
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistically improved PIM-1 membrane gas separation performance by PAF-1 incorporation and UV irradiation†
Rujing
Hou
 a,
Stefan J. D.
Smith
a,
Kristina
Konstas
b,
Cara M.
Doherty
b,
Christopher D.
Easton
b,
Jaesung
Park
c,
Heewook
Yoon
c,
Huanting
Wang
a,
Benny D.
Freeman
*ac and
Matthew R.
Hill
*ab
a,
Stefan J. D.
Smith
a,
Kristina
Konstas
b,
Cara M.
Doherty
b,
Christopher D.
Easton
b,
Jaesung
Park
c,
Heewook
Yoon
c,
Huanting
Wang
a,
Benny D.
Freeman
*ac and
Matthew R.
Hill
*ab
aMonash Centre for Membrane Innovation, Department of Chemical and Biological Engineering, Monash University, Clayton, VIC 3169, Australia. E-mail: matthew.hill@monash.edu; Benny.Freeman@monash.edu
bCSIRO, Manufacturing, Private Bag 10, Clayton South, VIC 3169, Australia. E-mail: Matthew.Hill@csiro.au
cJohn J. McKetta Jr. Department of Chemical Engineering, The University of Texas at Austin, 2501 Speedway, Austin, TX 78712, USA. E-mail: freeman@che.utexas.edu
First published on 15th March 2022
Abstract
Super-glassy polymer membranes have suffered from the trade-off relationship between permeability and selectivity for gas separation applications, despite the fact that membrane technology exhibits remarkable energy efficiency advantages over other separation methods. Polymers of intrinsic microporosity such as PIM-1 offer high fractional free volume (FFV) and intermediate gas selectivity, with permeability several orders of magnitude higher than conventional glassy polymers. The methods of producing mixed matrix membranes (MMM) by incorporating nanoparticles into a polymer matrix, or crosslinking, have been widely studied to improve membrane selectivity. While crosslinking and nanoparticle incorporation often increase selectivity or permeability, respectively, this is typically at the expense of the other, limiting transport properties to the Robeson upper bound. Porous aromatic frameworks such as PAF-1 have been shown to significantly increase the permeability of PIM membranes. Here, this nanoparticle additive is coupled with post UV irradiation treatment resulting in a membrane with both significantly improved membrane selectivity (i.e., 16-fold improvement for H2/CH4 selectivity, from 5.4 to 90) and high permeability (i.e., P(H2) = 4800 Barrer). Characterisation of the dual-enhanced membrane revealed that the synergetic performance is caused by a combination of the selective skin layer formed upon UV photo-oxidation with the additional permeable gas transport channels introduced to the bulk matrix by PAF-1. As a result of this dual-approach to membrane enhancement, the PIM-1 MMM exhibited better gas separation performance, surpassing the 2015 upper bounds for H2/N2 and H2/CH4 as well as 2008 upper bounds for H2/CO2 and CO2/CH4. Aging studies confirmed that PAF-1 addition, UV irradiation, and both modifications slowed physical aging rate compared to the pure PIM-1 membrane. The performance of this membrane was also investigated at a range of thicknesses, revealing its potential as a candidate for other membrane forms at scale.
Introduction
Due to its high energy efficiency and low instrument footprint area compared with traditional phase-change separation methodologies, membrane technology is garnering increased attention for gas separation. The current market-dominated membranes for industrial gas separation are mainly traditional glassy polymers, such as polyimides (PI), polysulfones (PSF), and cellulose acetate (CA). These materials usually show high selectivities (e.g., H2/CH4 = 56 for PSF) but low permeability (e.g., H2: 14 barrer for PSF) due to having a low fractional free volume (FFV).1 The invention of advanced glassy polymers with high FFV was driven by the need for highly permeable membranes. Polymers of Intrinsic Microporosity (PIMs), such as PIM-1, have a high FFV polymer structure because of a kinked and irregular polymer backbone through the contorted spiro-center (spiro-bisindane) site, which effectively disrupts polymer chain packing. PIM-1 usually possesses permeability several orders of magnitude higher than those of traditional polymers (e.g., P(H2): PIM-1 vs. PSF = 4900 vs. 14 barrer). Unfortunately, its selectivity (e.g., H2/CH4: PIM-1 vs. PSF = 4.8 vs. 56) needs improvement to meet industrial application requirements.1–5 The trade-off between permeability and selectivity was first examined in 1991 by Robeson using upper bound plots.6–8 Later, as a result of enormous efforts to improve membrane performance, the upper bounds were revised in 2008, 2015, and 2019 (for CO2/CH4 and CO2/N2).9–11 These efforts include polymer structure design (e.g., introducing Tröger's Base (TB)12–16 or triptycene moieties17,18); chemical19–21 or thermal crosslinking;22 UV crosslinking/oxidation;23–29 and adding nanoparticles into the polymer matrix to form Mixed Matrix Membranes (MMMs).30–34 Amongst these methods, crosslinking and UV oxidation are most effective at improving membrane selectivity, but with dramatic permeability loss.29 On the other hand, adding porous nanoparticles to form MMMs usually results in an improvement to membrane permeability, but is less effective at improving selectivity.35–37Porous aromatic frameworks (PAF-1) are formed by covalent bonds and with a short-range ordered, rigid, and three-dimensional open network. The highly porous structure has made PAF-1 a promising additive in MMMs to improve membrane permeability.31,35,38–45 For example, our previous work showed that 10 wt% loading of PAF-1 enhanced gas permeability (H2, N2, CH4, and CO2) of TPIM-2 membrane by 130–200%.45 Similarly, Smith et al. reported that PAF-1 containing thermally rearranged (TR) MMMs showed a 55-fold increased CO2 permeability without selectivity loss.36 Lau et al. also reported that the functionalized PAF-1 (PAF-1-Li6C60) containing PTMSP MMMs exhibited a 70% CO2 permeability enhancement and was associated with a slower physical aging rate relative to the pure PTMSP membrane (−9% vs. −74%).37 Our recent study also revealed that PAF-1 also works for conventional low FFV polyimide membrane.46 Despite these remarkable achievements, industrial applications will require further improvement to their relatively moderate selectivity.
UV crosslinking can effectively improve membrane selectivity. First reported in a US patent for a polyimide membrane with highly selective and stable properties,47 it has since been applied to many other polyimide membranes that contain aromatic ketone moieties (a photosensitizer) and benzylic methyl groups crosslinked through a hydrogen abstraction mechanism.23,24,27,28 In 2010, it was extended from these low permeable polyimide membranes to highly permeable PIMs membranes, which also effectively improved membrane selectivity as is the case for traditional polyimide polymers.48 For instance, H2/CO2 selectivity of PIM-1 membrane increased from 0.6 to 7.3 (11-fold up) in Chung et al.'s work.26 Similarly, H2/N2 and H2/CH4 selectivity of PIM-1 membrane increased from 9.8 to 37.4 and from 7.6 to 33.6 (upon 60 min UV exposure in air), respectively in Song's group work.29 Accordingly, the mechanism for the improved selectivity of PIM-1 membrane was revised as a photo-oxidation/chain scission effect due to its different polymer structure and reaction upon UV irradiation compared to low permeable polyimide membrane. Despite improved membrane selectivities for all the UV treated samples, their gas permeabilities were compromised by UV irradiation. For instance, the gas permeability of H2 decreased from 3731 to 452 barrer (an 88% drop upon 4 h UV irradiation at 254 nm) in Chung's group work and reduced from 3195 to 1427 barrer (a 55% drop upon 1 h UV exposure at 254 nm) in Song's group work, respectively.26,29
Given the distinguishing effects of nanoparticle addition and UV irradiation on membrane performance, this work has applied these two methodologies on PIM-1 membranes to obtain both high permeability and selectivity. Specifically, PIM-1 membrane was incorporated with PAF-1 nanoparticles to fabricate MMMs which were then irradiated with UV light. Despite the previous studies of either PIM-1/PAF-1 MMMs or UV treated PIM-1 membrane, this work is highlighted by the novelty of the synergistic effects between the advanced materials of PAF-1 and PIM-1 under UV exposure which contribute to a superior gas separation performance than that of the single functionalized PIM-1 membranes. The resulting membrane performance surpassed the 2015 upper bound for H2/CH4 and H2/N2, as well as 2008 upper bound for H2/CO2 and CO2/CH4 separation. A slower physical aging rate was also demonstrated. To elucidate the mechanism behind membrane performance, characterisations were conducted including Fourier-transform infrared spectroscopy (FT-IR), X-ray photoelectron spectroscopy (XPS), positron annihilation lifetime spectroscopy (PALS), scanning electron microscopy (SEM), and gel permeation chromatography (GPC).
Experimental section
Materials
Reagents 3,3,3′,3′-tetramethyl-1,1′-spirobiindane-5,5′,6,6′-tetraol (TTSBI, 99%, Sigma-Aldrich) and tetrafluoroterephthalonitrile (TFTPN, 99%, Sigma-Aldrich) were purified by methanol recrystallization and sublimation, respectively. Potassium carbonate (K2CO3) was dried under reduced pressure at 120 °C overnight. Other reagents and solvents including bis(1,5-cyclooctadiene)nickel(0), 2,2′-bipyridyl, 1,5-cyclooctadiene, tetrakis(4-bromophenyl)methane, anhydrous dimethylformamide (DMF), anhydrous mesitylene, calcium hydride (CaH2), hydrochloric acid (HCl, 37%), anhydrous chloroform (CHCl3), and tetrahydrofuran (THF), were used as received from Sigma-Aldrich without further purification. Deionized water (H2O) was used in PAF-1 synthesis as a washing agent.PIM-1 synthesis
PIM-1 polymer was synthesized as described in our previous work.2 Briefly, in a Schlenk tube, an equimolar ratio of TFTPN (51.6 mmol) and TTSBI (51.6 mmol) with excess K2CO3 (154.8 mmol) was stirred in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DMF:mesitylene solvent mixture (118 mL:118 mL) for 3 h at 160 °C under N2 atmosphere. The product was purified by precipitating PIM-1 from an excess volume of methanol and washing with dilute HCl (2 mol L−1) after redissolution into minimal chloroform. Yellow PIM-1 powder was obtained through repeated precipitation from methanol. The final product was dried under reduced pressure at 80 °C for 6 h. PIM-1 molecular weight was 242 kDa with a polydispersity of 3.7 (Table S1†). The molecular structure of PIM-1 is shown in Fig. 1a (inset).
:1 DMF:mesitylene solvent mixture (118 mL:118 mL) for 3 h at 160 °C under N2 atmosphere. The product was purified by precipitating PIM-1 from an excess volume of methanol and washing with dilute HCl (2 mol L−1) after redissolution into minimal chloroform. Yellow PIM-1 powder was obtained through repeated precipitation from methanol. The final product was dried under reduced pressure at 80 °C for 6 h. PIM-1 molecular weight was 242 kDa with a polydispersity of 3.7 (Table S1†). The molecular structure of PIM-1 is shown in Fig. 1a (inset).
 | ||
| Fig. 1 (a) P (PIM-1) membrane cross-sectional image. The inset at the bottom right is the PIM-1 molecular repeating unit. (b) M (PIM-1@PAF-1 MMM) membrane cross-sectional image. The inset at the bottom right is the PAF-1 repeating unit. | ||
PAF-1 synthesis
PAF-1 was synthesized according to Lau's work.39 Briefly, in a Schlenk tube, an equimolar ratio of 2,2′-bipyridyl (1.28 g, 8.18 mmol) and degassed bis(1,5-cyclooctadiene)nickel(0) (2.25 g, 8.18 mmol) were mixed and stirred in anhydrous DMF (120 mL) under argon, followed by adding 1,5-cyclooctadiene (1.05 mL, 8.32 mmol) dried over CaH2 under argon. The mixture was heated to 80 °C for 1 h, tetrakis(4-bromophenyl)methane (1.00 g, 1.57 mmol) was added under argon atmosphere, and the mixture was stirred overnight at 80 °C. After cooling to room temperature and thoroughly washing with CHCl3 (×8), THF (×8), and H2O (×8), in that order, we recovered an off-white powder with a surface area of 3440 m2 g−1 (Fig. S1†). The molecular structure of PAF-1 is shown in Fig. 1b (inset).Membrane fabrication
Membranes with a thickness of ∼80 ± 10 μm were fabricated by the solution casting method. These thick pure membranes were made by dissolving PIM-1 polymer (300 mg) into anhydrous chloroform (6.5 mL) with stirring for 24 h, followed by pouring the filtered solution (0.45 μm microfilters) into a Teflon dish (diameter: 7.5 cm) with a perforated aluminum foil cover to slowly evaporate the solvent. After 48 h evaporation, the formed membranes were soaked in methanol for 24 h to remove the residual casting solvent and then dried under reduced pressure at 80 °C for 6 h. MMMs with 10 wt% PAF-1 loading were fabricated in the same way as pure membranes except that 30 mg PAF-1 nanoparticles were added into the filtered PIM-1 (270 mg) solution and stirred for another 24 h. Both pure and MMM membranes demonstrated thermal stability up to 400 °C (Fig. S2†). The thin membrane (thickness of ∼1 μm) was fabricated by the spin coating method. Porous polyacrylonitrile (PAN) was applied as a substrate and cleaned with isopropanol before use. 10 wt% polydimethylsiloxane (PDMS) solution in hexane was coated on PAN as the gutter layer by spin coating at rotation speed 3000 rpm for 1 min. After being cured in the oven at 60 °C overnight, the PDMS surface was coated with 3 wt% PIM-1 solution at 3000 rpm for 1 min and then dried in the oven at 60 °C for 2 h. The pure membranes and MMMs were denoted as P and M, respectively.UV treatment on membranes
Both sides of the membrane were exposed in air under the UV light lamp (λ = 254 nm, 6 W with filter assembly, ENF-260C) at a distance of 1 cm for a controlled period (0.5, 1.5, 3.0, and 4.5 h). The UV-treated membranes with specific time sessions were labelled as P0.5, P1.5, P3.0, P4.5, and M0.5, M1.5, M3.0, M4.5 for pure membranes and MMMs, respectively.Characterization methods
Based on polystyrene standards (Pst), the molecular weight and polydispersity index (PDI) of the pure PIM-1 polymer and UV irradiated samples were measured through gel permeation chromatography (GPC) in tetrahydrofuran (THF) solvent. The surface area of PAF-1 was calculated by the BET method from nitrogen isotherms at 77 K, which were obtained using a Micromeritics ASAP 2420, with activation at 100 °C under vacuum (10−6 torr) for 24 h before analysis. Average fractional free volume, pore sizes, and the relative number of pores for samples were determined by positron annihilation lifetime spectroscopy (PALS) using Ortec fast–fast coincidence spectrometers under vacuum (5 × 10−6 torr). Thermal stability for samples was assessed on a Mettler Toledo TGA 2 STARe System thermogravimetric analyser from 50 °C to 800 °C at 10 °C min−1 under 50 mL min−1 nitrogen flow. Cross-sectional SEM images of membranes were taken through a JEOL JSM-7001 field emission scanning electron microscope (FESEM) with an accelerating voltage of 5 kV. Fourier transform infrared (FT-IR) spectra of all membrane samples were collected using a Thermo Scientific NICOLET 6700 FT-IR. Depth element analysis for membranes was performed by X-ray photoelectron spectroscopy (XPS) using an AXIS Nova spectrometer (Kratos Analytical Inc., Manchester, UK) with a monochromated Al Kα source at a power of 180 W (15 kV × 12 mA). Single gas measurements were performed on a home-built set-up following the constant volume and variable pressure method at 25 ± 1 °C. Mixed gas measurements were performed on a home-built set-up following the constant pressure and variable volume method at 35 °C. See the ESI† for further details.Results and discussion
Membrane initial performance
The effect of UV irradiation on membrane performance was investigated by measuring UV treated membrane gas permeability and ideal selectivity changes under different exposure times. The distance between UV source and sample, duration of UV irradiation, and UV intensity will affect the extent of UV modification. Therefore, the UV source (6 W) and distance between UV lamp and membranes (1 cm) were fixed to systematically investigate UV effects on membrane performance.UV irradiation effects were initially examined on pure PIM-1 membranes, as shown in Fig. 2. Fig. 2a demonstrates the continuing permeability drop with increasing UV irradiation time for all the studied gases (H2, N2, CH4, and CO2). For example, the relative permeability of H2 dropped from 1, to 0.70, 0.66, 0.38, and 0.19 at UV time from 0, to 0.5, 1.5, 3.0, and 4.5 h, respectively. A similar trend was observed on other gases, including N2, CH4, and CO2. In addition, a greater permeability drop for larger gases was recorded, with an increasing gas kinetic diameter order of H2, CO2, N2, and CH4. For instance, the largest gas studied, CH4, in line with the aforementioned H2, exhibited a 2–9 times greater permeability drop. Consequently, the reduced permeability loss for smaller gases (H2 and CO2) over larger gases (N2 and CH4) led to a continuous selectivity improvement for H2 over N2 and CH4 throughout the UV irradiation period from 0 to 4.5, as shown in Fig. 2b. For instance, H2/CH4 and H2/N2 selectivity increased from 5.4 to 127 and 8.6 to 109, respectively. On the other hand, CO2/CH4 and CO2/N2 selectivity continued increasing from 12.5 to 35 and from 19.8 to 30 at UV time from 0 to 3 h, then dropped to 19.5 and 16.8 at UV time 4.5 h, respectively. The reduced CO2 selectivity from UV time 3 to 4.5 h was due to the same amount of permeability loss for gases of N2 and CH4 as CO2 at UV 4.5 h as shown in Fig. 2a (99% loss for CO2, N2, and CH4 at UV time 4.5 h). The greater permeability loss of CO2 at UV time 4.5 h also contributed to the remarkable H2/CO2 selectivity enhancement, which was 0.4 at UV time 0, improving to 2.1 at UV time 3 h, and jumping to 6.5 at UV time 4.5 h (Fig. 2b, inset). The decreased membrane gas permeability and enhanced selectivity resulting from UV irradiation in this study were consistent with previous studies.26,29
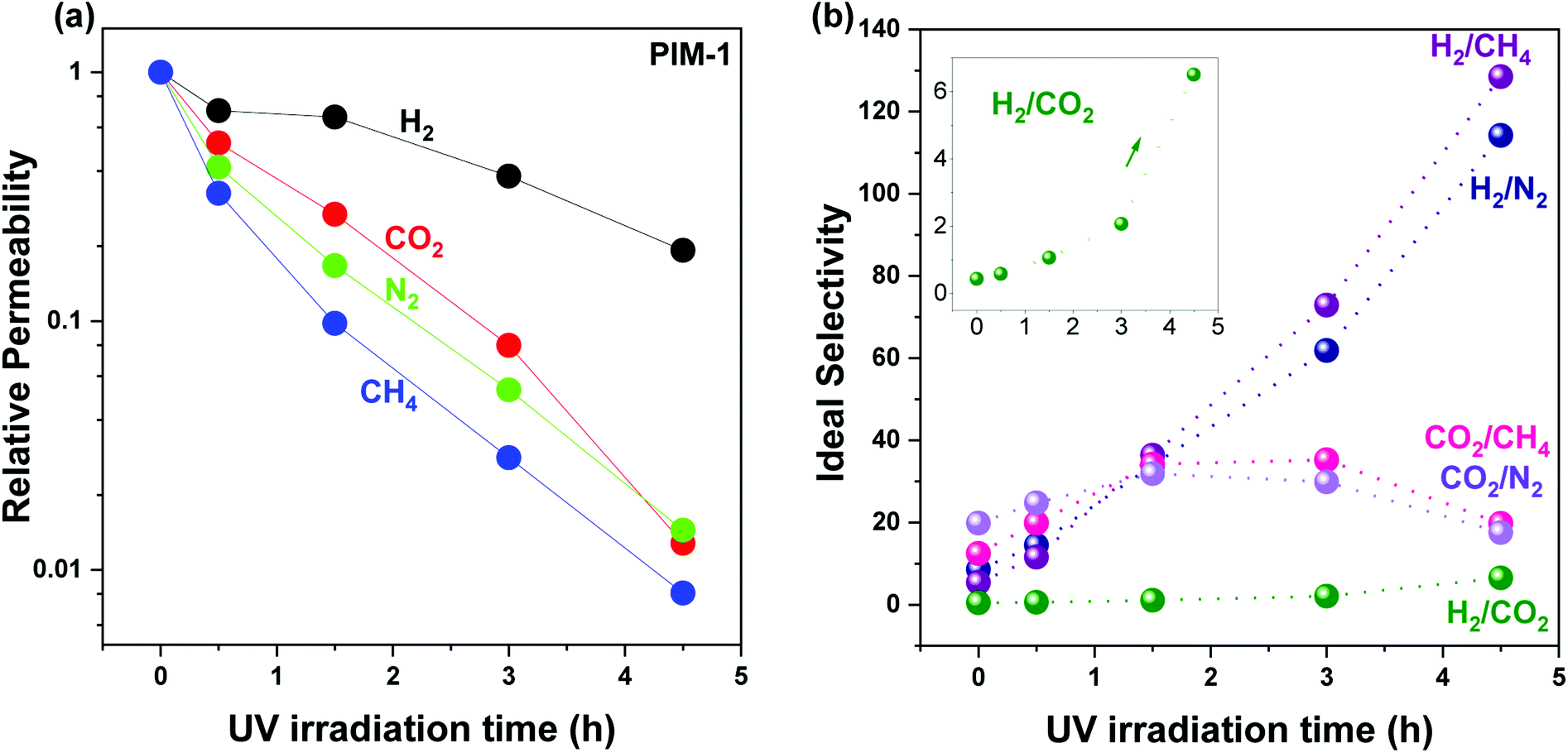 | ||
| Fig. 2 UV irradiation time effect on pure PIM-1 membrane (a) relative permeability change and (b) ideal selectivity change. The selectivity change of H2/CO2 is enlarged in the inseted figure in (b). Lines and arrows are drawn to guide the eye. Membrane permeability was measured twice for each gas at 25 ± 1 °C. The deviation is within ±10%. Individual data for each sample are given in Tables S2 and S3.† | ||
The PAF-1 effect on membrane was investigated by measuring changes in membrane morphology and performance (Fig. 1 and 3), comparing pure PIM-1 and PAF-1 containing MMMs. 2008, 2015, and 2019 upper bounds are included in Fig. 3 for purposes of comparison. Consistent with prior work, PAF-1 was found to be homogeneously dispersed in the PIM-1 matrix, and no non-selective voids were observed from the cross-sectional SEM images (Fig. 1b).2 As expected, the as-cast pure PIM-1 membrane demonstrated high permeability (e.g., P(H2) = 5300 barrer) but weak size sieving property (e.g., H2/CH4 = 5.4). After the addition of PAF-1 into the PIM-1 polymer matrix, the permeability of the small kinetic diameter H2 gas increased significantly, from 5300 to 7100 barrer, while larger gases such as N2, CH4, and CO2 remained relatively constant (from 620 to 640, 990 to 900, and 1200 to 1200 barrer, respectively). This behaviour led to a slightly increased H2 selectivity over CO2, N2, and CH4 (Fig. 3a–c, shaded area) which in return confirmed that no obvious defects were formed at the polymer-additive interface, consistent with the well-dispersed PAF-1 nanoparticle performance in PIM-1 polymer matrix (cross-sectional SEM image, Fig. 1b). We note the disparity between this work and Lau et al.'s previous PIM-1@PAF-1 work, which showed that H2, N2, and CH4 gas permeability increased significantly with the addition of PAF-1, from 1700 to 5500, 290 to 1200, and 500 to 2250 barrer, respectively.40 We ascribe this variance to the differing polymer synthesis procedures (this study vs. prior work: polycondensation reaction time: 3 h vs. 1 h; solvents: 1:1 volume ratio of DMF/mesitylene vs. 2:1 volume ratio of DMAC/toluene) and different membrane fabrication processes (with vs. without methanol soak, 80 °C, 6 h vs. 40 °C, 24 h membrane drying conditions). Budd's and Schwarz's work49,50 have suggested that trivial differences during the material polymerisation process, such as solvent choice/amount and reaction time, can have a significant effect on the resulting polymer conformation properties. Similar effects are known to result from variations in the membrane pre-treatment procedure undertaken immediately before gas testing.51 Consequently, different polymer architecture, membrane morphology, and polymer-additive interactions can be expected, and all these are known to influence membrane performance. Therefore, different gas permeability was also exhibited for the pure as-cast PIM-1 membrane (this work vs. prior work for P(H2): 5300 vs. 1700 barrer) as well as the aforementioned MMMs.
 | ||
| Fig. 3 PAF-1 incorporation and UV irradiation time effect on PIM-1-based membranes gas separation performance trade-off and their comparison with prior UV treated PIM-1 membranes and PIM-1 based MMMs of (a) H2versus CH4, (b) H2versus N2, (c) H2versus CO2, and (d) CO2versus CH4. Membrane permeability in this work was measured twice for each gas, at 25 ± 1 °C, and the deviation is within ±10%. Dotted/solid red and royal lines are drawn to guide the eye. Black upper bound curves of 2008, 2015, and 2019 are included. Individual data for each sample are given in Table S3.† | ||
UV treated MMM exhibited similar performance (highlighted in royal dotted lines in Fig. 3) to the UV treated pure PIM-1 membrane (highlighted in red dotted lines in Fig. 3), with continually decreased gas permeability and increased gas selectivity (H2 over N2, CH4, and CO2, Fig. 3a–c) with increasing UV irradiation time. For example, Fig. 3a shows H2 permeability of MMM decreased from 7100 barrer to 1500 barrer after UV irradiation 4.5 h, with a correspondingly increased H2/CH4 selectivity from 7.8 to 94. A similar trend was found for H2/N2 (Fig. 3b). Similar behavior was observed for CO2/CH4 selectivity (e.g., UV treated MMM) which initially increased from 13.7 to 39 after UV time 3 h and then decreased to 23 at UV time 4.5 h (Fig. 3d), due to the similar degree of reduced permeability for CO2 and CH4 (∼97–98% reduced, Table S2†) with longer UV exposure. Consequently, the larger gases' (CO2, N2, and CH4) transportation through the membrane were largely blocked (97–99% blocked, Table S2†), and the smallest (H2) gas diffused relatively freely through the membrane,26 which halted the increase in CO2 selectivity over N2 and CH4 (Fig. 3d) and continually increased H2/CO2 selectivity (Fig. 3c). Similar behaviour was also reported in Chung's previous work, where CO2/CH4 selectivity of PIM-1 membrane increased from 15.3 to 31.3 upon shorter UV exposure and then decreased to 23.8 upon longer UV irradiation.26 Conclusively, the simultaneous increase in membrane selectivity resulting from UV irradiation and the retained high gas permeability granted by the incorporated highly porous PAF-1 made the UV treated MMMs easily surpass 2015 H2/N2 and H2/CH4 upper bounds compared to the UV treated pure PIM-1 membrane (Fig. 3a and b). Both UV treated pure membrane and MMM surpassed the 2008 upper bounds for H2/CO2 and CO2/CH4 separation (Fig. 3c and d).
Synergistic effect from PAF-1 and UV irradiation
To investigate the synergistic function that results from PAF-1 and UV irradiation, we selected samples irradiated for 3 h with both desirable apparent permeability and selectivity (P3.0 and M3.0), accompanied by controlled non-UV treated pure (P) and MMM (M) samples for comparison. Before further discussion, we note that the permeability of the UV irradiated samples reported here is the apparent gas permeability, due to the asymmetric membrane structure (further details in Fig. 4). Apparent membrane permeability was used to compare with other previously reported UV treated PIM-1 and PIM-1 based MMMs membranes in Fig. 3 (Table S3†) as well as in the long-term property study shown in ESI (Fig. S10).†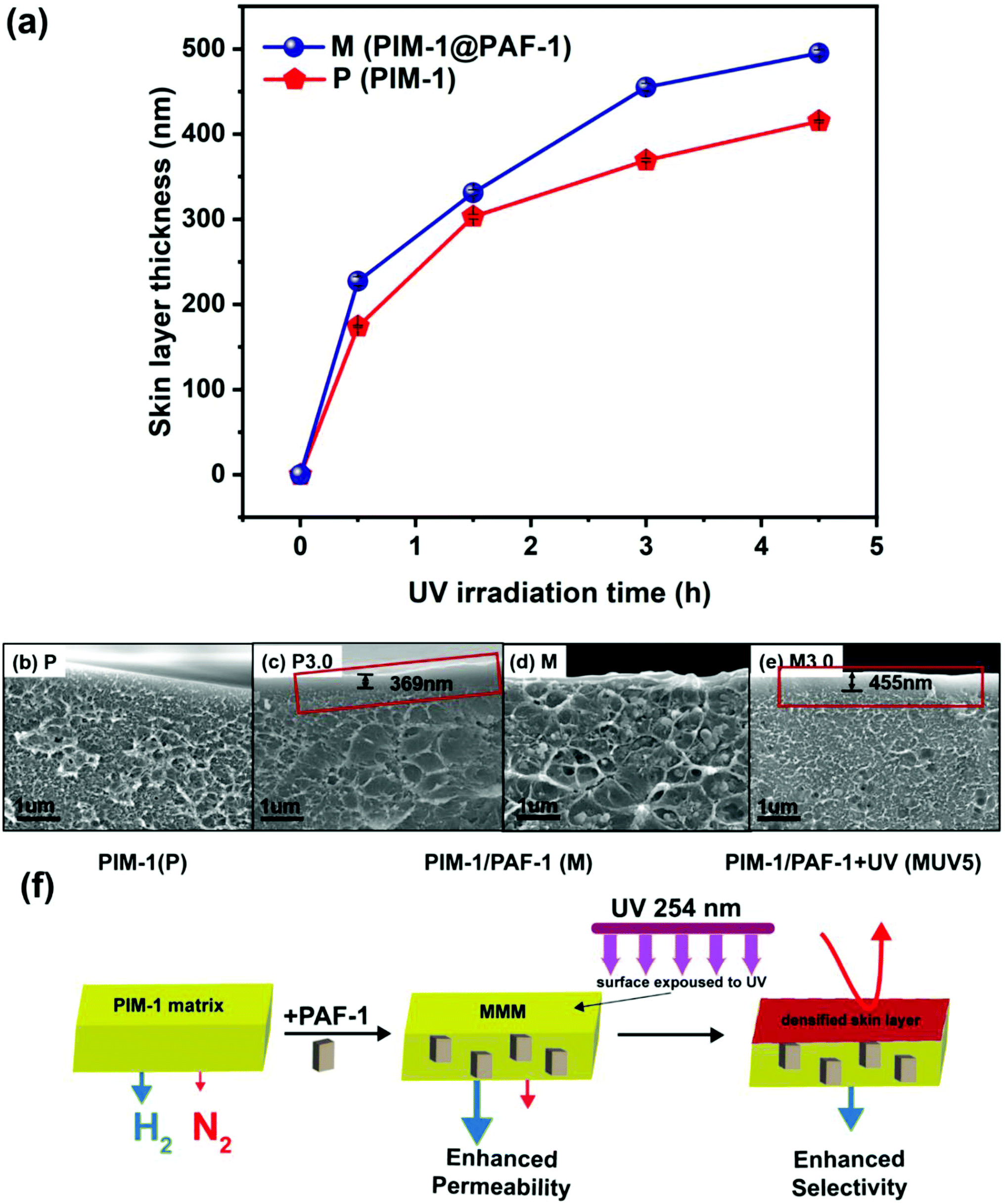 | ||
| Fig. 4 (a) Membrane surface skin layer thickness change based on UV irradiation time. Thickness was measured ten times for each UV irradiated sample and the deviation is within STDEV 6 nm. Red colour represents pure PIM-1 samples; royal colour represents MMMs (PIM-1@PAF-1). Cross-sectional SEM images of (b) P, (c) P3.0, (d) M, and (e) M3.0. (f) The effects of PAF-1 and UV irradiation on membrane morphology and performance change. The addition of porous PAF-1 contributes to the enhanced membrane permeability and densified skin layer resulting from UV irradiation is responsible for the improved membrane selectivity. | ||
Fig. 3 shows that porous PAF-1 had the primary role in improving H2 permeability, which increased from 5300 to 7100 barrer (highlighted by the red solid arrows from P to M in Fig. 3a–c), whereas UV irradiation mainly contributed to the significantly enhanced gas selectivity (Fig. 3a–d, highlighted with red solid arrows from P to P3.0, e.g., H2/CH4: from 5.4 to 72.7 in Fig. 3a), although accompanied by reduced gas permeability. In contrast, with the double functionalized membrane with both PAF-1 incorporation and UV irradiation (M3.0), a coupling effect was observed, which demonstrated both high selectivity and permeability and is highlighted by royal solid arrows from P to M3.0 (located between the single functionalized membranes in Fig. 3). For example, 16-fold and 12-fold increases in H2/CH4 selectivity (H2/CH4 = 90) were demonstrated for double functionalized M3.0 relative to the pure PIM-1 (P, H2/CH4 = 5.4) and the single functionalized MMM (M, H2/CH4 = 7.8). The high H2 permeability of M3.0 was also retained (4800 barrer) and was 2.4-fold higher relative to the corresponding P3.0 (2000 barrer), despite being lower than the pure PIM-1 (5300 barrer). Moreover, a synergistic effect was observed between the functions of PAF-1 and UV irradiation; that is, the porous PAF-1 property was not only responsible for the aforementioned retained permeability but also further improved membrane H2 selectivity when UV treatment (from 0 to 3 h) was applied. For instance, UV treated MMM (M3.0) had a 23% higher selectivity for H2/CH4 than the counterpart without PAF-1 (P3.0) (89.7 vs. 72.7) and 12% higher for H2/N2 (70.4 vs. 62.7), in Fig. 3a, b and Table S3.† The mechanism of this synergistic effect is discussed in detail in the next section. Consequently, the gas separation performance of the double functionalized membrane (M3.0) was superior to that of PIM-1 membranes reported in prior work with either UV treatment (cyan star symbols in Fig. 3) or nanoparticle incorporated MMMs (including PAF-1, grey star symbols in Fig. 3) by showing higher permeability, higher selectivity, or both.2,26,30,35,52 It is rare to see this behavior for MMMs in literature, as they usually possess high membrane permeability but only moderate selectivity.31 Furthermore, compared with the commercial polymer Matrimid, M3.0 not only surpassed H2 permeability by several orders of magnitude (e.g., P(H2) for M3.0 = 4800 vs. P(H2) for Matrimid = 27 barrer) but also performed with comparable H2 selectivity (e.g., H2/CH4 for M3.0 = 89.7 vs. H2/CH4 for Matrimid = 83.3).53 This superior membrane performance suggests many more opportunities for using UV irradiated PIM-1 MMMs in industrial applications.
Mechanism
To investigate the synergistic effects of PAF-1 and UV irradiation, membrane cross-sectional images (Fig. 4b–e and S4†) were obtained to visualize membrane surface morphology changes. As can be seen from Fig. 4c (highlighted in red frame), a thin dense layer was formed on the surface of pure PIM-1 membranes upon UV irradiation. This observation was consistent with Song's previous work, which illustrated that polymer chains were oxidized into shorter ones by the excited singlet oxygen and ozone resulting from UV irradiation in the presence of air, resulting in a tightly packed membrane surface acting as a selective layer (enhanced selectivity for UV treated membranes).29 The localized free volume on the PIM-1 surface acted as the micro-reactor for the photo-oxidative reaction. Consequently, with increasing UV exposure time (0, 0.5, 1.5, 3.0, and 4.5 h) on PIM-1, the dense skin layer thickness gradually increased from 0 to 174, 303, 369, and 415 nm (red line in Fig. 4a and SEM cross-sectional images in Fig. S4A–E†). This matched well with the continuingly increased H2 selectivity (over N2, CH4, and CO2) presented in Fig. 2b.Not limited to the pure PIM-1 membrane, MMMs containing PAF-1 presented a similar densified skin layer on the membrane surface (Fig. 4e, highlighted in red frame, details in Fig. S4A*–E*†) but slightly thicker than its counter-part pure PIM-1 under the same UV illumination conditions (from 0 to 227, 331, 455 and 495 nm, royal line in Fig. 4a). We attribute this to the high porosity property of PAF-1 (SA: 3435 m2 g−1) that allowed more oxygen to penetrate the membrane subsurface and enable further sub-surface photo-oxidization. This explains the synergistic effect resulting from PAF-1 and UV irradiation for further enhanced H2 selectivity that can be observed in Fig. 3a and b, compared with the counter-part UV treated pure PIM-1 membrane (e.g., H2/CH4 for M3.0 = 89.7 vs. H2/CH4 for P3.0 = 72.7). The synergistic effect occurred with UV irradiation from 0 to 3 h and disappeared at UV 4.5 h, possibly due to the trade-off function of PAF-1 between a thicker dense skin layer formation (contribution to the decreased permeability and increased selectivity) and its permanent gas transportation channels (contribution to the increased H2 permeability) in the bulk polymer matrix at UV 4.5 h. This does not affect the overall synergistic mechanism, as UV irradiation time ≤ 3 h yielded the optimal membrane performance. The membrane morphology changes upon PAF-1 nanoparticle incorporation and UV irradiation, as well as the effects on membrane performance, are summarized in Fig. 4f. This synergistic behaviour is more obvious in this work for PIM-1 than in our prior work for the PTMSP membrane.54 This is possibly due to their vastly different reaction rate with UV irradiation (3 h vs. 5 min) and slower rate give a chance to monitor the synergistic performance by recording membrane performance change.
Positronium Annihilation Lifetime Spectroscopy (PALS) helped to further elucidate the membrane pore sizes, their relative abundance (intensity, I%), and the average fractional free volume (FFV) associated with the membrane morphology changes that result from PAF-1 addition and UV irradiation on PIM-1 membranes. As can be seen in Fig. 5, the pure PIM-1 membrane has a bimodal pore size distribution at around 7 and 12 Å. With 3 h UV irradiation on the pure membrane (from P to P3.0, solid to the dotted red line and highlighted in red arrows), the decreased peak intensity at 12 Å led to an overall total FFV decline of 5% from P to P3.0 (Table 1), despite a slight shift to larger pores for both pore sizes. This drop in FFV was correlated to the decreased apparent membrane permeability upon UV exposure. The increased selectivity from P to P3.0 couldn't be identified by pore size change in PALS due to the low ratio (∼1%) of the dense skin layer formed on the bulk membrane surface. As a comparison, for homogenously dispersed highly porous PAF-1 (10 wt% loading and with SA = 3435 m2 g−1, Fig. S1†) in PIM-1 matrix, both pore sizes shifted to larger pore size (P to M, highlighted by black arrows in Fig. 5), and the FFV rose from P (total FFV = 29.6%) to M (total FFV = 37.3%, a Δ 26% increase, Table 1). The increased FFV from added PAF-1 was in line with the greatly enhanced H2 permeability for MMM (P to M, Fig. 3a–c). Despite the increase in FFV due to an increase in pore sizes and a considerable increase in intensity of the smaller pores, there was still an improvement in gas selectivity (P to M, Fig. 3a–c). This indicates that there may be a change in the interconnectivity between the small and large pores, due to the introduction of PAF-1. Upon further UV treatment of PIM-1@PAF-1 MMM from M to M3.0, both small and large pore sizes shifted smaller (highlighted by blue arrows in Fig. 5) and demonstrated a lower FFV but still higher than the UV treated pure PIM-1 membrane (e.g., total FFV: 28.1 (P3.0) < 33.4 (M3.0) < 37.3 (M), Table 1). The narrowed pore sizes from M to M3.0 matched with the considerably enhanced membrane selectivity demonstrated in Fig. 3 (e.g., H2/CH4 from 7.8 to 89.7). Despite the decreased total FFV from M to M3.0, the FFV was still higher than that of the P3.0 sample. This finding correlated to the coupling effect (Fig. 3a–c) that H2 permeability from M (7100 barrer) decreased to M3.0 (4800 barrer), but nonetheless higher than that of P3.0 (2040 barrer).
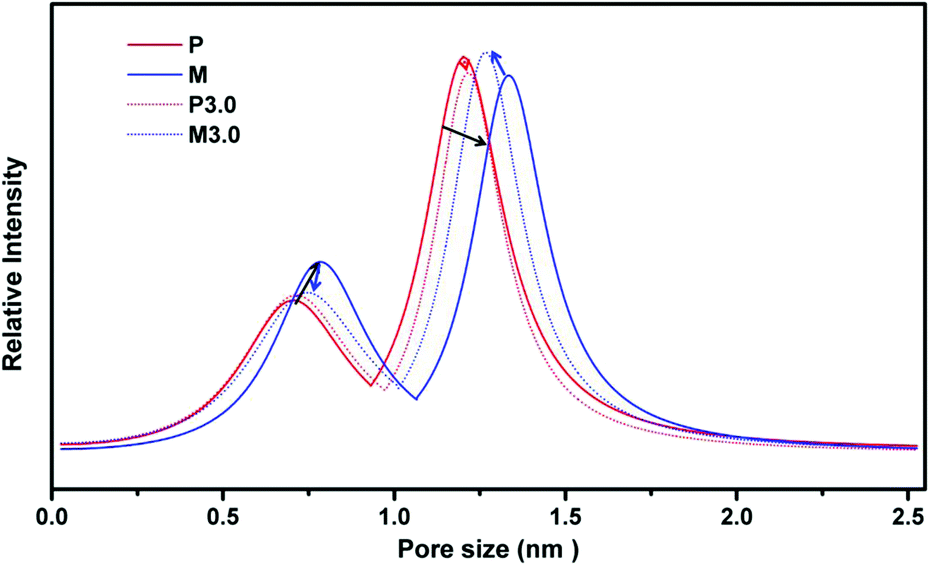 | ||
| Fig. 5 PALS for P (PIM-1), M (PIM-1@PAF-1), P3.0 (PIM-1 with 3 hours UV irradiation), M3.0 (PIM-1@PAF-1 with 3 hours UV irradiation). | ||
| FFV | FFV3 (%) | ± | FFV4 (%) | ± | Total FFV (%) | ± |
|---|---|---|---|---|---|---|
| a ±: deviation. | ||||||
| P | 2.18 | 0.40 | 27.5 | 1.7 | 29.6 | 2.1 |
| M | 3.64 | 0.46 | 33.6 | 1.8 | 37.3 | 2.3 |
| P3.0 | 2.32 | 0.74 | 25.8 | 2.8 | 28.1 | 3.5 |
| M3.0 | 2.59 | 0.46 | 30.8 | 1.8 | 33.4 | 2.2 |
Due to differences between the heterogeneous structure of UV irradiated samples (∼1% among overall membrane thickness) and the homogenous structure of PAF-1 dispersed membrane, a direct comparison of pore size change including these two factors need to be carefully considered. PALS (Fig. 5, Table 1) represents the bulk membrane properties, and any features due to UV exposures on the surface can not be separated from the bulk measurements. The overall FFV parameter was applied to determine average bulk membrane property changes with combined UV illumination and PAF-1 incorporation. PALS data were consistent with the corresponding apparent membrane performance.
In addition to the membrane morphology and average FFV changes observed from SEM (Fig. 4) and PALS (Fig. 5) characterisations, FT-IR spectra of membrane samples were also obtained to analyse membrane chemistry change as a function of PAF-1 and UV irradiation. No obvious change was found between PAF-1 loaded MMM and the pure PIM-1 in Fig. S5a,† possibly due to the low loading of PAF-1 in the polymer matrix and the fact that PAF-1 was embedded in the membrane rather than exposed on its surface. FT-IR spectra of PIM-1@PAF-1 MMM before and after UV treatment, Fig. 6, are representative of the UV irradiation effect on membrane surface chemistry. In addition to the representative peaks from PIM-1 polymer, including the nitrile group at 2238 cm−1, aromatic bending (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) at 1607 cm−1, C–H stretching from aromatic, methyl (CH3), and methylene (CH2) at 3055 cm−1, 2955 cm−1, 2930 cm−1, and 2840 cm−1, respectively, new oxidized groups emerged after UV exposure. These included the hydroxide (OH) group at 3300 cm−1 and the carbonyl group (CO) at 1730 cm−1, 1630 cm−1, and 1600 cm−1, all of which are in agreement with previous work.55 The broad peak of the carbonyl group was ascribed to the existence of various oxidation groups, including carboxylic acids, ketones, or aldehydes. UV irradiated samples also exhibited a newly formed nitroso group at 2207 cm−1 that resulted from the oxidization or dimerization of the nitrile (–CN) group in PIM-1. With increasing length of UV exposure, the intensity of these oxidized groups was reinforced accordingly, and the results matched well with the growing skin layer thickness in Fig. 4a and the correspondingly enhanced membrane H2 selectivity in Fig. 3a–c. The same oxidized groups appeared in pure PIM-1 upon UV irradiation, as seen in Fig. S5b.† This was consistent with Song's previously reported work and again affirmed the photo-oxidation mechanism. Conclusively, the synergistic effect of UV irradiation and PAF-1 addition on membrane performance showed that combining MMM and UV irradiation techniques can yield desirable membrane performance, with both high permeability and high selectivity. Furthermore, from the MMMs viewpoint, there are infinite combinations of polymers with porous nanoparticles that can be selected to target specific gas separation applications.
C) at 1607 cm−1, C–H stretching from aromatic, methyl (CH3), and methylene (CH2) at 3055 cm−1, 2955 cm−1, 2930 cm−1, and 2840 cm−1, respectively, new oxidized groups emerged after UV exposure. These included the hydroxide (OH) group at 3300 cm−1 and the carbonyl group (CO) at 1730 cm−1, 1630 cm−1, and 1600 cm−1, all of which are in agreement with previous work.55 The broad peak of the carbonyl group was ascribed to the existence of various oxidation groups, including carboxylic acids, ketones, or aldehydes. UV irradiated samples also exhibited a newly formed nitroso group at 2207 cm−1 that resulted from the oxidization or dimerization of the nitrile (–CN) group in PIM-1. With increasing length of UV exposure, the intensity of these oxidized groups was reinforced accordingly, and the results matched well with the growing skin layer thickness in Fig. 4a and the correspondingly enhanced membrane H2 selectivity in Fig. 3a–c. The same oxidized groups appeared in pure PIM-1 upon UV irradiation, as seen in Fig. S5b.† This was consistent with Song's previously reported work and again affirmed the photo-oxidation mechanism. Conclusively, the synergistic effect of UV irradiation and PAF-1 addition on membrane performance showed that combining MMM and UV irradiation techniques can yield desirable membrane performance, with both high permeability and high selectivity. Furthermore, from the MMMs viewpoint, there are infinite combinations of polymers with porous nanoparticles that can be selected to target specific gas separation applications.
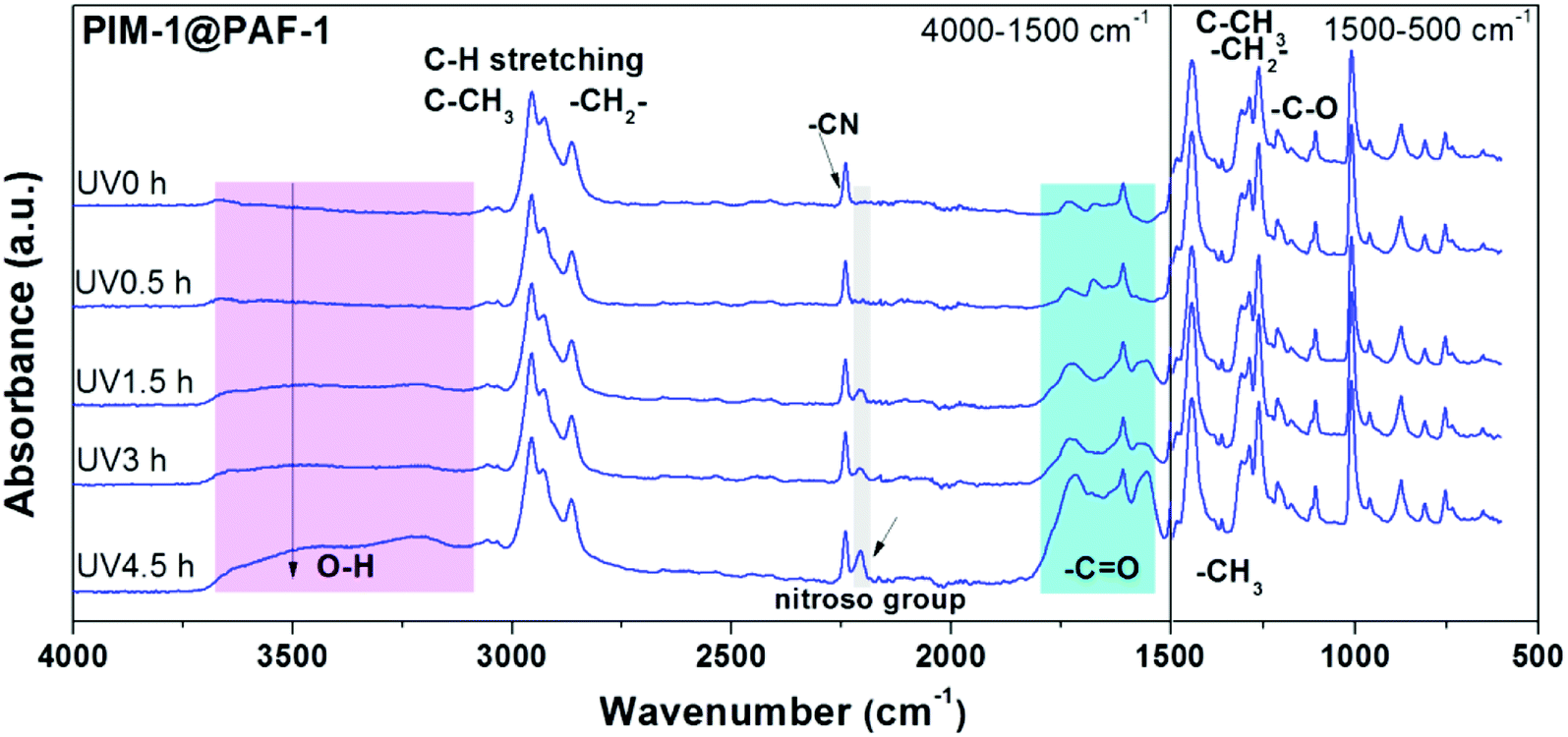 | ||
| Fig. 6 FT-IR spectra of PIM-1@PAF-1 MMM before and after UV irradiation with 0.5, 1.5, 3.0, and 4.5 h. | ||
XPS depth profiling experiments were employed as well to better understand the chemistry occurring at the surface and sub-surface of the UV-treated membranes. We compared four samples: the pure PIM-1 and PIM-1@PAF-1 samples, each with and without 4.5 h of UV treatment on both sides. The elemental quantification presented as atomic ratios was derived from survey spectra (Fig. 7), while insights into the carbon-based functional groups present were obtained from the fitting of high-resolution C 1s spectra (Fig. S6†).29,56 An example of the fit employed for the high-resolution C 1s spectra appears in Fig. S7.† Prior to etching, a significant increase in the O/C value can be observed for both UV treated membranes compared to the untreated membranes, confirming oxidation of the membrane surface with UV treatment. For example, M4.5 (MMM PIM-1@PAF-1 membrane with UV irradiation 4.5 h for both sides) and M (MMM PIM-1@PAF-1 without UV irradiation) had O/C values of 0.27 and 0.14 (expected value of 0.14), respectively, and the relative fractions of C derived from the fitting of high-resolution spectra support these results. An increase in component C5, assigned to O–CO, and component C3, assigned to C–O and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N, for the UV-treated samples were consistent with observations from FT-IR (Fig. 6). The increased O signal from the UV-treated samples can be observed at all time points of the etching, providing evidence that the UV treatment penetrates the membrane surface. We examined high-resolution N 1s spectra (Fig. S8†) of the samples to determine what impact, if any, UV-treatment had on the nitrile groups of the PIM-1 membrane, as suggested by the results from FT-IR (Fig. 6). A significant fraction of fitted spectra from the M4.5 and M samples was associated with nitrile (399.6 eV), as expected. An increase in the full width at half maximum of the component assigned to nitrile for sample M4.5 (1.3 eV) compared to M (0.95 eV) is consistent with an increase in the number of types of N-based functional groups contributing to this component. Nitroso groups have been reported in this region and higher binding energies (BEs), depending on the local environment of this functional group.57 A change in the component N2 at approximately 401.8 eV was observed with UV treatment. It is feasible that this component is associated with such groups, though N+ would also contribute intensity at this binding energy. Considering the non-specific nature of the UV treatment, which could result in the production of a wide range of different functional groups at the surface, it is difficult to provide definite assignments for the N 1s spectra based on the available information.
N, for the UV-treated samples were consistent with observations from FT-IR (Fig. 6). The increased O signal from the UV-treated samples can be observed at all time points of the etching, providing evidence that the UV treatment penetrates the membrane surface. We examined high-resolution N 1s spectra (Fig. S8†) of the samples to determine what impact, if any, UV-treatment had on the nitrile groups of the PIM-1 membrane, as suggested by the results from FT-IR (Fig. 6). A significant fraction of fitted spectra from the M4.5 and M samples was associated with nitrile (399.6 eV), as expected. An increase in the full width at half maximum of the component assigned to nitrile for sample M4.5 (1.3 eV) compared to M (0.95 eV) is consistent with an increase in the number of types of N-based functional groups contributing to this component. Nitroso groups have been reported in this region and higher binding energies (BEs), depending on the local environment of this functional group.57 A change in the component N2 at approximately 401.8 eV was observed with UV treatment. It is feasible that this component is associated with such groups, though N+ would also contribute intensity at this binding energy. Considering the non-specific nature of the UV treatment, which could result in the production of a wide range of different functional groups at the surface, it is difficult to provide definite assignments for the N 1s spectra based on the available information.
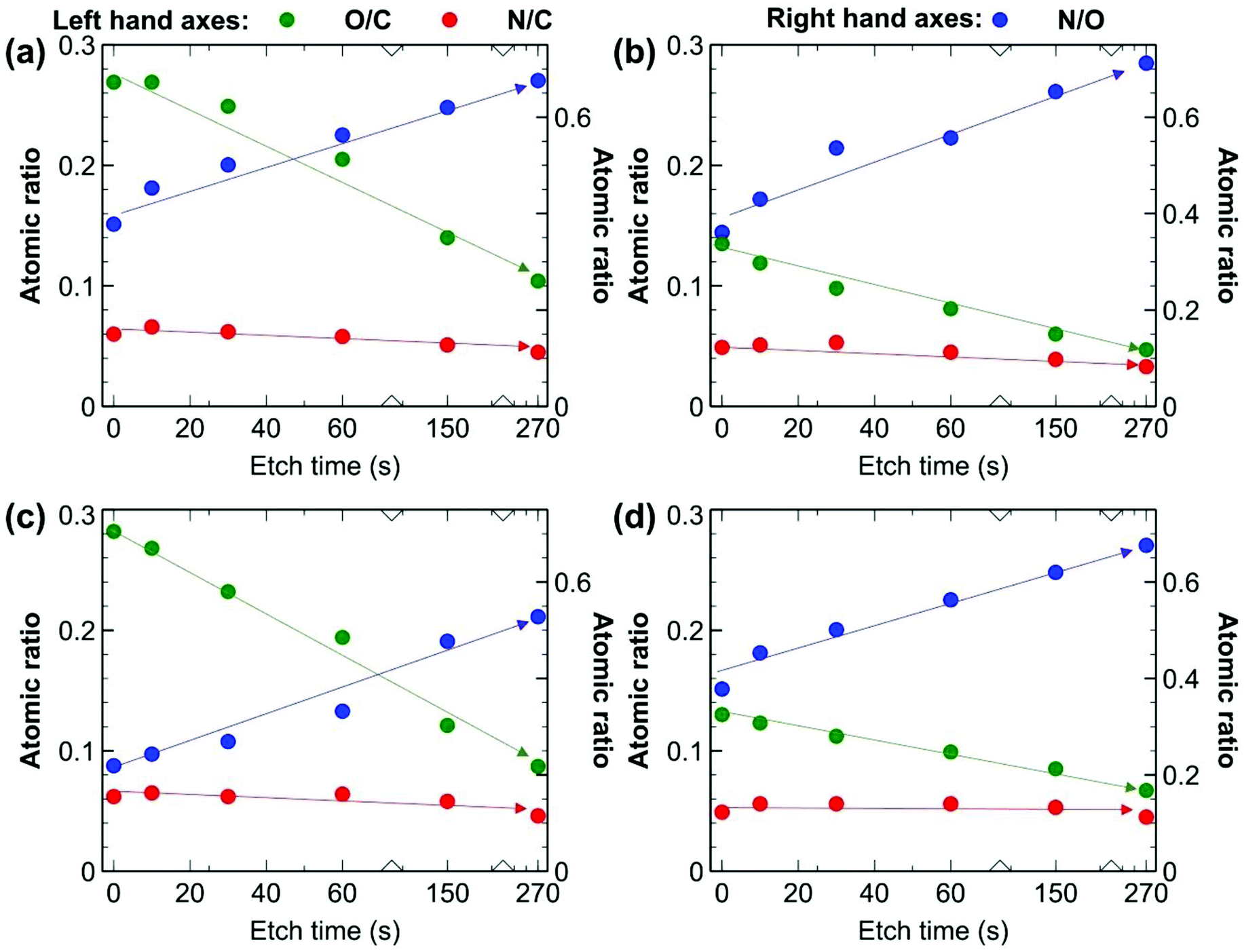 | ||
| Fig. 7 Elemental quantification derived from survey spectra expressed as atomic ratios from XPS depth profiling experiments for (a) M4.5, (b) M, (c) P4.5, (d) P. Although initial O/C values are higher at the beginning of the experiment for UV treated samples, indicating oxidation of the surface, N/C is roughly consistent throughout. Arrows and lines are included only to guide the eye and don't represent trendlines. | ||
The photo-oxidation mechanism was further supported by the reduced PIM-1 molecular weight (e.g., 242 kDa for Pvs.139 kDa for P3.0) and increased polydispersity (e.g., 3.7 for Pvs. 9.2 for P3.0) for UV irradiated samples (Table S1†). To check for any crosslinking between the newly formed oxidized groups, which can also lead to narrowed pore size and enhanced membrane selectivity, a gel fraction test was performed.23 No crosslinking was found as no remaining pieces (precipitation) were found in the commonly PIM-1 dissolved solvents of tetrahydrofuran (THF) or chloroform (CHCl3) (Fig. S9†).
Therefore, it is the photo-oxidation mechanism that contributed to the changes in membrane morphology and performance. Besides the aforementioned improvement in initial membrane performance, a slower physical aging rate was also demonstrated either by PAF-1 addition or by UV irradiation, or both. Details appear in ESI and Fig. S10.† Consistent with the single gas performance, the mixed gas performance of 50/50 CO2/CH4 also showed a similar synergistic effect. For instance, M3.0 exhibited a higher selectivity (66% up, 14 vs. 8.5) and retained permeability (6700 barrer) compared to that of the pure PIM-1 membrane. The lower selectivity of mixed gas (14) than that of single gas (39) performance is a typical phenomenon in gas mixture due to the competition of adsorption and diffusion compared to pure gas measurement conditions. Details appear in ESI and Table S4.†
Membrane thickness effect
To check membrane thickness effect on UV treated membrane performance, both thick membranes (∼10–1000 μm,58 in this work: ∼10 to 80 ± 5 μm) fabricated via a solution casting method and thin membranes (∼400 nm to 10 μm,58 in this work: ∼890 nm) made using a spin coating technique were given UV irradiation treatment. As seen in Fig. 8, solution casting-made thick membranes exhibit gradually increasing H2 selectivity along with decreasing membrane thickness, from 77 to 9 μm (black arrow in Fig. 8, e.g., H2/CH4P3.0,77 μm = 72.7 to H2/CH4P3.0,9 μm = 142.4). The same trend applies to the H2/N2 gas pair. On the other hand, CO2 gas pairs (CO2/N2 and CO2/CH4) exhibited reduced selectivity with decreasing membrane thickness (e.g., CO2/CH4P3.0,77 μm = 35.2 to CO2/CH4P3.0,9 μm = 26.4). This behaviour was similar to the trend observed for samples of similar thicknesses with increasing UV irradiation time, as shown in Fig. 2b and 3d. For instance, H2 selectivity kept increasing with increasing UV exposure time, whereas CO2 selectivity increased initially and then decreased with longer UV exposure. We ascribe this behavior to the gradually narrowed average size of membrane pores that results from increasing UV photo-oxidation effects, until the sieving point between H2 and CO2 is reached, terminating the increase in CO2 selectivity and even causing it to decrease. Similarly, with the same UV irradiation time, the densified skin layer ratio rises with a decrease in membrane thickness, and as a result, the average FFV of the entire membrane drops, demonstrating a similar gas selectivity trend. The consistent increase in H2/CO2 selectivity (from 2.1, 4.6, 4.8, and 5.4) as membrane thickness falls (from 77 to 47, 20, and 9 μm, black arrow) also supports this theory (highlighted by the enlarged inset in Fig. 8). A trade-off comparison of membrane performance based on various thicknesses is included in Fig. 3. On the other hand, as membrane thickness decreases further, the spin-coating fabricated thin membrane (∼890 nm) lost the remarkably enhanced gas selectivity performance that was present in thick membranes, perhaps because of contributions from the transition mesoporous intermediate layer beneath the densified layer.29 Unlike the thick membrane (>9 μm in this study)—which has a densified top skin layer, the intermediate mesoporous layer, and a bulky polymer as support—a thin (890 nm) membrane probably has only a surface skin layer, perhaps along with the mesoporous intermediate layer but without the bulky PIM-1 polymer, which leads to the drop in H2 and CO2 selectivity over N2 and CH4. This indicates membranes with various membrane thicknesses may have a different asymmetric membrane structure that directly affects the overall membrane performance. A proper membrane thickness is needed when UV irradiation apply to the membrane to avoid the mesoporous (pore size too large for gas separation) layer being the bottom/support layer.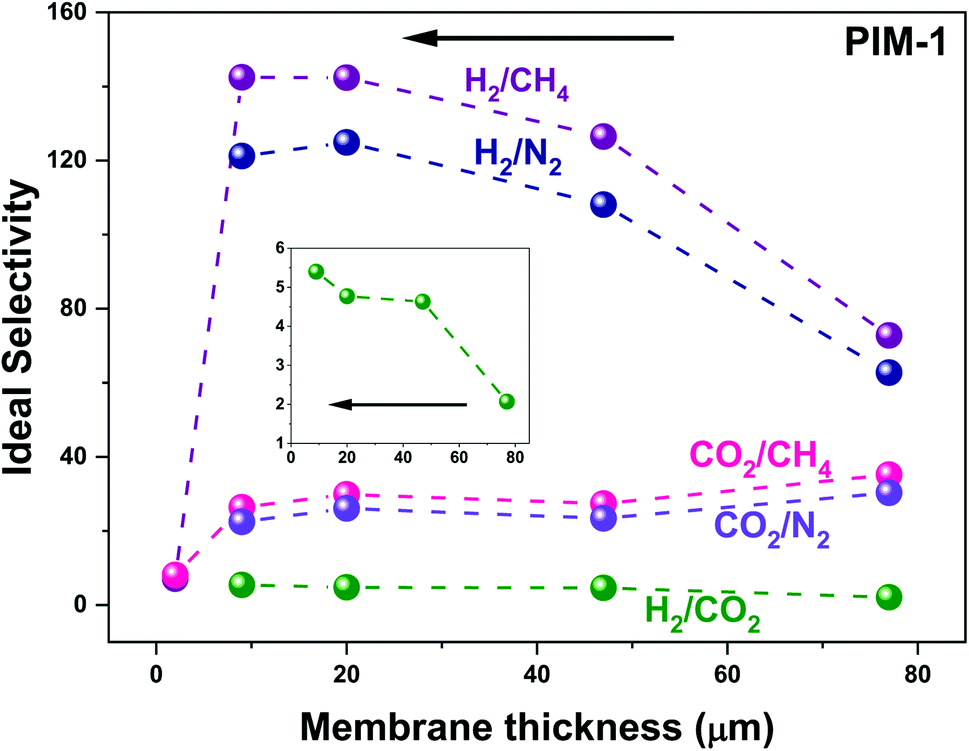 | ||
| Fig. 8 UV treated membranes selectivity change based on membrane thickness. Gas measurements operated at 25 ± 1 °C individual data can be found in Table S3.† | ||
Conclusion
Membranes with both high gas permeability and selectivity are pursued for gas separations. Here, favourable membrane performance, with both high H2 permeability and selectivity, was achieved under the synergistic function of PAF-1 and UV irradiation on the PIM-1 membrane. The incorporation of PAF-1 contributed to the largely enhanced PIM-1 membrane permeability, especially for such a small kinetic diameter gas as H2 (permeability increased from 5300 to 7100 barrer), thanks to the extra highly permeable gas transport channels provided by PAF-1. The densified selective skin layer formed on the membrane surface through photo-oxidation was responsible for the remarkably improved H2 selectivity (e.g., H2/CH4: from 7.8 to 89.7 and H2/N2: from 11.1 to 70.4). The simultaneously enhanced H2 permeability and selectivity enabled H2 separation performance superior to that of many recently reported polymers, including prior UV treated PIM-1 and PIM-1 based MMMs membranes with various additives. Dual-functionalized membrane performance also surpassed the 2015 upper bounds for both H2/CH4 and H2/N2 gas separation and the 2008 upper bounds for H2/CO2 and CO2/CH4 gas pairs. The slower aging rate of PIM-1 prepared with either PAF-1 addition or UV oxidation or a combination of the two as well as similar synergistic effect found in mixed gas performance makes them even more desirable for potential large-scale application. In addition, membrane selectivity can be easily regulated by adjusting UV irradiation time and membrane thickness, based on specific gas separation needs. The challenge of transferring the UV-treatment resulted high selectivity from thick to thin membrane demonstrated that the UV irradiation effect on membrane performance needs a proper thickness (no thinner than ∼1 μm). More studies related to membrane thickness are needed to transfer the synergistic effect observed in thick membranes to the industrially preferred thin membranes.Author contributions
Rujing Hou: conceptualization, investigation, writing – original draft, formal analysis, methodology. Stefan J. D. Smith: writing – review & editing, formal analysis, supervision. Kristina Konstas: resources, data curation, formal analysis. Cara M. Doherty: data curation, formal analysis. Christopher D. Easton: data curation, formal analysis. Jaesung Park: resources, formal analysis. Heewook Yoon: resources, formal analysis. Huanting Wang: writing – review & editing. Benny D. Freeman: project administration, conceptualization, writing – review & editing, supervision. Matthew R. Hill: project administration, conceptualization, writing – review & editing, supervision.Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors acknowledge the use of the instruments and scientific and technical assistance at the Monash Centre for Electron Microscopy, a Node of Microscopy Australia as well as TGA and FT-IR facilities from CSIRO. The authors acknowledge James Mardel for helping with FT-IR interpretation. CMD acknowledges her Veski Inspiring Women Fellowship for support.References
- V. Abetz, T. Brinkmann, M. Dijkstra, K. Ebert, D. Fritsch, K. Ohlrogge, D. Paul, K. V. Peinemann, S. Pereira-Nunes, N. Scharnagl and M. Schossig, Adv. Eng. Mater., 2006, 8, 328–358 CrossRef CAS.
- R. Hou, S. J. D. Smith, C. D. Wood, R. J. Mulder, C. H. Lau, H. Wang and M. R. Hill, ACS Appl. Mater. Interfaces, 2019, 11, 6502–6511 CrossRef CAS PubMed.
- P. M. Budd, K. J. Msayib, C. E. Tattershall, B. S. Ghanem, K. J. Reynolds, N. B. McKeown and D. Fritsch, J. Membr. Sci., 2005, 251, 263–269 CrossRef CAS.
- P. M. Budd, E. S. Elabas, B. S. Ghanem, S. Makhseed, N. B. McKeown, K. J. Msayib, C. E. Tattershall and D. Wang, Adv. Mater., 2004, 16, 456–459 CrossRef CAS.
- N. B. McKeown, P. M. Budd, K. J. Msayib, B. S. Ghanem, H. J. Kingston, C. E. Tattershall, S. Makhseed, K. J. Reynolds and D. Fritsch, Chem.–Eur. J., 2005, 11, 2610–2620 CrossRef CAS PubMed.
- B. D. Freeman, Macromolecules, 1999, 32, 375–380 CrossRef CAS.
- H. B. Park, J. Kamcev, L. Robeson, M. Elimelech and B. Freeman, Science, 2017, 356, 1138–1148 CrossRef PubMed.
- L. M. Robeson, J. Membr. Sci., 1991, 62, 165–185 CrossRef CAS.
- L. Robeson, J. Membr. Sci., 2008, 320, 390–400 CrossRef CAS.
- R. Swaidan, B. Ghanem and I. Pinnau, ACS Macro Lett., 2015, 4, 947–951 CrossRef CAS.
- B. Comesaña-Gándara, J. Chen, C. G. Bezzu, M. Carta, I. Rose, M.-C. Ferrari, E. Esposito, A. Fuoco, J. C. Jansen and N. B. McKeown, Energy Environ. Sci., 2019, 12, 2733–2740 RSC.
- S. Zhao, J. Liao, D. Li, X. Wang and N. Li, J. Membr. Sci., 2018, 566, 77–86 CrossRef CAS.
- R. Williams, L. A. Burt, E. Esposito, J. C. Jansen, E. Tocci, C. Rizzuto, M. Lanč, M. Carta and N. B. McKeown, J. Mater. Chem. A, 2018, 6, 5661–5667 RSC.
- E. Tocci, L. De Lorenzo, P. Bernardo, G. Clarizia, F. Bazzarelli, N. B. McKeown, M. Carta, R. Malpass-Evans, K. Friess, K. Pilnáček, M. Lanč, Y. P. Yampolskii, L. Strarannikova, V. Shantarovich, M. Mauri and J. C. Jansen, Macromolecules, 2014, 47, 7900–7916 CrossRef CAS.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, M. Lee, I. Rose and N. B. McKeown, Polym. Chem., 2014, 5, 5267–5272 RSC.
- M. Carta, R. Malpass-Evans, M. Croad, Y. Rogan, J. C. Jansen, P. Bernardo, F. Bazzarelli and N. B. McKeown, Science, 2013, 339, 303 CrossRef CAS PubMed.
- M. Carta, M. Croad, R. Malpass-Evans, J. C. Jansen, P. Bernardo, G. Clarizia, K. Friess, M. Lanč and N. B. McKeown, Adv. Mater., 2014, 26, 3526–3531 CrossRef CAS PubMed.
- R. Swaidan, M. Al-Saeedi, B. Ghanem, E. Litwiller and I. Pinnau, Macromolecules, 2014, 47, 5104–5114 CrossRef CAS.
- L. Shao, L. Liu, S.-X. Cheng, Y.-D. Huang and J. Ma, J. Membr. Sci., 2008, 312, 174–185 CrossRef CAS.
- K. Vanherck, A. Cano-Odena, G. Koeckelberghs, T. Dedroog and I. Vankelecom, J. Membr. Sci., 2010, 353, 135–143 CrossRef CAS.
- M. Z. Ahmad, H. Pelletier, V. Martin-Gil, R. Castro-Munoz and V. Fila, Membranes, 2018, 8, 67 CrossRef PubMed.
- F. Y. Li, Y. Xiao, T.-S. Chung and S. Kawi, Macromolecules, 2012, 45, 1427–1437 CrossRef CAS.
- Q. Liu, A. T. Shaver, Y. Chen, G. Miller, D. R. Paul, J. S. Riffle, J. E. McGrath and B. D. Freeman, Polymer, 2016, 87, 202–214 CrossRef CAS.
- J. R. Rowlett, Q. Liu, W. Zhang, J. D. Moon, M. E. Dose, J. S. Riffle, B. D. Freeman and J. E. McGrath, J. Mater. Chem. A, 2016, 4, 16047–16056 RSC.
- F. Y. Li and T.-S. Chung, Int. J. Hydrogen Energy, 2013, 38, 9786–9793 CrossRef CAS.
- F. Y. Li, Y. Xiao, Y. K. Ong and T.-S. Chung, Adv. Energy Mater., 2012, 2, 1456–1466 CrossRef CAS.
- M. Puertas-Bartolomé, M. E. Dose, P. Bosch, B. D. Freeman, J. E. McGrath, J. S. Riffle, A. E. Lozano, J. G. de la Campa and C. Álvarez, RSC Adv., 2017, 7, 55371–55381 RSC.
- M. S. McCaig and D. R. Paul, Polymer, 1999, 49, 7209–7225 CrossRef.
- Q. Song, S. Cao, P. Zavala-Rivera, L. P. Lu, W. Li, Y. Ji, S. A. Al-Muhtaseb, A. K. Cheetham and E. Sivaniah, Nat. Commun., 2013, 4, 1918 CrossRef PubMed.
- R. Castro-Muñoz, V. Fila and C. Dung, Chem. Eng. Commun., 2017, 204, 295–309 CrossRef.
- G. Dong, H. Li and V. Chen, J. Mater. Chem. A, 2013, 1, 4610–4630 RSC.
- Y. Zhang, X. Feng, S. Yuan, J. Zhou and B. Wang, Inorg. Chem. Front., 2016, 3, 896–909 RSC.
- R. Lin, B. Villacorta Hernandez, L. Ge and Z. Zhu, J. Mater. Chem. A, 2018, 6, 293–312 RSC.
- J. Dechnik, J. Gascon, C. J. Doonan, C. Janiak and C. J. Sumby, Angew. Chem., Int. Ed., 2017, 56, 9292–9310 CrossRef CAS PubMed.
- S. J. D. Smith, B. P. Ladewig, A. J. Hill, C. H. Lau and M. R. Hill, Sci. Rep., 2015, 5, 7823 CrossRef CAS PubMed.
- S. J. D. Smith, R. Hou, C. H. Lau, K. Konstas, M. Kitchin, G. Dong, J. Lee, W. H. Lee, J. G. Seong, Y. M. Lee and M. R. Hill, J. Membr. Sci., 2019, 585, 260–270 CrossRef CAS.
- C. H. Lau, K. Konstas, C. M. Doherty, S. Kanehashi, B. Ozcelik, S. E. Kentish, A. J. Hill and M. R. Hill, Chem. Mater., 2015, 27, 4756–4762 CrossRef CAS.
- Y. Cheng, Y. Ying, S. Japip, S.-D. Jiang, T.-S. Chung, S. Zhang and D. Zhao, Adv. Mater., 2018, 30, 1802401 CrossRef PubMed.
- C. H. Lau, P. T. Nguyen, M. R. Hill, A. W. Thornton, K. Konstas, C. M. Doherty, R. J. Mulder, L. Bourgeois, A. C. Y. Liu, D. J. Sprouster, J. P. Sullivan, T. J. Bastow, A. J. Hill, D. L. Gin and R. D. Noble, Angew. Chem., Int. Ed., 2014, 53, 5322–5326 CrossRef CAS PubMed.
- C. H. Lau, K. Konstas, A. Thornton, A. Liu, S. Mudie, D. Kennedy, S. Howard, A. Hill and M. Hill, Angew. Chem., Int. Ed. Engl., 2015, 54, 2669–2673 CrossRef CAS PubMed.
- C. H. Lau, X. Mulet, K. Konstas, C. M. Doherty, M.-A. Sani, F. Separovic, M. R. Hill and C. D. Wood, Angew. Chem., Int. Ed., 2016, 55, 1998–2001 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- S. J. D. Smith, R. Hou, K. Konstas, A. Akram, C. H. Lau and M. R. Hill, Acc. Chem. Res., 2020, 53, 1381–1388 CrossRef CAS PubMed.
- R. Hou, R. O'Loughlin, J. Ackroyd, Q. Liu, C. M. Doherty, H. Wang, M. R. Hill and S. J. D. Smith, Ind. Eng. Chem. Res., 2020, 59, 13773–13782 CrossRef CAS.
- R. Hou, B. S. Ghanem, S. J. D. Smith, C. M. Doherty, C. Setter, H. Wang, I. Pinnau and M. R. Hill, J. Mater. Chem. A, 2020, 8, 14713–14720 RSC.
- D. Wu, R. Hou, C. Yi, S. J. D. Smith, J. Fu, D. Ng, C. M. Doherty, R. J. Mulder, Z. Xie and M. R. Hill, Sep. Purif. Technol., 2021, 268, 118677 CrossRef CAS.
- R. A. Hayes and D. Hockessin, US Pat., 4717393, 1988 Search PubMed.
- C. Liu, S. T. Wilson and D. A. Lesch, US Pat., US 7758751B751, 2010 Search PubMed.
- A. B. Foster, M. Tamaddondar, J. M. Luque-Alled, W. J. Harrison, Z. Li, P. Gorgojo and P. M. Budd, Macromolecules, 2020, 53, 569–583 CrossRef CAS.
- H. R. Kricheldorf, N. Lomadze, D. Fritsch and G. Schwarz, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5344–5352 CrossRef CAS.
- M. L. Jue, C. S. McKay, B. A. McCool, M. G. Finn and R. P. Lively, Macromolecules, 2015, 48, 5780–5790 CrossRef CAS.
- C. H. Lau, K. Konstas, A. W. Thornton, A. C. Y. Liu, S. Mudie, D. F. Kennedy, S. C. Howard, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2015, 54, 2669–2673 CrossRef CAS PubMed.
- R. Castro-Muñoz and V. Fíla, Membranes, 2018, 8, 30 CrossRef PubMed.
- R. Hou, N. T. Eden, C. Fong, D. Acharya, C. M. Doherty, T. Gengenbach, K. Konstas, Z. Xie, B. D. Freeman and M. R. Hill, Ind. Eng. Chem. Res., 2021 DOI:10.1021/acs.iecr.1c03942.
- L. Hao, K.-S. Liao and T.-S. Chung, J. Mater. Chem. A, 2015, 3, 17273–17281 RSC.
- T. R. Gengenbach, G. H. Major, M. R. Linford and C. D. Easton, J. Vac. Sci. Technol., A, 2021, 39, 013204 CrossRef CAS.
- C. D. Batich and D. S. Donald, J. Am. Chem. Soc., 1984, 106, 2758–2761 CrossRef CAS.
- B. W. Rowe, B. D. Freeman and D. R. Paul, Polymer, 2009, 50, 5565–5575 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2ta00138a |
| This journal is © The Royal Society of Chemistry 2022 |