
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Li nucleation on the graphite anode under potential control in Li-ion batteries†
Arihant
Bhandari
 ab,
Chao
Peng
bc,
Jacek
Dziedzic
abd,
John R.
Owen
ab,
Denis
Kramer
bce and
Chris-Kriton
Skylaris
*ab
ab,
Chao
Peng
bc,
Jacek
Dziedzic
abd,
John R.
Owen
ab,
Denis
Kramer
bce and
Chris-Kriton
Skylaris
*ab
aSchool of Chemistry, University of Southampton, Highfield, Southampton SO17 1BJ, UK
bThe Faraday Institution, Quad One, Becquerel Avenue, Harwell Campus, Didcot, OX11 0RA, UK
cEngineering Sciences, University of Southampton, Southampton SO17 1BJ, UK
dFaculty of Applied Physics and Mathematics, Gdansk University of Technology, Gdansk 80-233, Poland
eHelmut-Schmidt-University, University of the Armed Forces, 22043 Hamburg, Germany. E-mail: C.Skylaris@soton.ac.uk
First published on 10th May 2022
Abstract
Application of Li-ion batteries in electric vehicles requires improved safety, increased lifetime and high charging rates. One of the most commonly used intercalation anode material for Li-ion batteries, graphite, is vulnerable to Li nucleation, a side reaction which competes with the intercalation process and leads to loss of reversible capacity of the battery, ageing and short-circuits. In this study, we deploy a combined grand canonical large-scale electronic density-functional theory (DFT) and Poisson–Boltzmann electrolyte theory to study the nucleation and growth of Li clusters on the graphite anode in the presence of its surrounding electrolyte environment at different applied voltages with respect to the Li metal reference electrode. We find the voltage below which the nucleation energy becomes negative (corresponding to Li nucleation becoming energetically favourable), the ‘potential of zero nucleation energy’ (UPZN). We observe a distinct minimum in the plots of UPZN as a function of the size of nucleated clusters. When the applied voltage on the graphite electrode is below the minimum value of UPZN, the nucleated clusters start growing unbounded on graphite electrode. This potential for cluster growth (UPCG) is found to be −0.12 V on the periodic basal plane of unlithiated graphite and −0.08 V on lithiated graphite. The corresponding potential for the zigzag edge termination is −0.06 V on unlithiated graphite and −0.04 V on lithiated graphite. Thus, the nucleation and cluster growth is favored on the zigzag edge termination of the graphite electrode as compared to the periodic basal plane and on the lithiated graphite as compared to the unlithiated graphite. We find that the surrounding environment plays a significant role and that nucleation is more likely to occur in electrolyte environment than that predicted from calculations in vacuum. We observe that the potentials obtained with grand canonical ensemble DFT method in electrolyte are close to experimentally available data. The study has profound implications for the nucleation, growth and control of metal dendrites in a battery cell.
1 Introduction
Li-ion batteries have revolutionized the portable electronics industry and are being intensively pursued for application in the electrification of the transport sector.1–3 Key requirements of Li-ion batteries for successful implementation in electric vehicles are fast charging, long lifetime and high safety. Graphite-based carbonaceous materials are one of the most widely used intercalation anode materials for Li-ion batteries since their inception.4 Thermodynamic and kinetic aspects of intercalation have been computationally studied for periodic graphite,5 edge-terminated graphite,6 and disordered carbon.7 The surface of graphite is susceptible to Li nucleation, a competing side-reaction which leads to a loss of reversible capacity. Moreover, Li nucleation followed by growth of dendrites can lead to a short-circuiting of the battery and is, hence, a critical safety challenge with Li-ion batteries.8 Li nucleation and dendrite growth also occurs on Li metal anodes.9,10In order to improve the performance of Li-ion batteries and gain more insight into this phenomenon, the process of Li nucleation has been extensively studied both experimentally and computationally in the recent years.11–13 Li nucleation is thermodynamically possible when the graphite electrode is at a lower potential (voltage) with respect to the Li/Li+ reference electrode.14 The voltage required for Li intercalation during a full charge of the graphite electrode corresponding to the phase transition from stage 2 to stage 1 graphite is around 0.065–0.085 V.15 When the overpotential for the Li intercalation is large enough, the voltage of the graphite electrode will drop below 0.0 V with respect to Li/Li+, and Li nucleation becomes thermodynamically possible.16 Large overpotential for Li intercalation can easily occur at high current density during fast charging or at low temperature due to the Arrhenius dependence of the charge transfer resistance.12 Detailed in situ experiments were performed by Gao et al. demonstrating the competition between Li intercalation and Li nucleation on graphite.13 Their observations were mostly limited to the kinetics of the two processes, with the observation of the intercalation of Li into the graphite until the graphite is fully lithiated, followed by nucleation of Li clusters at the graphite edges leading to dendrite formation and growth.
With the development of advanced atomistic methods, it is possible to gain more insight into the thermodynamics of the Li nucleation process.17 Fan et al. studied the adsorption of small Li clusters on graphene using canonical density functional theory calculations in vacuum (cDFTv) and found higher binding energy of Li clusters on graphite surface than on the (001) surface of Li metal, suggesting a possible cause for the initiation of Li nucleation.18 Liu et al. further studied the formation of Li clusters on graphene using cDFTv and found that the nucleation barrier is largely dependent on Li concentration ranging from over 3.2 eV at dilute concentrations to below 0.3 eV at high Li concentrations.19 Cui et al. also employed cDFTv to study the nucleation of small Li clusters with 2–6 Li atoms on graphene and found that the nucleation energy is positive and increases with the size of nucleated clusters, predominantly due to the lattice incompatibilities between the hexagonally-arranged carbon and the bcc structure of metallic Li.20 They further found a respective decrease in the nucleation energy on amorphous C, N-doped C, carboxylated C, hydroxylated C and epoxied C, showing favorable nucleation. Chen et al. also studied Li binding on heteroatom-doped carbon using cDFTv and found that O-doped and O/B-co-doped carbons exhibit high lithiophilicity.21 We note here that all these aforementioned studies of Li nucleation using cDFTv are on the basal plane of single layer graphene, without considering the effect of a finite edge termination of the basal plane. Several studies on Li intercalation have shown a large effect of the presence of a finite edge termination of the graphite basal plane.6,22 Therefore, it is also important to study Li nucleation at finitely terminated graphite edges.
In our previous work we used cDFTv to study the nucleation of larger clusters with 1–65 Li atoms on multi-layered graphite for the first time, while also considering the effect of the presence of a finite edge termination of graphite basal plane.23 We found that the nucleation energy barrier near the zigzag edge termination of the graphite is reduced by more than 1 eV relative to that on the periodic basal plane. We also found that chemical doping with nitrogen can increase the nucleation energy barrier and suppress Li nucleation. Most importantly, we observed a significant effect of the voltage on the nucleation process, with a decrease in the nucleation energy and energy barrier on lowering the potential. While these conclusions have significant impact on the understanding of Li nucleation, there are inherent limitations in state-of-the-art cDFTv methods.
cDFTv calculations are performed in ultra high vacuum conditions under a charge control regime, where the charge on the entire structural model is fixed to zero. This regime is schematically shown in Fig. 1a. Here the calculations are performed for an isolated neutral Li slab and an isolated neutral Lin|G system, where an n-atom Li cluster has been nucleated on graphite (G). The two systems are completely independent and do not interact electrically via a ‘wire’ that would allow transfer of electrons under the constraint on their electrochemical potentials. To overcome these limitations and to simulate electrochemical systems under experimental conditions, we have developed a model for grand canonical DFT calculations.24 Here, electrons can flow via an electrical connection between the working electrode and the reference electrode under potential control. The electrochemical potential of electrons at the working electrode (![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e0.gif) e−) can be set at a certain voltage (U) with respect to that at a reference electrode: e−(U) = refe−(U = 0) − e · U. Our model parameters have been calibrated according to the reduction potential of the Li metal electrode.24 As the number of electrons varies in response to the electrochemical potential, the electrode can have a net charge. The grand canonical calculations can be performed in vacuum (gcDFTv), as shown in Fig. 1b or in an implicit solvent-electrolyte environment (gcDFTe), shown in Fig. 1(c). In vacuum, the net charge on the electrode is neutralized by a uniform opposite background charge (‘jellium’). In a solvent environment, the charged electrode system is neutralized by a Poisson–Boltzmann electrolyte. The presence of a net charge on the electrode system leads to a shift in the electrolyte concentrations near the electrode surface, such that there is more of the oppositely charged ‘counter-ions’ than the ‘co-ions’ leading to the formation of double layers. The concentrations of the electrolyte ions reach their asymptotic bulk values far away from the surface. This experimentally observed phenomenon is implemented within the neutralization by electrolyte concentration shift (NECS) method.25 This method overcomes the limitations of the prevalent ‘jellium’ based approaches which introduce artificial opposite uniform background charge to neutralize the electrode charge.25 Our method works via the solution of a modified Poisson–Boltzmann equation using a highly-parallel, multigrid library DL_MG.26,27 The entire electrolyte model has been implemented in the order-N electronic total energy program (ONETEP), which allows large-scale DFT computations with the computational cost scaling linearly with the number of atoms (N).28 The model has been previously tested to accurately predict activity coefficients of LiPF6 in ethylene carbonate (EC),29 and the differential capacitance of graphite electrodes.24
e−) can be set at a certain voltage (U) with respect to that at a reference electrode: e−(U) = refe−(U = 0) − e · U. Our model parameters have been calibrated according to the reduction potential of the Li metal electrode.24 As the number of electrons varies in response to the electrochemical potential, the electrode can have a net charge. The grand canonical calculations can be performed in vacuum (gcDFTv), as shown in Fig. 1b or in an implicit solvent-electrolyte environment (gcDFTe), shown in Fig. 1(c). In vacuum, the net charge on the electrode is neutralized by a uniform opposite background charge (‘jellium’). In a solvent environment, the charged electrode system is neutralized by a Poisson–Boltzmann electrolyte. The presence of a net charge on the electrode system leads to a shift in the electrolyte concentrations near the electrode surface, such that there is more of the oppositely charged ‘counter-ions’ than the ‘co-ions’ leading to the formation of double layers. The concentrations of the electrolyte ions reach their asymptotic bulk values far away from the surface. This experimentally observed phenomenon is implemented within the neutralization by electrolyte concentration shift (NECS) method.25 This method overcomes the limitations of the prevalent ‘jellium’ based approaches which introduce artificial opposite uniform background charge to neutralize the electrode charge.25 Our method works via the solution of a modified Poisson–Boltzmann equation using a highly-parallel, multigrid library DL_MG.26,27 The entire electrolyte model has been implemented in the order-N electronic total energy program (ONETEP), which allows large-scale DFT computations with the computational cost scaling linearly with the number of atoms (N).28 The model has been previously tested to accurately predict activity coefficients of LiPF6 in ethylene carbonate (EC),29 and the differential capacitance of graphite electrodes.24
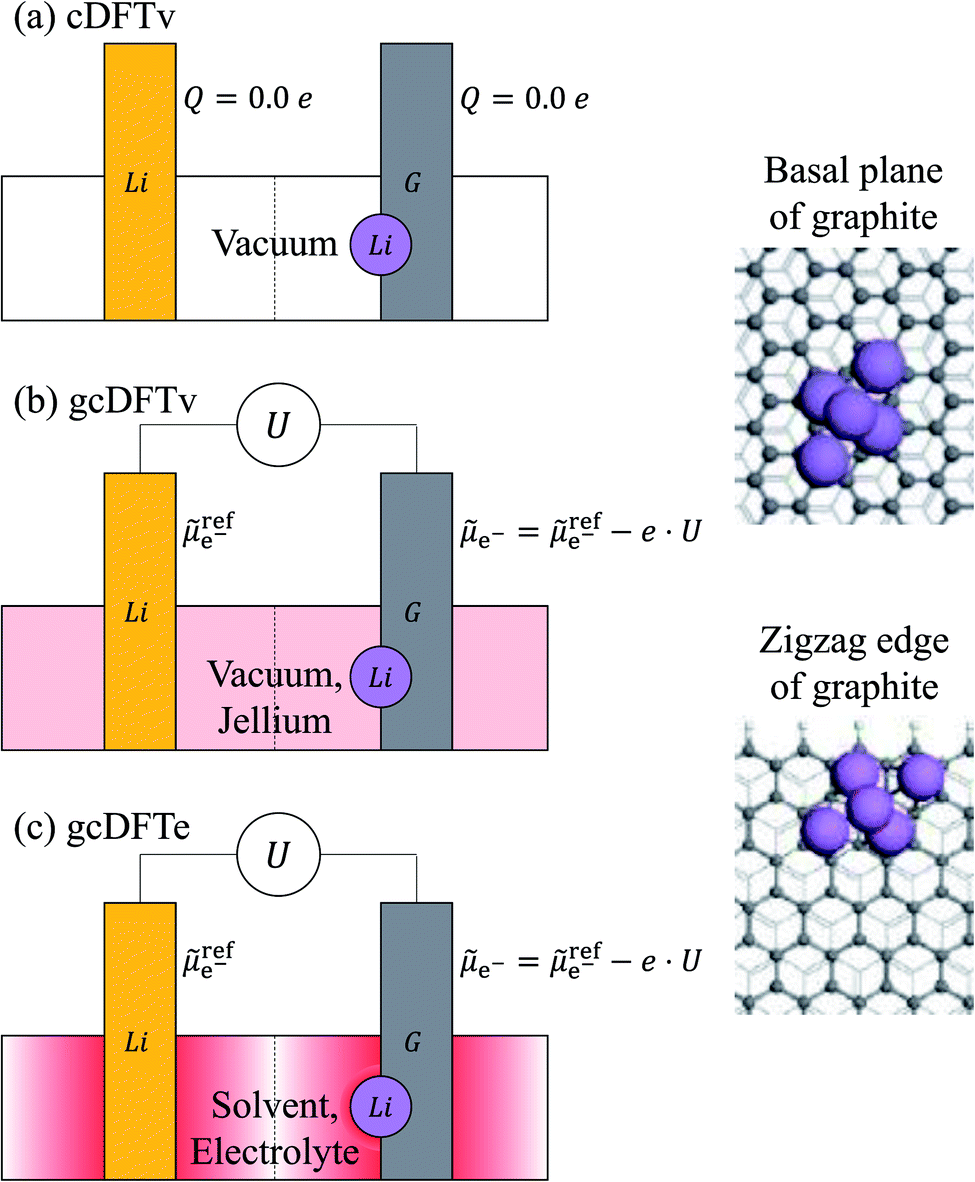 | ||
Fig. 1 Schematic of a Li nucleation process in different regimes: (a) zero charge regime using canonical DFT calculations in vacuum (cDFTv). The total charge on each of the electrodes is constrained to be zero, while there is no constraint on the electrochemical potentials of electrons at the two electrodes. (b) Potential control regime using grand canonical DFT calculations in vacuum (gcDFTv). The net charge on the electrode is neutralized by a uniform opposite background charge (‘jellium’). (c) Potential control regime using grand canonical DFT calculations in electrolyte (gcDFTe). In a solvent environment, the charged electrode system is neutralized by a Poisson–Boltzmann electrolyte. In grand canonical methods (b and c), the applied potential (U) controls the electrochemical potential of electrons (![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e0.gif) e−) at the graphite electrode (G) with respect to that at the Li reference electrode (ref)e−. e−) at the graphite electrode (G) with respect to that at the Li reference electrode (ref)e−. | ||
In this paper we extend our previous work with cDFTv,23 to apply our newly developed grand canonical model to study Li nucleation on graphite electrodes in realistic electrochemical conditions, in order to answer the following questions: (1) What voltage marks the onset of Li nucleation? (2) What is the critical voltage below which the nucleated Li clusters start growing spontaneously? (3) How does the presence of environment affect the nucleation energetics? (4) Where on graphite do the Li clusters prefer to nucleate? (5) What is the effect of the degree of lithiation of graphite on the nucleation process? We now present our theory (Sec. 2), computational details (Sec. 3), results and discussion (Sec. 4), followed by conclusions (Sec. 5).
2 Theory
The Li nucleation reaction on graphite can be written as:Reference electrode:nLi → nLi+(sol.) + ne−
Working electrode:nLi+(sol.) + ne− + G → Lin|G
Overall reaction:nLi + G → Lin|G
The Li+ ions move through the solution, while the electrons (e−) travel through the outer electrical circuit. G represents the graphite and Lin|G represents an n-atom Li cluster nucleated on a graphite working electrode. The nucleation energy is the change in free energy due the reaction (ΔΩ). For an electrochemical cell, the ionic path is usually thermodynamically reversible but the electronic path may be interrupted by a potentiometer, in which case the potential difference (U) on the potentiometer gives an experimental value for the nucleation energy (ΔΩ = −n · e · U, where e is the elementary charge). Conversely, in a potential controlled experiment, the voltage U is applied which results in the nucleation of a Li cluster assuming thermodynamic equilibrium. Similarly, in the present study we could expect the nucleation energy (ΔΩ(U)) to indicate whether a cluster of a given size n will form at a voltage (U). The nucleation energy (ΔΩ(U)) is calculated as:
| ΔΩ(U) = ΩLin|G(U) − ΩG(U) − n·refLi(U = 0), | (1) |
refLi is the electrochemical potential of Li at the reference electrode, which is calculated from the change in the grand free energy (ΩrefLi) of the Li reference electrode slab with respect to the number of Li atoms in the slab (nrefLi): | (2) |
3 Computational details
We perform simulations for Li nucleation using the ONETEP linear-scaling density functional theory (DFT) program,28,30 where the computational cost scales linearly with the number of atoms as opposed to the cubic scaling of conventional DFT. The simulations under potential control are performed using the grand canonical model for electrochemistry simulations,24 utilizing the neutralization by electrolyte concentration shift (NECS) method for a modified Poisson–Boltzmann equation (P–BE).25,29 The P–BE is solved using a bespoke, highly parallel, multigrid solver DL_MG.26,27The kinetic energy cutoff for the basis set, which consists of psinc functions and is equivalent to plane waves, is 1000.0 eV.31 The radius of the localized orbitals called as the non-orthogonal generalized Wannier functions (NGWFs) is set to 8.0 a0 for all calculations.32 The convergence threshold for the electronic energy is 10−6 eV and that for the NGWF rms gradient is 5 × 10−7 a.u. The convergence threshold on the number of electrons in the grand canonical ensemble is 10−6 electrons per atom. For van der Waals interactions, a D2 dispersion correction has been used.33 In the grand canonical calculations the electrochemical potential of electrons in the Li reference electrode is set to −1.39 eV (cf.Table 1 of ref. 24) corresponding to the experimental value, while we set the applied voltage of the graphite electrode at U = −0.1, 0.0, 0.1 V with respect to the Li metal reference electrode. The structural geometries for Li clusters with 1–65 atoms and graphite electrodes have been obtained from AIMD simulations in our previous work using cDFTv as shown in Fig. 1, S3 and S5 of ref. 23. The chemical formula of structural models and size of simulation cell are summarized in Table 1.
| Graphite model (G) | Chemical formula (Lin|G, 1 ≤ n ≤ 65) | X (Å) | Y (Å) | Z (Å) |
|---|---|---|---|---|
| Unlithiated basal plane | Lin|C512 | 34.17 | 19.73 | 100.03 |
| Lithiated basal plane | Lin|Li100C900 | 37.38 | 21.58 | 100.00 |
| Unlithiated zigzag edge | Lin|C648H36 | 53.41 | 22.19 | 67.73 |
| Lithiated zigzag edge | Lin|Li102C972H54 | 57.38 | 22.42 | 67.77 |
The size of the graphite model is much larger than the number of atoms (n) in the Li cluster, so that the interaction between periodic images is negligible. The equilibrium distance of Li clusters from the graphite electrode is further optimized in electrolyte environment for each applied voltage by minimizing the grand free energy of the system as a function of the distance of the Li cluster from the graphite surface.
The surrounding environment is described by an implicit 1.0 M LiPF6 electrolyte in ethylene carbonate solvent (EC). The dielectric permittivity of the EC solvent is described using the soft-sphere model,34 and varies smoothly from 1.0 near the electrode to that of the bulk solvent which is set to 90.7 as per the experimental value at 298.15 K.35 For an even closer approximation of the environment in Li-ion batteries, one can consider using the dielectric constant of a mixture of solvents.36 The soft-sphere radius for the transition of the dielectric permittivity is calibrated from the reduction potential of standard reference electrodes (cf. Tables 2 and 3 of ref. 24). Correspondingly, soft-sphere radii of 3.048 a0 for Li, 3.907 a0 for C and 2.644 a0 for H are used. The solute–solvent cavitation, dispersion and repulsion interactions are taken as proportional to the solvent accessible surface area, with the proportionality constant being the scaled surface tension of the solvent.37 The surface tension of EC is taken to be 0.0506 N m−1 as per the experimental value.38 The accessibility function of the implicit electrolyte is also described via a soft-sphere model,29 varying from 0.0 near the electrode to 1.0 in the bulk electrolyte. The radius of transition spheres is set by considering the total size of the solute surrounded by a solvation shell. The solute size is described by isoradii of the electronic density of isolated atoms at an isovalue of 0.001 e/a30. A solvation shell radius of 3.0 a0 is used. The values of these parameters have been previously calibrated to yield realistic activity coefficients of LiPF6 in EC.29
We calculate the nucleation energy for Li clusters with 1–65 atoms on the graphite electrode at different voltages (U = −0.1, 0.0, 0.1 V) with respect to the Li reference electrode. From the trend of the variation of the nucleation energy with voltage, we find the voltage which would give a zero nucleation energy. This potential of zero nucleation energy (UPZN) is then plotted as a function of the size of the cluster. The minimum in the potential of zero nucleation energy is the critical voltage for cluster growth, below which the nucleated clusters start growing unbounded on graphite. Regarding the location of nucleation on graphite, we consider Li nucleation on the periodic basal plane (far from the edge), as well as close to the zigzag edge termination of the basal plane. We further consider the two end cases where graphite is fully unlithiated and fully lithiated.
4 Results and discussion
4.1 Li reference chemical potential (![[small mu, Greek, tilde]](https://www.rsc.org/images/entities/h3_i_char_e0e0.gif) refLi)
refLi)
For the Li reference electrode, we consider (100) slabs of Li metal with 11, 13, 15 layers at an applied voltage of U = 0.0 V and compute the grand free energy using the grand canonical DFT method in electrolyte (gcDFTe).24 The electrolyte is 1 M LiPF6 in ethylene carbonate (EC) solvent. From the change in grand free energy with the number of Li atoms, we compute the chemical potential of Li at the reference electrode (refLi) using eqn (2). We also consider a grand canonical calculation in vacuum (gcDFTv), where the charge of the electrode is neutralized by an opposite uniform background charge (‘jellium’). The computed values of (refLi) are shown in Table 2. We see a large difference in the computed values of (refLi) with the choice of environment. The values in vacuum are higher by approximately half an electron volt (eV) than those in EC/1 M LiPF6, which is more representative of the actual environment in Li-ion batteries. The computed chemical potential of Li at the reference electrode (refLi) feeds into the calculation of the nucleation energy of Li clusters at the graphite electrode viaeqn (1).
refLi) using eqn (2)
| Environment | Vacuum, jellium | EC, 1 M LiPF6 |
|---|---|---|
| (refLi)(U = 0) (eV) |
−200.6018 | −201.0472 |
4.2 Nucleation energy (ΔΩ)
The nucleation energy (ΔΩ) calculated from eqn (1) informs whether the formation of a cluster of a particular size (n) is thermodynamically feasible. A negative nucleation energy suggests a thermodynamically feasible nucleation, while a positive nucleation energy indicates that nucleation is not feasible. In Fig. 2, we show the nucleation energy in the grand canonical ensemble (ΔΩ) computed for Li clusters of 1–65 atoms on the graphite electrode. The simulations are performed under potential control at different voltages (U = −0.1, 0.0, 0.1 V) with respect to the Li reference electrode using the grand canonical ensemble DFT method.24 We have considered both lithiated and unlithiated graphite electrodes, while considering nucleation on the periodic basal plane, as well as near the finite zigzag edge termination of graphite. The nucleation energy is calculated using the grand canonical DFT method in an electrolyte solution (gcDFTe), as well as in vacuum (gcDFTv). In vacuum, the surface charge on the electrode is neutralized by an opposite uniform background charge (‘jellium’). In implicit solvent, the charged electrode is neutralized by the excess of oppositely charged electrolyte near the surface via the NECS method.25 The bulk concentration of LiPF6 electrolyte is 1 M.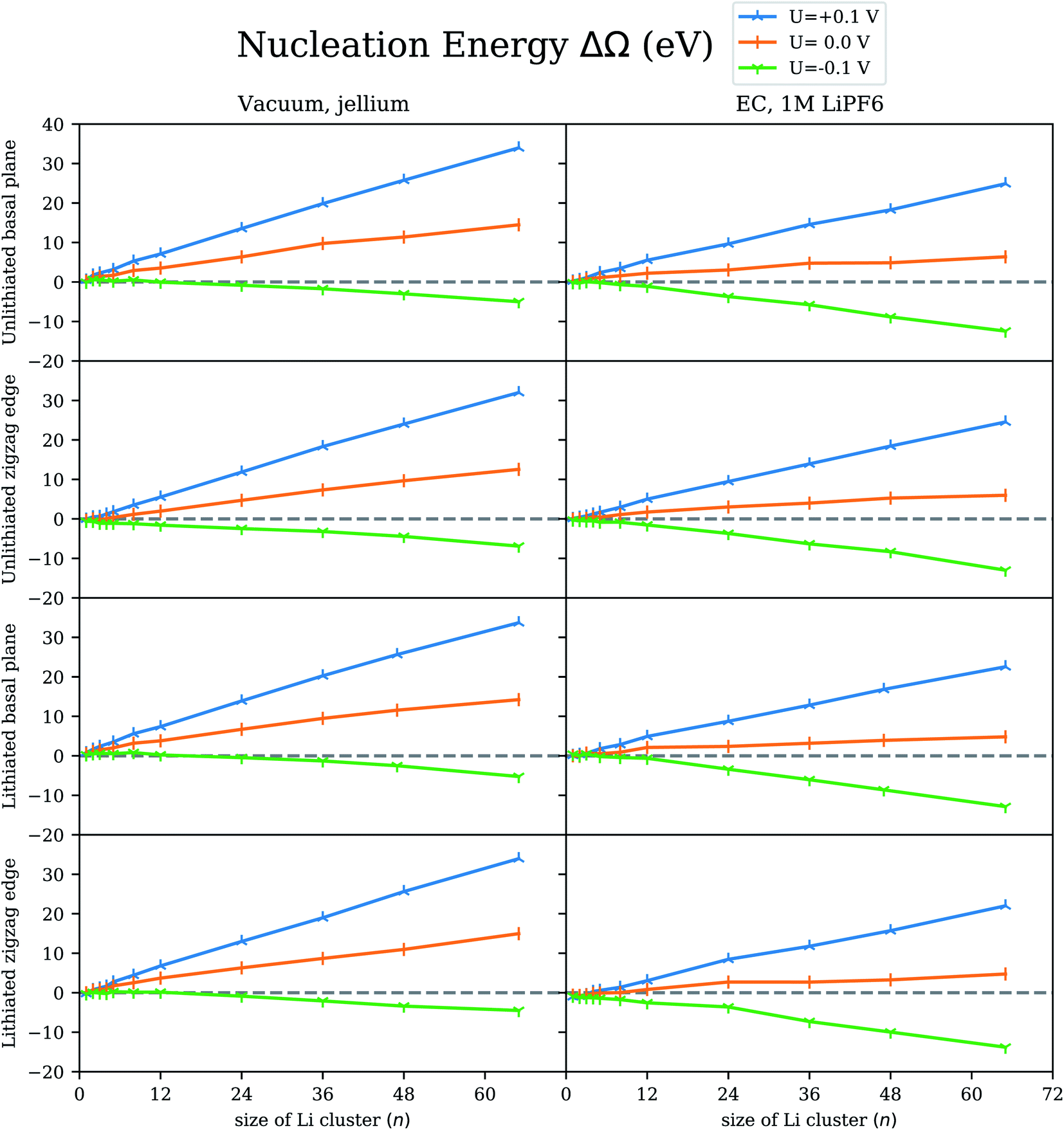 | ||
| Fig. 2 The computed nucleation energy (ΔΩ) for Li clusters with 1–65 atoms on the graphite electrode at voltages U = −0.1, 0.0, 0.1 V with respect to the Li reference electrode using the grand canonical ensemble DFT method.24 We have considered both lithiated and unlithiated graphite electrodes, while considering nucleation on the periodic basal plane as well as near the finite zigzag edge termination of graphite. The nucleation energy is calculated in vacuum as well as in ethylene carbonate (EC) solvent environments. Two types of neutralization schemes are considered: an opposite uniform background charge (‘jellium’) and neutralization by electrolyte concentration shift (NECS).25 The electrolyte is 1 M LiPF6. | ||
As a general observation for large clusters, the nucleation energy for Li clusters on the graphite electrode at voltages U ≥ 0.0 V with respect to the Li reference electrode is positive and monotonically increasing. This suggests that nucleation is generally unlikely to occur at non-negative voltages (U ≥ 0.0 V). The nucleation energy on a graphite electrode at a voltage, U = −0.1 V with respect to the Li reference electrode becomes negative at least beyond a certain cluster size for all the cases considered, suggesting that Li nucleation is possible on a graphite electrode at a negative voltage with respect to the Li reference electrode. To find the voltage below which nucleation energy becomes negative, we compute the potential of zero nucleation energy, which is described next.
4.3 Potential of zero nucleation energy (UPZN)
The potential of zero nucleation energy (UPZN) is the voltage for which the nucleation energy (ΔΩ) computed from eqn (1) becomes zero. In other words, it is the voltage below which the formation of a Li cluster of a particular size becomes thermodynamically feasible. For each cluster size, the potential of zero nucleation energy is calculated by interpolating the calculated nucleation energy (ΔΩ) at the three potential values of U = −0.1, 0.0, 0.1 V to obtain UPZN where the nucleation energy, ΔΩ = 0. This potential of zero nucleation energy (UPZN) is plotted in Fig. 3 for all the clusters with 1–65 atoms in all considered situations. The nucleation of a cluster of a particular size (n) occurs when the applied voltage at the graphite electrode is below the value of UPZN(n). Comparing different situations, we see a significant difference between the results in vacuum with jellium as compared to the electrolyte environment, the latter being more representative of the actual environment in Li-ion batteries. The UPZN is higher in the electrolyte environment than in vacuum, suggesting that the Li nucleation is more likely to occur in the electrolyte environment, than what would be predicted from vacuum calculations. From a comparison between the basal plane and zigzag edge, we see that UPZN is higher near the zigzag edge than on the basal plane, suggesting the nucleation to be more likely on the zigzag edge termination of the graphite electrode than the periodic basal plane. The value of UPZN(n) gives the necessary condition on the voltage for nucleation of a particular cluster, which leads to the next question as to what the critical voltage for cluster growth is. | ||
| Fig. 3 The potential of zero nucleation energy (UPZN) as a function of the size of the Li cluster (n), obtained from interpolating the nucleation energy computed for potentials (U = −0.1, 0.0, 0.1 V) in Fig. 2. The left panel shows the UPZN computed from the grand canonical DFT method in vacuum (gcDFTv), while the right panel shows the same from the grand canonical DFT method in electrolyte (gcDFTe). The electrolyte is 1 M LiPF6 in EC solvent. | ||
4.4 Potential for cluster growth (UPCG)
In each case of Fig. 3, the UPZN values represent the threshold applied potential below which the nucleation of a Li cluster of size n is possible. For n = 1, we find this potential close to zero in many cases and even at positive potentials under some conditions. Proceeding to larger n values, we see a decrease of the UPZN with cluster size. This suggests that subsequent cluster growth requires successively larger (more negative) overpotentials until the minimum UPZN is reached. Beyond this cluster size, the UPZN increases so that spontaneous growth can occur without an increased overpotential, and in some cases even at reduced overpotentials. Thus we can define a potential for cluster growth, | (3) |
| Ensemble | Canonical | Grand canonical | Experiments | ||
|---|---|---|---|---|---|
| Environment | Vacuum (cDFTv) | Vacuum, jellium (gcDFTv) | EC, 1 M LiPF6 (gcDFTe) | EC/DEC (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 1 M LiPF6 :1), 1 M LiPF6 |
|
| System | |||||
| Unlithiated graphite | Basal plane | −0.65 V (ref. 23) | −0.20 V | −0.12 V | −0.15 V to −0.04 V (ref. 13) |
| Zigzag edge | −0.31 V (ref. 23) | −0.07 V | −0.06 V | ||
| Lithiated graphite | Basal plane | N.A. | −0.18 V | −0.08 V | |
| Zigzag edge | −0.47 V (ref. 23) | −0.14 V | −0.04 V | ||
4.5 Electrostatic potential and charge density
The effect of the environment can be more evident from the plots of electrostatic potential and charge density. We investigate the case of a 5-atom Li cluster on graphite surface at U = −0.1 V and plot the total electrostatic potential and charge density in a plane cutting through the Li cluster on the periodic basal plane in Fig. 4 and on the zigzag edge termination of the graphite in Fig. 5. The total electrostatic potential of the system in vacuum with jellium is diffuse and extends all over the simulation cell. In the presence of electrolyte, the total electrostatic potential is effectively screened at the interface so that it decays rapidly as it should in bulk electrolyte. The total charge density is shown in the right panels. In vacuum, the negatively charged electrode system is neutralized by the uniform positively charged jellium which extends all over the simulation cell. In electrolyte environment, the negatively charged electrode system is neutralized by the build up of positively charged ions near the interface, forming a double layer and the electrolyte charge density decays to zero far away in bulk electrolyte. The total charge density integrates to zero in the simulation cell in all cases, which is a necessary requirement for the convergence of the electrostatic potential in periodic boundary conditions.25 The net charge on the electrode system is shown in Table 4 for all cases. The net charge on the surrounding medium (jellium or electrolyte) is the opposite of this charge on the electrode system. The magnitude of the charging of the electrode system is much larger in electrolyte environment than what occurs in vacuum with jellium. All these observations highlight the limitations of conventional in-vacuum models based on jellium neutralization and show the realistic description with our advanced electrolyte model.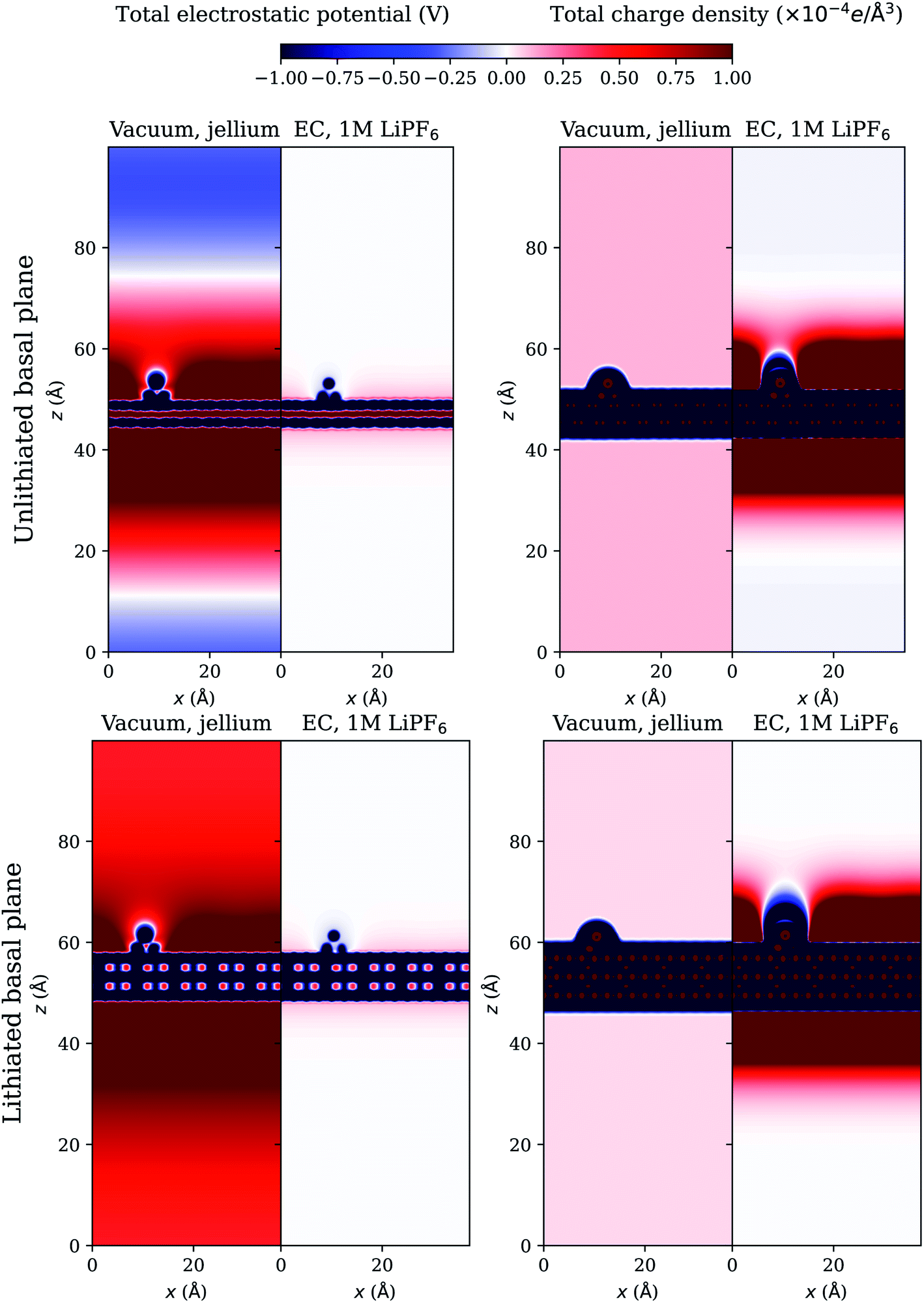 | ||
| Fig. 4 Total electrostatic potential (left) and charge density (right) in a plane cutting through a 5-atom Li cluster nucleated on the periodic basal plane of the graphite electrode at U = −0.1 V, computed using the grand canonical DFT method. | ||
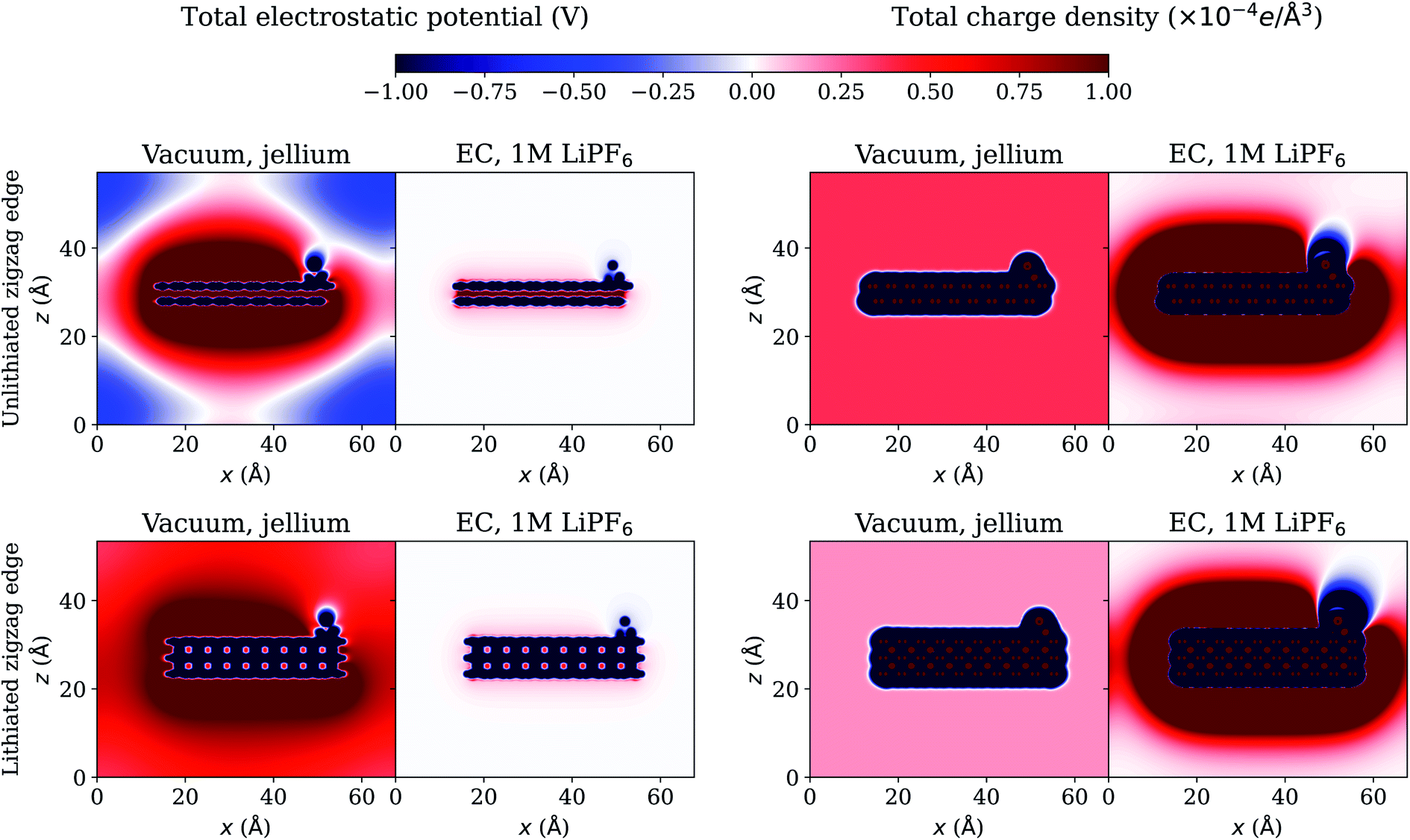 | ||
| Fig. 5 Total electrostatic potential (left) and charge density (right) in a plane cutting through a 5-atom Li cluster nucleated near the zigzag edge termination of the graphite electrode at U = −0.1 V, computed using the grand canonical DFT method. | ||
| Environment | Vacuum, jellium (gcDFTv) | EC, 1 M LiPF6 (gcDFTv) | |
|---|---|---|---|
| System | |||
| Unlithiated graphite | Basal plane | −0.73e | −12.75e |
| Zigzag edge | −2.96e | −23.67e | |
| Lithiated graphite | Basal plane | −0.41e | −11.58e |
| Zigzag edge | −1.45e | −19.96e | |
5 Conclusions
We studied the thermodynamics of the nucleation of Li clusters of 1–65 atoms and their growth on graphite electrodes at an applied voltage with respect to the Li reference electrode, using a combined grand canonical large scale density-functional and Poisson–Boltzmann Theory. We computed the nucleation energy of Li clusters for different values of the applied voltage, U = −0.1, 0.0, 0.1 V, with respect to the Li reference electrode. We find that the nucleation energy is positive and monotonically increasing at U = 0.0 V and U = 0.1 V, while it achieves a negative value beyond a certain cluster size for a negative potential U = −0.1 V. A negative value of the nucleation energy would mean an energetically favourable nucleation process. To find the voltage below which the nucleation energy would become negative, we compute the potential of zero nucleation energy (UPZN) for each cluster size (n). The lowest value of UPZN(n) with respect to the Li reference electrode marks the potential for cluster growth on graphite electrode, below which the Li clusters would grow unbounded on graphite. We have considered the variation with the state of charge, location of nucleation, and the influence of the surrounding electrolyte environment, reaching the following observations:1. The grand canonical method in electrolyte (gcDFTe) is a significant advancement over conventional canonical DFT in vacuum (cDFTv). For instance, the potential for cluster growth computed with gcDFTe method on the zigzag edge is −0.06 V on unlithiated graphite and −0.04 V on lithiated graphite. This is close to experimentally observed values of −0.04 V to −0.15 V.13 A corresponding value from cDFTv method is −0.31 V on unlithiated graphite and −0.47 on lithiated graphite.23
2. The consideration of the environment plays an important role in quantifying the thermodynamics of the process. The magnitude of the potential of cluster growth is found to lower be in an electrolyte environment (gcDFTe) compared to vacuum (gcDFTv) for all cases. E.g. the potential for cluster growth on the zigzag edge of lithiated graphite in vacuum is −0.14 V as compared to −0.04 V in electrolyte environment. Thus, the nucleation process is more likely to happen in electrolyte environments than what could be predicted from calculations in vacuum.
3. Nucleation is preferred on the zigzag edge termination rather than the basal plane of graphite. E.g. the potential for cluster growth on the unlithiated graphite basal plane is −0.12 V while that on the zigzag edge termination is −0.06 V with respect to the Li reference electrode.
4. Nucleation is preferred on the lithiated graphite as compared to the unlithiated graphite. The potential for cluster growth at the zigzag edge is −0.06 V on unlithiated graphite as compared to −0.04 V on the lithiated graphite, while the corresponding value on the basal plane is −0.12 V on unlithiated as compared to −0.08 V on lithiated graphite.
The above observations have profound implications for the occurrence, avoidance and control of dendrite growth in a battery cell through ab initio simulations. The resulting thermodynamic parameters will represent the very first step of subsequent kinetic analyses for in silico predictions of important results before experimental verification, leading to rapid discovery of new electrolytes and charging procedures to improve safety and lifetime of future cells.
The technologically-important insights obtained from this study to understand the issue of Li nucleation in Li-ion batteries, which is vital for controlling degradation, have been made possible by the development of methods for atomistic simulations of electrodes in an electrolyte environment. The study also demonstrates the usefulness of the grand canonical DFT method in electrolyte (gcDFTe) which is a significant advancement over conventional canonical DFT in vacuum (cDFTv). The gcDFTe method is generally applicable and could be used to model other technologically relevant electrochemical systems such as fuel cells and electrolyzers. The grand canonical method can also be used for molecular dynamics simulations. However, even with a linear-scaling code like ONETEP, which overcomes the length-scale limitations of conventional DFT, we are still limited by the very-short time-scales possible with ab initio molecular dynamics. Tackling the time-scale problem would require further developments that could combine more empirical quantum atomistic methods with machine-learning approaches.
Data availability
The data that supports the findings of this study is available within the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out with the funding from the Faraday Institution (https://www.Faraday.ac.uk; EP/S003053/1), grant numbers FIRG003 and FIRG025. The calculations presented in this work were performed on the Iridis5 supercomputer of the University of Southampton, the Michael supercomputer of the Faraday Institution, the Young supercomputer at the UCL and the ARCHER2 UK National Supercomputing Service (https://www.archer2.ac.uk; via EPSRC grant: EP/P022030/1). We acknowledge the United Kingdom Materials and Molecular Modelling Hub for computational resources, partially funded by EPSRC (EP/P020194/1).References
- J. B. Goodenough and K.-S. Park, The Li-Ion Rechargeable Battery: A Perspective, J. Am. Chem. Soc., 2013, 135, 1167–1176 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Challenges in the development of advanced Li-ion batteries: a review, Energy Environ. Sci., 2011, 4, 3243–3262 RSC.
- J. B. Goodenough and Y. Kim, Challenges for rechargeable Li batteries, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- H. Zhang, Y. Yang, D. Ren, L. Wang and X. He, Graphite as anode materials: Fundamental mechanism, recent progress and advances, Energy Storage Mater., 2021, 36, 147–170 CrossRef.
- K. Persson, Y. Hinuma, Y. S. Meng, A. Van der Ven and G. Ceder, Thermodynamic and kinetic properties of the Li-graphite system from first-principles calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 125416 CrossRef.
- C. Peng, M. P. Mercer, C. K. Skylaris and D. Kramer, Lithium intercalation edge effects and doping implications for graphite anodes, J. Mater. Chem. A, 2020, 8, 7947–7955 RSC.
- J. X. Huang, G. Csányi, J. B. Zhao, J. Cheng and V. L. Deringer, First-principles study of alkali-metal intercalation in disordered carbon anode materials, J. Mater. Chem. A, 2019, 7, 19070–19080 RSC.
- J. Vetter, P. Novák, M. R. Wagner, C. Veit, K. C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, Ageing mechanisms in lithium-ion batteries, J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- X. Qin, M. Shao and P. B. Balbuena, Elucidating mechanisms of Li plating on Li anodes of lithium-based batteries, Electrochim. Acta, 2018, 284, 485–494 CrossRef CAS.
- J. Jiao, G. Lai, L. Zhao, J. Lu, Q. Li, X. Xu, Y. Jiang, Y. B. He, C. Ouyang, F. Pan, H. Li and J. Zheng, Self-Healing Mechanism of Lithium in Lithium Metal, Adv. Sci., 2022, 9(12), 2105574 CrossRef CAS PubMed.
- D. Hu, L. Chen, J. Tian, Y. Su, N. Li, G. Chen, Y. Hu, Y. Dou, S. Chen and F. Wu, Research Progress of Lithium Plating on Graphite Anode in Lithium-Ion Batteries, Chin. J. Chem., 2021, 39, 165–173 CrossRef CAS.
- T. Waldmann, B. I. Hogg and M. Wohlfahrt-Mehrens, Li plating as unwanted side reaction in commercial Li-ion cells – A review, J. Power Sources, 2018, 384, 107–124 CrossRef CAS.
- T. Gao, Y. Han, D. Fraggedakis, S. Das, T. Zhou, C. N. Yeh, S. Xu, W. C. Chueh, J. Li and M. Z. Bazant, Interplay of Lithium Intercalation and Plating on a Single Graphite Particle, Joule, 2021, 5, 393–414 CrossRef CAS.
- P. Arora, M. Doyle and R. E. White, Mathematical Modeling of the Lithium Deposition Overcharge Reaction in Lithium-Ion Batteries Using Carbon-Based Negative Electrodes, J. Electrochem. Soc., 1999, 146, 3543–3553 CrossRef CAS.
- N. Legrand, B. Knosp, P. Desprez, F. Lapicque and S. Raël, Physical characterization of the charging process of a Li-ion battery and prediction of Li plating by electrochemical modelling, J. Power Sources, 2014, 245, 208–216 CrossRef CAS.
- C. Uhlmann, J. Illig, M. Ender, R. Schuster and E. Ivers-Tiffée, In situ detection of lithium metal plating on graphite in experimental cells, J. Power Sources, 2015, 279, 428–438 CrossRef CAS.
- L. M. Morgan, et al., Pushing the boundaries of lithium battery research with atomistic modelling on different scales, Prog. Energy, 2022, 4, 012002 CrossRef.
- X. Fan, W. T. Zheng, J. L. Kuo and D. J. Singh, Adsorption of single li and the formation of small li clusters on graphene for the anode of lithium-ion batteries, ACS Appl. Mater. Interfaces, 2013, 5, 7793–7797 CrossRef CAS PubMed.
- M. Liu, A. Kutana, Y. Liu and B. I. Yakobson, First-Principles Studies of Li Nucleation on Graphene, J. Phys. Chem. Lett., 2014, 5, 1225–1229 CrossRef CAS PubMed.
- J. Cui, S. Yao, M. Ihsan-ul haq, J. Wu and J.-k. Kim, Correlation between Li Plating Behavior and Surface Characteristics of Carbon Matrix toward Stable Li Metal Anodes, Adv. Energy Mater., 2019, 9(1), 1802777 CrossRef.
- X. Chen, X. R. Chen, T. Z. Hou, B. Q. Li, X. B. Cheng, R. Zhang and Q. Zhang, Lithiophilicity chemistry of heteroatom-doped carbon to guide uniform lithium nucleation in lithium metal anodes, Sci. Adv., 2019, 5(2), eaau7728 CrossRef CAS PubMed.
- E. G. Leggesse, C.-L. Chen and J.-C. Jiang, Lithium diffusion in graphene and graphite: Effect of edge morphology, Carbon, 2016, 103, 209–216 CrossRef CAS.
- C. Peng, A. Bhandari, J. Dziedzic, J. R. Owen, C.-K. Skylaris and D. Kramer, Mechanism of Li nucleation at graphite anodes and mitigation strategies, J. Mater. Chem. A, 2021, 9, 16798–16804 RSC.
- A. Bhandari, C. Peng, J. Dziedzic, L. Anton, J. R. Owen, D. Kramer and C.-K. Skylaris, Electrochemistry from first-principles in the grand canonical ensemble, J. Chem. Phys., 2021, 155, 024114 CrossRef CAS PubMed.
- A. Bhandari, L. Anton, J. Dziedzic, C. Peng, D. Kramer and C.-K. Skylaris, Electronic structure calculations in electrolyte solutions: Methods for neutralization of extended charged interfaces, J. Chem. Phys., 2020, 153, 124101 CrossRef CAS PubMed.
- J. C. Womack, L. Anton, J. Dziedzic, P. J. Hasnip, M. I. J. Probert and C.-K. D. L. _M. G. Skylaris, A Parallel Multigrid Poisson and Poisson–Boltzmann Solver for Electronic Structure Calculations in Vacuum and Solution, J. Chem. Theory Comput., 2018, 14, 1412–1432 CrossRef CAS PubMed.
- http://www.dlmg.org .
- J. C. A. Prentice, et al., The ONETEP linear-scaling density functional theory program, J. Chem. Phys., 2020, 152, 174111 CrossRef CAS PubMed.
- J. Dziedzic, A. Bhandari, L. Anton, C. Peng, J. C. Womack, M. Famili, D. Kramer and C.-K. Skylaris, Practical Approach to Large-Scale Electronic Structure Calculations in Electrolyte Solutions via Continuum-Embedded Linear-Scaling Density Functional Theory, J. Phys. Chem. C, 2020, 124, 7860–7872 CrossRef CAS.
- C.-K. Skylaris, P. D. Haynes, A. A. Mostofi and M. C. Payne, Introducing ONETEP: Linear-scaling density functional simulations on parallel computers, J. Chem. Phys., 2005, 122, 084119 CrossRef PubMed.
- A. A. Mostofi, P. D. Haynes, C.-K. Skylaris and M. C. Payne, Preconditioned iterative minimization for linear-scaling electronic structure calculations, J. Chem. Phys., 2003, 119, 8842–8848 CrossRef CAS.
- C.-K. Skylaris, A. A. Mostofi, P. D. Haynes, O. Diéguez and M. C. Payne, Nonorthogonal generalized Wannier function pseudopotential plane-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 035119 CrossRef.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- G. Bramley, M.-T. Nguyen, V.-A. Glezakou, R. Rousseau and C.-K. Skylaris, Reconciling Work Functions and Adsorption Enthalpies for Implicit Solvent Models: A Pt (111)/Water Interface Case Study, J. Chem. Theory Comput., 2020, 16, 2703–2715 CrossRef CAS PubMed.
- D. S. Hall, J. Self and J. R. Dahn, Dielectric Constants for Quantum Chemistry and Li-Ion Batteries: Solvent Blends of Ethylene Carbonate and Ethyl Methyl Carbonate, J. Phys. Chem. C, 2015, 119, 22322–22330 CrossRef CAS.
- N. Yao, X. Chen, X. Shen, R. Zhang, Z. H. Fu, X. X. Ma, X. Q. Zhang, B. Q. Li and Q. Zhang, An Atomic Insight into the Chemical Origin and Variation of the Dielectric Constant in Liquid Electrolytes, Angew. Chem., Int. Ed., 2021, 60, 21473–21478 CrossRef CAS PubMed.
- J. Dziedzic, H. H. Helal, C.-K. K. Skylaris, A. A. Mostofi and M. C. Payne, Minimal parameter implicit solvent model for ab initio electronic-structure calculations, Epl, 2011, 95, 43001 CrossRef.
- R. Naejus, C. Damas, D. Lemordant, R. Coudert and P. Willmann, Excess thermodynamic properties of the ethylene carbonate-trifluoroethyl methyl carbonate and propylene carbonate-trifluoroethyl methyl carbonate systems at T = (298.15 or 315.15) K, J. Chem. Thermodyn., 2002, 34, 795–806 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Files for all calculations are attached for reference and reproduction of results. Fig. S1: Enlarged plot of Fig. 2 for small Li clusters (1 ≤ n ≤ 12). See https://doi.org/10.1039/d2ta02420a |
| This journal is © The Royal Society of Chemistry 2022 |