
Integrating recyclable polymers into thermoelectric devices for green electronics†
 *ab
Samantha Faye Duran
Solco,
a
Xian Yi
Tan,
ad
Qiang
Zhu,
a
Qingyu
Yan,
d
Ady
Suwardi
*ac
and
Zibiao
Li
*abc
*ab
Samantha Faye Duran
Solco,
a
Xian Yi
Tan,
ad
Qiang
Zhu,
a
Qingyu
Yan,
d
Ady
Suwardi
*ac
and
Zibiao
Li
*abc
Abstract
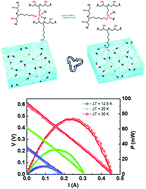
Electronic waste (e-waste) recycling is one of the central frameworks of the circular economy. However, most e-wastes consist of both organic and inorganic components, which significantly limits their clean separation and recycling for repurposing. Herein, we demonstrate the use of a recyclable polymer (vitrimer) as the encapsulation matrix to construct a recyclable thermoelectric device. An epoxy vitrimer containing dynamic silyl ether linkage was employed as the device encapsulation to provide mechanical support, conformability to surfaces, and more importantly, reprocessability. Benefiting from these features, the resultant vitrimer encapsulated thermoelectric device not only showed an enhanced power generation ability relative to the parent device, but also exhibited new alluring characteristics, such as strong mechanical properties, operability under deformed conditions, clean separation capability, and recyclability. Remarkably, the refabricated device retains its power generation performance, demonstrating the reliability of this method. The strategy reported here can be generally applied to other electronic devices, therefore contributing towards sustainable and circular utilization of resources.
- This article is part of the themed collections: Polymer Upcycling, A collection of papers from RSC journals on chemistry and the circular economy, 2023 Journal of Materials Chemistry A Lunar New Year collection and Journal of Materials Chemistry A Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

