
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Promise and reality of organic electrodes from materials design and charge storage perspective
Amitava
Banerjee†
 *a,
Nabil
Khossossi†
b,
Wei
Luo
b and
Rajeev
Ahuja
*bc
*a,
Nabil
Khossossi†
b,
Wei
Luo
b and
Rajeev
Ahuja
*bc
aDepartment of Metallurgical and Materials Engineering, Indian Institute of Technology Jodhpur, Rajasthan 342030, India. E-mail: amitava@iitj.ac.in
bCondensed Matter Theory Group, Materials Theory Division, Department of Physics and Astronomy, Uppsala University, Box 516, 75120 Uppsala, Sweden. E-mail: rajeev.ahuja@physics.uu.se
cDepartment of Physics, Indian Institute of Technology Ropar, Rupnagar 140001, Punjab, India
First published on 17th June 2022
Abstract
Organic electrode materials are becoming increasingly important as they reduce the C-footprint as well as the production cost of currently used and studied rechargeable batteries. With increasing demand for high-energy-density devices, over the past few decades, various innovative new materials based on the fundamental structure–property relationships and molecular design have been explored to enable high-capacity next-generation battery chemistries. One critical dimension that catalyzes this study is the building up of an in-depth understanding of the structure–property relationship and mechanism of alkali ion batteries. In this review, we present a critical overview of the progress in the technical feasibility of organic battery electrodes for use in long-term and large-scale electrical energy-storage devices based on the materials designing, working mechanisms, performance, and battery safety. Specifically, we discuss the underlying alkali ion storage mechanisms in specific organic batteries, which could provide the designing requirements to overcome the limitations of organic batteries. We also discuss the promising future research directions in the field of alkali ion organic batteries, especially multivalent organic batteries along with monovalent alkali ion organic batteries.
1. Introduction
Organic electrode materials have drawn significant attention in the last few years in the search of suitable electrode materials to meet the requirements of the rapidly growing markets of consumer electronics, electric vehicles, and grid integration of renewable energy.1 Organic electrodes are endowed with several merits over conventional transition metal based inorganic electrodes, such as sustainable production with low carbon footprint, low cost, excellent stability, high flexibility, as well as resource sustainability.2–5 In terms of sustainability, many organic electrode materials can be synthesized from biomass using green-chemistry techniques that have negligible environmental foot print. For example, the organic electrode dilithium rhodizonate (Li2C6O6) can be prepared from the natural sugar myo-inositol, which exists in corn plants in the hexaphosphate form.6 Another useful redox active molecule, polyquinone can be prepared by the polycondensation of malic acid, which is a natural product found in apples.7 On the other hand, the diversity of organic functional groups and flexibility of organic molecules allow the molecular level modifications of organic electrodes to alter their alkali ion storage capacity.1,8–11The history of organic electrode materials (Fig. 1) can be traced back to as early as the 1960s, when a tricarbonyl compound was first used as a cathode for lithium-ion batteries (LIBs).12 However, a rapid decay in capacity due to the high solubility of small carbonyl compounds in aprotic electrolytes limited their further development. Subsequently, in the 1970s, many chemists explored conductive polymers as LIB electrode materials due to their low solubility in electrolytes.13–16 In this category, the most widely studied conductive polymers are polyacetylene, polyaniline, polypyrrole, polythiophene, and poly(p-phenylene).13,16,17 In the late 1980s, coin-type batteries18 with polyaniline cathodes and LiAl alloy anodes were introduced but quickly disappeared from the market as they never showed their ideal performance. The main concern with the conductive polymers is low doping level limits, typically below 50%, which means in the charging/discharging process less than half of the redox-active groups participate. Due to this reason the practical capacities usually do not exceed 150 mA h g−1, which motivated the search for alternative chemistry for cathode materials.13,16 In the development of alternative chemistry to obtain high capacity cathode materials, attention was paid to organosulfur compounds where the S–S bond formation (cleavage) takes place during the charge (discharge) process. Organosulfur compounds exhibit high capacities.19 But there are other concerns, such as the sluggish kinetics of S–S bond formation in the charging process, poor cycling stability, and a serious shuttle effect (where dissolved species diffuse between the anode and cathode).20 Along with this development, electrode materials based on nitriles, electron-poor molecules with highly reversible redox chemistry, were also investigated.21 Besides the above-mentioned materials, organic radicals were tested as a promising organic electrode material due to their attractive rapid redox compatibility (where bond rearrangements during charging and discharging are almost nil).22 In fact, they possessed a higher electron-transfer rate compared to organosulfur compounds; for example, in nitroxyl, the self-exchange electron transfer (RNO+ + RNO˙ ⇌ RNO˙ + RNO+) rate constant is ∼10−1 cm s−1, which is higher than those of organosulfur compounds (∼10−8 cm s−1).23,24 However, after 1991, the research and development on organic electrode materials received less attention due to the successful commercialization of inorganic electrode materials-based (cathode: LiCoO2; anode: graphite) high voltage LIBs,25 and only a few studies were published up to early 2000. From 2002 organic electrode materials again began to gain more attention, when organic radicals were also reported as high voltage cathodes for LIBs.22 The search for organic electrode materials got a boost around 2008, when Armand and Tarascon7 pointed out the environmental importance and bright prospect of organic electrode materials. In the last few years, along with the advancement of Li-chemistry, search for organic materials is also extended beyond the Li chemistry, such as other mono-valent (e.g. K), divalent (e.g. Mg, Ca, Zn) and trivalent cation (e.g. Al) chemistry.26,27 Although there are many reviews of organic electrode materials,2,16,20,28–30 it is noteworthy that a comprehensive review focusing on the alkali ion storage mechanism along with the structural design strategies to improve the electrochemical performance of various metal-ion batteries in parallel to the practical requirements is still missing. In this review, we first present the general charge storage mechanism of organic electrode materials. We then discuss more insight into the materials chemistry specific mechanism, and the strategies applied to increase the electrochemical performance (such as output voltage, capacity, cycling stability, and rate performance), and the designing requirements to further enhance the performance to reach the practical application goal. We also discuss the key challenges of existing organic electrode materials and the future prospects of this field beyond the monovalent alkali ion organic batteries.
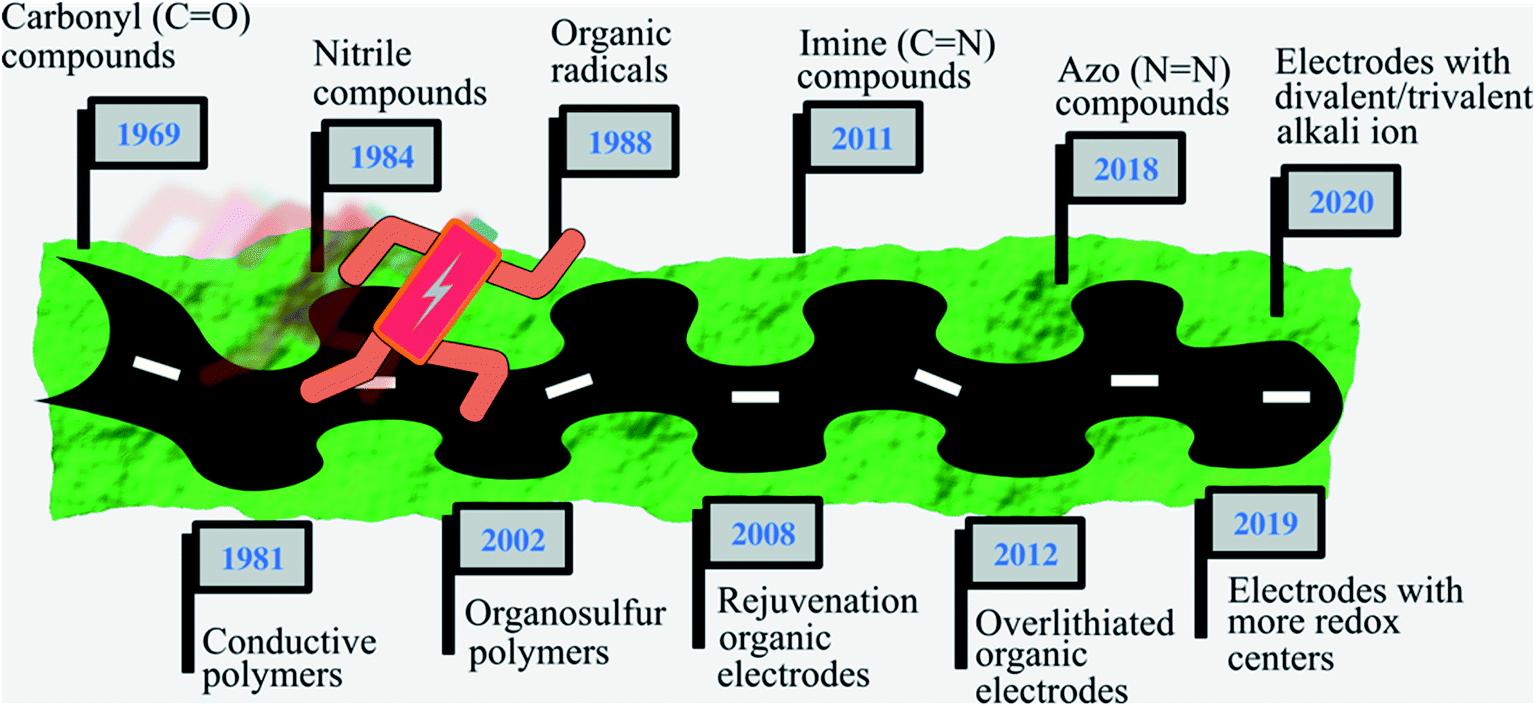 | ||
| Fig. 1 Schematic of the historical development of organic electrodes. Here each year represents the reporting6,7,12,14,19,21,22,31–35 of new developments either in the form of new chemistry or new redox mechanisms. | ||
2. General charge/discharge mechanism
In general, the charge/discharge mechanism of organic electrodes is based on the redox reactions of the electrode materials, where the charge state of the electroactive groups changes. In contrast to transition metal-based electrodes, usually an organic polymer is processed in the uncharged form.28 Due to this reason, initially the cathode is oxidized and anode is reduced. The general charge/discharge process is shown in Fig. 2, where the electrodes are placed in an ion-conducting electrolyte and an ion-permeable membrane is used to separate them. This ion-conducting electrolyte plays an important role in both charge and discharge processes. In the charge process, electrolyte ions are compensated by the charge formed in the electrode, whereas in the discharge process, the counter ions migrate from the polymer into the electrolyte. There are few fundamental differences between inorganic and organic electrode materials. During the charge/discharge process, an inorganic electrode undergoes alloying or conversion reactions; as a result, the electrode experiences a large structural change.36 On the other hand, the redox reactions of most of the organic electrodes manifest very minimal structural changes with limited bond rearrangement.37,38 But there are some exceptions as well in the case of organic materials, e.g. disodium rhodizonate and Na2(2,5-dihydroxy-1,4-benzoquinone) show relatively large structural changes during the charge/discharge process.39–41 Another important difference between inorganic and organic electrode materials is the characteristic of the counterion. In the latter, the redox reaction chemistry is not influenced by the radius and charge of the counterions, which is a great advantage of organic electrode materials, so that the same organic electrodes could be used in different aqueous or non-aqueous metal-ion batteries. For example, perylene dianhydride is utilized as an electrode for Li+, Na+, K+, Mg2+, and Ca2+ counter ions, where the radius and charge of the counter ions are different.42–45 Based on the charge-state change of the active ion, overall the organic electrode materials are classified into three different categories:28 n-type, p-type and bipolar type. The general mechanism of the redox chemistry of various organic electrode materials is shown in Fig. 3. The redox reaction in the n-type electrode takes place between the neutral and negatively charged states, whereas in the p-type electrode the reaction occurs between the neutral and positively charged states. Mostly n-type organics are first reduced and then they bind with metal counterions. Conversely, p-type organics are first oxidized before binding with the anions from the electrolyte. Organic electrodes belonging to the bipolar type can be oxidized or reduced first depending on the applied voltage. It is noteworthy that the particular redox chemistry of many organic electrodes is still unclear and in-depth investigation is needed. For instance, during the lithiation process some organic materials (n-type and bipolar type) are believed to exist as radical intermediates, but it is very difficult to identify these types of intermediates due to their poor stability in batteries.46 Also, the reversibility of the over-lithiation process is still under debate.34,47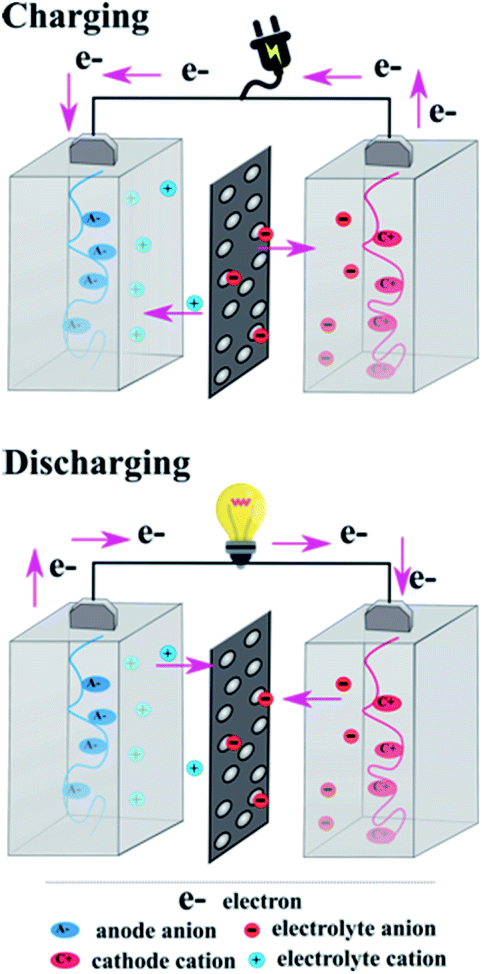 | ||
| Fig. 2 Schematic representation of the general charge–discharge process in all organic batteries. | ||
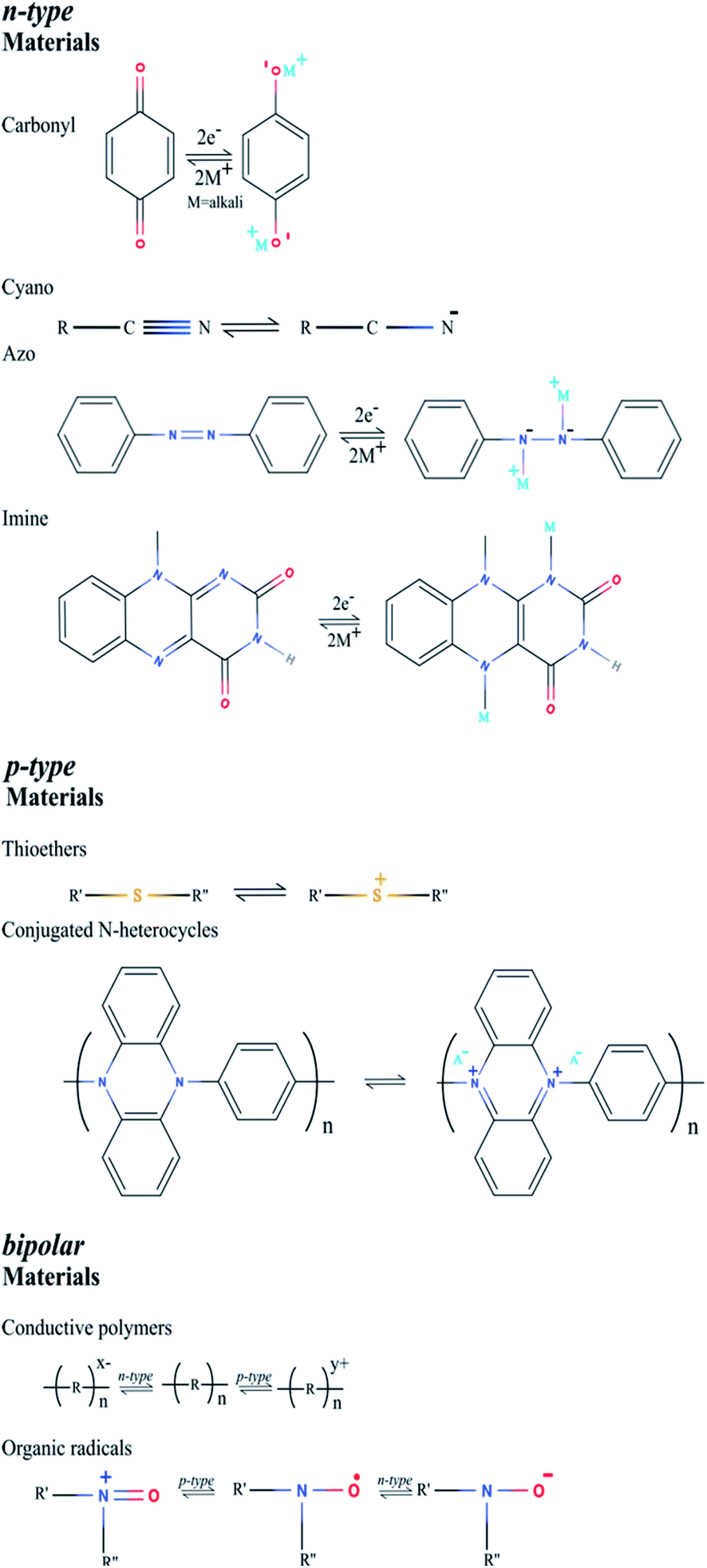 | ||
| Fig. 3 General mechanism of the redox chemistry of various organic electrode materials. | ||
3. Classification of organic electrode materials
3.1 Carbonyl group
In the context of energy storage, carbonyl compounds were first explored sporadically in the 1970s and 80s but they were studied intensively over the past few decades.48–52 One of the reasons could be that chemists can extract them by a variety of synthetic means as they are one of the common organic functionalities. Also, they can show oxidative stability as well as reversible reduction capability. The major structural changes (which are eventually the driving force of their electrochemical mechanism) that occur upon reduction are (A) the vicinal carbonyls form stable enolates, (B) whereas aromatic carbonyl groups disperse the negative charge over the delocalized system. Depending on their practical working potentials, they can serve as cathodes as well as anodes. As electrode materials, carbonyl compounds (quinones, carboxylates, anhydrides, imides, and ketones) are mostly used in the form of small molecules and non-conjugated polymers. A detailed review about carbonyls for battery applications including polymers as well as small molecules is available.53 Here we will first summarize the potentiality as well as the inferiority of the small molecule form then the non-conjugated polymer form.3.1.1.1 Fusing multiple carbonyl groups in the compound. Zou et al.55 found that increasing the number of aromatic rings with the carbonyl group could enhance the solubility and capacity of the small molecule, quinone. The tetrahydrohexaquinone synthesized by them retained 54.1% and 26.5% of its theoretical capacity (628 mA h g−1), respectively, at 200 mA g−1 and 800 mA g−1. Although the presence of large aromatic structures decreases the solubility (compared to its parent anthraquinone) and increases the yield of theoretical capacity, the presence of redox groups in close proximity could also cause electrostatic repulsion during reduction. This could lead to weak crystal packing and, finally, dissolution and capacity fading. Eventually, Zou et al.55 observed 41% loss of initial capacity after 40 cycles.
3.1.1.2 Functionalization of quinones with ionic groups. Addition of ionic groups is another strategy to improve the stability as well as the working potential of the electrode. Mono and disodium sulfate anthraquinones produce a high specific capacity, respectively, on the order of 130 and 150 mA h g−1 at 0.2C.59 Also, the presence of an additional electron-withdrawing group in disodium sulfate anthraquinone helps to provide better cycling stability, ∼92% after 100 cycles at 0.1C.59 Yokoji and co-workers60 observed a 600 meV increase in the reduction potential of benzoquinones by employing a fluorinated electron-withdrawing group. They also observed a slight increase in stability, which could be due to the stable intermediate formation but it did not fully suppress the rapid capacity fading issue due to dissolution. Kim et al.61 provided a DFT based design strategy to understand the effect of halogen substitution on quinone. Their designed tetra-chloroquinone for the sodium-ion battery cathode exhibited a capacity of 150 mA h g−1 at 10 mA g−1 with high voltage plateaus at 2.6 and 2.9 V vs. Na/Na+. Unfortunately, it lost 95% of its initial capacity after 20 cycles. The positive effect of ionic group functionalization on voltage is impressive as it also enhances the stability slightly in a few cases. But it also has a negative effect on theoretical capacity due to the addition of inactive mass to the compound without enhancing its electron acceptance capability. Therefore, a clear trade-off is necessary between the theoretical capacity and reduction potential to apply this strategy.
3.1.1.3 Entrapping within the insoluble substrate. The cycling stability of the small molecule could be improved by impregnating the redox small molecule within an insoluble substrate, such as various carbon materials, SiO2 nanoparticles, etc. as shown in Fig. 4. The thought behind this strategy is that strong interaction (which could be physical, chemical, or both) between the substrate and the organic molecule could reduce the dissolution. Li et al.57 applied this strategy by impregnating bis-naphthoquinone within mesoporous carbon CMK-3 to improve the cycling stability of bis-naphthoquinone. The observed capacity was ∼100% of its theoretical capacity at 0.1C and it retained ∼66% of the initial capacity after 50 cycles. This strategy no doubt improves the cycling stability and active material usage but cannot relieve the dissolution problem completely. Another example of this strategy is the lumiflavine (LF) composite with single-walled carbon nanotubes (SWCNTs), where pi–pi interaction (physical interaction) occurs between the aromatic structured LF and nanotubes.56 This approach could be extended to anthraquinone also. The composite with 50 wt% of LF retains a high capacity of 99.7% after 100 cycles but increasing the LF loading beyond 60 wt% causes poor cycle performance, which may be due to the lower pi–pi interaction with excess LF on SWCNTs. Therefore, one of the drawbacks of this strategy is the limited loading of active mass, which certainly limits the energy density. In addition to carbon materials, Lin et al. employed SiO2 nanoparticles to make a composite with poly(2,2,6,6-tetramethylpiperidin-1-oxyl-4-yl methacrylate) (PTMA).58 Although this is not an example of a single molecule, we are only discussing this example here to emphasize the similar strategy. Here PTMA is grafted covalently on the nanoparticles. Thanks to this strong (chemical) interaction, which controlled the dissolution of active PTMA, it showed a long cycle life of 300 cycles. So, impregnating (grafting) organic molecules on conductive insoluble substrates could enhance the capacity and cycling stability, which may need to improve further for practical use. A combination of this strategy with other unique strategies will be required to deal with the solubility issue of small molecule cathodes.
 | ||
| Fig. 4 (a) Schematic of the electrochemical redox mechanism of bis-naphthoquinone and electrode reaction of the bis-naphthoquinone/CMK-3 nanocomposite, (b) cycling performance and the corresponding coulombic efficiency of the bis-naphthoquinone/CMK-3 nanocomposite at a rate of 0.1C, (c) capacity retention of LF-SWNT electrodes and pristine LF electrodes, (d) pi–pi interaction of the organic nanolayer of LF on SWNTs, (e) synthetic route of PTMA-based brush/SiO2 composites. The figures are reproduced from ref. 56–58 with permission. © Royal Society of Chemistry, John Wiley and Sons, Elsevier. | ||
3.1.1.4 Partial replacement of carbonyl to thiocarbonyl. It was observed that partial replacement of the carbonyl group in anthraquinone by a thio group (from thiocarbonyl) could enhance its cycling stability and working potential.62 This material retained 33.8% of the initial capacity after 40 cycles and showed two discharge plateaus at 2.7 and 2.1 V vs. Li/Li+. Although it improved the cycling stability, working potential, and charge carrier mobility of anthraquinone, the stability is far from ideal due to the side reaction and dissolution.
3.1.1.5 Introducing nitrogen-containing heterocyclics. It is another way to improve the voltage of organic carbonyl cathodes without having a negative effect on the theoretical capacity. Shimizu et al.63 investigated this effect on anthraquinone and phenanthrenequinone in lithium-ion batteries as shown in Fig. 5. They found that introducing “N” at 1, 4, 5, and 8 positions enhances the reduction potential of phenanthrenequinone from 2.52 V to 2.73 V and 2.94 V vs. Li/Li+. But the capacity fadeout issue of those molecules still persists. Therefore, combining this strategy with other strategies, such as porous carbon entrapping or the addition of an ionic group (EWG) could improve the small molecule electrode performance.
 | ||
| Fig. 5 The average voltages of nitrogen-containing heterocyclics, anthraquinone and phenanthrenequinone. | ||
3.1.1.6 Crystalline nanostructure. Besides the molecular engineering approach, this is another method to tune the cycling stability and electrochemical performance of the small organic molecule. Crystalline nanowires of croconic acid disodium salt (CADS) not only overcome the barriers of low electronic conductivity of CADS and lithiation-induced strain but also exhibit much better electrochemical performance than their crystal bulk material and microwire counterpart.64 As shown in Fig. 6, the crystalline nanowire could retain ∼100% of its initial capacity after 110 cycles at 0.2C.
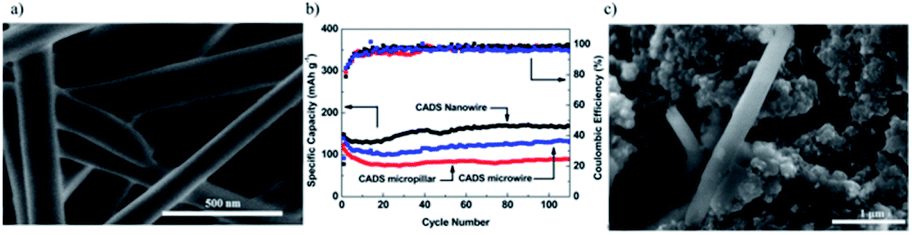 | ||
| Fig. 6 (a) SEM images of CADS nanowires, (b) cycle life of CADS micropillars, CADS microwires, and CADS nanowires, (c) SEM images of CADS nanowires after 100 cycles. The figures are reproduced from ref. 64 with permission. © American Chemical Society. | ||
3.1.1.7 Aromaticity tuning. To improve the voltage of the small organic molecule cathodes, Wu and co-workers65 studied the correlation between the aromaticity (electron delocalization) and the discharge potential of carbonyl-containing polycyclic aromatics. Their study revealed that high discharge potential could be achieved if the aromaticity increases after lithiation. This aromaticity change is expressed by the average change of Clar sextet numbers (ΔC2Li):
| ΔC2Li = ΔC/(0.5 × ΔLi) |
 | ||
| Fig. 7 Correlation between the change in aromaticity and the reduction potential of the small molecule anthracene with carbonyl groups where the arrow signifies the migration of Clar sextets. The figure is reproduced from ref. 65 with permission. © Royal Society of Chemistry. | ||
3.1.1.8 Impact of phase change. Phase transformation during the charge–discharge cycle could also affect the capacity of organic materials. For example, during the sodiation process the alpha-phase of disodium rhodizonate (Na2C6O6) transforms into the gamma-phase, but the reverse phase transformation during desodiation is kinetically suppressed as observed by Bao et al.39 This irreversible phase transformation results in the low practical capacity of Na2C6O6. Bao and co-workers revealed that without molecular design, reducing the particle size and optimizing the electrolyte could deliver a capacity value (484 mA h g−1) very close to the theoretical capacity (501 mA h g−1, 4Na). A similar report of capacity fadeout and cycling instability on dilithium rhodizonate (Li2C6O6) due to crystal structure change was published by Kim and co-workers.41 Therefore, not only molecular design but also crystal structure design and electrode morphology are crucial factors in enhancing the performance of carbonyl systems.
Simple quinone compounds usually show serious capacity fading and as a consequence generate a shuttle effect. This is due to the organic species dissolution in the electrolyte. As we noticed, several strategies are applied to tackle this problem, such as molecular design, loading with advanced carbon mass, and phase change. In addition to these, there are other possible strategies such as increasing the viscosity of the electrolyte, increasing the polarity of the salt, and polymerization. In the following section, we will show how polymerization of the small electrode molecule could prevent the dissolution of the electrode material.
3.1.2.1 Position of the carbonyl group: ortho vs. para. Within the various molecular engineering approaches, Gottis et al.67 introduced a smart way to increase the voltage of carbonyl group-containing molecules without adding EWGs or electronegative atoms. So, this approach will not affect the theoretical capacity of the parent molecule. The authors found that the presence of carbonyl groups in the ortho position of the lithiated enolates delivers a voltage of 300 mV over its para position (as shown in Fig. 8(a)) due to the favourable coulombic interaction. Also, they observed that this enolate retains ∼100% of its initial capacity after 30 cycles.
 | ||
| Fig. 8 (a) Charge–discharge curves of dihydroxyterephthaloyl derivatives with two –OLi in para and ortho positions, (b) SEM image of the N-doped C-coated sphere of lithium terephthalate, (c) the cycling performance of the bulk lithium terephthalate (PTALW), porous spray-dried lithium terephthalate (PTALS) and carbon-coated spray-dried lithium terephthalate (PTALS6). The figures are reproduced from ref. 66 with permission. © Royal Society of Chemistry. | ||
3.1.2.2 Electrode processing methods. The electrochemical performance of the carboxylate group molecules could be enhanced by changing the processing methods. Zhang et al.66 reported that synthesized lithium terephthalate porous microspheres (via a spray drying method) with an N-doped carbon layer coating could improve not only the cycling stability and rate capability of the resultant electrode but also the electronic conductivity and diffusion of lithium ions. At the lowest rate of 0.05C, it gives the highest specific capacity of 259 mA h g−1, and 67.9% of this capacity is retained after 50 cycles (Fig. 8). Zhang et al.66 observed that these performance values are higher compared to standard electrode processing and formulations.
3.1.2.3 Conjugated carboxylates: linear vs. cyclic. Lee and co-workers observed an interesting effect of conjugation in linear as well as cyclic carboxylate compounds.68 In the case of the cyclic system, dilithium terephthalate, Li2TP, during discharge, shows one plateau at 0.81 V and another sloping plateau, from 0.8 to 0.0 V. It delivers a specific capacity (522 mA h g−1 at 30 mA g−1) ∼73% higher than its theoretical capacity based on one lithium insertion per carboxylate group. The thiophene derivative of this family, dilithium thiophene-2,5-dicarboxylate, Li2TDC, also delivers a very high specific capacity of 850 mA h g−1. However, its linear system, Li2C6H6O4, does not deliver the excess capacity. By performing the C13 isotope labeling experiment, Lee et al.68 concluded that the excess capacity is coming from the internal alkene of the cyclic system, which can accommodate the extra lithium ions. But the linear system breaks the conjugation of the structure. The charge–discharge mechanism of these molecules with the corresponding reduction potentials is shown in Fig. 9. Based on this mechanism, the “super-lithiated” compound was also explored,47 where the obtained capacity was very high. But its long sloping discharge plateau limited its application in practical use, so the charge storage was limited. Therefore “conjugation” could be an efficient way to improve the capacity of small molecule electrodes.
 | ||
| Fig. 9 (a) Mechanism of extra capacity in conjugated carboxylates of the cyclic Li2TP system compared to the linear Li2C6H6O4 system, (b) extended conjugation of Li2TP to the 2,6-naphthalene dicarboxylate molecule and SBDC to SSDC, (c) lithium-intercalated crystal structure of 2,6-Naph(COOLi)2 and the structure of Li-doped naphthalene rings with π-stacking (intercalated Li = orange, Li = yellow, O = red, C = blue, and H = white), (d) cycling performance of K2TP at 1 A g−1, (e) schematic diagrams for the proposed electrochemical reactions during sodiation/desodiation of PTCDA. The figures are reproduced from ref. 45, 72, 73 and 76 with permission. © Royal Society of Chemistry, American Chemical Society, WILEY-VCH. | ||
3.1.2.4 Extension of conjugation. We mentioned in the previous section that conjugation could improve the capacity of small molecules. Now we will see the effect on the performance if we extend the conjugation. It is observed that the extension of electronic conjugation between the carboxylate can increase the rate capability of 2,6-naphthalene dicarboxylate compared to Li2TP.69 It gives a specific capacity of 200 mA h g−1 at 0.1C and 176 mA h g−1 at 1C and retains ∼65.3% capacity after 50 cycles at 1C. Perylene tetracarboxylate is another example in this context of achieving high rate capability, by extending the aromatic core between carboxylate groups.70 This compound could achieve 95% of its theoretical capacity (233 mA h g−1) at 1.25C and could retain ∼47% of it at 1.25C after 100 cycles. Therefore, the authors improved the electrode rate capabilities by using a large aromatic core at the cost of cycling stability. In this effort, another possibility is formation of a highly crystalline electrode, 2,6-naphthalene dicarboxylate dilithium. Here the metal–organic framework (Fig. 9) helps to pack the aromatic ring closely, which eventually enhances the electronic and ionic conductivity of the electrode. The metal–organic framework of this compound retains 100% capacity after 10 cycles at 0.1C.71,72 However, the number of cycles in the experiment is too low to comment on the high cycle stability.
Extended π conjugation is also important to enhance the intermolecular interactions and layer-by-layer packing, which could be a fast route for alkali ion diffusion between two layers as well as charge transport. Introducing a carbon–carbon double bond linkage in sodium benzene-dicarboxylate (SBDC) could extend its π-conjugated system to form a sodium 4,4′-stilbene-dicarboxylate (SSDC) compound73 (Fig. 9). SBDC exhibits a specific capacity of 260 mA h g−1 at 50 mA g−1 and 72 mA h g−1 at 10 A g−1 and SSDC shows 192 mA h g−1 at 50 mA g−1 and 22 mA h g−1 at 10 A g−1 73. The cyclability performance under a high current density of 1 A g−1 showed that the capacity is still higher than 112 mA h g−1 even after 400 cycles with retention higher than 70%. These results imply that the extension of the π-conjugated system is an efficient strategy to improve the high rate performance.
3.1.2.5 Degree of deprotonation. In the effort to enhance the cycle stability, the degree of deprotonation is another alternative way. For example, the degree of deprotonation of the carboxylic acid (COOH) groups in 4,4′-biphenyldicarboxylate (bpdc) sodium salts affects their electrochemical performance and corresponding reaction mechanisms.74 Choi et al.74 observed that bpdc needs to be fully deprotonated to exhibit a promising electrochemical performance with a reversible capacity of about 200 mA h g−1 at ca. 0.5 V vs. Na/Na+, stable cycle performance over 150 cycles, and excellent rate performance of 100 mA h g−1 even at a 20C rate.74 The electrochemical performance is better in the fully deprotonated bpdc-sodium salt than in the partially deprotonated bpdc-sodium salt and complete deprotonation is required for good stability during cycling.
3.1.2.6 Choice of counter ion. It is observed that the choice of the counter ion itself is also important to tune the cycle stability of the small molecule electrode. For instance, the carboxylate derivative, benzene diacrylate as an anode for the sodium-ion battery shows the capacity fading problem. It delivers a specific capacity of 177.7 mA h g−1 at 0.025C with a 91% coulombic efficiency, which decreases to 40 mA h g−1 after 40 cycles.75 This decrease in capacity is due to the dissolution of the material in the electrolyte. But its lithiated form does not show severe capacity fading. The authors suspected that this is because the lithium salts easily form a polymer network, which restricts the dissolution of the active material.
3.1.2.7 Salification. In this strategy, organic materials form organic salts by introducing high-polarity groups to enhance the cycling stability of the materials by limiting their dissolution in the aprotic electrolyte. Organic salts are mostly used in metal-ion batteries, such as Li2TP, a widely considered anode in LIBs.10 Similarly, Na2TP and K2TP are used for SIBs and KIBs. In fact, the high polarity of –COOK in K2TP was responsible for the high capacity retention of 94.6% over 500 cycles at 1 A g−1 with a coulombic efficiency of 100% (Fig. 9(d)).76,77
3.1.3.1 Capacity enhancement without tuning unsaturated bonds. As an organic semiconductor-based electrode material, perylene-3,4,9,10-tetracarboxylic dianhydride (PTCDA) has been investigated for both Na and Li-ion batteries.45,78,79 Here we will discuss another strategy to enhance the capacity without affecting the unsaturated bond. Along with acetylene black, NaPTCDA yields a specific capacity (361 mA h g−1) higher than its theoretical capacity (273 mA h g−1).78 This extra capacity-gain could suggest a similar mechanism to that observed in the case of Li and aromatic anodes, an additional reaction between the unsaturated carbon and alkali metal. Surprisingly, the same mechanism is not observed in NaPTCDA. The authors suggested that the extra capacity is attributed to the formation of a solid electrolyte interphase (SEI) layer resulting from the irreversible reaction with the electrolyte.78 Wang et al. showed that the PTCDA electrode could retain a reversible capacity of 250.5 mA h g−1 and coulombic efficiency of 100% after 140 cycles, corresponding to a capacity retention ratio of 40.4%.78 On the other hand, Wei et al. showed that PTCDA could adjust the amount of Na-insertion by fixing the discharge voltage as shown in Fig. 9. At the discharge voltage of 0.01 V, PTCDA could insert 15 Na ions and delivered a capacity of 1017 mA h g−1.45 Authors exhibited high electrochemical reversibility in forming Na2−xPTCDA (0 ≤ x < 2) only without any modification commercial PTCDA sub-micrometer rods.45 PTCDA is a prime candidate for the SIB anode compared to most SIB anodes, including hard carbon, alloys, and metal oxides or sulfides. But the cycling stability needs to improve further for it to be a highly promising practical anode material for SIBs.
3.1.3.2 3D hybrid structures for retention of capacity. Yuan et al.80 suggested nanoengineered ultralight cathode materials based on a PTCDA and graphene aerogel composite, namely, the PGC40 composite cathode, where PTCDA molecules were confined in a 3D hybrid architecture. This composite cathode material involves a two-step redox reaction and delivers a specific capacity of 202 mA h g−1 at 50 mA g−1 and 64 mA h g−1 at a high current density of 2000 mA g−1. Also, it still exhibited a reversible capacity of 78 mA h g−1 after 500 cycles even at a high current density of 1000 mA g−1 with a capacity retention of 66%, which is far better than the pristine PTCDA. These enhanced electrochemical properties of PGC40 come from its special 3D structure, which not only improves the conductivity of both electrons and Li-ions, but also effectively prohibits the dissolution of the active material into the electrolyte.
3.1.3.3 Increasing the molecular weight. Wang et al.81 studied four anhydride organic compounds to reveal the essential reason for the rapid capacity attenuation. They found that the reason was the loss of active material due to the formation of an easily dissolvable dilithium compound during the discharge reaction. Out of the four anhydride compounds of different molecular weights, the lowest molecular weight compound showed faster attenuation of the discharge capacity and poorer electrochemical cycle performance. This observation implies that the increasing molecular weight of the active material could reduce its dissolution in the electrolyte.
 | ||
| Fig. 10 (a) Reversible and irreversible processes in the diimide system, (b)charge–discharge voltage profile of PDI and its Br derivatives. The figures are reproduced from ref. 82 with permission. © WILEY-VCH. | ||
3.1.4.1 Influence of the active ion. Disodium pyromellitic diimidate shows poor capacity retention with a steady decrease in capacity during cycling.83 Interestingly, a similar effect is not observed in the case of its Li-homologue. Brandell and co-workers suggested that this decomposition occurs due to the highly concentrated negative charge on the molecules during cycling instead of the solubility of the active material in the electrolyte.84 This kind of negative charge accumulation is not observed in the case of its Li-homologue as the high-affinity of Li+ ions can shield and well compensate the negative charge.
3.1.4.2 Effect of substituents and functionalization. Different substituent groups are used to enhance the voltage of naphthalene diimides (NDIs). Vadehra and coworkers showed the effect of this strategy on NDIs with and without functionalization of diimide nitrogens with hexyl groups.85 In both cases, they observed a change in voltage. Out of all considered substituents (Me2N, F, CN), cyanide could increase the voltage from 2.5 V to 2.9 V. But it was also reported that with hexyl functionalization NDIs lose the capacity rapidly due to dissolution. This work also reported that the performance of unsubstituted NDIs was poor due to an unfavorable crystal packing. The performance could be increased further by reducing the domain size of the active material and increasing the homogeneity. Bhosale et al. showed that the performance of hydrazine-treated benzoic acid-functionalized NDIs could be improved compared to the untreated one.86 It was observed that hydrazine increases the conductivity and rate capabilities with 85 mA h g−1 specific capacity at 1C and 68 mA h g−1 at 10C with 88% capacity retention after 200 cycles at 5C. Therefore, this hydrazine treatment could be a useful strategy to improve the rate capabilities of other NDI analogues having high theoretical capacities.
3.1.4.3 Without excess mass. Cycling stability could be increased without the addition of excess mass in terms of substituents or functionalization. Chen et al. showed that designing a triangular-shaped NDI could lower the solubility and enhance the cycling stability with 60% retention of capacity after 300 cycles at 10C.87 Also, it delivers a specific capacity of 146.4 mA h g−1 at 0.1C and 58.1 mA h g−1 at 100C. Benefits of these triangular arrangements are fast diffusion of lithium-ion through the triangular channel (which ensures high rate performance), and high electronic conductivity due to the electronic coupling of the redox unit. The triangular-shaped NDI is slightly soluble in the neutral and reduced states, which could be improved by replacing NDI with the less soluble perylene diimide (PDI).
3.1.4.4 Molecular engineering to shape the discharge curve. A stable voltage output of organic molecules could be achieved by tuning the shape of the discharge curve by molecular engineering. Banda et al. discovered how the dihedral angles in PDI could affect the shape of the discharge curve.82 They increased the dihedral angles by increasing the number of Br substituents in the molecule. It was observed that (Fig. 10) PDI with one and two Br displays two discharge plateaus, whereas PDI with three and four Br shows one discharge plateau.
3.1.5.1 Suppression of dissolution by di-imide polymers. Arylene di-imide incorporated non-conjugated polymeric materials have been studied as cathodes. In an effort to design a flexible and free-standing binder-free cathode, Wu and co-workers performed an in situ polymerization of pyromellitic dianhydride with ethylenediamine and SWCNTs to form polyimide.88 This material exhibits good rate capabilities with a high Csp of 226 mA h g−1 at 0.1C and 120 mA h g−1 at 20C and retains 85% of its initial capacity after 200 cycles at 0.5C due to its insolubility in the electrolyte. Wang et al. studied a series of different non-conjugated arylene di-imide polymers with pyromellitic, NDI, and PDI cores with different lengths of diamine spacers.8 A decrease in Csp was observed with increase in the alkyl spacer length from propyl to butyl with the PDI core. Most importantly, it was reported that the cycling stability increases by increasing the size of the arylene core. The capacity fading issue, especially dissolution could be suppressed by using arylene di-imide polymers with N-atoms. But the cycling stability still needs to improve to serve the practical purpose. An interesting example of this non-conjugated polymer class is a dendronized polymer. Kim and co-workers synthesized a dendronized polymer with anthraquinone groups pendant to the dendrons as a cathode.89 Although it exhibits high capacity retention (∼90–95% after 100 cycles at 0.5C). But electrical conductivity is an issue that may be caused by large polymer domains that are electrically isolated from the conductive pathway of the carbon black particles.
The non-conjugated polymer form is also considered to improve the anode performance. For example, 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA)-derived polyimide has been proposed as the anode material for aqueous rechargeable lithium-ion or sodium-ion batteries.90 The polymeric nature undoubtedly aids in the cycling stability by preventing dissolution. In the case of a lithium electrolyte, it has a 160 mA h g−1Csp at 100 mA g−1 and as a full cell with a LiCoO2 cathode, it delivers 71 mA h g−1Csp with an operating potential of 1.12 V. With a sodium electrolyte, the Csp is 165 mA h g−1, and in a full device with NaVPO4 as the cathode, the Csp is 40 mA h g−1. It is also observed that the capacity decay is faster in the NaVPO4F electrode, ∼30% capacity drop within 20 cycles.
In general, non-conjugated polymers are used less often as anode materials for metal-ion batteries. This is because the backbone is susceptible to decomposition at low voltages and therefore, they are mostly used as aqueous battery anodes.
3.1.6.1 Polymerization with and without sulfur. Chloroanilic acid and dilithium chloranilate, when polymerized with sulfur, exhibit, respectively, 214 mA h g−1 and 247 mA h g−1Csp at 50 mA g−1, whereas without polymerization, they exhibit 119 mA h g−1 and 193 mA h g−1Csp at 50 mA g−1, respectively.91 It is also observed that the polymerization increases the cycling stability, e.g. the lithiated derivative retained 90% of the capacity after 1500 cycles at 500 mA g−1. In a follow-up study, Song and co-workers polymerized different isomers of anthraquinone with either sulfur or by condensation polymerization.92 Out of all synthesized polymers, they observed that the 1,4-isomer of anthraquinone formed by condensation polymerization is most promising with 263 mA h g−1Csp at 0.2C and 98.3% capacity retention after 100 cycles at 0.2C. Poly(anthraquinonyl sulfide) (PAQS) shows similar cycling stability and 98.4% capacity retention after 100 cycles at 0.2C. But the discharge capacity is low, 213.8 mA h g−1Csp at 0.2C. They also reported93 that poly(benzoquinonyl sulfide) as a sodium-ion battery cathode, has 268 mA h g−1Csp at 50 mA g−1 and 68% capacity retention after 100 cycles at 500 mA g−1. Therefore, these reports suggest that polymerization with sulfur is an attractive strategy to design stable and high-performance organic cathodes.
3.1.6.2 Donor–acceptor copolymers. Liang and co-workers introduced donor–acceptor copolymers for use in lithium-ion battery electrodes.94 Donor–acceptor copolymers such as poly{[N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl]-alt-5,5′-(2,2′-bithiophene)} or (P(NDI2OD-T2)) and poly{[N,N′-bis(2-octyldodecyl)-1,4,5,8-naphthalenedicarboximide-2,6-diyl]-alt-5,5′-[2,2′-(1,2-ethanediyl)bithiophene]} or (P(NDI2OD-TET)) have also been studied as ultrafast lithium-ion battery cathodes. The authors observed 96% capacity retention after 3000 cycles for P(NDI2OD-T2). Low theoretical capacities of 54.2 and 52.7 mA h g−1 were observed for P(NDI2OD-T2) and P(NDI2OD-TET), respectively. They also observed that interruption of polymer conjugation with a saturated ethylene linker lowers the rate capability. Therefore, the main drawback here is Csp, which could be enhanced by removing the solubilizing alkyl chains.
3.2 Porous organic polymers
Organic electrode materials have attracted considerable interest mainly owing to their outstanding features, namely remarkable electrochemical capabilities, low costs as well as versatility of the geometrical structure. Notably, the conjugated molecules have been recently promoted to serve as prospective electrodes for upcoming advanced rechargeable batteries with high-performance electrochemical properties.9,95 In this regard porous organic polymers, abbreviated as POPs, relying on rigid covalent lattices providing high stability with wide pores allowing easier ion transfer from the electrolyte, represent exciting prospects for rechargeable battery electrodes.96,97 Furthermore, the functional POPs displayed enhanced Li/Na/K+ conductivity with enormous prospects for serving solid-state electrolyte materials. In this section, we will discuss the recent developments in the application of porous organic polymers in various advanced batteries, including K-ion and Li–S batteries. Table 1 summarizes the electrochemical performances of select POP electrodes and gives a comparison with other electrodes.| Rechargeable battery type | Selected electrodes | S BET (m2 g−1) | Capacity (mA h g−1) | Current energy density | Long-term performance | Voltage (V) |
|---|---|---|---|---|---|---|
| a —: SBET data are not available in the literature. | ||||||
| Li-ion batteries | Li2TP69 | — | 200 | 0.1C | 50 cycles | 0.88 |
| Benzoic-NDI86 | — | 85 | 1C | 200 cycles | 2.9 | |
| mp-COF-graft-PS106 | 14.8 | 425 | 250 mA g−1 | 500 cycles | 1.7–3.0 | |
| DTP-ANDI-COF@CNTs107 | 478 | 67 | 2.4C | 700 cycles | 1.5–3.5 | |
| NT-COF99 | 1276 | 124 | 10 mA g−1 | 100 cycles | 2.7 | |
| TFPB-COF99 | 458 | 968 | 100 mA g−1 | 300 cycles | 0.005–3 | |
| E-TFPB-COF/MnO2 (ref. 100) | 345 | 1359 | 100 mA g−1 | 300 cycles | 0.005–3 | |
| ADALS35 | — | 190 | 0.5C | 100 cycles | 0.61–1.3 | |
| PBQS93 | — | 275 | 50 mA g−1 | 1000 cycles | 1.5–4.0 | |
| Na-ion batteries | SBDC73 | — | 260 | 50 | 400 cycles | 0.18–2.5 |
| SSDC73 | — | 192 | 50 | 400 cycles | 0.1–2.5 | |
| PTCDA78 | — | 361 | 25 mA g−1 | 140 cycles | 0–3.0 | |
| CTF-HUSTs109 | 764 | 467 | 50 mA g−1 | 500 cycles | 0.05–2.0 | |
| TFPB–TAPT COF108 | 120 | 246 | 30 mA g−1 | 500 cycles | 0.001–3.0 | |
| ANDASS141 | — | 170 | 0.2C | 1000 cycles | 0.5–2.5 | |
| PBQS93 | — | 268 | 50 mA g−1 | 100 cycles | 1.0–3.5 | |
| K-ion batteries | PyBT CMP121 | 493 | 428 | 30 mA g−1 | 500 cycles | 0.1–3 |
| SHPNC122 | 693–1798 | 318 | 25 mA g−1 | 370 cycles | 0.01–3 | |
| Li–sulfur batteries | CTF-1 (ref. 123) | 789 | 848 | 0.2C | 50 cycles | 1.1–3.0 |
| Por-COF124 | 1095 | 633 | 0.5C | 200 cycles | 1.8–2.7 | |
We have already mentioned that the implementation of organic materials as cathode batteries has some advantages such as low-cost and chemically tuneable characteristics, but they still face some disadvantages, most notably, dissolution of the redox-active material in the electrolytes, suppression of the electrolyte-ion transport in the cathodes, as well as very lower conductivity.30,101,102 To date, POP materials as battery cathodes offer significant strengths as they feature well-ordered open pores for ion transport from the electrolyte, small volume expansion upon the insertion of Na+, as well as robust stability.73,103,104 Notwithstanding the fact that POPs provide several benefits as cathode materials in Li-ion rechargeable batteries, they mostly suffer from low redox potentials and low conductivity thus constraining their applicability in LIBs.105
To tackle the above concerns, a variety of approaches have been investigated, such as the introduction of redox-active monomers, insertion of redox-active modules by means of post-synthetic functionalization of POP materials, boosting their conductivity behavior, and introducing various active materials to hybridize POP materials.
 | ||
| Fig. 11 (a) Chemical configuration of mp-COF & mp-COF-graft-PS structures; (b) DTP-ANDI-COF structure, inset: redox process of NDI; (c) NT-COF structure, inset: CV of NT-COF in the presence and absence of light; (d) TFPB-COF structure; inset: schematic-illustration of the chemical-exfoliation of COFs. Figures are reproduced from ref. 98–100 with permission. © 2018 WILEY-VCH, 2019 WILEY-VCH, 2020 WILEY-VCH. | ||
On the other hand, Na-ion batteries based on COFs as battery electrodes have also been investigated in recent years. K. Wang et al.109 have introduced a new strategy to construct covalent triazine frameworks (CTF-HUSTs) through the polycondensation method with many interesting properties such as layered structures with a wide inter-layer distance of about 3.9 Å, large surface areas, and adjustable functions. Particularly, CTF-HUST-4 has been revealed to be a prospective electrode material with an excellent discharge capacity of approximately 467 mA h g−1 for sodium-ion batteries.109 Furthermore, Pradhan et al. have reported a new COF material made up of 1,3,5-tris-(4-formylphenyl)benzene (TFPB) and 1,3,5 tris-(4-amino phenyl)-triazine (TAPT) with C3–C3 symmetrical topology,108 denoted as TFPB–TAPT COF, as a sustainable anode material for sodium-ion batteries. As shown in Fig. 12, the TFPB–TAPT COF revealed an outstanding electrochemical performance including a good reversible capacity of 246 mA h g−1, excellent cycle stability up to 500 cycles and a very high Na+-storage capability at several current rates owing to its wide-open ordered porous framework.108 The outcomes of this study suggest that the redox-active layered 2D TFPB–TAPT COF shows high promise not only as an anode for SIBs but also for diverse alkali metal batteries.
 | ||
| Fig. 12 The electrochemical efficiency of TFPB–TAPT COF organic anode materials in sodium-ion batteries. (a) The model for reversible Na+-storage upon charging, (b) charge and discharge process profiles for three different cycles, and (c) rate efficiency at various current rates. Figures are reproduced from ref. 108 with permission. © 2016 Royal Society of Chemistry. | ||
Conjugated microporous polymers (CMPs),117,118 which serve as a strong candidate for addressing the growing environmental and energy related concerns, have been extensively probed for various applications. As a consequence of the widespread π-conjugation throughout the polymer skeleton, outstanding physico-chemical stability, highly porous nature along with increased surface area, CMPs have emerged as prospective high-energy storage materials.119,120 Jiang et al. successfully synthesized two groups of CMPs with distinct configurations (Fig. 13(a and b)), and investigated their possible use as negative electrodes for K-ion batteries.121 Some influencing factors such as the electronic structure and the lowest unoccupied molecular orbitals of the polymer skeleton have a great impact on the performance of the K-ion batteries. The de-localized LUMO not only enables remarkable redox activity due to a high degree of charge de-localization across the polymer skeleton, but also simultaneously decreases the charge density of redox-active sites. Consequently, K-ion batteries with PyBT-CMPs have shown increased charge de-localization, and small electronic bandgaps, and have achieved a large specific capacity of about 428 mA h g−1 along with smooth cycling stability as depicted in Fig. 13(c and d). A strategy for effective conception of CMPs to serve as anodes of high-performance K-ion batteries has been presented within the context of this study.121
 | ||
| Fig. 13 (a and b) The polymer structures of both benzene-containing and benzothiadiazole-containing conjugated microporous polymers. (c) The electrochemical performance of PyBT at a current density between 30 and 500 mA g−1. And (d) the cycling stability/coulombic performance of PyBT at 50 mA g−1. The figures are reproduced from ref. 121 with permission. © 2019, American Chemical Society. | ||
More recently, a new hierarchically porous nitrogen-doped carbon (SHPNC) has been synthesized via a cocoon-silk chemistry technique as a potential anode material for batteries by Luo and co-workers.122 Notably, both the hierarchically porous nature and the high n-doping within the SHPNC enable a high rate of ion as well as electron transport, serving a significant purpose in buffering K-ion volume changes, and further extending the electrochemically binding sites. Accordingly, the capacity versus cycling number at varying charge–discharge current rates have been probed within a potential voltage window of about 0.01–3.00 V versus K/K+. Additionally, as the current density drops to 25 mA g−1, roughly 93% of the discharge capacity can be recovered with a reversible specific capacity of about 318 mA h g−1 achieved over 200 cycles, which indicates the high stability of SHPNC as an anode for KIBs.
Liao et al.123 used COFs for the first time as host materials for S-containing cathodes to address the problem caused by the loss of soluble lithium polysulfide intermediates that occur in the discharge process.123 The composite covalent triazine-based framework (CTF-1) was made up with the melt – diffusion technique. A high capacity of about 848 mA h g−1 at a current density of 0.2C has been achieved. Nevertheless, a very small sulfur charge of 34% has been achieved in CTF-1. Furthermore, CTF as the host for the S-cathode shows capacity retention of 64% after 50 cycles at charge/discharge performance of 0.1C.123 Subsequently, for the purpose of increasing the sulfur loading and cycle stability, they designed another type of COFs, a 2D porous porphyrin-based COF (Por-COF) with a comparatively large pore volume and small pore size distribution. Por-COF as a host-material for sulfur storage in LiSBs has demonstrated a superior capacity of 670 mA h g−1 at charge/discharge rates of 1.0C with high cycle stability.124
3.3 Organometallic polymers
Organometallic polymers having organic binders and transition metal ions exhibit bifunctional characteristics of both organic and metal components. They possess distinct electrochemical features such as high capacity and active metal sites of inorganic components. Moreover, most of these materials are not soluble in the electrolyte. Therefore, organo-metallic polymers could serve as potential electrodes for rechargeable batteries, mainly Li- and Na-ion batteries and high-performance Li–S batteries. At present, a wide range of organometallic polymers have been explored as battery electrodes, and they are mainly divided into following groups: metal–organic frameworks, porphyrins, phthalocyanines, ferrocene, and so on.125,126 MOFs consist of porous materials featuring a uniform structure made up of metal-ions or clusters, acting as joints, connected by multi-directional organic ligands, which serve as linkers in the lattice structure. They feature improved characteristics such as high porosity, wide surface area, and numerous metal-centers, therefore, they have been widely employed for electrochemical energy-based technologies.126,127 Over the years since 2006, MOFs have been consistently presented128 and extensively explored for rechargeable battery applications. In 2013, Satoshi et al. reported the ionic conductivity and transport of MOF materials for batteries and fuel-cells.129 Recently, Pang et al. reviewed MOF-based materials as a potential candidate in energy storage technologies with fascinating electrochemical performances for metal-ion and advanced Li–S batteries.130 Wang and co-workers have suggested a new strategy to use transition-metal-based MOFs and their derivatives as potential anode materials for Li-ion batteries due to their adjustable structure, large surface area, and highly porous structure.131 The transition metal in MOFs is susceptible to inducing many different valence variations in redox reactions, thereby leading to increased capacities. Furthermore, MOFs could serve as host materials for sulfur-cathodes by virtue of their tuneable porosity. Also, MOFs as battery electrodes have the ability to sieve the ions which is advantageous for uniform deposition of Li-ion thereby preventing dendrite growth in lithium metal anodes. The porphyrin complexes involve the bonding of metal ions in the middle together with the four nitrogen atoms in the hetero-cycles. In this process, the metal-ions act as electron-acceptors while the nitrogen atoms act as electron-donors and thus display redox activity132 as shown in Fig. 14. Within alkali metal-ion batteries, the porphyrin is subjected to a transformation from anti-aromatic into aromatic during the redox processes.132 Likewise, phthalocyanines consist of a macro-cyclic structure with up to eight N-atoms and eight C-atoms. The metal-ions and N-atoms in the middle can absorb lithium-polysulfide, thereby inhibiting the shuttling effect in Li–S batteries. Additionally, ferrocene compounds consist of dicyclopentadiene and ferrous-ions (hereinafter referred to as Fe2+).133 Accordingly, Fe2+ can be oxidized and inversely reduced in metal-ion batteries. The ferrocene in Li–S batteries can also act as a containment medium for the lithium-polysulfide species owing to its outstanding flexibility, reversibility, and fast diffusion.133 | ||
| Fig. 14 Redox process of antiaromatic dimesitylnorcorrole nickel(II) complex (NiNC) with 16 pi electrons. Reproduced with permission from ref. 132. © 2014, Wiley-VCH. | ||
Metal–organic frameworks (MOFs), consisting of either metal ions or clusters linked to an organic binder via coordinate bonds, have gained considerable attention in the field of electrochemical energy storage based on their high-porosity structure and pseudo-capacitive capabilities upon the inclusion of redox-active metal cores.134,135 The MOFs have been extensively implemented for electrochemical energy storage, either as MOFs or MOF-derived carbonaceous materials.136 Notwithstanding recent progress,135,136 their applicability in metal-ion and Li–S batteries has been limited due to the low electrical conductivity and poor stability of pristine MOFs.137–139 Therefore, further investigation and characterization of highly stable MOFs as electrode materials with both high-power density and increased energy density, as well as improved cycling stability and enhanced electrical conductivity for convenient uses, is essential, and it remains a major challenge.
3.4 Azo based salts
Organic battery electrode materials show promise for sustainable and eco-friendly secondary batteries owing to the light-weight, low-cost, durability, and recyclability of organic-based materials. Nevertheless, conventional organic-based electrodes are negatively affected by their low cycling stability and limited power density. In recent years, a new organic-chemical material in which the azo-group (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–) is part of the molecular structure was widely investigated as an organic battery electrode for large-scale electrochemical storage applications. Luo et al. reported a class of organic anode materials that contain azo-functional groups, namely azobenzene-4,4′-dicarboxylic acid lithium salt (ADALS) having an azo-group in the middle of the conjugated structure for lithium and sodium-ion batteries.35 ADALS has achieved a high capacity of about 190 mA h g−1 at an energy density of 0.5C with up to 90%, 71%, and 56% capacity retention as the current density is raised to 2, 10, and 20C, respectively. Additionally, ADALS retains about 89% of its starting capacity even after 5000 cycles at a current energy density of 20C with a very low capacity decay factor of 0.0023% by cycle, which represents the highest performance among all organic-based anode materials.35
N–) is part of the molecular structure was widely investigated as an organic battery electrode for large-scale electrochemical storage applications. Luo et al. reported a class of organic anode materials that contain azo-functional groups, namely azobenzene-4,4′-dicarboxylic acid lithium salt (ADALS) having an azo-group in the middle of the conjugated structure for lithium and sodium-ion batteries.35 ADALS has achieved a high capacity of about 190 mA h g−1 at an energy density of 0.5C with up to 90%, 71%, and 56% capacity retention as the current density is raised to 2, 10, and 20C, respectively. Additionally, ADALS retains about 89% of its starting capacity even after 5000 cycles at a current energy density of 20C with a very low capacity decay factor of 0.0023% by cycle, which represents the highest performance among all organic-based anode materials.35
Azo compounds as a family of organic anode materials have also demonstrated potential performance for Na-ion batteries.140 Luo et al. have also reported an organic anode material based on azobenzene-4,4′-dicarboxylic acid sodium salt (ADASS) as a new strategy to improve the electrochemical performance of Na-ion batteries.141 ADASS organic anode showed a good reversible capacity of about 170 mA h g−1 at current energy density of 0.2C and retains about 66% and 58% reversible capacities as the current density increases to 10C (1000 cycles) and 20C (2000 cycles), respectively, showing outstanding high-rate capability and long cycle life. Furthermore, it maintains a reversible capacity of 98 mA h g−1 for 2000-cycles at 20C and achieves a low capacity decay rate of 0.0067% by cycle. This is attributed to the presence of a carboxyl group which prevents the azo-benzene from dissolving in the liquid electrolyte.141 Based on energy density, high rate capability and cycle-life, the azo-group materials as battery electrodes surpass many other organic electrodes available for Na-ion batteries, exclusively those based on carbonyl-groups (CO), including Na-terephthalate77,142 and benzo-quinone derivatives,143 as well as imine compounds (CN), namely Schiff's base derivatives.144
3.5 Organic radical polymers
Recently, great attention has been paid to organic radical polymers (ORPs), which have led to several outstanding advances in different areas, including electrochemical energy storage applications. The synthesis techniques currently employed for their development have been widely surveyed.128,140,145–147 The majority of ORPs are made up of two main components: a robust radical pendant and a backbone polymer. In order to be suitable for use in rechargeable batteries, ORPs require long-term cycling stability, insolubility, and affinity in the electrolyte, as well as very high-energy-density.Firstly, the long-term cycling stability of ORP cathodes in full cells remains to be fully explored to evaluate their practical applicability. In this context, one of the most important and challenging criteria to meet in molecular conception consists of the fact that the presence of unpaired electrons in the polymer have to be sufficiently non-reactive in the electrolyte. The representative structures of the radical counterparts are the alicyclic-nitroxyls (such as TEMPO and PROXYL). For instance, the unpaired electrons of the TEMPO framework are capable of sustaining for over a year within an aprotic medium which contains 1.0 M of LiPF6.148
In addition, the geometrical structure of the backbone polymer in the ORPs has an impact on the insolubility and affinity in the electrolyte. In order to avoid contact of the radical polymer with the other electrode, the polymer requires to be insoluble in the electrolyte and to have good affinity for the electrolyte in order to ensure good ionic conductivity. So far, a wide range of backbone polymers, including polymethacrylate,149 polystyrene,150 polynorbornene,151,152 polyether,153 polyacetylene,154 and cellulose,155 have been identified and widely reviewed. Also, the polymer to be employed should be dense in the thick electrode in order to provide a high enough capacity. From the perspective of processability and structural stability, polymethacrylate and polyether have proven to be the highly desirable backbone for ORPs.
Furthermore, the energy density of a rechargeable battery can be determined by the product of the voltage profile and the specific capacity. The voltage profile of ORPs is mainly related to redox reactions. RPs undergo two types of redox reactions, namely the p-type reaction between a radical and a cation and the n-type reaction between a radical and an anion.149,156 While both reactions are appropriate as alternatives to the conventional Li-TMO reaction, the potential voltage of the p-type reaction typically tends to be greater than that of the n-type reaction. Hence, employing a p-type reaction is preferable to achieve high energy density.
4. Future outlook: multivalent alkali ion and dissolution issue
Despite the various scientific achievements, there is still a long way to go before realizing the large-scale applications of organic electrode materials in metal-ion batteries. As discussed above, the implementation of organic electrode materials in rechargeable batteries has been expected to reduce the energy-storage price as well as provide long-term durability.128 More specifically, multi-valent metal–organic batteries have also recently attracted significant interest owing to their large availability as well as the high volumetric energy density of the multi-valent metals, such as Al, Mg, and Ca.157–159 Besides their geo-political abundance over Li-atom, they also have higher volumetric-energy density by virtue of their multi electronic redox features.160,161 Furthermore, the electrodes with multi-valent metals show lower susceptibility to dendrite-growth,162 and this is due to the significantly shorter diffusion profile of multi-valent metals.163 Relative to other active monovalent metals, the multi-valent metals are less reactive in air, thereby substantially reducing the manufacturing cost and could provide better battery stability–safety.164,165 Considering the availability of both organic electrode materials and multi-valent metals, the metal–organic batteries could stand as a sustainable and affordable high-performance rechargeable battery, suitable for large-scale electrochemical energy storage systems.Despite the fact that over ∼30 years, these organic batteries have been studied, still significant interest in the practical implementations of organic electrode materials is hindered mainly owing to their low electrical/ionic conductivity, their dissolution into the electrolyte, their small potential–voltage and their limited specific capacity. For this purpose, several alternative techniques have been derived for improving the electrochemical performance of organic electrode materials in rechargeable batteries through the structural design, polymerization, and amorphization of organic electrode materials. Such approaches were shown to dramatically boost the performance of organic electrode materials in multi-valent metal–organic batteries.166–168 In addition to the above-discussed strategies, this review prescribes the following guidelines, which could help not only to understand but also to overcome the potential drawbacks of organic batteries of monovalent as well as multivalent alkali ions:
Firstly, the combination of computational design and successful synthesis of high-efficiency organic electrode materials having a high-operating potential voltage with extended lifetime capability remains a primary concern. Ideal organic electrodes require a moderate specific capacity of more than 200 mA h g−1, a wide operating potential voltage of over 2.0 V, and an extended lifetime of greater than 1000 cycles in order to be suitable for use in large-scale energy-storage systems.169 A prominent approach involves reducing the molecular weight of the active materials as well as increasing the concentration of redox active sites in the organic electrode surface. Furthermore, the synthesis approaches that are carried out shall be eco-friendly and highly scalable and cost-effective. From the existing literature overview, the highest mean performance for organic electrode materials is apparently exhibited by the carbonyl-based group materials owing to their strongly reversible multi-electron redox features,170 although lowering their solubility as well as increasing their structural strength needs to be addressed properly. Polymerization and formation of alkali ion salts could be effective in improving the cycling performance of organic cathodes. Increase of molecular weight could lower the dissolution, but at the same time it actually decreases the theoretical capacity of the cathode material. Therefore, future research should focus not only on designing the organic cathode materials with lower equivalent molecular weight, but also on developing suitable solid electrolytes with high ionic conductivity and low solubility of organic cathode materials. Furthermore, organic cathode materials possess lower redox potentials with a lower stability window compared to conventional inorganic cathodes, which again lowers the energy density. Here, structural modification and molecular functionalization of organic cathode materials could be performed to further improve their redox potential and provide a competitive energy density. It is noteworthy that recent advancements in theoretical modeling and materials informatics such as machine learning, artificial intelligence (AI) kernel and data mining can not only trigger and narrow down the high-throughput search process more effectively but also suggest potential candidates for synthesis.
Secondly, a deeper insight into the electrochemical storage behavior of organic electrode materials in multi-valent metal–organic batteries needs to be provided. The operation principle of organic materials and electrodes in metal–organic batteries is much more challenging as compared to that in LIBs. For instance, the stored ions on the electrodes (cathode: AlCl4−, AlCl2+ and anode: Al3+) can be different in the multi-valent metal–organic batteries with chlorine containing electrolytes compared to typical inorganic systems for aluminum batteries.171 Both the structural flexibility and poor inter-molecular interactions provided by organic based materials ensure the reversible diffusion and storage of bulky aluminum carrier ions without undergoing significant structural modifications, as in the case of majority of inorganic-based materials.172,173 In addition, organic electrodes typically sustain a storage process through “ionic-coordination” thereby significantly contrasting with the slow kinetics provided by certain inorganic-based compounds resulting from their severe coulombic interactions with Al-carrier ions.174 An exciting development over the past few years has been the rise of a broad class of organic materials for high-performance Al-organic batteries.175–178 Notably, quinones are remarkable for their increased specific capacity, enhanced redox potential and multielectron reactivity. Apart from the para- and/or ortho-quinone, the electronic integration of the contiguous carbonyl groups leads to the creation of negative enolates, which can be conjugated with Al ions to maintain the structure.179,180 The aim of the most recent quinone studies was to increase the number of carbonyl groups within the framework.181 However, studies have revealed that the 2 or 3 adjacent carbonyl groups could be chelated by only a single bulky Al-carrier ion, thus restricting the efficient application of the active sites as well as hindering their electrochemical performance.182 In particular, the nitrogen-containing hetero-aromatic materials are potentially implicated in the reversible redox reactions with Al-carrier ions, although they exhibit a comparatively limited working potential with a poor battery efficiency.183 Regardless of all these efforts, so far there are no available reports regarding the development of organic cathode materials with more than one synergistic active core for Al-organic based batteries, as well as exploring the related redox-reactions is still not fully accomplished. Within this framework, heterocyclic conjugated polymer nano-architectonics which entails molecular engineering of active organic molecules with both CO and/or CN redox active sites along with π-conjugation within a framework has the potential184 to yield a fundamentally novel perspective for designing and developing new innovative electrode-based materials for Al–organic batteries along with enhanced capacitance, long-term cyclability, and superior energy density. Consequently, further investigation efforts are expected to shift towards more sophisticated operando approaches for better insight into the structural modifications of organic electrodes in multi-valent metal–organic batteries.
Thirdly, addition of large amounts (∼30–60 wt%) of conductive carbon is considered to balance the low electrical conductivity of organic electrode materials. Addition of a large amount of conductive carbon also lowers the energy density. Therefore, designing organic electrodes with less than 30 wt% of conductive carbon is one of the bottlenecks for successful realization of organic batteries. At the same time, it is also important to consider as future research focus on the thermal stability of the organic materials, specially the electrode consists of small organic molecules.
In short, research activities of organic cathode materials should focus on the high mass loadings, solid electrolyte design, minimal electrolyte usage, higher potential window, and easy and affordable large-scale production for their commercialization.
5. Conclusions
In summary, this review provides an overview of the most recently explored organic electrode materials for promising mono- and multi-valent alkali-ion based organic batteries with reference to the materials chemistry and the storage mechanisms of alkali metal-ions. Organic electrode materials have attracted considerable focus from the rechargeable battery research community over the last 10–15 years, as evidenced by the wide range of publications on the subject mirrored in this review.Capabilities of organic batteries could frequently exceed the ones achieved using inorganic materials. One feature that is perhaps less debated about with regard to organic materials is that they possess a smaller specific gravity range from 1.5 to 2.2 g cm−3 relative to approximately 4.5 g cm−3 in the case of LiCoO2. Substantial use of conductive additives in the organic batteries could lower the energy density, whereas covalent bonding in the organic molecules could also provide stability. So, there are various facts that could either enhance or suppress the efficiency of organic batteries. This review article discussed possible strategies such as fusing multiple carbonyl groups, introducing nitrogen-containing heterocyclics, crystalline nanostructures, tuning of the aromaticity, electrode processing methods, extension of conjugation, degree of deprotonation, choice of counterion, enhancing the molecular weight, the effect of substituents and functionalization, molecular engineering, polymerization, etc. to address the critical issues related to organic electrodes, i.e. dissolution, capacity fading, lower energy density, and electrical conductivity. To conclude, we believe that organic electrode materials still have suitable physical and chemical properties to be implemented in large-scale high-efficiency rechargeable batteries, if we can overcome the aforementioned problems. Here, we have critically addressed each strategic descriptor to show its effect on the successful realization of organic batteries. It is expected that this study will bring more insight into this area and will refuel the worldwide attention on sustainable energy storage studies, especially to move from academia to practical applications in the foreseeable future.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. B. acknowledges the Indian Institute of Technology Jodhpur Seed grant. N. K., W. L. and R. A. acknowledge the Swedish Research Council (VR-2016-06014 & VR-2020-04410) and J. Gust. Richert stiftelse, Sweden (2021-00665) for financial support. SNIC, HPC2N and IITJ-supercomputer are acknowledged for providing computing facility to visualize the structures. We also acknowledge Dr Deobrat Singh for collecting a few references to part of the carbonyl section of the manuscript titled, “Promise and reality of organic electrodes in alkali ion organic batteries: based on the perspective of the alkali ion storage mechanisms”.References
- R. B. Araujo, A. Banerjee, P. Panigrahi, L. Yang, M. Strømme, M. Sjödin, C. M. Araujo and R. Ahuja, J. Mater. Chem. A, 2017, 5, 4430–4454 RSC.
- S. Lee, G. Kwon, K. Ku, K. Yoon, S. K. Jung, H. D. Lim and K. Kang, Adv. Mater., 2018, 30(42), 1704682 CrossRef PubMed.
- Y. Lu and J. Chen, Nat. Rev. Chem., 2020, 4(3), 127–142 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22(3), 587–603 CrossRef CAS.
- J. R. Owen, T. Le Gall, K. H. Reiman and M. C. Grossel, J. Power Sources, 2003, 119, 316–320 Search PubMed.
- H. Chen, M. Armand, G. Demailly, F. Dolhem, P. Poizot and J. M. Tarascon, ChemSusChem, 2008, 1(4), 348–355 CrossRef CAS PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451(7179), 652–657 CrossRef CAS PubMed.
- H. G. Wang, S. Yuan, D. L. Ma, X. L. Huang, F. L. Meng and X. B. Zhang, Adv. Energy Mater., 2014, 4(15), 1400554 CrossRef.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2011, 11, 19–29 CrossRef PubMed.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribière, P. Poizot and J. M. Tarascon, Nat. Mater., 2009, 8, 120–125 CrossRef CAS PubMed.
- K. Nakahara, J. Iriyama, S. Iwasa, M. Suguro, M. Satoh and E. J. Cairns, J. Power Sources, 2007, 165(1), 398–402 CrossRef CAS.
- D. L. Williams, J. J. Byrne and J. S. Driscoll, J. Electrochem. Soc., 1969, 116(1), 2 CrossRef CAS.
- P. Novák, K. Müller, K. S. V. Santhanam and O. Haas, Chem. Rev., 1997, 97(1), 207–282 CrossRef PubMed.
- D. MacInnes, M. A. Druy, P. J. Nigrey, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1981,(7), 317–319 RSC.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977,(16), 578–580 RSC.
- X. Jia, Y. Ge, L. Shao, C. Wang and G. G. Wallace, ACS Sustainable Chem. Eng., 2019, 7(17), 14321–14340 CrossRef CAS.
- R. B. Araujo, A. Banerjee, P. Panigrahi, L. Yang, M. Sjödin, M. Strømme, C. M. Araujo and R. Ahuja, Phys. Chem. Chem. Phys., 2017, 19(4), 3307–3314 RSC.
- T. Matsunaga, H. Daifuku, T. Nakajima and T. Kawagoe, Polym. Adv. Technol., 1990, 1(1), 33–39 CrossRef CAS.
- S. J. Visco and L. C. DeJonghe, J. Electrochem. Soc., 1988, 135(12), 2905 CrossRef CAS.
- D. Y. Wang, W. Guo and Y. Fu, Acc. Chem. Res., 2019, 52(8), 2290–2300 CrossRef CAS PubMed.
- S. Tobishima, J. Yamaki and A. Yamaji, J. Electrochem. Soc., 1984, 131(1), 57 CrossRef CAS.
- K. Nakahara, S. Iwasa, M. Satoh, Y. Morioka, J. Iriyama, M. Suguro and E. Hasegawa, Chem. Phys. Lett., 2002, 359(5–6), 351–354 CrossRef CAS.
- T. Suga, Y. J. Pu, K. Oyaizu and H. Nishide, Bull. Chem. Soc. Jpn., 2004, 77(12), 2203–2204 CrossRef CAS.
- M. Liu, S. J. Visco and L. C. De Jonghe, J. Electrochem. Soc., 1990, 137(3), 750 CrossRef CAS.
- R. Van Noorden, Nature, 2014, 507(7490), 26–28 CrossRef CAS PubMed.
- T. B. Schon, B. T. McAllister, P. F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45(22), 6345–6404 RSC.
- R. B. Araujo, A. Banerjee and R. Ahuja, J. Phys. Chem. C, 2017, 121(26), 14027–14036 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116(16), 9438–9484 CrossRef CAS PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1(4), 739–768 CrossRef CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6(8), 2280–2301 RSC.
- Y. Lu, X. Hou, L. Miao, L. Li, R. Shi, L. Liu and J. Chen, Angew. Chem., 2019, 131(21), 7094–7098 CrossRef.
- A. Ponrouch, J. Bitenc, R. Dominko, N. Lindahl, P. Johansson and M. R. Palacin, Energy Storage Mater., 2019, 20, 253–262 CrossRef.
- T. Matsunaga, T. Kubota, T. Sugimoto and M. Satoh, Chem. Lett., 2011, 40(7), 750–752 CrossRef CAS.
- X. Han, G. Qing, J. Sun and T. Sun, Angew. Chem., Int. Ed., 2012, 51(21), 5147–5151 CrossRef CAS PubMed.
- C. Luo, O. Borodin, X. Ji, S. Hou, K. J. Gaskell, X. Fan, J. Chen, T. Deng, R. Wang, J. Jiang and C. Wang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(9), 2004–2009 CrossRef CAS PubMed.
- J. Meng, H. Guo, C. Niu, Y. Zhao, L. Xu, Q. Li and L. Mai, Joule, 2017, 1(3), 522–547 CrossRef CAS.
- A. Vizintin, J. Bitenc, A. Kopač Lautar, K. Pirnat, J. Grdadolnik, J. Stare, A. Randon-Vitanova and R. Dominko, Nat. Commun., 2018, 9(1), 1–7 CrossRef CAS PubMed.
- Y. Liang and Y. Yao, Joule, 2018, 2(9), 1690–1706 CrossRef CAS.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2(11), 861–868 CrossRef CAS.
- X. Wu, S. Jin, Z. Zhang, L. Jiang, L. Mu, Y. S. Hu, H. Li, X. Chen, M. Armand, L. Chen and X. Huang, Sci. Adv., 2015, 1(8), e1500330 CrossRef PubMed.
- H. Kim, D. H. Seo, G. Yoon, W. A. Goddard, Y. S. Lee, W. S. Yoon and K. Kang, J. Phys. Chem. Lett., 2014, 5(17), 3086–3092 CrossRef CAS PubMed.
- I. A. Rodríguez-Pérez, Y. Yuan, C. Bommier, X. Wang, L. Ma, D. P. Leonard, M. M. Lerner, R. G. Carter, T. Wu, P. A. Greaney, J. Lu and X. Ji, J. Am. Chem. Soc., 2017, 139(37), 13031–13037 CrossRef PubMed.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef CAS.
- X. Han, C. Chang, L. Yuan, T. Sun and J. Sun, Adv. Mater., 2007, 19(12), 1616–1621 Search PubMed.
- W. Luo, M. Allen, V. Raju and X. Ji, Adv. Energy Mater., 2014, 4(15), 1400554 CrossRef.
- A. J. Wain, G. G. Wildgoose, C. G. R. Heald, L. Jiang, T. G. J. Jones and R. G. Compton, J. Phys. Chem. B, 2005, 109(9), 3971–3978 CrossRef CAS PubMed.
- S. Renault, V. A. Oltean, C. M. Araujo, A. Grigoriev, K. Edström and D. Brandell, Chem. Mater., 2016, 28(6), 1920–1926 CrossRef CAS.
- J. S. Foos, S. M. Erker and L. M. Rembetsy, J. Electrochem. Soc., 1986, 133(4), 836 CrossRef CAS.
- B. L. Funt and P. M. Hoang, in Electrochemical Society Extended Abstracts, 1984, vol. 84–1 Search PubMed.
- T. Boschi, R. Pappa, G. Pistoia and M. Tocci, J. Electroanal. Chem., 1984, 176(1–2), 235–242 CrossRef CAS.
- T. Ohzuku, H. Wakamatsu, Z. Takehara and S. Yoshizawa, Electrochim. Acta, 1979, 24(6), 723–726 CrossRef CAS.
- H. Alt, H. Binder, A. Köhling and G. Sandstede, Electrochim. Acta, 1972, 17(5), 873–887 CrossRef CAS.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5, 1402034 CrossRef.
- T. B. Reddy, Linden's Handbook of Batteries, McGraw-Hill, New York, 4th edn, 2011 Search PubMed.
- Q. Zou, W. Wang, A. Wang, Z. Yu and K. Yuan, Mater. Lett., 2014, 117, 290–293 CrossRef CAS.
- M. Lee, J. Hong, H. Kim, H. D. Lim, S. B. Cho, K. Kang and C. B. Park, Adv. Mater., 2014, 26(16), 2558–2565 CrossRef CAS PubMed.
- H. Li, W. Duan, Q. Zhao, F. Cheng, J. Liang and J. Chen, Inorg. Chem. Front., 2014, 1(2), 193–199 RSC.
- H. C. Lin, C. C. Li and J. T. Lee, J. Power Sources, 2011, 196(19), 8098–8103 CrossRef CAS.
- W. Wan, H. Lee, X. Yu, C. Wang, K. W. Nam, X. Q. Yang and H. Zhou, RSC Adv., 2014, 4(38), 19878–19882 RSC.
- T. Yokoji, H. Matsubara and M. Satoh, J. Mater. Chem. A, 2014, 2, 19347–19354 RSC.
- H. Kim, J. E. Kwon, B. Lee, J. Hong, M. Lee, S. Y. Park and K. Kang, Chem. Mater., 2015, 27, 7258–7264 CrossRef CAS.
- A. Iordache, V. Maurel, J. M. Mouesca, J. Pécaut, L. Dubois and T. Gutel, J. Power Sources, 2014, 267, 553–559 CrossRef CAS.
- A. Shimizu, Y. Tsujii, H. Kuramoto, T. Nokami, Y. Inatomi, N. Hojo and J. I. Yoshida, Energy Technol., 2014, 2, 155–158 CrossRef CAS.
- C. Luo, R. Huang, R. Kevorkyants, M. Pavanello, H. He and C. Wang, Nano Lett., 2014, 14(3), 1596–1602 CrossRef CAS PubMed.
- D. Wu, Z. Xie, Z. Zhou, P. Shen and Z. Chen, J. Mater. Chem. A, 2015, 3, 19137–19143 RSC.
- H. Zhang, Q. Deng, A. Zhou, X. Liu and J. Li, J. Mater. Chem. A, 2014, 2(16), 5696–5702 RSC.
- S. Gottis, A. L. Barres, F. Dolhem and P. Poizot, ACS Appl. Mater. Interfaces, 2014, 6(14), 10870–10876 CrossRef CAS PubMed.
- H. H. Lee, Y. Park, K. H. Shin, K. T. Lee and S. Y. Hong, ACS Appl. Mater. Interfaces, 2014, 6(21), 19118–19126 CrossRef CAS PubMed.
- L. Fédèle, F. Sauvage, J. Bois, J.-M. Tarascon and M. Bécuwe, J. Electrochem. Soc., 2014, 161(1), A46 CrossRef.
- L. Fédèle, F. Sauvage and M. Bécuwe, J. Mater. Chem. A, 2014, 2(43), 18225–18228 RSC.
- T. Yasuda and N. Ogihara, Chem. Commun., 2014, 50(78), 11565–11567 RSC.
- N. Ogihara, T. Yasuda, Y. Kishida, T. Ohsuna, K. Miyamoto and N. Ohba, Angew. Chem., 2014, 126(43), 11651–11656 CrossRef.
- C. Wang, Y. Xu, Y. Fang, M. Zhou, L. Liang, S. Singh, H. Zhao, A. Schober and Y. Lei, J. Am. Chem. Soc., 2015, 137(8), 3124–3130 CrossRef CAS PubMed.
- A. Choi, Y. K. Kim, T. K. Kim, M. S. Kwon, K. T. Lee and H. R. Moon, J. Mater. Chem. A, 2014, 2, 14986–14993 RSC.
- V. A. Mihali, S. Renault, L. Nyholm and D. Brandell, RSC Adv., 2014, 4, 38004–38011 RSC.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, Energy Environ. Sci., 2017, 10(2), 552–557 RSC.
- Y. Park, D. S. Shin, S. H. Woo, N. S. Choi, K. H. Shin, S. M. Oh, K. T. Lee and S. Y. Hong, Adv. Mater., 2012, 24(26), 3562–3567 CrossRef CAS PubMed.
- H. G. Wang, S. Yuan, Z. Si and X. B. Zhang, Energy Environ. Sci., 2015, 8, 3160–3165 RSC.
- M. Veerababu, U. V. Varadaraju and R. Kothandaraman, Int. J. Hydrogen Energy, 2015, 40, 14925–14931 CrossRef CAS.
- C. Yuan, Q. Wu, Q. Li, Q. Duan, Y. Li and H. G. Wang, ACS Sustainable Chem. Eng., 2018, 6, 8392–8399 CrossRef CAS.
- J. Wang, X. Wang, H. Li, X. Yang and Y. Zhang, J. Electroanal. Chem., 2016, 773, 22–26 CrossRef CAS.
- H. Banda, D. Damien, K. Nagarajan, A. Raj, M. Hariharan and M. M. Shaijumon, Adv. Energy Mater., 2017, 7, 1701316 CrossRef.
- R. Rajagopalan, Y. Tang, C. Jia, X. Ji and H. Wang, Energy Environ. Sci., 2020, 13, 1568–1592 RSC.
- S. Renault, V. A. Mihali, K. Edström and D. Brandell, Electrochem. Commun., 2014, 45, 52–55 CrossRef CAS.
- G. S. Vadehra, R. P. Maloney, M. A. Garcia-Garibay and B. Dunn, Chem. Mater., 2014, 26, 7151–7157 CrossRef CAS.
- M. E. Bhosale and K. Krishnamoorthy, Chem. Mater., 2015, 27, 2121–2126 CrossRef CAS.
- D. Chen, A.-J. Avestro, Z. Chen, J. Sun, S. Wang, M. Xiao, Z. Erno, M. M. Algaradah, M. S. Nassar, K. Amine, Y. Meng, J. Fraser Stoddart, D. Chen, A. Avestro, J. Sun, Z. Erno, Z. Chen, K. Amine, S. Wang, M. Xiao, Y. Meng, M. M. Algaradah and M. S. Nassar, Adv. Mater., 2015, 27, 2907–2912 CrossRef CAS PubMed.
- H. Wu, S. A. Shevlin, Q. Meng, W. Guo, Y. Meng, K. Lu, Z. Wei and Z. Guo, Adv. Mater., 2014, 26, 3338–3343 CrossRef CAS PubMed.
- J. Kim, J. Kim, J. Lee, H. K. Song and C. Yang, ChemElectroChem, 2014, 1(10), 1618–1622 CrossRef CAS.
- H. Qin, Z. P. Song, H. Zhan and Y. H. Zhou, J. Power Sources, 2014, 249, 367–372 CrossRef CAS.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7(12), 4077–4086 RSC.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 13947–13951 CrossRef CAS PubMed.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2(9), 1500124 CrossRef PubMed.
- Y. Liang, Z. Chen, Y. Jing, Y. Rong, A. Facchetti and Y. Yao, J. Am. Chem. Soc., 2015, 137, 4956–4959 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334(6058), 928–935 CrossRef CAS PubMed.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42(20), 8012–8031 RSC.
- Z. Cheng, H. Pan, H. Zhong, Z. Xiao, X. Li and R. Wang, Adv. Funct. Mater., 2018, 28(38), 1707597 CrossRef.
- X. Liu, C. F. Liu, W. Y. Lai and W. Huang, Adv. Mater. Technol., 2020, 5, 2000154 CrossRef CAS.
- J. Lv, Y. X. Tan, J. Xie, R. Yang, M. Yu, S. Sun, M. De Li, D. Yuan and Y. Wang, Angew. Chem., Int. Ed., 2018, 57, 12716–12720 CrossRef CAS PubMed.
- X. Chen, Y. Li, L. Wang, Y. Xu, A. Nie, Q. Li, F. Wu, W. Sun, X. Zhang, R. Vajtai, P. M. Ajayan, L. Chen and Y. Wang, Adv. Mater., 2019, 31(29), 1901640 CrossRef PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458(7235), 190–193 CrossRef CAS PubMed.
- P. S. Herle, B. Ellis, N. Coombs and L. F. Nazar, Nat. Mater., 2004, 3(3), 147–152 CrossRef CAS PubMed.
- X. Zhan, Z. Chen and Q. Zhang, J. Mater. Chem. A, 2017, 5(28), 14463–14479 RSC.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357(6352), 673–676 CrossRef CAS PubMed.
- G. Zhou, E. Paek, G. S. Hwang and A. Manthiram, Nat. Commun., 2015, 6(1), 1–11 Search PubMed.
- Y. Wu, Z. Zhang, S. Bandow and K. Awaga, Bull. Chem. Soc. Jpn., 2017, 90(12), 1382–1387 CrossRef CAS.
- F. Xu, S. Jin, H. Zhong, D. Wu, X. Yang, X. Chen, H. Wei, R. Fu and D. Jiang, Sci. Rep., 2015, 5(1), 1–6 Search PubMed.
- B. C. Patra, S. K. Das, A. Ghosh, A. K. Raj, P. Moitra, M. Addicoat, S. Mitra, A. Bhaumik, S. Bhattacharya and A. Pradhan, J. Mater. Chem. A, 2018, 6(34), 16655–16663 RSC.
- K. Wang, L. M. Yang, X. Wang, L. Guo, G. Cheng, C. Zhang, S. Jin, B. Tan and A. Cooper, Angew. Chem., Int. Ed., 2017, 56(45), 14149–14153 CrossRef CAS PubMed.
- L. Fan, Y. Hu, A. M. Rao, J. Zhou, Z. Hou, C. Wang and B. Lu, Small Methods, 2021, 5, 2101131 CrossRef CAS PubMed.
- S. Liu, L. Kang and S. C. Jun, Adv. Mater., 2021, 33, 2004689 CrossRef CAS PubMed.
- J. Ge, L. Fan, A. M. Rao, J. Zhou and B. Lu, Nat. Sustain., 2021, 5(3), 225–234 CrossRef.
- J. Y. Hwang, S. T. Myung and Y. K. Sun, Adv. Funct. Mater., 2018, 28, 1802938 CrossRef.
- C. Han, K. Han, X. Wang, C. Wang, Q. Li, J. Meng, X. Xu, Q. He, W. Luo, L. Wu and L. Mai, Nanoscale, 2018, 10, 6820–6826 RSC.
- X. Lu, D. Zhang, J. Zhong, L. Wang, L. Jiang, Q. Liu, G. Shao, D. Fu, J. Teng and W. Yang, Chem. Eng. J., 2022, 432, 134416 CrossRef CAS.
- Y. Meng, C. Nie, W. Guo, D. Liu, Y. Chen, Z. Ju and Q. Zhuang, Mater. Today Energy, 2022, 25, 100982 CrossRef CAS.
- H. Ma, Y. Chen, X. Li and B. Li, Adv. Funct. Mater., 2021, 31, 2101861 CrossRef CAS.
- K. Amin, N. Ashraf, L. Mao, C. F. J. Faul and Z. Wei, Nano Energy, 2021, 85, 105958 CrossRef CAS.
- M. Gamal Mohamed, T. Hassan Mansoure, M. Mohamed Samy, Y. Takashi, A. A. K. Mohammed, T. Ahamad, S. M. Alshehri, J. Kim, B. M. Matsagar, K. C.-W. Wu and S.-W. Kuo, Mol, 2022, 27, 2025 CrossRef PubMed.
- S. Luo, Z. Zeng, H. Wang, W. Xiong, B. Song, C. Zhou, A. Duan, X. Tan, Q. He, G. Zeng, Z. Liu and R. Xiao, Prog. Polym. Sci., 2021, 115, 101374 CrossRef CAS.
- C. Zhang, Y. Qiao, P. Xiong, W. Ma, P. Bai, X. Wang, Q. Li, J. Zhao, Y. Xu, Y. Chen, J. H. Zeng, F. Wang, Y. Xu and J. X. Jiang, ACS Nano, 2019, 13, 745–754 CrossRef CAS PubMed.
- H. Luo, M. Chen, J. Cao, M. Zhang, S. Tan, L. Wang, J. Zhong, H. Deng, J. Zhu and B. Lu, Nano-Micro Lett., 2020, 12, 1–13 CrossRef PubMed.
- H. Liao, H. Ding, B. Li, X. Ai and C. Wang, J. Mater. Chem. A, 2014, 2(23), 8854–8858 RSC.
- H. Liao, H. Wang, H. Ding, X. Meng, H. Xu, B. Wang, X. Ai and C. Wang, J. Mater. Chem. A, 2016, 4(19), 7416–7421 RSC.
- H. Zhang, Y. Gao, X. H. Liu, Z. Yang, X. X. He, L. Li, Y. Qiao, W. H. Chen, R. H. Zeng, Y. Wang and S. L. Chou, Adv. Funct. Mater., 2022, 32, 2107718 CrossRef CAS.
- D. Y. Wang, R. Liu, W. Guo, G. Li and Y. Fu, Coord. Chem. Rev., 2021, 429, 213650 CrossRef CAS.
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21(4), 1555–1565 CrossRef CAS PubMed.
- F. Xie, W. Hu, L. Ding, K. Tian, Z. Wu and L. Li, Polym. Chem., 2017, 8, 6106–6111 RSC.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46(11), 2376–2384 CrossRef CAS PubMed.
- M. Du, Q. Li, Y. Zhao, C. Sen Liu and H. Pang, Coord. Chem. Rev., 2020, 416, 213341 CrossRef CAS.
- T. Li, Y. Bai, Y. Wang, H. Xu and H. Jin, Coord. Chem. Rev., 2020, 410, 213221 CrossRef CAS.
- J. Y. Shin, T. Yamada, H. Yoshikawa, K. Awaga and H. Shinokubo, Angew. Chem., Int. Ed., 2014, 53, 3096–3101 CrossRef CAS PubMed.
- J. L. Qin, B. Q. Li, J. Q. Huang, L. Kong, X. Chen, H. J. Peng, J. Xie, R. Liu and Q. Zhang, Mater. Chem. Front., 2019, 3, 615–619 RSC.
- R. Zhao, Z. Liang, R. Zou and Q. Xu, Joule, 2018, 2, 2235–2259 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D. S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- X. Li, S. Zheng, L. Jin, Y. Li, P. Geng, H. Xue, H. Pang and Q. Xu, Adv. Energy Mater., 2018, 8, 1800716 CrossRef.
- W. Zhu, A. Li, Z. Wang, J. Yang and Y. Xu, Small, 2021, 17, 2006424 CrossRef CAS PubMed.
- S. Bai, X. Liu, K. Zhu, S. Wu and H. Zhou, Nat. Energy, 2016, 1, 1–6 Search PubMed.
- Y. Zheng, S. Zheng, H. Xue and H. Pang, J. Mater. Chem. A, 2019, 7, 3469–3491 RSC.
- K. A. Hansen and J. P. Blinco, Polym. Chem., 2018, 9(13), 1479–1516 RSC.
- C. Luo, G. L. Xu, X. Ji, S. Hou, L. Chen, F. Wang, J. Jiang, Z. Chen, Y. Ren, K. Amine and C. Wang, Angew. Chem., Int. Ed., 2018, 57(11), 2879–2883 CrossRef CAS PubMed.
- L. Zhao, J. Zhao, Y. S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2(8), 962–965 CrossRef CAS.
- C. Luo, J. Wang, X. Fan, Y. Zhu, F. Han, L. Suo and C. Wang, Nano Energy, 2015, 13, 537–545 CrossRef CAS.
- M. López-Herraiz, E. Castillo-Martínez, J. Carretero-González, J. Carrasco, T. Rojo and M. Armand, Energy Environ. Sci., 2015, 8(11), 3233–3241 RSC.
- H. Roghani-Mamaqani, V. Haddadi-Asl and M. Salami-Kalajahi, Polym. Rev., 2012, 52(2), 142–188 CrossRef CAS.
- C. Hua, A. Rawal, T. B. Faust, P. D. Southon, R. Babarao, J. M. Hook and D. M. D’Alessandro, J. Mater. Chem. A, 2014, 2(31), 12466–12474 RSC.
- Y. Xie, K. Zhang, Y. Yamauchi, K. Oyaizu and Z. Jia, Mater. Horiz., 2021, 8(3), 803–829 RSC.
- K. Nakahara, K. Oyaizu and H. Nishide, Chem. Lett., 2011, 40, 222–227 CrossRef CAS.
- H. Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50(2–3), 827–831 CrossRef CAS.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23(6), 751–754 CrossRef CAS PubMed.
- J. Qu, T. Katsumata, M. Satoh, J. Wada and T. Masuda, Polymer, 2009, 50(2), 391–396 CrossRef CAS.
- T. Suga, H. Konishi and H. Nishide, Chem. Commun., 2007,(17), 1730–1732 RSC.
- K. Oyaizu, T. Suga, K. Yoshimura and H. Nishide, Macromolecules, 2008, 41(18), 6646–6652 CrossRef CAS.
- K. Oyaizu, T. Sukegawa and H. Nishide, Chem. Lett., 2011, 40(2), 184–185 CrossRef CAS.
- J. Qu, R. Morita, M. Satoh, J. Wada, F. Terakura, K. Mizoguchi, N. Ogata and T. Masuda, Chem.–Eur. J., 2008, 14(11), 3250–3259 CrossRef CAS PubMed.
- H. Hishide and T. Suga, Electrochem. Soc. Interface, 2005, 14, 32–36 CrossRef.
- C. Li, C. C. Hou, L. Chen, S. Kaskel and Q. Xu, EnergyChem, 2021, 3, 100049 CrossRef CAS.
- R. Cang, Y. Song, K. Ye, K. Zhu, J. Yan, J. Yin, G. Wang and D. Cao, J. Electroanal. Chem., 2020, 861, 113967 CrossRef CAS.
- N. Khossossi, D. Singh, A. Banerjee, W. Luo, I. Essaoudi, A. Ainane and R. Ahuja, ACS Appl. Energy Mater., 2021, 4, 7900–7910 CrossRef CAS.
- J. Chen, Q. Zhu, L. Jiang, R. Liu, Y. Yang, M. Tang, J. Wang, H. Wang and L. Guo, Angew. Chem., Int. Ed., 2021, 60, 5794–5799 CrossRef CAS PubMed.
- F. Wu, H. Yang, Y. Bai and C. Wu, Adv. Mater., 2019, 31, 1806510 CrossRef PubMed.
- D. Y. Wang, C. Y. Wei, M. C. Lin, C. J. Pan, H. L. Chou, H. A. Chen, M. Gong, Y. Wu, C. Yuan, M. Angell, Y. J. Hsieh, Y. H. Chen, C. Y. Wen, C. W. Chen, B. J. Hwang, C. C. Chen and H. Dai, Nat. Commun., 2017, 8(1), 1–7 CrossRef PubMed.
- M. Jäckle, K. Helmbrecht, M. Smits, D. Stottmeister and A. Groß, Energy Environ. Sci., 2018, 11, 3400–3407 RSC.
- K. Zhang, K. O. Kirlikovali, J. M. Suh, J. W. Choi, H. W. Jang, R. S. Varma, O. K. Farha and M. Shokouhimehr, ACS Appl. Energy Mater., 2020, 3, 6019–6035 CrossRef CAS.
- T. T. Wei, P. Peng, S. Y. Qi, Y. R. Zhu and T. F. Yi, J. Energy Chem., 2021, 57, 169–188 CrossRef.
- B. Pan, J. Huang, Z. Feng, L. Zeng, M. He, L. Zhang, J. T. Vaughey, M. J. Bedzyk, P. Fenter, Z. Zhang, A. K. Burrell and C. Liao, Adv. Energy Mater., 2016, 6, 1600140 CrossRef.
- Y. Wang, Z. Liu, C. Wang, Y. Hu, H. Lin, W. Kong, J. Ma and Z. Jin, Energy Storage Mater., 2020, 26, 494–502 CrossRef.
- D. Lu, H. Liu, T. Huang, Z. Xu, L. Ma, P. Yang, P. Qiang, F. Zhang and D. Wu, J. Mater. Chem. A, 2018, 6, 17297–17302 RSC.
- M. Mao, C. Luo, T. P. Pollard, S. Hou, T. Gao, X. Fan, C. Cui, J. Yue, Y. Tong, G. Yang, T. Deng, M. Zhang, J. Ma, L. Suo, O. Borodin and C. Wang, Angew. Chem., 2019, 131, 17984–17990 CrossRef.
- B. Häupler, A. Wild and U. S. Schubert, Adv. Energy Mater., 2015, 5(11), 1402034 CrossRef.
- H. Yang, H. Li, J. Li, Z. Sun, K. He, H. M. Cheng and F. Li, Angew. Chem., Int. Ed., 2019, 58, 11978–11996 CrossRef CAS PubMed.
- J. Zou, K. Fan, Y. Chen, W. Hu and C. Wang, Coord. Chem. Rev., 2022, 458, 214431 CrossRef CAS.
- C. Wang, Energy Environ. Mater., 2020, 3, 441–452 CrossRef CAS.
- K. Qin, J. Huang, K. Holguin and C. Luo, Energy Environ. Sci., 2020, 13, 3950–3992 RSC.
- X. Peng, Y. Xie, A. Baktash, J. Tang, T. Lin, X. Huang, Y. Hu, Z. Jia, D. J. Searles, Y. Yamauchi, L. Wang and B. Luo, Angew. Chem., Int. Ed., 2022, e202203646 CAS.
- D. J. Kim, D. J. Yoo, M. T. Otley, A. Prokofjevs, C. Pezzato, M. Owczarek, S. J. Lee, J. W. Choi and J. F. Stoddart, Nat. Energy, 2018, 4, 51–59 CrossRef.
- J. Chen, Q. Zhu, L. Jiang, R. Liu, Y. Yang, M. Tang, J. Wang, H. Wang and L. Guo, Angew. Chem., Int. Ed., 2021, 60, 5794–5799 CrossRef CAS PubMed.
- F. Guo, Z. Huang, M. Wang, W. L. Song, A. Lv, X. Han, J. Tu and S. Jiao, Energy Storage Mater., 2020, 33, 250–257 CrossRef.
- J. Zhou, X. Yu, J. Zhou and B. Lu, Energy Storage Mater., 2020, 31, 58–63 CrossRef.
- J. Bitenc, N. Lindahl, A. Vizintin, M. E. Abdelhamid, R. Dominko and P. Johansson, Energy Storage Mater., 2020, 24, 379–383 CrossRef.
- Y. Li, L. Liu, Y. Lu, R. Shi, Y. Ma, Z. Yan, K. Zhang and J. Chen, Adv. Funct. Mater., 2021, 31, 2102063 CrossRef CAS.
- Y. T. Kao, S. B. Patil, C. Y. An, S. K. Huang, J. C. Lin, T. S. Lee, Y. C. Lee, H. L. Chou, C. W. Chen, Y. J. Chang, Y. H. Lai and D. Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 25853–25860 CrossRef CAS PubMed.
- M. Mao, C. Luo, T. P. Pollard, S. Hou, T. Gao, X. Fan, C. Cui, J. Yue, Y. Tong, G. Yang, T. Deng, M. Zhang, J. Ma, L. Suo, O. Borodin and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 17820–17826 CrossRef CAS PubMed.
- J. Kim, J. H. Kim and K. Ariga, Joule, 2017, 1, 739–768 CrossRef CAS.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |