
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel molecular synthesis route to Li2S loaded carbon fibers for lithium–sulfur batteries
Veronika
Brune
 a,
Christoph
Bohr
a,
Tim
Ludwig
a,
Michael
Wilhelm
a,
Sebastian Daniel
Hirt
b,
Thomas
Fischer
a,
Sebastian
Wennig
b,
Bernd
Oberschachtsiek
b,
Arun
Ichangi
a and
Sanjay
Mathur
*a
a,
Christoph
Bohr
a,
Tim
Ludwig
a,
Michael
Wilhelm
a,
Sebastian Daniel
Hirt
b,
Thomas
Fischer
a,
Sebastian
Wennig
b,
Bernd
Oberschachtsiek
b,
Arun
Ichangi
a and
Sanjay
Mathur
*a
aInstitute of Inorganic Chemistry, University of Cologne, Greinstraße 6, D-50939 Cologne, Germany. E-mail: sanjay.mathur@uni-koeln.de
bThe Fuel Cell Research Center, ZBT Duisburg, Carl-Benz-Straße 201, D-47057 Duisburg, Germany
First published on 5th April 2022
Abstract
The synthesis of a novel air-stable molecular precursor (LiSC2H4)2NMe enables the formation of the desired 1D lithium sulfide (Li2S) via the electrospinning method under ambient conditions. The solubility of the precursor in polar solvents combined with a common polymer (PVP) allowed a suitable spinning solution to obtain ideal green Li2S loaded fibers. 3D fiber mats of the calcined homogeneous 1D electrospun Li2S/C fibers were characterized by electron microscopy and X-ray powder diffraction analysis. Direct integration of Li2S in an electronically conductive carbon matrix as the cathode obviates the need of elemental lithium as the anode, which is a great advantage against the reported lithium–sulfur batteries. An initial capacity of about 870 mA h g−1 at C/20, a capacity retention of 73% after 100 cycles at C/10 and a capacity of about 400 mA h g−1 at 1C were observed for the presented system.
Introduction
The global rapid consumption of fossil fuels is directly linked to huge CO2 emissions. Therefore, energy obtained from renewable sources like wind and solar is crucial for a sustainable energy supply.1,2In this context, large-scale energy storage will play a key role in future energy technologies.3 In general, batteries as electrochemical storage devices for different applications should meet several requirements of safety, high energy and power density, fast charging capability, sustainability and natural abundance of raw materials.1 Among the innumerable energy-storage technologies, the lithium–sulfur battery has emerged as a promising alternative because of its high theoretical gravimetric energy density (about 2600 W h kg−1).4 Furthermore, sulfur is highly abundant, economically priced and non-toxic.5–7 However, the low electrical conductivity of sulfur and lithium sulfide leads to limited high rate capability.8 Additionally, the polysulfide shuttle effect and the volume change of about 80% during cell operation are further challenges of this battery system that have prevented its commercialization so far.9–11 The volume change leads to mechanical stress within the cathode, which can result in electrode cracking and electrode pulverization as well.12 The shuttle effect involves the dissolution of higher-ordered polysulfides (Li2S4–Li2S8) in the electrolyte, their migration to the anode, and their electrochemical reduction on the anode's surface, which result in the formation of an additional passive layer composed of solid lithium sulfides such as Li2S and Li2S2. This leads to an increased cell resistance. Lower-ordered lithium polysulfides diffuse back to the cathode, provoking chemical reactions with the cathode resulting in a decreased coulombic efficiency.10 Finally, the shuttle effect causes a loss of the active material at the cathode, resulting in capacity fading.13,14
Applying elemental sulfur as the cathode material requires the deployment of elemental lithium as the anode. Lithium anodes can cause serious safety concerns since lithium dendrites can grow through the separator leading to short-circuits which can result in a thermal runaway.15
Recently, lithium sulfide (Li2S) has received much attention as an electrochemically active cathode material in lithium–sulfur batteries because a lithium-free anode can be used and thus a higher level of safety can be ensured.15 Li2S also has a higher melting point than elemental sulfur, enabling the synthesis of advanced materials at higher temperatures. Therefore, Li2S establishes various strategies for composite formation and surface modification to enhance the electrical conductivity of the cathode. A common method for achieving this purpose is the embedding of the active material in a carbon matrix.16,17
The synthesis of hierarchically porous 1D structures by electrospinning methods reveals an alternative synthetic approach to embed the active material in a flexible, freestanding, and conductive carbon matrix to accelerate the electronic and ionic transfer of the active material for improving the cycling stability.18 This synthesis enables the formation of 1D-fibers with varying diameters owing to the influence of changing process parameters such as the viscosity of spinning solution, voltage or kind of collector among others.19–21 Porous carbon nanofibers tend to be an ideal matrix due to their low weight, high electronic conductivity and adjustable structural properties, meaning pore volume, pore size and specific surface area.16,22,23 Furthermore, carbon fibers can mitigate polysulfide dissolution by enhanced chemical adsorption of intermediate species and therefore reduce the shuttle effect significantly.19,24–27 A simple synthetic access to lithium sulfide cathodes is hampered by the moisture sensitivity and reactivity of lithium.28 So far, the electrospinning of Li2S has been limited to inert electrospinning devices or the use of Li2SO4 as the precursor, which can be reduced to Li2S by a carbon source and hydrogen, respectively.29,30 Li2SO4 has the advantages of non-inert handling due to its high stability against oxygen and moisture. As a drawback, the solubility of Li2SO4 is almost exclusive to aqueous solutions, which is a limiting factor in the choice of polymer for the electrospinning process.
By using different precursors instead of Li2SO4, Ye et al. achieved Li2S/C fibers with high mass loading (72 wt%) by electrospinning a Li2S3/PVP precursor solution under inert conditions. The disadvantage of this synthesis route is that this precursor and material must be always under inert conditions, compared to our here presented molecular precursor.31
Using synthesized compounds to combine lithium and sulfur already at a molecular level shows the advantages of preformed Li–S bonds and the opportunity to shield and protect the sensitive Li–S unit against oxidation by introducing a suitable ligand. Therefore, we combined the dianionic, tridentate SNS ligand system ((HSC2H4)2NMe) with a suitable lithium source (butyllithium). The stabilizing properties and clear thermal decomposition of this ligand system have already been reported in molecular precursor synthesis before.32
To overcome the current limitations in the synthesis of Li2S electrospun fibers, a molecular approach towards a stable synthesized precursor will be presented in the following, which, to the best of our knowledge, has not been reported before. This molecular precursor enables the electrospinning of lithium sulfide-based fibers under ambient conditions from a single precursor solution, which is not limited to only aqueous solutions, ensuring a simple preparation of Li2S/C nanofibers. The scheme in Fig. 1 shows the battery configuration with Li2S loaded fibers as the cathode material, which have been provided by a novel synthetic approach, and will be described within this work.
 | ||
| Fig. 1 Illustration of the battery configuration with I-Li/PVP fibers as the cathode material in a lithium free anode. | ||
Results & discussion
Complexes of the tridentate ligand N-methyldiethanethiolamine (I) with lithium-delivered molecular precursors of formula (LiSC2H4)2NMe were successfully synthesized and characterized. The N-methyldiethanethiolamine was synthesized according to a process described in the literature before (Scheme 1a) and isolated by distillation under reduced pressure as a colorless oil.32–34 To deprotonate the thiol function of the isolated compound I, an acid–base reaction of n-butyllithium or lithium alcoholate was performed in n-heptane to produce the lithium derivative N-methyldiethanethiolatoamine-lithium (LiSC2H4)2NMe (I-Li) (Scheme 1b) as a colorless solid. The synthesized compound is air-stable and soluble in protic solvents such as alcohols or water as well as in dimethylsulfoxide (DMSO). The appropriate solubility combined with good stability reveals that the compound is an ideal precursor for a simple preparation of lithium sulfide materials.14,35,36 The comfortable handling under ambient conditions and high degrees of freedom regarding the choice of solvents make this molecular compound an ideal precursor for material processing methods.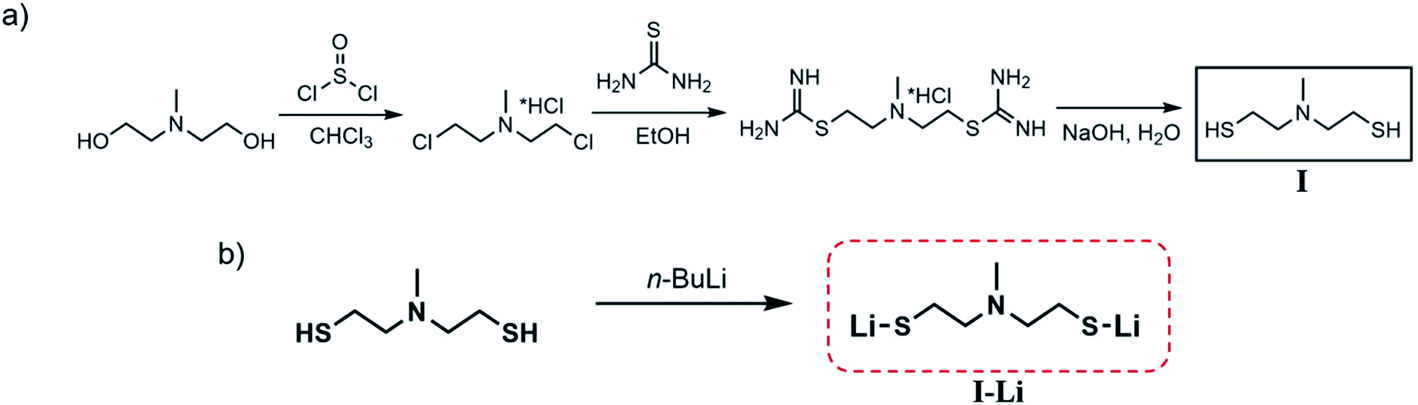 | ||
| Scheme 1 (a) General synthesis route to N-methyldiethanethiolamine (HSC2H4)2NMe (I) modified from ref. 32 and (b) synthesis of N-methyldiethanethiolatoamine-lithium (LiSC2H4)2NMe (I-Li). | ||
The new compound was analyzed by multi-nuclear magnetic resonance (NMR) spectroscopy using a combination of 1D and 2D NMR experiments (1H, 13C, 1H1H COSY, 1H13C HSQC, 1H13C HMBC). For I, three different proton signals were detected with an integrative ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:8 (Fig. 2b) which is attributed to the symmetry of I along with the methyl function and the nitrogen, showing the possible free rotation around the CH2 axis.
:3:8 (Fig. 2b) which is attributed to the symmetry of I along with the methyl function and the nitrogen, showing the possible free rotation around the CH2 axis.
 | ||
| Fig. 2 1H NMR of (a) (LiSC2H4)2NMe I-Li and (b) (HSC2H4)2NMe I. | ||
The signals of both ethanethiol chains appear as one signal with an equal chemical shift located at 2.58 ppm. The two acidic thiol protons resulted in a broad signal at 1.74 ppm and the methyl group was found at 2.24 ppm. The NMR spectroscopic studies of compound I were in agreement with the literature34 and corroborated the proposed structure of the ligand in solution (Fig. 2b). The deprotonation of I by treatment with butyllithium resulted in the lithium derivative I-Li (Fig. 2a) showing separated CH2 protons for the ethanethiol chains in NMR analysis, resulting from the hindered rotation around the C–C-axis. In total 11 protons were detected, confirming the deprotonation of pre-synthesized ligand I and successful proton exchange by the desired alkali metal.
To investigate the thermal decomposition behavior of I-Li, thermogravimetric measurements were performed under an inert atmosphere (Fig. 3a). The multi-step decomposition showed a total weight loss of 72% at temperatures up to 800 °C, resulting in the formation of crystalline residue of lithium sulfide (Li2S). The decomposition of molecular precursor sets in at around 100 °C and a nearly complete decomposition was observed at around 350 °C. At this point, Li2S was already formed in an amorphous phase, which was confirmed by XRD measurements.37,38 At elevated temperatures, beginning at 650 °C, conversion to the crystalline cubic phase of Li2S takes place.
 | ||
| Fig. 3 (a) Thermogravimetric measurements of the synthesized precursor (LiSC2H4)2NMe I-Li up to 800 °C under a nitrogen atmosphere; (b) XRD pattern of the decomposed compound I-Li to the corresponding lithium sulfide Li2S (ref. PDF #01-26-1188). | ||
X-ray diffraction investigations of the decomposition residue obtained at 800 °C under a nitrogen atmosphere confirmed the formation of the desired crystalline Li2S phase (Fig. 3b). The simple thermal decomposition to phase pure Li2S in combination with the comfortable operation, good solubility in protic solutions and air stability qualified the synthesized precursor I-Li as an efficient molecular source for the fabrication of Li2S loaded carbon-based 1D materials.
Therefore, (LiSC2H4)2NMe–PVP aligned fiber mats were produced from ethanolic solutions of I-Li with polyvinylpyrrolidone (PVP) by the electrospinning process (Fig. 4).
 | ||
| Fig. 4 Electrospinning of the I-Li/PVP solution with a drum collector. | ||
Calcination of electrospun fibers was performed by applying a ramping of 2 °C min−1 up to 280 °C (holding time 2 h) under ambient conditions, to stabilize the fibrous structure by partly oxidizing,39 followed by a temperature increase of 2 °C min−1 up to 700 °C (holding time 5 h) under a nitrogen atmosphere to finalize the carbonization process and to produce crystalline Li2S/C-fiber mats (I-Li/C_45%, Fig. 5a), respectively. The desired product formation was confirmed by XRD measurements of the calcined specimen (Fig. 5b).
 | ||
| Fig. 5 Electrospun I-Li fibers containing 45% Li2S in their calcined form. (a) SEM image of the electrospun green I-Li fibers (I-Li/PVP_58%), (b) XRD of I-Li/C_45% (ref. PDF #01-26-1188) and (c) TEM image of the calcined electrospun I-Li fibers (I-Li/C_45%). | ||
A precursor solution with an I-Li to PVP ratio of 1:1.5 was used to yield an appropriate viscosity for electrospinning which is mandatory to produce homogeneous fibers. A rotary drum collector was chosen since electrospinning onto a stationary collector led to low fiber adhesion towards the aluminum foil with bundling or aggregation effects. The aligned composite fiber (I-Li/PVP_58%) contained a (LiSC2H4)2NMe mass fraction of 58 wt%. After the calcination process, a lithium sulfide/conductive carbon composite fiber mat with a total Li2S fraction of 45 wt% (I-Li/C_45%) was obtained which was determined by thermogravimetric measurements.
TEM analysis of I-Li/C_45% displayed a 3D fiber network out of 1D electrospun fibers (Fig. 5c).39 The porosity of the fiber structure leads to a high surface-to-volume ratio and therefore to an intensive contact between the active material and electrolyte, which results in fast lithium-ion diffusion enabling higher charge and discharge rates.
Electrodes made from I-Li/C_45% fibers, PVP as the binder and carbon black as the conductive additive were characterized electrochemically.
Cyclic voltammetry (Fig. 6a) was used to investigate the electrochemical behavior of I-Li/C_45% fibers. In the first cycle, two oxidation peaks located at 2740 mV vs. Li/Li+ and at 3710 mV vs. Li/Li+ were observed. In the literature, the first peak at 2751 mV vs. Li/Li+ is attributed to side reactions or the formation of sulfur from Li2S or polysulfides.40,41 The broad peak between 3170 and 3900 mV vs. Li/Li+ with a maximum at 3708 mV vs. Li/Li+ is assigned to the oxidation of Li2S to lithium polysulfides and sulfur, respectively. Usually, the oxidation of Li2S to elemental sulfur is located between 2200 and 2600 mV vs. Li/Li+ depending on particle size and active material loading.42,43 The size of the crystalline Li2S material was calculated using Scherrer's equation at around 10 nm. However, for the activation of a fresh Li2S cathode, a higher potential within the first cycle is required.44 The overpotential was necessary for phase nucleation of polysulfides, the removal of surface passivation44 and successive further oxidation to elemental sulfur.43 This phenomenon has been reported for electrodes containing PVP, due to the interaction of lithium ions from Li2S with the oxygen functions of the PVP matrix.45 A significant current flow was still observed between 3900 and 4000 mV vs. Li/Li+, which can be assigned to side reactions such as electrolyte decomposition. For the oxidation reaction, a capacity of 1456 mA h g−1 was calculated from the CV. This capacity is 290 mA h g−1 higher than the theoretical capacity of Li2S. Presumably, this can be explained by side reactions such as electrolyte decomposition and the shuttle mechanism, respectively. Three cathodic peaks located at 2355, 2111 and 2025 mV vs. Li/Li+ were observed, which were assigned to the reduction of sulfur. At 2355 mV vs. Li/Li+ the reaction of elemental sulfur (S8 ring) with two lithium ions forming long-chain polysulfide Li2S8 has been observed.42 The reduction peak at 2111 mV vs. Li/Li+ is not characteristic for all Li2S cathodes. However, it can be assigned to the further reduction of the long-chained Li2S8 to the shorter-chained polysulfide Li2S4.46 The electrochemical conversion of Li2S4 to Li2S2 and Li2S occurred at 2025 mV vs. Li/Li+. The calculated capacity for the reduction was 863 mA h g−1 giving a coulombic efficiency of 59.2%. A low coulombic efficiency in the first cycle was already described in the literature and can be explained by the formation of passivation layers as well as by the dissolution of polysulfides in the electrolyte.47 For the second cycle, the cut-off oxidation potential was set to 3000 mV vs. Li/Li+. Two anodic peaks at 2307 and 2407 mV vs. Li/Li+ were detected. The peak at 2307 mV vs. Li/Li+ corresponded to the formation of short-chain polysulfides and the latter peak was assigned to the formation of long-chain polysulfides and sulfur, respectively.48,49 The 1705 mV vs. Li/Li+ lower oxidation potential of the second cycle was identified as a common phenomenon of Li2S cathodes.50 The polysulfides, which were formed in the first cycle, dissolved in the electrolyte and catalyzed the oxidation of Li2S to S8 leading to a lower oxidation potential.51
 | ||
| Fig. 6 (a) Cyclic voltammogram of I-Li/C_45%; (b) voltage profile of the first cycle measured at C/20; (c) specific capacity retention of I-Li/C_45% at C/10 when cycled between 2600 and 1850 mV and (d) discharge capacities and coulombic efficiencies of I-Li/C_45% at different C-rates; each data point represents the mean value of five cycles at the specific current from three different cells. | ||
The reductive peaks showed comparable potentials as identified in the first cycle, which was consistent with the literature.52–54 Additionally, cycling experiments were performed to evaluate the capacity, capacity retention and high-rate capability (Fig. 6b–d).
The initial cycle was performed at C/20 and the voltage profile is shown in Fig. 6b.
During the charging process, a small plateau at 2696 mV was observed, which seemed to be corresponding to the peak at 2740 mV vs. Li/Li+ in the first cycle of the CV. The voltage shift resulted from the cell setup (CV: three-electrode configurations, cycling: two-electrode configuration) as well as the different current flow. As seen in Fig. 6a the cell voltage rose after the plateau drastically. However, the voltage seemed to run into a plateau at about 3600 mV instead of reaching the set cut-off voltage of 3900 mV. This leads to the presumption that the polysulfide shuttle had a strong influence on cell voltage. The shuttle caused a constant voltage by generating polysulfides which were reduced and re-oxidized at the electrode.55 Additionally, electrolyte decomposition at a voltage higher than 3500 mV vs. Li/Li+ was more and more likely. Hence, the charging process was stopped after 21 h.
The further cycles were recorded at C/10 (Fig. 6c).
A capacity of 702 mA h g−1 and capacity retention of 73% after 100 cycles were observed. The mean coulombic efficiency was 96%. The reason for the limited coulombic efficiency could be the side reactions caused by the polysulfide shuttle.56 In comparison with the Li2S–C–PVP material of Liu et al.45 the material showed a slightly lower initial capacity. However, after 50 cycles the capacity of Liu's material reached 460 mA h g−1 instead of the 554 mA h g−1 for I-Li/C_45%.45 Perhaps, this can be explained by the better structural integrity of our material.
Additionally, the high-rate capability was measured by applying different currents (Fig. 6d). A reduced capacity at higher C-rates is a commonly known property of lithium–sulfur batteries57 and can be explained by the slow kinetics of the polysulfide redox reaction and the very low electrical conductivity of the lithium–sulfur species.58 The presented material showed at a rate of 1C at least 56% of the capacity at 0.1C. This promising result can be explained by the good electrical connection between the particles and of the particles with the current collector, respectively.
Conclusion
Herein, we demonstrated a novel molecular approach to lithium sulfide for battery application. Characterization of the synthesized molecular precursor (LiSC2H4)2NMe provides an extraordinary control of the desired material formation. Good solubility in various solvents enables simple handling and an innovative preoperational fabrication carbon embedded Li2S. Ethanolic solution of both compounds, polymer and precursor, united in the same spinning solution makes the preparation of a suitable electrospinning system possible, which resulted in homogeneous Li2S/C-based fiber mats. Calcination of the electrospun fibers provides Li2S loaded fiber mats, which in the case of Li2S/C show a capacity retention of 73% over 100 cycles and a capacity of about 400 mA h g−1 at 1C. For further application of electrospun Li2S loaded carbon fibers the usage of PVP with lower molecular weight will allow higher Li2S loading through optimized PVP/I-Li ratios in the spinning solution. This will have influence on the electrochemical performance of battery cells.The use of the as-prepared molecular precursor and the application of the so obtained Li2S/C as the cathode material enable lithium metal free lithium–sulfur batteries with a higher safety level.
Introducing heavier alkali metals to the discussed molecular precursor system shows first promising results for the synthesis of (MISC2H4)2NMe (MI = Na, K) compounds and from that the preparation of Na2S and K2S materials, which is still under investigation.
Experimental
All manipulations of air- and moisture-sensitive materials were carried out under a nitrogen atmosphere using stock-type all-glass assemblies. All solvents and reactants were used without further purification, except otherwise mentioned. n-Butyl lithium (2.5 M in n-hexane solution) and polyvinylpyrrolidone (PVP, 1300000 g mol−1) were purchased from Acros Organics. NMR spectra were recorded on a Bruker Avance II 300 spectrometer; chemical shifts are quoted in parts per million relative to external TMS (1H and 13C). Thermal analysis was performed on a TGA/DSC 1 (Mettler-Toledo GmbH, Germany) in a nitrogen atmosphere (25 mL min−1) at a rate of 10 °C min−1 from 30 to 800 °C. For the determination of the Li2S amount in the final product, synthetic air was used at a rate of 10 °C min−1 from 30 to 800 °C. This temperature was held for 5 h. The sample weight was in the range of 5–20 mg. Room temperature powder X-ray diffraction (XRPD) was conducted on an STOE-STADI MP diffractometer, operating in transmission mode using Mo Kα (λ = 0.71073 Å) radiation. Measured peak patterns were compared to reference powder diffraction files (PDF). The crystallite size of the produced Li2S in electrospun fibers was calculated from the line broadening analysis of the most intense diffraction peak using Scherrer's equation; D¼Kλ/(βcosθ), where D is the coherently diffracting domain size (average crystallite size), K is the shape factor (0.94), λ is the wavelength of the X-ray radiation, β is the full width at half maximum (FWHM) measured in radians and θ is the Bragg angle. The size and morphology of the obtained fibers were analyzed using an FEI Strata Dual Beam 235 focused ion beam coupled scanning electron microscope (FIB-SEM) and a JEOL JEM-2200FS transmission electron microscope (TEM). A FuG electronics GmbH high voltage power supply and a Landgraf Laborsysteme LA-100 syringe pump were used for electrospinning. As spinnerets, 21G (inner diameter 0.5 mm) blunt needles were applied. The synthesis of (HSC2H4)2NMe I has been described before.32
Synthesis of N-methyl-diethanethiolamine-lithium (LiSC2H4)2NMe (I-Li)
12.60 mL (31.20 mmol) of 2.5 M n-butyl lithium in n-hexane were added to a liquid nitrogen-cooled solution of N-methyl-diethanethiolamine (2.39 g, 15.60 mmol) in 10 mL heptane and stirred for 30 min at room temperature. The resulting colorless powder was washed several times with n-heptane. Afterward, the solvent was removed under reduced pressure. The product was isolated as a colorless solid in a yield of 94% (2.39 g, 14.60 mmol).1 H NMR (D 2 O, 300 MHz, RT, ppm): 2.37 (s, 3H, –NCH 3 ), 2.61 (m, 4H, –NCH 2 CH 2 S–), 2.66 (m, 4H, –SCH 2 CH 2 N–).
13 C NMR (D 2 O, 75 MHz, RT, ppm): 20.4 (–SCH 2 CH 2 N–), 40.4 (–NCH 3 ), 62.4 (–SCH 2 CH 2 N–).
Preparation of the composite metal sulfide/conductive carbon fiber mats
In a typical electrospinning process, the corresponding spinning solution (see Table 1) was fed with a feed rate of 10 μL min−1. The distance between the needle and rotary drum collector was kept constant at 15 cm and a high voltage of 15 kV was applied. The process was conducted at room temperature with relative humidity below 35%.| Sample ID | Precursor concentration [mol L−1] | PVP concentration [mol L−1] |
|---|---|---|
| I-Li/PVP_58% | 0.46 | 4.29 × 10−5 |
The obtained green fibers were calcined in a tubular furnace with a temperature program of 1 °C min−1 up to 280 °C (holding for 2 h) under ambient conditions, followed by a temperature increase of 1 °C min−1 up to 700 °C (holding for 3 h) under a nitrogen atmosphere. All handling after calcination was performed under strictly inert conditions.
Electrode preparation and electrochemical testing
Cathode preparation and assembly of test cells were done in an argon-filled glovebox (MBraun, Germany). The contents of oxygen and moisture were below 2.0 ppm. First, the synthesized active material (I-Li/C_45%) was homogenized and mixed with carbon black (Super P C65, Timcal) using a mortar. PVP (PVP K90, AppliChem) was dissolved in N-methyl-2-pyrrolidone (NMP, Acros Organics) to form a binder solution. Afterward, the mixture of active material and carbon black was added to the binder solution. The suspension was homogenized by magnetic stirring to form a slurry. The ratio of the active material, carbon black and PVP was set to 80:5:15. The corresponding slurry was cast on a non-woven carbon (Sigracet 28AA, SGL Carbon SE) using a doctor blade. Subsequently, the electrodes were dried at 90 °C under vacuum for 16 hours. Finally, the electrodes with a diameter of 13 mm (coin cells) and 12 mm (Swagelok® T-cells) were punched out. The loading of Li2S was about 0.5 mg cm−2.
A solution of 1.0 M lithium bis(trifluoromethane sulfonyl)imide (LiTFSI, Sigma Aldrich) and 0.2 M lithium nitrate (LiNO3, Alfa Aesar) in 1,3-dioxolane (DOL, Sigma Aldrich) and dimethoxyethane (DME, Sigma Aldrich) (1:1, v/v) served as the electrolyte for the electrochemical investigation.
The charge/discharge experiments of the Li2S electrodes were performed in coin cells (CR 2032, Hohsen Corp.) with a BasyTec CTS-Lab XXL. Lithium metal (Alfa Aesar, thickness: 380 μm) served as the counter electrode. A glass fiber filter (GF/D, Whatman) soaked with 100 μl electrolyte was used as the separator. The formation of the cells was done at 0.05C between 3900 and 1850 mV. Further cycles were recorded between 2600 and 1850 mV by applying various currents which correspond to C rates between C/10 and 2C. The charge/discharge measurements were carried out as triple determination, and the average values of capacities are shown as representative results.
Cyclic voltammograms (CV) were recorded with a Gamry Interface 5000E potentiostat in Swagelok® T-cells using a three-electrode configuration. The CVs of Li2S electrodes were recorded between 1800 and 4000 mV vs. Li/Li+ (initial cycle) and between 1800 and 3000 mV vs. Li/Li+ (second cycle), respectively, at a scan rate of 50 μV s−1. Lithium metal was used as the counter and reference electrode and polypropylene fleeces (FS2190, Freudenberg) soaked with electrolyte 300 μl were used as the separator.
Author contributions
The conceptualization, formal analysis and synthetic work, investigation, visualization, and writing were carried out at the University of Cologne by V. Brune, C. Bohr and T. Ludwig; M. Wilhelm contributed to the synthetic work and formal analysis of the Li2S loaded material; C. Bohr and M. Wilhelm realized SEM measurements. T. Fischer performed TEM measurements and the corresponding analysis of the produced materials. For review of the writing and editing A. Ichangi was responsible. The scientific supervision, editing of the text and project administration were accomplished by S. Mathur. Electrochemical measurements and analytics of the obtained data were realized by S. Hirt, S. Wennig and B. Oberschachtsiek from ZBT. All authors discussed the results and commented on the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for the financial and infrastructural support provided by the University of Cologne. This work was funded by the Federal Ministry for Economic Affairs and Energy based on a decision of the German Parliament, Project IGF 15EWN.References
- X. Fan, J. Yue, F. Han, J. Chen, T. Deng, X. Zhou, S. Hou and C. Wang, ACS Nano, 2018, 12, 3360–3368 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- R. C. Longo, L. E. Camacho-Forero and P. B. Balbuena, J. Chem. Phys., 2020, 152, 014701 CrossRef CAS PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- Y. Xiao, S. H. Lee and Y. K. Sun, Adv. Energy Mater., 2017, 7, 1601329 CrossRef.
- C. Van Pham, L. Liu, B. Britton, M. Walter, S. Holdcroft and S. Thiele, Sustainable Energy Fuels, 2020, 4, 1180–1190 RSC.
- J. Yan, W. Li, R. Wang, P. Feng, M. Jiang, J. Han, S. Cao, Z. Zhang, K. Wang and K. Jiang, ACS Energy Lett., 2020, 5, 1307–1315 CrossRef CAS.
- J. Huo and Y. Wang, Int. J. Electrochem. Sci., 2020, 15, 1529–1538 CrossRef CAS.
- X. Fang and H. Peng, Small, 2015, 11, 1488–1511 CrossRef CAS PubMed.
- S. F. Ng, M. Y. L. Lau and W. J. Ong, Adv. Mater., 2021, 2008654, 1–54 Search PubMed.
- Y. C. Ho and S. H. Chung, Chem. Eng. J., 2021, 422, 130363 CrossRef CAS.
- A. Manthiram, Y. Fu, S. H. Chung, C. Zu and Y. S. Su, Chem. Rev., 2014, 114, 11751–11787 CrossRef CAS PubMed.
- Z. Sun, M. Xiao, S. Wang, D. Han and S. Song, J. Mater. Chem. A, 2014, 2, 9280–9286 RSC.
- X. Zhang, H. Xie, C. S. Kim, K. Zaghib, A. Mauger and C. M. Julien, Mater. Sci. Eng., R, 2017, 121, 1–29 CrossRef.
- D. Su, D. Zhou, C. Wang and G. Wang, Adv. Funct. Mater., 2018, 28, 1–23 Search PubMed.
- J. Liu, Z. Li, B. Jia, J. Zhu, W. Zhu, J. Li, H. Pan, B. Zheng, L. Chen and G. Pezzotti, et al. , J. Mater. Chem. A, 2020, 8, 6303–6310 RSC.
- Z. Tong, L. Huang, W. Lei, H. Zhang and S. Zhang, J. Energy Chem., 2021, 54, 254–273 CrossRef.
- X. Ge, S. Liu, M. Qiao, Y. Du, Y. Li, J. Bao and X. Zhou, Angew. Chem., Int. Ed., 2019, 58, 14578–14583 CrossRef CAS PubMed.
- T. Pakki, E. H. Mohan, N. Y. Hebalkar, J. Adduru, S. V. Bulusu, A. Srinivasan, K. M. Mantravadi and N. R. Tata, J. Mater. Sci., 2019, 54, 9075–9087 CrossRef CAS.
- S. Cavaliere, S. Subianto, I. Savych, D. J. Jones and J. Rozière, Energy Environ. Sci., 2011, 4, 4761–4785 RSC.
- G. Sun, L. Sun, H. Xie and J. Liu, Nanomaterials, 2016, 6, 1–30 Search PubMed.
- C. H. Chang, S. H. Chung and A. Manthiram, Mater. Horiz., 2017, 4, 249–258 RSC.
- P. Strubel, S. Thieme, T. Biemelt, A. Helmer, M. Oschatz, J. Brückner, H. Althues and S. Kaskel, Adv. Funct. Mater., 2015, 25, 287–297 CrossRef CAS.
- W. Ren, W. Ma, X. Jin, S. Zhang and B. Tang, ChemSusChem, 2019, 12, 2447–2456 CrossRef CAS PubMed.
- Q. Cheng, W. Xu, S. Qin, S. Das, T. Jin, A. Li, A. C. Li, B. Qie, P. Yao and H. Zhai, et al. , Angew. Chem., Int. Ed., 2019, 58, 5557–5561 CrossRef CAS PubMed.
- D. Liu, C. Zhang, G. Zhou, W. Lv, G. Ling, L. Zhi and Q. H. Yang, Adv. Sci., 2018, 5, 1700270 CrossRef PubMed.
- M. Liu, N. Deng, J. Ju, L. Fan, L. Wang, Z. Li, H. Zhao, G. Yang, W. Kang and J. Yan, et al. , Adv. Funct. Mater., 2019, 29, 1–34 Search PubMed.
- K. Han, J. Shen, C. M. Hayner, H. Ye, M. C. Kung and H. H. Kung, J. Power Sources, 2014, 251, 331–337 CrossRef CAS.
- M. Yu, Z. Wang, Y. Wang, Y. Dong and J. Qiu, Adv. Energy Mater., 2017, 7, 1700018 CrossRef.
- Z. Li, S. Zhang, C. Zhang, K. Ueno, T. Yasuda, R. Tatara, K. Dokko and M. Watanabe, Nanoscale, 2015, 7, 14385–14392 RSC.
- F. Ye, Y. Hou, M. Liu, W. Li, X. Yang, Y. Qiu, L. Zhou, H. Li, Y. Xu and Y. Zhang, Nanoscale, 2015, 7, 9472–9476 RSC.
- V. Brune, C. Hegemann and S. Mathur, Inorg. Chem., 2019, 58, 9922–9934 CrossRef CAS PubMed.
- J. Harley-Mason, J. Chem. Soc., 1947, 74, 320–322 RSC.
- M. Sun, C. Hong and C. Pan, J. Am. Chem. Soc., 2012, 20581–20584 CrossRef CAS PubMed.
- A. Fotouhi, D. Auger, L. O'Neill, T. Cleaver and S. Walus, Energies, 2017, 10, 1937 CrossRef.
- K. Patel, J. Undergrad. Res., 2016, 9, 39–42 Search PubMed.
- X. Meng, D. J. Comstock, T. T. Fister and J. W. Elam, ACS Nano, 2014, 8, 10963–10972 CrossRef CAS PubMed.
- F. Ye, M. Liu, X. Yan, J. Li, Z. Pan, H. Li and Y. Zhang, Small, 2018, 14, 1–7 Search PubMed.
- J. W. Jung, C. L. Lee, S. Yu and I. D. Kim, J. Mater. Chem. A, 2016, 4, 703–750 RSC.
- M. Yu, Z. Wang, Y. Wang, Y. Dong and J. Qiu, Adv. Energy Mater., 2017, 7, 1700018 CrossRef.
- X. Chen, L. Peng, L. Yuan, R. Zeng, J. Xiang, W. Chen, K. Yuan, J. Chen, Y. Huang and J. Xie, J. Energy Chem., 2019, 37, 111–116 CrossRef.
- X. Huang, Z. Wang, R. Knibbe, B. Luo, S. A. Ahad, D. Sun and L. Wang, Energy Technol., 2019, 7, 1801001 Search PubMed.
- Y. Yang, G. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef CAS PubMed.
- Y. Jung and B. Kang, Phys. Chem. Chem. Phys., 2016, 18, 21500–21507 RSC.
- J. Liu, H. Nara, T. Yokoshima, T. Momma and T. Osaka, J. Power Sources, 2015, 273, 1136–1141 CrossRef CAS.
- K. Cai, M.-K. Song, E. J. Cairns and Y. Zhang, Nano Lett., 2012, 12, 6474–6479 CrossRef CAS PubMed.
- Z. W. Seh, J. H. Yu, W. Li, P.-C. Hsu, H. Wang, Y. Sun, H. Yao, Q. Zhang and Y. Cui, Nat. Commun., 2014, 5, 5017 CrossRef CAS PubMed.
- B.-J. Lee, H.-Y. Park, D.-S. Yang, T.-H. Kang, S. Hwang and J.-S. Yu, J. Electrochem. Soc., 2019, 166, A5244–A5251 CrossRef CAS.
- Y. Fu and A. Manthiram, J. Phys. Chem. C, 2012, 116, 8910–8915 CrossRef CAS.
- Y. Yang, G. Zheng, S. Misra, J. Nelson, M. F. Toney and Y. Cui, J. Am. Chem. Soc., 2012, 134, 15387–15394 CrossRef CAS PubMed.
- Y. Fu, Y. S. Su and A. Manthiram, Adv. Energy Mater., 2014, 4, 1–5 Search PubMed.
- K. Cai, M.-K. Song, E. J. Cairns and Y. Zhang, Nano Lett., 2012, 12, 6474–6479 CrossRef CAS PubMed.
- M. Yu, Z. Wang, Y. Wang, Y. Dong and J. Qiu, Adv. Energy Mater., 2017, 7, 1700018 CrossRef.
- Y. Chen, S. Lu, J. Zhou, W. Qin and X. Wu, Adv. Funct. Mater., 2017, 27, 1700987 CrossRef.
- Y. Diao, K. Xie, S. Xiong and X. Hong, J. Power Sources, 2013, 235, 181–186 CrossRef CAS.
- M. Wild, L. O'Neill, T. Zhang, R. Purkayastha, G. Minton, M. Marinescu and G. J. Offer, Energy Environ. Sci., 2015, 8, 3477–3494 RSC.
- T. Zhang, M. Marinescu, S. Walus, P. Kovacik and G. J. Offer, J. Electrochem. Soc., 2018, 165, A6001–A6004 CrossRef CAS.
- Y. Tao, Y. Wei, Y. Liu, J. Wang, W. Qiao, L. Ling and D. Long, Energy Environ. Sci., 2016, 9, 3230–3239 RSC.
| This journal is © The Royal Society of Chemistry 2022 |