2 capture we turned to X-ray micro computed tomography (μCT), Fig. 2. A 3D render of the NaNO3–MgO_B system after 5 hours of exposure to CO2 at 330 °C, NaNO3–MgO_B5hours-CO2, is shown in Fig. 2a. From the 3D render, it is clear that NaNO3 no longer fills the entire volume of the groove, thus confirming a volumetric loss of NaNO3 during carbonation. Moreover, the surface of the remaining volume of NaNO3 forms a concave meniscus, which is an indication that the molecules of the liquid (molten NaNO3) show stronger adhesion to the wall material (MgO) than to each other. A quantification of the volumetric NaNO3 loss was performed by segmentation of MgO and NaNO3 using their X-ray contrast. The volume fraction of NaNO3 inside the groove is calculated by summing up the number of voxels occupied by NaNO3 and dividing it by the total number of voxels inside the groove. MgCO3 and NaNO3 have a similar X-ray contrast, which hindered differentiation of these two materials. Therefore, the volume of MgCO3 is included in the volume of NaNO3, however it has a negligible contribution as the volume of MgCO3 is significantly smaller than that of NaNO3. Fig. 2b and c plot the volume fraction of NaNO3 in the groove as a function of, respectively, time and depth for the sample NaNO3–MgO_B. Our results plotted in Fig. 2b show that the loss of NaNO3 during carbonation accelerates (volume fraction of NaNO3 is 100% for the as-prepared sample and decreases to 99 vol% after 1 hour, while after 7 hours of carbonation there is 0 vol% NaNO3 in the groove of a depth of 122 μm). The accelerated loss is further evident through a second order polynomial fit to the data, as shown by the dashed line in Fig. 2b. Considering a fixed carbonation time of 5 hours, we observe that in shallower grooves the volume fraction of NaNO3 is lower compared to deeper grooves, pointing to a faster volumetric loss of NaNO3 in shallower grooves (Fig. 2c). The experiments are carried out at 330 °C (below the boiling point of NaNO3 of 380 °C) and the loss of NaNO3 is ascribed to its evaporation (the vapor pressure of NaNO3 at 330 °C is 3 mPa).22 Raman micro-spectroscopy of the NaNO3-filled grooves revealed that NaNO3 did not change its phase during carbonation (where evaporation has occurred).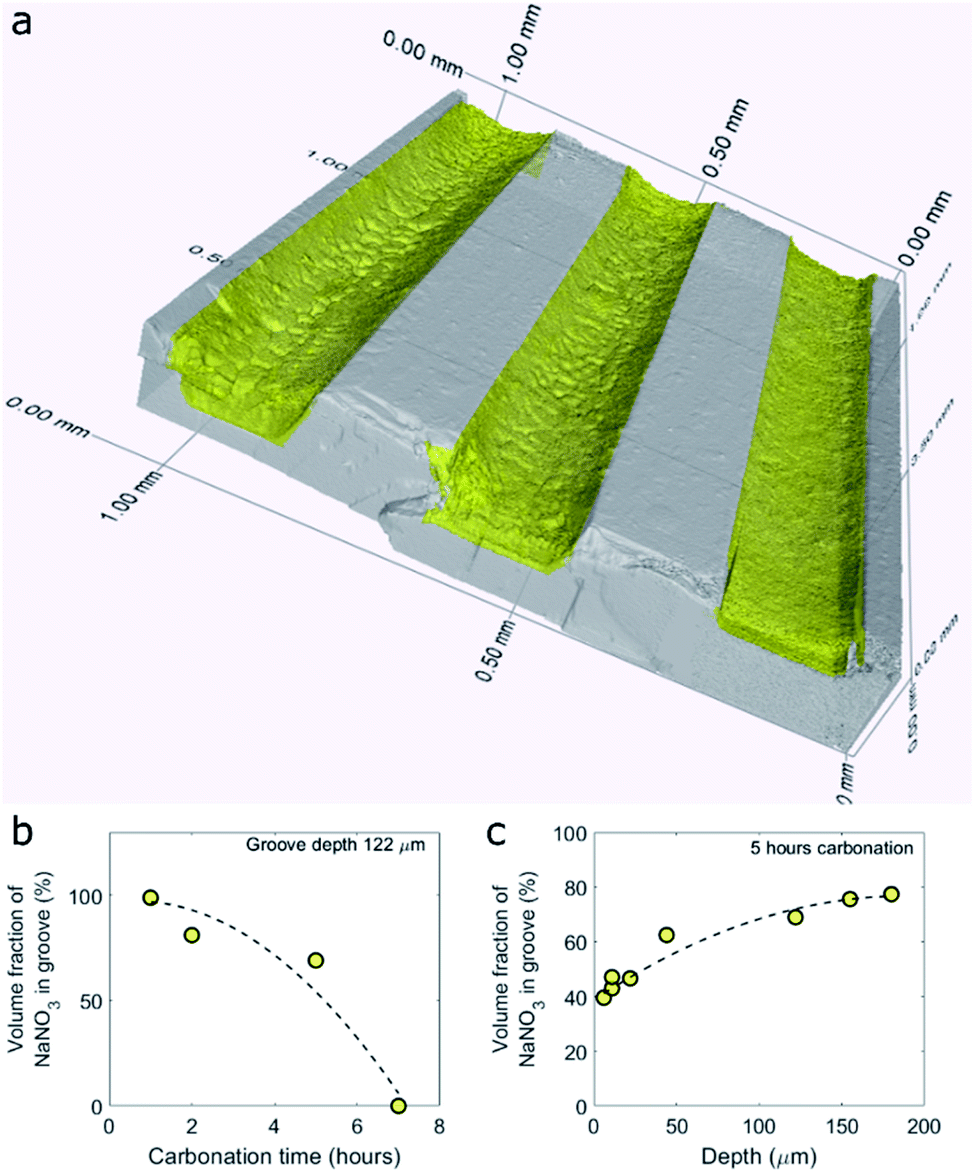 | ||
| Fig. 2 Quantification of the volume fraction of NaNO3 in the grooves of a MgO(100) single crystal after carbonation using 3D X-ray micro computed tomography. (a) 3D render of three grooves in the NaNO3 filled MgO sample (NaNO3–MgO_B, groove depths of dgrooves = 84, 44, 22 μm) showing a volumetric loss of NaNO3 after 5 hours of carbonation at 330 °C in a CO2 rich atmosphere (CO2 flow of 80 ml min−1 with a purge flow of N2 25 ml min−1) i.e. NaNO3–MgO_B5hours-CO2. Volume fraction of NaNO3 in the grooves as a function of (b) carbonation time (groove with a depth of 122 μm) and (c) volume fraction as function of groove depth after 5 hours of carbonation at 330 °C in CO2. Second order polynomial fits are plotted as dashed lines. | ||
The accelerated loss of NaNO3 as function of time and the faster loss of NaNO3 in shallower grooves is very likely due to a higher surface to volume ratio in such configurations leading in turn to a higher rate of evaporation per volume NaNO3.23,24 The loss of the alkali metal salt promoter has also been observed in conventional powder samples. For example, Chen et al. observed that the mass ratio (measured by inductively coupled plasma – optical emission spectrometry ICP-OES) of alkali metal ions (i.e., Li+,K+, Na+) to Mg2+ decreased during cycling and was also attributed to the evaporation of the alkali metal salt.21 The faster loss of NaNO3 in the model systems used here when compared to powder samples21 is very likely due to the high surface to volume ratio of the promoter in our model systems and larger capillary forces in the powder sample.25 After having established a loss of NaNO3 with carbonation time, we focus in the following on elucidating the role of NaNO3 in accelerating the rate of MgCO3 formation. To this end, we characterized in more detail the MgCO3 formed in samples that have been exposed to CO2 (76 vol%) for 5 hours or less, as the samples that were exposed for longer carbonation times had lost most of the NaNO3.
Identifying the location of MgCO3 growth in the NaNO3–MgO model system
The location of MgCO3 formed in the NaNO3–MgO model systems after their exposure to CO2 is identified by Raman micro-spectroscopy, SEM energy dispersive X-ray spectroscopy (EDX), and plasma FIB-SEM EDX. The Raman spectra of two locations and EDX maps of a mechanically cleaved cross-section of one of the grooves in the NaNO3–MgO_A5hours-CO2 sample are provided in Fig. 3. Fig. 3a illustrates the spots in which Raman spectra were collected (spot size of ca. 0.6–1.8 μm) and the area used for EDX analysis. In the illustration, the phases MgO, MgCO3, NaNO3 are highlighted in grey, blue and yellow colours, respectively, based on Raman spectra and EDX analysis. The Raman spectrum collected in a location in which NaNO3 is present (colored yellow in Fig. 3a), shows bands at 101, 186, 724, 1067 and 1385 cm−1 that are characteristic of NO3− in NaNO3, Fig. 3b.15,26–29 The most intense band at 1067 cm−1 is attributed to symmetric stretching vibrations, while the bands at 101 cm−1, 186 cm−1, 724 cm−1 and 1385 cm−1 are due to asymmetric stretching vibrations of NO3−.29 A representative spectrum acquired close to the wall of the groove (blue coloured region in Fig. 3a) exhibit bands associated with NO3− and two additional bands that are due to MgCO3.30,31 These additional Raman bands at 740 cm−1 and 1094 cm−1 are assigned to bending and symmetric stretching vibrations of CO32−, respectively.15,30,31 Bands due to NaNO3 are observed in the spectrum acquired in the (blue) region that has been assigned to MgCO3 because the size of the MgCO3 particle (width ca. 1–2 μm) is of approximately the same size as the laser spot and within the lateral precision of the sample stage. Ex situ Raman micro-spectroscopy of samples that have been exposed to varying carbonation times showed no noticeable phase change of NaNO3 with carbonation time, Fig. S4,† confirming the phase stability of NaNO3 under the conditions investigated here. The region of the cross-section used for EDX analysis is shown in the overview SEM image in Fig. 3c and with a higher magnification in Fig. 3d. The respective EDX elemental maps are given in Fig. 3e–h. Comparing the magnified SEM image (Fig. 3d) with the elemental maps, it is clear that the structure observed at the wall of the groove contains Mg, C, O, while Na is absent, providing further evidence that the particle structure formed at the wall of the groove is indeed composed of MgCO3, in agreement with Raman micro-spectroscopy analysis.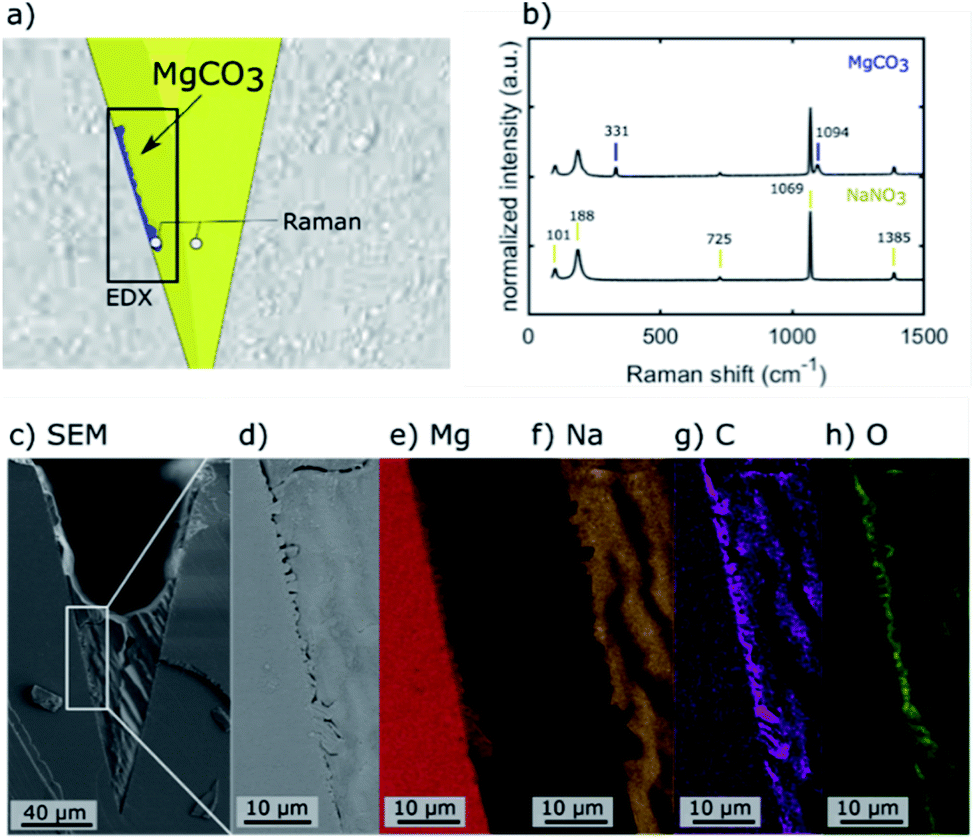 | ||
| Fig. 3 EDX analysis and Raman micro-spectroscopy of a NaNO3 filled groove (NaNO3–MgO_A5hours-CO2) after its exposure to CO2 for 5 hours using a mechanically cleaved cross-section. (a) Illustration showing the location of the region used for EDX analysis and the two spots at which Raman spectra were acquired. (b) Raman spectra acquired at the two spots indicated in (a), i.e. inside the NaNO3 promoter and at the wall of the groove where MgCO3 was formed. (c) SEM image showing the entire cross-section including the NaNO3 remaining at the bottom of the groove. (d–h) magnified SEM image and EDX elemental maps of Mg, Na, C, and O of the region highlighted in (a). | ||
Subsequently we used plasma FIB-SEM to obtain a smooth cross-section of a groove allowing for high quality stacks of SEM images of cross-sections through a groove (ca. 300 μm × 200 μm). This is shown schematically in Fig. 4a and b. A series of SEM micrographs taken after different times of plasma FIB milling along the length of the grooves are shown in Fig. 4c–f. Note that slice #1, shown in Fig. 4c, is at the same location as Fig. 3d (lower magnification) and the blue colour is used to visually ease the identification of the location of MgCO3. In the ESI S5,† we demonstrate how we use EDX analysis (Na, Mg, O) to identify the location of MgCO3 and use this information to colour the SEM images in Fig. 4 with blue to highlight the structures that are composed of MgCO3. Three additional slices were milled by plasma FIB to provide 3D insight into the structure and location of MgCO3. Importantly, these additional FIB cuts demonstrate that (i) MgCO3 grows at the NaNO3/MgO interface, i.e. at the wall of the groove, and (ii) MgCO3 grows both close, but also far away from the triple phase boundary (TPB). These observations are in agreement with earlier reports indicating that MgCO3 grows at the buried NaNO3/MgO interface.15,16 Indeed, our results establish that the TPB is not the primary location for MgCO3 nucleation and growth, as we do not see a higher fraction of MgCO3 particles at the TPB compared to other locations of the NaNO3/MgO interface. Additional analysis by SEM provides more evidence that MgCO3 nucleates and grows both close and far away from the TPB (vide infra).
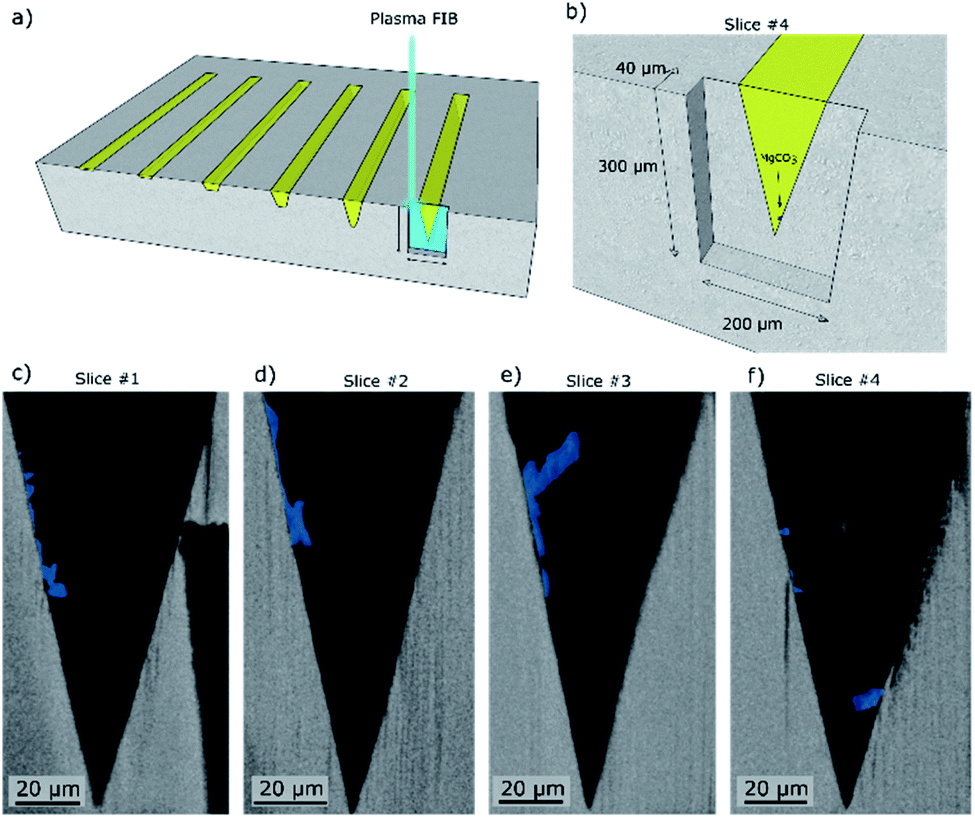 | ||
| Fig. 4 Scanning electron micrographs of cross-sections obtained by plasma focused ion beam (FIB) milling taken at four different distances within the groove length (mechanically cleaved followed by broad ion beam (BIB) cross-section polishing). (a) The turquoise box marks the region of the model structure that was FIB milled. (b) Close up showing the location of slice #4 given in (c). (c–f) Series of cross-sectional SEM images whereby each slice is taken at a specific distance from the mechanical cleaved cross section of the sample NaNO3–MgO_A5hours-CO2. The MgCO3 phase is highlighted in blue for better visibility based on EDX analysis (ESI Fig. S2†). Slices #1–4 were taken at distances of ca. 0 μm, 5 μm, 10 μm and 40 μm from the mechanically cleaved surface, respectively. | ||
Morphology and location of MgCO3 growth in the NaNO3–MgO model system
We gained further insight into the morphology and location of formation of MgCO3 that is formed during carbonation by SEM (Fig. 5 and S6–S8†). Prior to the morphological analysis, the sample was rinsed in water for 1 minute to remove NaNO3 exposing the MgCO3 grown at the bottom of the grooves (NaNO3–MgO_Bxhours-CO2 where the subscript x refer to carbonation time).9,15 Representative examples of MgCO3 particles grown after 0.5, 1, 2, and 5 hours of carbonation (groove with a depth of dgroove = 44 μm) are shown in Fig. 5a–d. Additionally, an overview of the MgCO3 particles grown after 5 hours at the bottom of deep and shallow grooves are shown in Fig. S6–S7.† | ||
| Fig. 5 Growth of MgCO3 crystals with time when exposed to CO2 at 330 °C (sample NaNO3–MgO_Bxhours-CO2 where the subscript x corresponds to the carbonation time ranging between 0.5 hour and 5 hours). Prior to imaging/analysis the samples are rinsed in H2O to remove NaNO3. (a–d) SEM images taken at the bottom of the MgO groove (groove depth 44 μm) after, respectively, 0.5 hour, 1 hour, 2 hours and 5 hours of exposure to CO2. (e) Mean particle size of MgCO3 formed on the bottom of the grooves of depth 155 μm as a function of carbonation time (error bars show the standard deviation of the measured particle diameter based on more than 100 particles per data point). (f) Exemplary particle size distribution in the groove of depth 155 μm after 5 hours of carbonation (normal distribution function was used for fitting). | ||
The mean particle size of MgCO3 as a function of carbonation time (groove depth 44 μm, NaNO3–MgO_Bxhours-CO2, x = 0.5, 1, 2, 5 hours) is plotted in Fig. 5e and the particle size distribution after 5 hours of carbonation at 330 °C in CO2 is given in Fig. 5f (groove depth 44 μm, NaNO3–MgO_B5hours-CO2). The number of particles (MgCO3 particles as those observed in Fig. 5a–d are counted as one particle) is plotted as a function of groove depth (at the flat bottom) and carbonation time in Fig. S8.† Notice, only particles located at the flat bottom of the grooves in MgO_B are counted and since the bottom area is smaller in deeper grooves, the number of particles is normalized by the area of the bottom of the grooves. The CO2 capture performance of AMS-promoted MgO powder samples can be quantified for example by its weight change in a TGA experiment, however, the weight changes (per gram of sample) in the model system are too small to be detected. To quantify the amount of absorbed CO2, instead we determined the fraction of the MgO surface (at the bottom of the groove) that is covered by MgCO3 in Fig. S7† and the volume of MgCO3 formed by profilometry below.
The examples of the MgCO3 particles presented in Fig. 5 and S6–S7† show that MgCO3 grows as individual crystals/particles and in some cases the individual crystals/particles have merged to form larger agglomerates covering a large fraction of the surface of MgO, Fig. S6.† The MgCO3 particle grown after 0.5 hour of carbonation, Fig. 5, has a diameter of approximately 3 μm and consist of three smaller crystallites. Increasing the carbonation time to 5 hours leads to an increasing number of small crystallites, but also the further growth of existing crystallites, Fig. 5b–d. Overall, the shape of the individual particles formed in our model systems is very similar to the 2D MgCO3 islands formed on MgO(100) promoted with NaNO3.15 To obtain a full 3D representation of the formed MgCO3 crystal, we performed additional focused ion beam (FIB) SEM tomography measurement of MgCO3 grown on MgO(100) single crystals promoted with NaNO3 after 5 hours of CO2 exposure (Fig. 6). The shape of the crystals/particles formed on the laser ablated surface, Fig. 5, point to a nucleation and crystal growth type process from a supersaturated solution of [Mg2+…CO32−] in agreement with literature.10,14–16 While MgCO3 forms through a nucleation and growth type process in both the laser ablated MgO sample, Fig. 5, and MgO(100), Fig. 6, there are still some variations in the growth habit between these two systems. For example, each MgCO3 particle grown on the ablated MgO surface is composed of a comparatively large number of small crystallites and there is a more extensive out-of-plane 3D growth (crystal thickness of up to 3 μm, see below in Fig. 7) of the MgCO3 crystals on the ablated MgO surface as compared to the MgCO3 crystals grown on the surface of MgO(100) single crystals (crystal thickness in the range 0.2–2 μm), in agreement with our previous work.15 The mean particle size (in-plane) after 5 hours of exposure to CO2 is 9 ± 1 μm on the ablated surface in comparison to a size of 10–50 μm on the MgO(100) single crystal surface. The morphology of the MgCO3 formed on the ablated MgO surface can be described as “radiating dendrites” in contrast to the “sector plate” morphology on MgO(100). To rationalize the differences in morphology of the MgCO3 formed on these two model systems it is worth looking at the growth habits of ice crystals and ice analog crystals. Their growth habits have been mapped with respect to the level of supersaturation. For example, the crystal growth of Na2SiF6 from an aqueous solution e.g. can be induced by evaporation of the solution, whereby the supersaturation level is controlled by the rate of evaporation.25 Under such conditions, sodium fluorosilicate crystals exhibit a “sector plate” morphology for lower levels of supersaturation while when increasing the degree of supersaturation “radiating dendrite” morphologies are observed. Assuming that there is a similar link between the degree of supersaturation and the growth habit for MgCO3, we can infer that the laser ablated MgO surface provides an environment with a higher supersaturation level (radiating dendrite morphology with a larger number of small crystals) compared to a flat MgO(100) surface (sector plate morphology with a few large crystallites). The higher level of supersaturation of [Mg2+…CO32−] ionic pairs is likely a result of the more defective nature of the laser ablated surface promoting in turn the dissolution of MgO, yielding [Mg2+…O2−] ionic pairs which are the precursors to carbonate ionic pairs, as dissolution preferentially occurs on surface defects.32
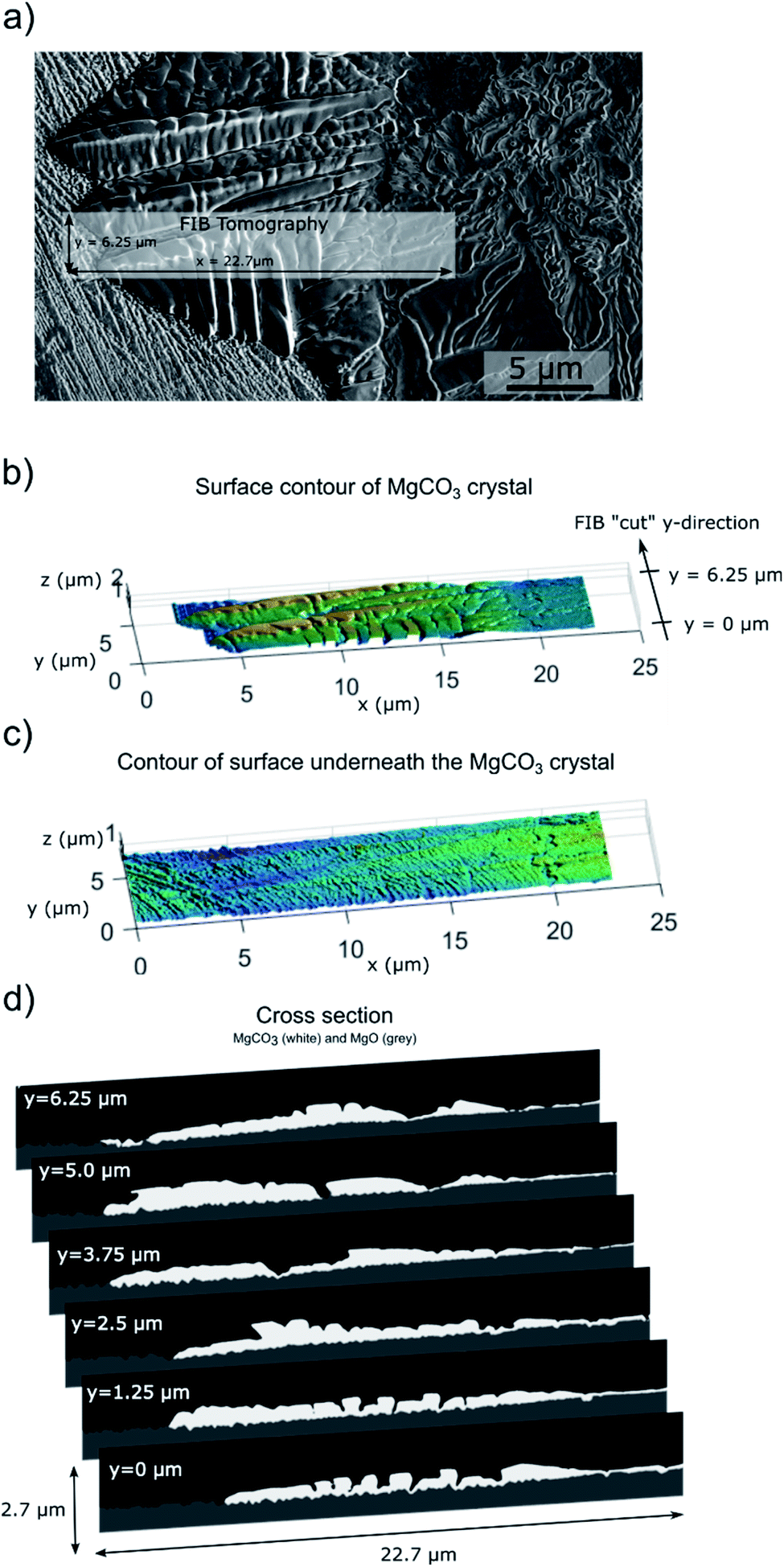 | ||
| Fig. 6 FIB-SEM tomography of a MgCO3 crystal grown on the surface of MgO(100) promoted with NaNO3 after 5 hours of exposure to CO2i.e. NaNO3–MgO(100)5hours-CO2. Prior to the tomographic measurement the sample was rinsed in water. (a) SEM Image of the MgCO3 crystal grown on a MgO(100) single crystal (the grey box shows the area that was used for the tomographic measurement; resolution in x, y, z is, respectively, 10 nm, 25 nm, and =10 nm). (b and c) Surface contours of MgCO3 and MgO are determined by segmenting MgCO3 and MgO through a combination of thresholding by grey-level (using the backscattered electron, BSE, signal) and a directional search for grey-level edges. (d) Segmentation of MgCO3 and MgO in a cross-section (window size [x,z] = 22.7 × 2.7 μm) along the y-direction. Each cross section is taken at a y-spacing of 25 nm and is shown at selected positions, i.e. y = 0, 1.25, 2.5, 3.75, 5, and 6.25 μm. | ||
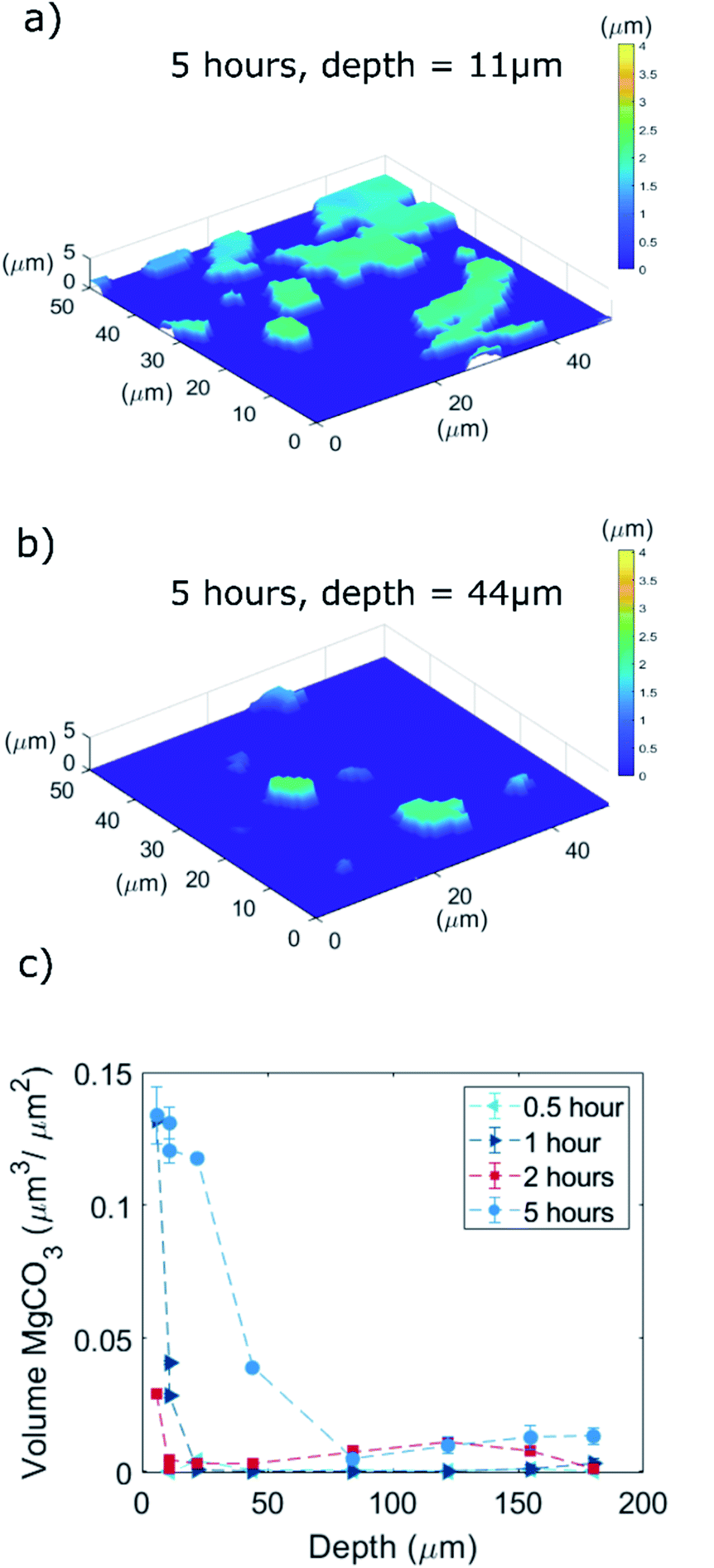 | ||
| Fig. 7 Quantification of the volume of the MgCO3 crystals by 3D profilometry measurements after removal of NaNO3 by rinsing with H2O. The volume is calculated by the sum of “voxels” of MgCO3, i.e. the voxels that are at a height z > 0 μm with respect to the flat MgO surface at the bottom of the groove (z = 0 μm, roughness subtracted) multiplied by the dimension of the pixel in the xy direction (0.69 × 0.69 μm2). As the area at the bottom of a groove is smaller in deeper grooves, the MgCO3 volume is normalized by the area of the bottom part of the groove. Note that the optical profile measurements rely on the reflection of light at the surface. Hence, only MgCO3 grown at the bottom of the groove can be quantified, as the steep angle of the groove wall with respect to the incoming light minimizes reflection, which hinders quantification of the volume of MgCO3 grown on the walls of the groove. (a and b) MgCO3 formation after 5 hours of exposure to CO2 in grooves with a depth of 11 μm and 44 μm, respectively. (c) Total volume of MgCO3 crystals formed at the bottom of the grooves normalized by the area of the bottom of the groove as a function of the depth of the groove (dgroove = 6, 11, 11, 22, 44, 84, 122, 155, 180 μm) and carbonation time (0.5, 1, 2, 5 hours). | ||
As described above, the locations of MgCO3 formation are found both close and far away from the TPB. Indeed, we observe a homogenous distribution of MgCO3 particles at the NaNO3/MgO interface, Fig. S7,† with no preferential growth at the TPB. This observation is in agreement with the location of MgCO3 particles seen in cross-sections of the grooves (Fig. 4). In addition, Fig. S6† shows that the number of MgCO3 particles per surface area of MgO is independent of depth and time (note we count the structures visualized in Fig. 5a–d that comprise several smaller crystallites as one particle). For example, on average ca. 2000 MgCO3 particles are found per mm2 MgO in grooves with a depth of 11 μm. The same number of MgCO3 particles per surface area MgO were also counted in grooves with a depth of 180 μm (Fig. S6a†). Assuming that each particle arises from a single nucleation event, these numbers confirm that MgCO3 nucleation occurs with the same frequency under a thin and thick coating of NaNO on MgO3, i.e., relatively close and rather far away from the TPB. Since the morphology of the MgCO3 crystals indicate crystal growth from a supersaturated solution and we observe a density of nucleation sites that is independent of time and depth we can conclude that the critical level of supersaturation that is required for the formation crystalline magnesium carbonate is reached in both the shallow and deep grooves (at least locally and within the timeframe studied here). To summarize, our results show that the location of MgCO3 nucleation is at the NaNO3/MgO interface and the density of nucleation sites is independent of the groove depth (i.e. the height/thickness of the promoter covering MgO). In other words, for the groove depths studied here the dissolution and diffusion of CO2 to the reaction interface is not rate limiting for the nucleation step.
Lastly, we explored the volumetric growth of the MgCO3 crystals in more detail and as a function of time and the thickness of the NaNO3 coating to assess whether diffusion of CO2 through the promoter is rate limiting during the growth step.
3D visualization and quantification of the volume of MgCO3 formed
To visualize and quantify the volume of the magnesium carbonate formed through CO2 absorption at the bottom of the grooves in the NaNO3–MgO_B model system, we used 3D optical profilometry (Fig. 7). The MgCO3 formed appeared at heights with z > 0 μm with respect to the originally flat MgO surface (z = 0 μm). Fig. 6a and b visualize a series of MgCO3 particles at the bottom of two different grooves with depths of, respectively, 11 and 44 μm (the samples were exposed to CO2 for 5 hours and rinsed with water prior to the measurements, NaNO3–MgOB-5hours-CO2). We observe that in the first 2 hours of carbonation the MgCO3 structures grow with equal rates in all three dimensions i.e. after 2 hours of carbonation the particles have a width and thickness of 2 μm. Subsequently, MgCO3 grows largely in a 2 dimensional fashion, i.e. parallel to the MgO plane (i.e. after 5 hours of carbonation MgCO3 has a thickness of ca. 2 μm and lateral dimension of 10 μm) leading to the formation of “2D islands” that in some cases merge to form larger agglomerates. This observation is in agreement with a previous study that reported the formation of 2D islands of MgCO3 (thickness in the range of 0.2–2 μm and lateral dimension in the range 10–50 μm) after 5 hours of carbonation of a NaNO3-promoted single crystals of MgO(100).15 From our profilometry results acquired at the bottom of various grooves, it is clear that shallower grooves (e.g. depth of 11 μm) contain MgCO3 particles that have a larger volume (when normalized by the available surface area of MgO at the bottom of the groove) than those formed in deeper grooves (e.g. of depth 44 μm). This is in agreement with our SEM results (Fig. S9†) revealing that the bottom of shallow grooves are almost fully covered by MgCO3 whereas the bottom of deeper grooves shows a significantly lower coverage of MgCO3. Thus, a thinner coating of NaNO3 facilitates the further growth of MgCO3. This is in contrast to the observations made for the nucleation step which was found to be independent of the coating thickness.The volume (per unit area of the bottom section of the groove) of the MgCO3 particles formed at the bottom of grooves as function of the groove depth after exposure to CO2 for 0.5, 1, 2, and 5 hours is shown in Fig. 7c. Considering the sample that has been exposed to CO2 for 5 hours, the MgCO3 formed at the bottom of the shallow groove (dgroove = 6 μm) has a total volume of 0.13 μm3 μm−2 compared to a total MgCO3 volume of 0.013 μm3 μm−2, i.e. 10 times lower, in the deepest groove (dgroove = 180 μm). The same trend is apparent for the samples that have been exposed to CO2 for either 1 or 2 hours, while no apparent trend between MgCO3 volume and groove depths is observed for a CO2 exposure of 0.5 hour, presumably because at these short carbonation times crystal growth is still limited (instead nucleation dominates) and CO2 diffusion has limited effects on the nucleation step. To summarize, in Fig. 7c we observe a clear trend of an accelerated growth of MgCO3 in shallow grooves compared to deep grooves, which is attributed to a shorter diffusion path length of CO2 to the reaction interface in such systems, yielding in turn a higher density of ionic pairs of magnesium carbonate. It is conceivable that the faster evaporation rate of NaNO3 as observed in shallower grooves also contributes to a faster precipitation of MgCO3.
Experimental
Laser ablation of grooves in MgO
MgO single crystals (MgO(100), Alineason, ≥99.95% 10 mm × 10 mm × 0.5 mm) were used as purchased. The fabrication of grooves of varying depths in the MgO single crystals was carried out by ultra-short pulse (USP) laser ablation generating a defined geometry of high surface smoothness. A frequency-doubled Amphos 200 laser source of 515 nm wavelength, 800 fs pulse duration, and a power of up to 120 W was used. The laser beam was set to circular polarization using a quarter-waveplate and coupled into a galvo scanner, where the MgO crystal was placed on a high-precision xyz-stage. For all samples we used a 2.5D strategy i.e. removal of material layer-by-layer, see ESI Fig. S1† for illustration of the ablation process. For one ablated layer, the laser ablates the surface in parallel lines with a line distance of lb = 5 μm. In the next layer, the sample was rotated by γlayer = 90° thus ablates perpendicular to the previous lines and achieving a cross hatching pattern. To reach the desired groove depth the number of ablated layers was adjusted. For MgO_A, the laser pulse repetition rate was set to frep = 400 kHz, the laser scan speed was vs. = 400 mm s−1 and an average power of 1 W used at a focal diameter of dfocus = 9 μm. To avoid re-deposition of ablated MgO, a flow of pressurized air mitigated the accumulation of debris during ablation. A subsequent laser cleaning step removed re-deposited MgO on the walls by ablation of the grooves once more with 0.3 W average power. Finally, the samples were cleaned in ethanol by ultra-sonication for 10 minutes. For the MgO_B samples we used a comparable system using a Trumpf TruMicro 5270 laser (frep = 800 fs pulse duration). The ablation process was slightly modified by using a larger focal diameter of dfocus = 16 μm and using a laser pulse repetition rate of frep = 600 kHz and a laser scan speed of vs. = 500 mm s−1 (2 W of average power). Further details on the laser ablation process and its optimization for single crystal MgO(100), we refer to ref. 33.Coating laser-ablated MgO(100) with NaNO3 and carbonation experiments
To complete the preparation of the NaNO3–MgO model system, the laser-ablated single crystal MgO samples containing grooves of different depths were filled with the promoter NaNO3. To fill the grooves, approximately 1 g of crystalline sodium nitrate (NaNO3, Sigma-Aldrich, ≥99.5%) powder was placed as-received in an Al2O3 crucible with a diameter of 10 mm. The MgO crystal was placed with the grooves facing down towards the loosely packed NaNO3 powder. The crucible with the MgO crystal was placed for 2 minutes on a hot plate (surface temperature of 375 °C) and subsequently cooled down to 25 °C at ambient conditions. This procedure yielded a sample with crystalline NaNO3 filling the grooves of the MgO single crystal with some excess salt. To remove the excess promoter NaNO3, the sample was polished with sandpaper (2601 Sianor J, Grit 410, Sia Abrasives). For carbonation, the sample is placed into a thermogravimetric analyzer (TGA) and exposed to a flow of CO2 (80 ml min−1 was used with a purge flow of N2 25 ml min−1) at 330 °C for a duration between 30 minutes to 24 hours with heating and cooling rates of 10 °C min−1.3D X-ray micro computed tomography
For X-ray micro computed tomography (CT) measurements, the NaNO3–MgO groove samples (5 mm × 5 mm × 0.5 mm) were cleaved mechanically into smaller pieces (1–2 mm × 10 mm × 0.5 mm) to reach a high resolution. CT measurements were carried out using an Easy Tom XL from RX Solutions with the X-ray tube operating at 80 kV and 30 μA current. The CT scans were performed using a Varian PaxScan 2520DX detector. The voxel size of the scans was ca. 1.5 μm.Raman micro spectroscopy
Raman spectra were acquired with a Thermo Scientific Instrument equipped with a 455 nm laser and a spot size of 1.8 μm and 0.6 μm for a 10- and 100-fold magnification, respectively. Spectra were collected in the range of 100–3500 cm−1 with a spectral resolution of 0.96 cm−1 and a laser power of 3 mW. Each spectrum consisted of five repetitions with a total integration time of 25 seconds.Plasma FIB-SEM and SEM
Large cross-sections of the MgO single crystal containing grooves (ca. 300 μm × 200 μm) for scanning electron microscopy (SEM) and energy dispersive X-ray spectroscopy (EDX) were prepared through plasma focused ion beam (FIB) milling and acquired by a Tescan Fera3. The microstructure and particle size of the MgCO3 product formed on the surface of MgO (after removal of NaNO3) was characterized by SEM using a FEI Quanta 200F.FIB-SEM tomography
3D FIB-SEM tomography of a MgCO3 crystal grown on NaNO3-promoted MgO after 5 hours of exposure to CO2 is performed on a FIB-SEM Helios 5 UX. The imaging resolution was 10 × 25 × 10 nm3. SEM images of the backscattered electrons (BSE) signal using the through the lens detector (TLD) were acquired. Prior to the tomographic measurement, a “T”-shaped fiducial marker was cut to enable alignment of the stack of images. For data analysis, registration was performed using the Stackreg plugin (https://bigwww.epfl.ch/thevenaz/stackreg/) for ImageJ. “Salt and pepper” noise in the SEM 3D image is removed using Matlab's medfilt3, which performs median filtering in three dimensions. We used a setting of medfilt(A,9,9,9), where each voxel in the 3D image “A” contains the median value of the 9 by-9-by-9 neighbourhood around the corresponding voxel in A. Subsequently, segmentation is performed using the BSE signal based on a combination of thresholding using the grey-level and directional search for grey-level edges.3D surface profiling
The topography of the surface of the bottom of the MgO grooves, after the removal of NaNO3, was measured using a Sensofar S Neox 3D vertical scanning interferometer. All measurements were acquired using a EPI 20X objective with a pixel size 0.128 μm × 0.128 μm and a lateral resolution of 0.01 μm. The volume of the MgCO3 particles formed at the flat bottom of the grooves (sample MgO_B) was determined by summing up the “voxels” of MgCO3. The voxels correspond to the height, z, with respect to the bottom of the MgO groove (z = 0), multiplied by the pixel size (0.69 μm × 0.69 μm). The volume of MgCO3 formed was determined on the entire bottom area of each groove and since the area of the bottom of a groove depends on the depth of the groove, the volume of MgCO3 is normalized by the surveyed area.Conclusion
In this work, we fabricated a model system of a MgO-based CO2 sorbent, i.e. a MgO(100) single crystal containing grooves of defined geometry that are filled with a molten salt promoter. These well-defined model structures allowed us to identify the location of MgCO3 nucleation and growth, i.e. the (buried) NaNO3/MgO interface or the triple phase boundary (TPB), extract indications of the prevailing level of supersaturation and assess the possibility of diffusive limitations to the nucleation and growth process. SEM image analysis demonstrates clearly that MgCO3 crystals grow at the NaNO3/MgO interface and not inside the melt. Importantly, there was no preference of MgCO3 nucleation at the TPB compared to the buried NaNO3/MgO interface. There is no observable CO2 diffusion limitation on the nucleation step indicating that all grooves (deep and shallow) reach the critical supersaturation for nucleation within the first 30 minutes of carbonation.Comparing the growth of crystalline MgCO3 in shallow (6 μm) and deep (180 μm) grooves shows that the growth rate of MgCO3 is approximately an order of magnitude higher in shallow grooves compared to that in deep grooves (after 5 hours of exposure to CO2). We attribute this observation to the shorter diffusion path of CO2 from the gas phase to the reaction interface indicating that the dissolution and transport of CO2 is the rate-limiting step for the growth of MgCO3. However, it is conceivable that the increased rate of NaNO3 evaporation in shallower grooves also contributes to a faster precipitation of MgCO3. In summary, our findings indicate that an effective MgO-based sorbent should contain a homogenous, conformal coating of an alkali metal salt promoter to increase the area of the promoter-MgO interface. To yield rapid MgCO3 growth kinetics (i.e. a high rate of CO2 uptake), the salt coating should be thin enough to avoid diffusive limitations and MgO should be highly porous (micro- to mesoporous) with subsequent coating (infiltration) as to maximize the interfacial contact between the salt and the oxide surface.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program grant agreement No. 819573 and from the Swiss National Science Foundation (SNSF, 200020_156015). The authors thank the Scientific Centre for Optical and Electron Microscopy (ScopeM) of ETH Zürich for providing access to electron microscopy. We further thank Prof. Konrad Wegener for access to a 3D surface profiling microscope, and Lukas Seeholzer for equipment support. We acknowledge the Philippe Thévenaz, Biomedical Imaging Group, Swiss Federal Institute of Technology Lausanne for the ImageJ plugin for the recursive alignment of images for FIB-SEM tomography.References
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- M. Haaf, R. Anantharaman, S. Roussanaly, J. Ströhle and B. Epple, Resour., Conserv. Recycl., 2020, 162, 104973 CrossRef.
- D. P. Hanak, S. Michalski and V. Manovic, Energy Convers. Manage., 2018, 177, 428–452 CrossRef CAS.
- M. Bhagiyalakshmi, J. Y. Lee and H. T. Jang, Int. J. Greenhouse Gas Control, 2010, 4, 51–56 CrossRef CAS.
- K. K. Han, Y. Zhou, W. G. Lin and J. H. Zhu, Microporous Mesoporous Mater., 2013, 169, 112–119 CrossRef CAS.
- W. Gao, T. Zhou and Q. Wang, Chem. Eng. J., 2018, 336, 710–720 CrossRef CAS.
- W. J. Liu, H. Jiang, K. Tian, Y. W. Ding and H. Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- X. Zhao, G. Ji, W. Liu, X. He, E. J. Anthony and M. Zhao, Chem. Eng. J., 2018, 332, 216–226 CrossRef CAS.
- K. Zhang, X. S. Li, W. Z. Li, A. Rohatgi, Y. Duan, P. Singh, L. Li and D. L. King, Adv. Mater. Interfaces, 2014, 1, 1–6 CrossRef.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- A. T. Vu, Y. Park, P. R. Jeon and C. H. Lee, Chem. Eng. J., 2014, 258, 254–264 CrossRef CAS.
- K. Zhang, X. S. Li, Y. Duan, D. L. King, P. Singh and L. Li, Int. J. Greenhouse Gas Control, 2013, 12, 351–358 CrossRef CAS.
- W. Gao, J. Xiao, Q. Wang, S. Li, M. A. Vasiliades, L. Huang, Y. Gao, Q. Jiang, Y. Niu, B. Zhang, Y. Liu, H. He and A. M. Efstathiou, Adv. Mater., 2022, 34, 2106677 CrossRef CAS PubMed.
- H. Jeon, M. L. T. Triviño, S. Hwang, J. H. Moon, J. Yoo and J. G. Seo, J. CO2 Util., 2020, 39, 101153 CrossRef CAS.
- A. H. Bork, M. Rekhtina, E. Willinger, P. Castro-Fernandez, J. Drnec, P. M. Abdala and C. R. Müller, Proc. Natl. Acad. Sci., 2021, 118, 26 CrossRef PubMed.
- S. I. Jo, Y. I. An, K. Y. Kim, S. Y. Choi, J. S. Kwak, K. R. Oh and Y. U. Kwon, Phys. Chem. Chem. Phys., 2017, 19, 6224–6232 RSC.
- G. J. Janz and N. P. Bansal, J. Phys. Chem. Ref. Data, 1982, 11, 505–693 CrossRef CAS.
- H. Lee, M. L. T. Trivinìo, S. Hwang, S. H. Kwon, S. G. Lee, J. H. Moon, J. Yoo and J. G. Seo, ACS Appl. Mater. Interfaces, 2018, 10, 2414–2422 CrossRef CAS PubMed.
- F. L. P. Veenstra, N. Ackerl, A. J. Martín and J. Pérez-Ramírez, Chem, 2020, 6, 1707–1722 CAS.
- N. Ackerl, A. H. Bork, R. Hauert, E. Müller and M. Rottmar, Appl. Surf. Sci., 2021, 545, 149020 CrossRef CAS.
- J. Chen, L. Duan, F. Donat and C. R. Müller, ACS Sustainable Chem. Eng., 2021, 9, 6659–6672 CrossRef CAS.
- V. I. Glazov, G. P. Dukhanin and M. K. Dkhaibe, Russ. J. Appl. Chem., 2003, 76, 1405–1407 CrossRef CAS.
- I. Langmuir, Phys. Rev., 1918, 12, 368 CrossRef CAS.
- F. Dalla Barba, J. Wang and F. Picano, Phys. Fluids, 2021, 33, 1–5 Search PubMed.
- M. J. Krasiński and J. Prywer, J. Cryst. Growth, 2007, 303, 105–109 CrossRef.
- M. D. Fontana, K. Ben Mabrouk and T. H. Kauffmann, 4th IMEKO TC19 Symp. Environ. Instrum. Meas. 2013 Prot. Environ. Clim. Chang. Pollut. Control, 2013, pp. 27–29 Search PubMed.
- H. J. Tong, J. P. Reid, J. L. Dong and Y. H. Zhang, J. Phys. Chem. A, 2010, 114, 12237–12243 CrossRef CAS PubMed.
- J. Y. Yu, Y. Zhang, G. Zeng, C. M. Zheng, Y. Liu and Y. H. Zhang, J. Phys. Chem. B, 2012, 116, 1642–1650 CrossRef CAS PubMed.
- D. L. Rousseau, R. E. Miller and G. E. Leroi, J. Chem. Phys., 1968, 48, 3409–3413 CrossRef CAS.
- P. Gillet, Am. Mineral., 1993, 78, 1328–1331 CAS.
- Q. Williams, B. Collerson and E. Knittle, Am. Mineral., 1992, 77, 1158–1165 CAS.
- J. A. Mejias, A. J. Berry, K. Refson and D. G. Fraser, Chem. Phys. Lett., 1999, 314, 558–563 CrossRef CAS.
- N. Ackerl, PhD Thesis, ETH Zurich, Zurich, Switzerland, 2020.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02897b |
| This journal is © The Royal Society of Chemistry 2022 |