
Hydrogels for underwater adhesion: adhesion mechanism, design strategies and applications
 *a
*a
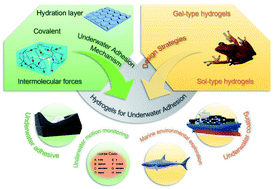
Abstract
A hydrogel is a polymer material with a porous network structure, which allows the hydrogel to swell rapidly underwater without dissolving. This unique property of hydrogels lays the foundation for their application in underwater adhesion. However, due to the existence of the hydration layer, the interaction between the hydrogel and the adherend surface is seriously hindered, and the underwater adhesion ability of the hydrogel is severely weakened. In the past few decades, various hydrogels for underwater adhesion have been developed, among which the underwater adhesion mechanisms of natural organisms have provided a steady stream of inspiration for researchers to design underwater adhesion hydrogels. In this review, we first summarize the adhesion mechanism and design strategies of underwater adhesion hydrogels, and then introduce the currently common experimental methods to test the adhesion strength and adhesion toughness of underwater adhesion hydrogels. According to the development trend in recent years, we summarize the common application fields of underwater adhesion hydrogels. Finally, we present our views on the challenges of hydrogels for underwater adhesion, and provide an outlook on future research directions. This review provides a comprehensive overview of underwater adhesion hydrogels, which will provide rational guidelines for the design and fabrication of underwater adhesion hydrogels.
- This article is part of the themed collections: 2023 Journal of Materials Chemistry A Lunar New Year collection and 2022 Journal of Materials Chemistry A Most Popular Articles
 Please wait while we load your content...
Please wait while we load your content...

