
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crosslinked xylose-based polyester as a bio-derived and degradable solid polymer electrolyte for Li+-ion conduction†
Matthew
Oshinowo
 ,
James R.
Runge
,
Marco
Piccini
,
Frank
Marken
and
Antoine
Buchard
*
,
James R.
Runge
,
Marco
Piccini
,
Frank
Marken
and
Antoine
Buchard
*
Centre for Sustainable and Circular Technologies, Department of Chemistry, University of Bath, Bath BA2 7AY, UK. E-mail: a.buchard@bath.ac.uk
First published on 25th February 2022
Abstract
A novel crosslinked polyester derived from D-xylose and 10-undecenoic acid (from castor oil) has been investigated as a new material for solid polymer electrolyte (SPE) applications. Acyclic diene metathesis polymerisation of a bio-derived monomer, followed by crosslinking with 2,2-(ethylenedioxy)diethanethiol (to impart film properties and mechanical strength) and incorporation of lithium bis(trifluoromethanesulfonyl)imide (LiTFSI), resulted in flexible and transparent SPE films. The materials exhibited Tg's between −25 °C and 13 °C and thermal stability up to 234 °C. Ionic conductivity was measured as a function of molar mass, crosslinking density and salt molarity which were optimised to achieve an ionic conductivity as high as 1.0 × 10−5 S cm−1 at 60 °C. A high lithium transference number of 0.84 was also achieved and electrochemical stability up to 3.88 V was demonstrated. Revealing 33% of the xylofuranose core OH groups via ketal deprotection resulted in a semi-crystalline polymer whose crystallinity was disrupted by incorporation of LiTFSI. The resulting SPE material offered a small, yet non-significant, improvement of ionic conductivity (3.5 × 10−5 S cm−1vs. 1.0 × 10−5 S cm−1 at 60 °C).
Introduction
Lithium-ion batteries (LIBs) have become more and more synonymous with our everyday lives, as exemplified by the exponential rise in the number of electric vehicles in use over the past decade, which passed 10 million in 2020.1 As a consequence, the global production capacity of LIBs is soaring, having reached 455 GW h in 2020.2 Accordingly, there has never been a greater need for the development of novel battery materials from sustainable feedstocks, not least the electrolyte component, which is responsible for transporting the charge-carrying Li+ cations between the electrodes. The liquid electrolyte component of current LIB cells will inevitably soon be replaced by superior solid-state electrolytes, due to the many shortfalls of liquid electrolytes.3 Most notably, their poor safety record is a major concern and can be attributed to their high flammability and poor electrochemical and mechanical stability.4–6 Inorganic solid-state electrolytes, such as Li7La3Zr2O12 ‘garnet-type’ materials, are the forerunner solid-state electrolyte with expected mass-market adoption by the end of the decade due to their significant safety benefits, mechanical stability, low cost and high ionic conductivity.7 However, solid polymer electrolytes (SPEs) are an alternative solid-state electrolyte contender with similar merits.8 SPEs also offer the additional benefits of transparency and flexibility which may allow their use in more specialised applications where inorganic electrolytes might not be applicable (e.g. wearable devices). Moreover, the ability to synthesise specialised polymers from renewable, bio-sourced feedstocks is also attractive if we are to design next-generation devices with sustainability in mind.Amongst the various polymer chemistries investigated for SPE application, those comprising of polyethylene oxide (PEO) with various lithium salts have undoubtedly received the most interest, however recent studies of similar polyethers and polyacetals have also been reported by Balsara and Coates.9–13 Despite high ionic conductivity (ca. 10−3 S cm−1 at 70 °C) above the melting temperature of PEO, poor room temperature performance, high crystallinity and low lithium transference numbers (typically around 0.2) of PEO-based SPEs remain a limitation to their practical implementation.14 Polyesters (e.g., Miller)15 and polycarbonates (e.g., Tominaga,16 Brandell,17 and Mecerreyes18,19) have also been studied for SPE applications, as well as more sophisticated materials such inorganic/polymer composites,20–22 block co-polymers,23–30 single-ion conducting polymers24,31,32 and crosslinked polymers.33–36 Among the latter category, Jang, Kim and co-workers recently reported impressive ionic conductivity close to the practical requirement (ca. 10−3 S cm−1 at room temperature) from a fluorosulfonylimide methacrylic-based crosslinked SPE.33
However, in most of these cases, these polymers are derived from petrochemicals and there has been much less research concerning the application of bio-derived polymers in SPEs, although some examples can be found and are well summarised in reviews from Singh et al.37 and from Lizundia and Kundu.38 Natural biopolymers extracted directly from biomass (such as cellulose, starch or lignin) are designed by nature to have rigid properties to add structure to plants, meaning that their polymer properties are not typically aligned with those expected to yield high ionic conductivity such as low crystallinity (ideally amorphous) and low glass transition temperatures (Tg). Cellulose (in paper form, for example) is better utilised as a biopolymer separating membrane due to its high porosity and nanostructure.39 Alternatively, integration of the biopolymer nanofibers as structural elements with other polymers or liquids can afford SPE or gel polymer electrolyte (GPE) materials.40,41 The direct chemical modification of methyl cellulose to incorporate pendant lithium sulfonate groups was used by Lee and An in 2020 to produce an SPE with impressive ionic conductivity (10−3 S cm−1).42 There are essentially no reported examples of commercially relevant synthetic polymers synthesised from bio-derived monomers (e.g. polyethylene furanoate (PEF), bio-polybutylene succinate (bioPBS), bio-polyethylene terephthalate (bioPET) or bio-polyurethanes (bioPUs)) being tested in SPE applications for similar reasons, with the exception of poly(lactic acid) (PLA). Whilst this route can offer more control over the final polymer properties via the choice of the monomer, these polymers are typically designed to replicate the material properties of traditional plastics and post-polymerisation functionalisation can be difficult. PLA, for example, is a semi-crystalline polyester with a Tg of 60–65 °C that has yet to be applied directly as an SPE due to challenges in its chemical modification, such as decreasing the glass transition to a suitably low enough temperature for ion mobility. Rather, PLA-based SPE examples have been found as blends with petrochemically derived ionic liquids43 or with polymers with lower Tg's such as poly(methyl methacrylate)44 and PEO.45,46
Herein, we report a novel SPE material utilising a polyester boasting 92 wt% biobased content in combination with the commonly used lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) salt as the source of lithium ions. The polyester, first reported by our group last year,47 is derived from D-xylose (a renewable monosaccharide) and 10-undecenoic acid (a castor oil derivative) and has been crosslinked with a small amount of 2,2′-(ethylenedioxy)diethanethiol (a dithiol resembling PEO) to render SPE membranes. After optimising the molar mass, crosslinking density and salt molarity of the SPEs, electrochemical measurements of the ionic conductivity, lithium transference number and electrochemical stability window were conducted, in addition to thermal, hydrolytic and mechanical stability studies. We hope that this initial study will open the door to a new class of bio-derived SPE materials with opportunity for future investigations and optimisations.
Results and discussion
Monomer synthesis and polymerisation
The monomer was synthesised according to the procedure previously reported by our group (Fig. 1).47 Polymers of varying molar mass were prepared via the acyclic diene metathesis (ADMET) polymerisation of monomer 1 (Table 1). | ||
| Fig. 1 Scheme of the synthetic route to crosslinked solid polymer electrolytes derived from D-xylose and 10-undecenoic acid. The nomenclature classifies SPEs as 1–4 according to their molar mass (1 = oligomeric, 2 = low, 3 = medium, 4 = high), a–f according to crosslinker equivalents (a = 0.0, b = 0.05, c = 0.1, d = 0.2, e = 0.5, f = 1.0) and 0–100 according to the amount (mol%) of LiTFSI present. | ||
| Entry | Catalyst identityb | Catalyst (mol%) | Moderatorc (mol%) | Conversiond (%) | M n,theo (kg mol−1) | M n,SEC [ĐM]f (kg mol−1) |
|---|---|---|---|---|---|---|
| a Polymerisations were carried out at 90 °C in the absence of solvent with overhead mechanical stirring under a dynamic vacuum (ca. 1 mbar) for a duration of 20 hours unless otherwise stated. b G-II = Grubbs 2nd generation catalyst, HG-II = Hoveyda–Grubbs 2nd generation catalyst. c Molar mass moderator = methyl 10-undecenoate. d Calculated by comparison of the relative integration of the terminal alkene signals (4.90 and 5.75 ppm) of 1 and internal alkene signals (5.25–5.40 ppm) of poly(1) in the 1H NMR spectrum of the crude polymer. e See ESI eqn (S1) for calculation of theoretical molar mass. f Calculated by size-exclusion chromatography (SEC) methods relative to polystyrene standards in THF, ĐM = MW/Mn. g Polymerisation performed with magnetic stirring. h Polymerisation performed at 80 °C. | ||||||
| 1 | G-II | 0.2 | 2.0 | 86 | 3.1 | 2.9 [1.26] |
| 2 | G-II | 1.0 | 20.0 | 100 | 2.9 | 11.1 [1.64] |
| 3g | G-II | 5.0 | 0.0 | 100 | 10.4 | 12.8 [2.34] |
| 4 | G-II | 0.5 | 4.5 | 100 | 10.4 | 23.3 [1.96] |
| 5 | G-II | 0.5 | 2.0 | 100 | 20.3 | 24.9 [2.06] |
| 6 | G-II | 0.5 | 0.75 | 100 | 40.1 | 51.4 [3.55] |
| 7h | HG-II | 2.0 | 7.0 | 100 | 6.0 | 22.0 [1.79] |
| 8 | HG-II | 0.5 | 7.0 | 100 | 7.1 | 22.6 [1.85] |
| 9 | HG-II | 1.0 | 5.0 | 100 | 8.7 | 36.0 [1.84] |
2nd generation Grubbs or 2nd generation Hoveyda–Grubbs catalysts were used with methyl 10-undecenoate being employed as an end-capping molar mass moderator. Whilst the moderator was effective at decreasing the amount of precious ruthenium-based Grubbs catalyst required for lower molar mass polymers, the molar masses were typically much higher than the theoretical targets. However, the effect of the moderator is still significant as the molar masses determined by size-exclusion chromatography (SEC) are much lower than the theoretical molar masses calculated for polymerisation in its absence. Difficulty in achieving homogeneous stirring and sufficiently low vacuum with the overhead stirring setup may also have affected the polymerisations. Notably, incomplete conversion of the monomer resulted in formation of oligomer species (Table 1 entry 1; degree of polymerisation = 6), which proved useful when investigating low molecular mass SPE films.
Initial crosslinking investigation
Poly(1) was primarily identified as a possible SPE candidate owing to its amorphous nature and relatively low Tg (e.g., −31 °C, Mn = 5.1 kg mol−1, see ESI Fig. S12†) which are considered to be important SPE metrics for the promotion of segmental motion. Moreover, the presence of seven potential Li+-ion coordinating oxygen atoms per repeat unit, in addition to an unsaturation in the polymer backbone, amenable to functionalisation, were appealing. Existing as a sticky viscous liquid in its unmodified form, poly(1) clearly required some modification to make it a potential SPE material. Nevertheless, SPE 3a-70 was prepared from poly(1) (Mn 24.9 kg mol−1) and LiTFSI (70 mol%) via solution casting from THF (see Fig. 1 caption for nomenclature). Although the resulting material had some film-like properties, it remained sticky and was not a self-standing film, therefore demonstrating the need for crosslinking. The salt had a notable effect on the Tg of the polymer, increasing it from −15 °C to −3 °C (determined by differential scanning calorimetry (DSC)).Following this, an investigation into the crosslinking of poly(1) to produce self-standing SPE films was carried out. 2,2′-(ethylenedioxy)diethanethiol (EDDET) was selected as the crosslinker of choice due to its close resemblance to the molecular structure of PEO (Fig. 1). Moreover, the crosslinker is relatively long and flexible and the resulting SPEs should retain some flexibility. The two thiol groups of EDDET allow for facile crosslinking of poly(1) through the alkene bonds in the polymer chains, via the thiol–ene “click” reaction under UV irradiation (λ = 365 nm), using Irgacure 819 (phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide) as a photoinitiator. To demonstrate the reactivity of the olefin with EDDET, poly(1) (Mn 13.1 kg mol−1) was stirred in neat EDDET (10 equivalents) for 2 hours in the presence of Irgacure 819 (0.5 equivalents) under UV irradiation (365 nm). The reaction was performed at 70 °C to allow dissolution of the polymer in EDDET. Before precipitation from methanol, an aliquot was taken and analysed by 1H NMR spectroscopy, which showed 83% conversion of alkene bonds as seen by the reduction in the integration of the signals at 5.37–5.29 ppm (ESI Fig. S1†). FTIR spectroscopy showed the complete disappearance of the S–H stretching vibration observed for neat EDDET at 2556 cm−1 (ESI Fig. S3†) which suggested that under the conditions used, EDDET effectively crosslinked poly(1), leaving no pendant thiol groups. Thermal analysis of this sample revealed a Tg at −35 °C by DSC, lower than the polymer prior to crosslinking (−22 °C). This result, though unexpected, could perhaps be attributed to residual thiol remaining in the sample and acting as a plasticiser. The thermal degradation profile, determined by TGA analysis (ESI Fig. S20†), exhibited two decomposition processes with maxima at 297 °C and 452 °C, with total mass loss of 85% at 600 °C. Decomposition at the former temperature was responsible for the majority of the mass loss and closely matches that previously reported by our group for poly(1).47 Therefore, the second decomposition was attributed to that of the PEO-like crosslinks.
Formation of crosslinked SPE films
Due to the insolubility of poly(1-EDDET), LiTFSI was incorporated before the crosslinking of poly(1). After thorough mixing of the polymer, crosslinker, photoinitiator and salt in THF, brown self-standing transparent SPE films were obtained via solvent casting and subsequent UV irradiation (Fig. 2A). Wide-angle X-ray scattering (WAXS) analysis was carried out on selected SPE samples. The absence of crystallinity detected in the films (determined from the absence of sharp peaks in the WAXS chart) is strong evidence of complete dissociation of LiTFSI within the crosslinked polymer matrix, leaving no clusters of crystalline salt. This highlights the strong Li+-coordinating ability of the oxygen atoms in the sugar which are able to fully dissociate the salt despite the absence of functionality in the long aliphatic chains of the polymer backbone. | ||
| Fig. 2 Photo of representative SPE films of thickness 200–400 μm (A); charts displaying the conductivities of poly(1-EDDET) based SPE films at 60 °C as (B) a function of poly(1) molar mass (salt molarity = 70–72 mol%, crosslinker equivalents = 1.0), and (C) a function of crosslinker equivalents (poly(1) Mn = 24.4–24.9 kg mol−1, salt molarity = 70–72 mol%). | ||
The irradiation time required for completion of the crosslinking was investigated by shining UV light on a sample in the absence of LiTFSI and measuring the Tg of aliquots at regular intervals. The results suggest that crosslinking is complete in as little as 20 minutes as there is no change in the Tg after an initial increase from −24 °C in the film prior to irradiation, to −18 °C from 20 minutes onwards (ESI Fig. S13†). To ensure consistency across samples with different crosslinking amounts, an irradiation time of 90 minutes was implemented for all later experiments.
Electrochemical characterisation
In all cases, Nyquist plots obtained from electrochemical impedance spectroscopy (EIS) measurements of the SPEs showed characteristic (partial) semi-circles followed by a diagonal line, representative of the bulk resistance (Rb) and Warburg diffusion, respectively (ESI Fig. S6†). As such, the data was fitted to a Randles circuit model which allowed the determination of the ionic conductivity.14 Initial characterisations of the SPE films by EIS displayed significant hysteresis in the conductivity when repeating measurements. Therefore, SPE 1f-100 (ESI Table S4,† entry 8) was subjected to EIS measurements at regular intervals to determine the time required for the SPE to properly anneal to the electrode surfaces. The resulting plot (ESI Fig. S7†) displays a plateau after ca. 12 hours, therefore clearly demonstrating the necessity to allow an overnight temperature equilibration of the sample in the cell.Sample optimisation
The optimum composition parameters for these SPE films, which would afford the highest conductivity (see Table 2 for selected tabulated data), was next investigated. Four classes of poly(1) molar masses (Mn) were used for SPE preparation, including: oligomeric (2.9 kg mol−1; 1f), low (11.1–12.8 kg mol−1; 2e and 2f), medium (24.4–24.9 kg mol−1; 3a–3f) and high (51.4 kg mol−1; 4f). Firstly, the effect of the molar mass of poly(1) prior to crosslinking was studied, keeping the salt molarity at 70 mol%.‡ While a large excess of EDDET was used previously during SPE formation, then removed during the precipitation step, the number of equivalents was lowered to 1.0 to prevent an excess of residual EDDET in the final SPE.§ The conductivity data (Fig. 2B) for the molar mass study displays a trend of exponential decay in the ionic conductivity with increasing molar mass which matches previously reported trends in the literature.48 This is to be expected as increasing the molar mass results in the electrolyte deviating further from a liquid state in which the ions have the most mobility. Fig. 2B clearly demonstrates the importance of molar mass as there is a ca. 100-fold decrease in the ionic conductivity at 60 °C when the molar mass is increased from 3 to 25 kg mol−1.| Entry | SPE reference | M n,SEC (kg mol−1) | Crosslinker equivalentsd | Salt mol%b [wt%]c | T g (°C) | Conductivityf (S cm−1) | E a (kJ mol−1) | A (S cm−1) |
|---|---|---|---|---|---|---|---|---|
| a Refers to molar mass of poly(1) prior to crosslinking. b Salt mol% calculated as a percentage of moles of salt relative to moles of polymer repeat units. c Salt wt% is in reference to the polymer mass prior to crosslinking. d Calculated as ratio of moles of EDDET to moles of polymer repeat units. e Obtained from the second heating cycle on the DSC thermogram. f Normalised to 60 °C using linear regression of an Arrhenius plot of 1000/T vs. log(σ). g Calculated from linear regression fitting of Arrhenius conductivity data to the VTF equation. | ||||||||
| 1 | 1f-72 | 2.9 | 1.0 | 72 [42] | −25 | 7.5 × 10−6 | 13.3 | 1.0 × 100 |
| 2 | 2e-70 | 11.1 | 0.5 | 70 [41] | −6 | 7.3 × 10−6 | 7.9 | 2.6 × 10−2 |
| 3 | 2f-70 | 12.8 | 1.0 | 70 [41] | 9 | 4.2 × 10−6 | 7.9 | 5.2 × 10−3 |
| 4 | 3a-70 | 24.9 | 0.0 | 70 [41] | −3 | 1.9 × 10−5 | 7.5 | 9.2 × 10−2 |
| 5 | 3b-0 | 24.7 | 0.05 | 0 | −18 | — | — | — |
| 6 | 3b-10 | 24.7 | 0.05 | 10 [6] | −12 | 4.2 × 10−6 | 9.0 | 3.0 × 10−4 |
| 7 | 3b-25 | 24.7 | 0.05 | 25 [15] | −7 | 1.7 × 10−6 | 10.0 | 4.7 × 10−2 |
| 8 | 3b-50 | 24.7 | 0.05 | 50 [29] | −3 | 4.8 × 10−6 | 8.8 | 5.7 × 10−2 |
| 9 | 3b-70 | 24.9 | 0.05 | 70 [41] | 7 | 1.0 × 10−5 | 9.5 | 6.7 × 10−1 |
| 10 | 3b-100 | 24.7 | 0.05 | 100 [58] | 13 | 4.7 × 10−6 | 7.7 | 6.3 × 10−2 |
| 11 | 3c-70 | 24.4 | 0.1 | 70 [41] | 10 | 3.2 × 10−6 | 9.8 | 4.4 × 10−1 |
| 12 | 3d-70 | 24.9 | 0.2 | 70 [41] | −9 | 2.1 × 10−6 | 10.9 | 1.2 × 10−1 |
| 13 | 3f-70 | 24.9 | 1.0 | 70 [41] | 4 | 1.0 × 10−7 | 4.5 | 1.8 × 10−5 |
| 14 | 4f-70 | 51.4 | 1.0 | 70 [41] | 6 | 1.0 × 10−7 | 6.8 | 2.8 × 10−4 |
| 15 | 5b(depr)-70 | 8.7 | 0.05 | 70 [42] | −21 | 3.5 × 10−5 | 9.5 | 2.2 × 10−1 |
The effect of crosslinking density was then investigated by crosslinking medium molar mass polymers with decreasing equivalents of EDDET (Fig. 2C; samples 3a–3f). A similar and expected trend was observed whereby decreasing the amount of crosslinking resulted in higher ionic conductivity as crosslinking binds the polymer chains together and further deviates from the liquid state. With 0.05 equivalents of EDDET and 70 mol% of salt (3b-70), an ionic conductivity of 1.0 × 10−5 S cm−1 was achieved at 60 °C (Table 2, entry 9). When fewer than 0.05 equivalents were used (3a-70), the resulting films had poor mechanical integrity and more closely resembled poly(1) (a viscous liquid). As the conductivity rapidly declines above 0.05 equivalents of EDDET, it is therefore clear that a balanced crosslinking density must be achieved in these SPEs: high enough to impart mechanical strength to the films but also not so high as to hinder the electrochemical performance. In fact, the ionic conductivity of the aforementioned SPE 3a-70 (prepared without crosslinking) was determined to be 1.9 × 10−5 S cm−1 at 60 °C (Table 2, entry 4). Such similar conductivities suggest that very low crosslinking densities do not hinder conductivity but remain essential for mechanical integrity.
The final effect investigated was that of salt molarity, whilst maintaining the same polymer molar mass and crosslinking with 0.05 equivalents of EDDET. The effect of increasing the amount of LiTFSI on both the ionic conductivity and the Tg is shown in Fig. 3A. The presence of LiTFSI does not seem to have a plasticising effect on the polymer. Rather, as more salt is added, the SPE continues to become more rigid as demonstrated by the increase in Tg from −18 to +13 °C. Contrary to other polymers which exhibit a lower Tg due to the plasticising effect of LiTFSI (e.g. polyethylene carbonate),49 SPEs 3b-x behave more similarly to PEO-based SPEs in which incorporation of LiTFSI increases the Tg.50 However, in the case of PEO, the increase in Tg has been attributed to the increased number of crystalline regions induced by LiTFSI. In the case of SPEs 3b-x, the increase in Tg might be more akin to early studies of polyethylene succinate by Watanabe51 and Shriver52 in which the increase was attributed to Li+-induced crosslinking. Despite the increased rigidity, an increase in ionic conductivity is observed up to 70 mol% of salt, after which there is a decline. This demonstrates the competing effects of increased charge carriers and polymer chain flexibility. As the salt molarity is increased to 70 mol%, the presence of more charge carriers may be enough to outcompete the decreased polymer chain mobility (segmental motion), one of the main ion transport mechanisms. Beyond 70 mol%, the decreased polymer chain mobility may outcompete the extra charge carriers and the conductivity is reduced. It is also worth noting that in the extreme case of high salt molarity (100 mol%), a second minor Tg at −6 °C is observed in addition to the expected Tg at 13 °C (see Fig. S15†). One possible explanation is that such high salt concentration, domains of undissociated LiTFSI remain in the polymer matrix, which act locally as plasticisers, accounting for the additional lower Tg. WAXS analysis of such samples was performed but no LiTFSI crystalline domains were detected.
 | ||
Fig. 3 Characterisation data for SPEs 3b-x (Mn,SEC = 27.4–27.9 kg mol−1, 0.05 equivalents of EDDET), with varying amount of LiTFSI salt; (A) conductivity (normalised to 60 °C) and Tg as a function of salt molarity; (B) Arrhenius plot showing the temperature dependence of the ionic conductivities of SPEs 3b-x; (C) Signals in FTIR spectrum representing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration. O stretching vibration. | ||
Like in PEO-based SPEs, increasing the amount of lithium salt in SPEs 3b-x increases the ionic conductivity up to a maximum before decreasing (Fig. 3A). In PEO systems, peak ionic conductivity occurs at around 8 mol% of salt (52 wt%),13,50 compared to 70 mol% (41 wt%) for 3b. Similar wt% but different mol% values are due to the very large molecular mass of the monomer repeat unit (494.67 g mol−1) in poly(1) compared to that of PEO (44.05 g mol−1).
A salt molarity study was also carried out on a poly(1) sample of 2.9 kg mol−1 with 1.0 EDDET equivalents. Interestingly, this study also demonstrated that similar salt molarity of 72 mol% was the optimal amount, although the Tg values obtained from these samples were erratic and displayed no clear trend (see ESI Table S4 and Fig. S10†).
Analysis of the FTIR spectra obtained from samples of 3b with increasing salt molarities (Fig. 3C) clearly shows a broadening and shift to lower wavenumber of the ester CO stretching vibrational peak. The stretching frequency ranges from 1739 cm−1 for poly(1-EDDET) and SPEs with low salt concentrations to as low as 1716 cm−1 in 3b-100. In accordance with literature reports of carbonate- and ester-based SPEs,53,54 this implies that the carbonyl oxygen is a major coordination site of the Li+ ions. However, it is difficult to determine the extent of coordination of Li+ ions to the ether oxygens of the EDDET crosslinkers based on the FTIR spectra of the SPE samples (e.g., ESI Fig. S4†). Whilst the ether C–O stretch appears as a strong peak at 1100 cm−1 for EDDET, that spectral region is crowded for poly(1-EDDET) and its resulting SPEs due to the presence of other C–O bonds in the xylose core, as well as other vibrations in the same region (e.g., CC bend).
Knowing the optimal salt (70 mol%) and crosslinking (0.05 equivalents) amounts, we then set about making an ‘optimised’ SPE from an oligomeric (∼3 kg mol−1) or low molar mass (∼12 kg mol−1) sample of poly(1) which should theoretically exhibit the highest conductivity. However, when low EDDET equivalents (0.05 and 0.10) were used to crosslink oligomeric and low molar mass polymers with 70 mol% of salt, the resulting films had no mechanical integrity. However, a robust film was obtained using 0.5 EDDET equivalents to yield SPE 2e-70 which gave an almost identical conductivity to 1f-72 (7.3 × 10−6 and 7.5 × 10−6, respectively).
At this point it is worth noting that the three highest conductivities achieved so far are very similar, exhibited by 1f-72, 2e-70 and 3b-70 (between 7.3 × 10−6 and 1.0 × 10−5 S cm−1). In other words, the same outcome can be achieved, in terms of ionic conductivity, by combination of oligomer with high crosslinking (1f-72), low molar mass with medium crosslinking (2e-70), and medium molar mass with low crosslinking (3b-70). Whilst there could be a mechanical benefit or improved cation transference for 1f-72, it is recognised that from a sustainability standpoint, this option is less favourable. This is because an oligomer is obtained either from a polymerisation with <100% conversion, adding more catalyst equivalents or more molar mass moderator equivalents. In the latter case, although methyl 10-undecenoate is the industrial precursor to 10-undecenoic acid, it is still an additional synthetic reagent.¶ Higher molar mass polymers with a lower degree of crosslinking are therefore favourable due to less catalyst, moderator, EDDET and photoinitiator being required, thus resulting in an SPE with a greater amount of bio-derived atoms. With this in mind, SPE 1f-72 was omitted and 2e-70 and 3b-70 were taken forward for further characterisation and analysis.
Mechanical strength
The tensile strength and Young's modulus were briefly investigated as a measure of mechanical strength. Whilst batteries are not typically subjected to physical deformation, the Young's modulus can still be useful for demonstrating the benefit of SPEs over traditional liquid electrolytes. For example, SPEs offer batteries an increased protection against short-circuiting due to impact or suppression of lithium dendrite growth, as reported in 2014 by Coates with crosslinked SPEs based on PEO and polyethylene.55Although the SPEs were mechanically robust enough to be punched into disk shapes, cutting of the films into the required shapes for uniaxial tensile strength testing resulted in edge weaknesses that were sometimes the source of breakage upon applied force. As a result, little correlation was found between the amount of crosslinking and the Young's modulus, determined from the initial linear region (ESI Fig. S18†). Nevertheless, samples 3b-70, 3c-70 and 3d-70 (differing only by the amount of crosslinking) exhibited a linear, elastic region up until the point of breaking with Young's moduli in the range of 1–4 MPa. Although these are much smaller than those reported for SPEs engineered to be high-modulus (e.g., 1 GPa for crosslinked PEO/polystyrene nanostructured SPEs),56 this testing is still a useful demonstration of the advantage of using crosslinked films as opposed to traditional liquid electrolytes, viscous liquid polymers such as poly(1) or even polymer gel electrolytes.
Vogel–Tammann–Fulcher (VTF) fitting
To gain a deeper insight into the ion transport mechanism and try to isolate the effects of charge carrier concentration and polymer segmental motion, a VTF fitting was applied to the Arrhenius conductivity data of representative samples. The following version of the VTF equation was applied, which omits the temperature dependence on the pre-exponential factor, A:
In this equation, A is related to charge carrier concentration, R is the universal gas constant and T0 is the ‘Vogel temperature’ equal to 50 K below the Tg. The T–T0 parameter is generally accepted as a measure of polymer segmental motion at a given temperature as the Vogel temperature refers to a point of zero configurational entropy (segmental motion) for polymer electrolytes.23,57,58 The data was fitted two ways: direct (non-linear) fitting of σ vs. T–T0, and a linear regression of ln(σ) vs. 1000/(T–T0) (e.g., Fig. 4A), both of which gave very similar results. The results based on the linear regression are reported in the final two columns of Table 2. Unfortunately, little relationship could be found between A, Ea and the other parameters (σ, salt molarity, Tg and EDDET equivalents), thus leaving the door open for future study of ion transport mechanisms of these SPE systems.
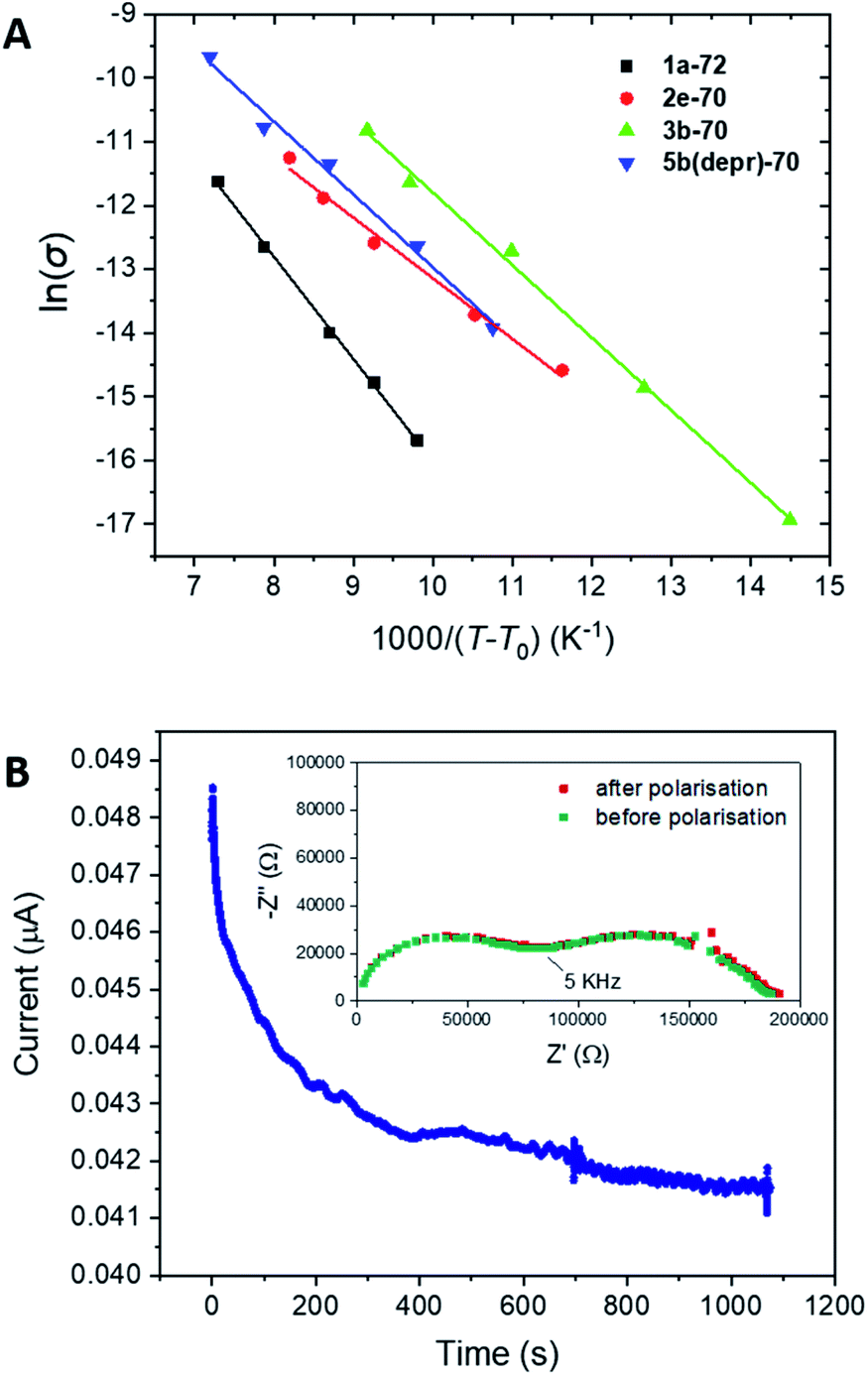 | ||
| Fig. 4 (A) VTF fitting for selected SPE samples (T0 = Tg – 50 K); (B) representative chronoamperometry and Nyquist plot (inlayed) obtained for the determination of t+ for 2e-70. A 10 mV applied polarization voltage was used for the chronoamperometry in a symmetrical Li|SPE|Li cell. The Nyquist plot was obtained by EIS recorded in the frequency range of 0.1 Hz to 1 MHz and the annotated frequency corresponds to the resistance at the intersecting circles. | ||
Transference number
The lithium transference number of 2e-70 was determined using the commonly employed Bruce–Vincent method of combined DC polarisation chronoamperometry and EIS measurements in a symmetrical Li|SPE|Li cell.59 Compared to the Nyquist plots obtained from the measurement of samples with blocking SS electrodes, a second semi-circle of larger resistance was observed when utilising lithium electrodes the size of the second semi-circle increased over time and then stabilised after leaving the cell at 70 °C overnight. This can likely be attributed to formation of a solid electrolyte interface (SEI) layer on the lithium surface. Upon application of a small (10 mV) potential, steady state current was achieved relatively quickly with very small changes in current on the nA scale (Fig. 4B). There was almost no change in the Nyquist plot obtained directly before and after DC polarisation, suggesting that the sample is stable over the duration of the experiment. A t+ value of 0.84 ± 0.01 was obtained for 2e-70, which is markedly higher than those achieved by PEO-based SPEs, therefore highlighting a major benefit of this novel class of bio-derived SPEs. Although the polymer has not been optimised to have a high t+ value (like in the case of single-ion conducting polymers where t+ can approach unity),60 it is possible the crosslinked polymer matrix favours movement of smaller Li+ ions over bulky TFSI− ions.SPE stability studies
The electrochemical stability of the SPEs was determined by linear sweep voltammetry (LSV; Fig. 5A). SPE 2e-70 displayed anodic stability up to 3.88 V (vs. Li/Li+) which is comparable to PEO-based SPEs which also suffer oxidative decomposition above 4 V,61 although lower than the 4.2 V stability requirement for practical application.62 More experiments are required to determine the origin of the low anodic stability, however it may be due to electrochemical processes involving the vinyl ether end groups of the polymer or residual photo-initiating species present in the sample. | ||
| Fig. 5 (A) LSV of 2e-70 obtained at 25 °C with a scan rate of 1 mV s−1 from 0–6 V (vs. Li/Li+); (B) TGA trace poly(1) (Mn,SEC = 13.1 kg mol−1) crosslinked with 10 equivalents of EDDET (black) and representative SPE samples (Td1 and Td2 refer to temperatures for the maximum of the derivative peaks); (C) percentage mass loss of 3b-70 after 72 hours in the hydrolytic conditions listed. | ||
The thermal stability, as determined by TGA, of representative SPE samples with different compositions is shown in Fig. 5B. Like poly(1-EDDET), the SPEs exhibited two main degradation temperatures. 1f-72 and 3b-70 displayed similar thermal degradation profiles, with a first degradation step with maxima at 213 and 219 °C, and a second degradation step at 424 and 410 °C, respectively. The first step occurs roughly 70 °C lower than poly(1-EDDET), implying that at elevated temperatures the presence of Lewis acidic Li+ ions facilitates the decomposition of the polymer, likely via a ring-opening pathway of the sugar moieties. Nevertheless, thermal stability over 200 °C is acceptable with regards to practical battery applications. 2e-70 performed almost identically to 3b-70 but with slightly less mass loss at lower temperatures. Despite the lower molar mass, the increased crosslinking may protect the sugar units from ring-opening. It is also noteworthy that the amount of residual char of the SPEs at 600 °C was greater in comparison to poly(1-EDDET) due to the presence of LiTFSI, and the relative amount of residual char at 600 °C correlates to the overall wt% of LiTFSI in the SPE.
Polyester-based SPEs may help to address environmental concerns about the persistence of electronic waste coupled with the adventitious leakage of liquid electrolytes. Due to cleavable ester bonds, aliphatic polyesters can be degraded under controlled conditions whilst providing non-leaking materials for electrolyte applications. As such, 3b-70 was subjected to a range of aqueous conditions for 3 days to observe the extent of the polymer degradation (Fig. 5C). As the crosslinked polymer is insoluble in the SEC solvent, the amount of degradation was determined by mass loss. Unsurprisingly, the chart in Fig. 5C shows that greater degradation occurred when using higher HCl or NaOH concentrations and when performed at a higher temperature. Moreover, NaOHaq was shown to achieve greater degradation than HClaq under all conditions, with a maximum mass loss of 53% after 3 days at a concentration of 1.0 M at 50 °C. This is promising as it demonstrates the degradability of the SPE under relatively controlled, mild conditions. However, although crosslinking prevents recyclability of the polymer back to the monomer, chemical recycling by cross metathesis of the CC bonds with ethylene could yield a reusable oligomeric network.63
Effect of xylose OH deprotection
The effect of revealing the OH groups on the xylose core protected by ketal groups on the ionic conductivity was also briefly investigated. Deprotection of the ketal groups of poly(1) to reveal hydroxyl groups was achieved via acid hydrolysis following previously reported literature procedures (Fig. 6).47,64 The degree of deprotection was monitored by 1H NMR spectroscopy by relative integration of the methylene protons on the 10-undecenoic acid chain (m, 4H, 2.34–2.29 ppm) against the isopropylidene methyl protons (s, 3H, 1.52 ppm) as according to previous methods (ESI Fig. S2†).47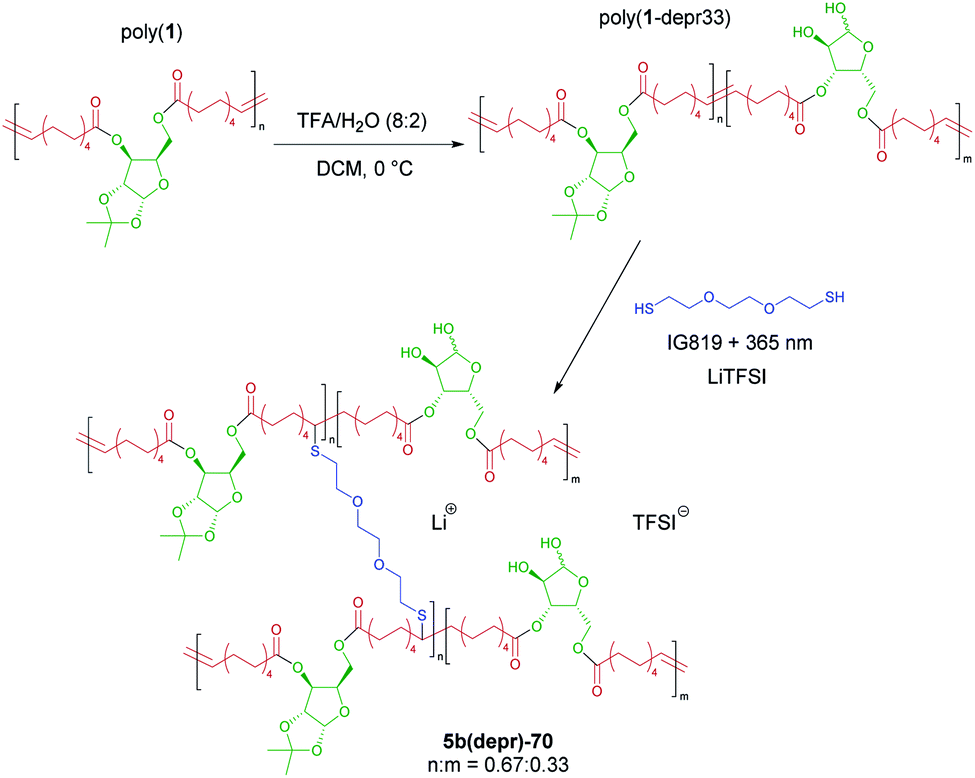 | ||
| Fig. 6 Synthetic scheme representing partial OH deprotection of poly(1) followed by crosslinking and salt incorporation to yield deprotected SPEs. Poly(1-depr33) is represented in which 33% OH deprotection was achieved. TFA = trifluoroacetic acid, IG819 = Irgacure 819. | ||
Initial monitoring experiments showed that deprotection increased over time yielding a polymer with up to 96% ketal groups deprotected achieved after 24 hours. Polymers with >70% deprotected ketal groups maintained their solubility in THF when dissolved immediately after precipitation from methanol. However, once dried under vacuum at 70 °C the polymers became insoluble in various solvents including THF, CHCl3, acetone, DMSO and water. This behaviour suggests that revealing the OH groups may cause the formation of an extensive hydrogen-bonding network between the polymer chains.
To attempt to retain solubility in THF for facile SPE preparation, a polymer with 20–40% deprotected ketal groups was targeted. After 2 hours, a polymer with 33% deprotected ketal groups (poly(1-depr33)) was achieved in an 86% yield. Poly(1-depr33) proved soluble in THF with gentle heating even after drying at 70 °C. SEC analysis showed that the material remained polymeric after deprotection, however, a decrease in Mn from 38 kg mol−1 to 8.7 kg mol−1 was noted. This loss in molar mass may be explained by hydrolysis of the ester bonds in the polymer chain. DSC analysis of the deprotected polymer showed the material to be semi-crystalline with a Tg of −21 °C and a Tm of 21 °C (ESI Fig. S16†). The semi-crystalline behaviour may be attributed to hydrogen bonding involving the newly revealed OH groups generating crystalline regions within the polymer.
SPE 5b(depr)-70 was then prepared from poly(1-depr33) with 70 mol% LiTFSI and 0.05 EDDET equivalents using the same procedure. Interestingly, the resulting material retained a Tg of −21 °C but the addition of salt was enough to completely disrupt crystallinity and form an amorphous SPE film with no Tm (ESI Fig. S17†). The peak conductivity achieved by 5b(depr)-70 turned out to be higher than that achieved by the other SPEs, reaching 3.5 × 10−5 S cm−1 at 60 °C. However, this system also showed a unexpected behaviour whereby the conductivity decreased from its peak to 1.4 × 10−5 S cm−1 after the SPE was maintained at 60 °C for 5 hours after an overnight equilibration. This behaviour could perhaps be explained by the formation of new H-bonds over time which hinder ionic mobility. Moreover, determination of t+ by the Bruce–Vincent method was not successful as the system did not reach a steady state of current, therefore suggesting that the chemical nature of the system is dynamic once the OH groups are revealed. With regards to thermal stability, 5a-(depr)70 performed better than oligomeric 1f-72 but was comparable to 2e-70 and 3b-70.
Discussion
Overall, although not state-of-the-art, the performance of this crosslinked polyester system is comparable to other crosslinked SPE systems reported in the literature which sacrifice electrochemical performance for improved mechanical stability. For example, Opris and co-workers recently synthesised polysiloxanes which were also crosslinked with EDDET that resulted in ionic conductivity (4.8 × 10−5 S cm−1 at 60 °C) on the same order of magnitude as those reported in this study.65 Mindemark and co-workers also recently reported ionic conductivity on the order of 10−5 S cm−1 at 60 °C for a crosslinked polyester-polycarbonate.66 Amongst other polyesters, Zhang and co-workers, for example, reported the highest ionic conductivity (4 × 10−3 S cm−1 at 85 °C) in 2017 with a polycaprolactone-based SPE.67 However, in their case the material also employed a polyacrylonitrile skeleton framework and succinonitrile as a filler molecule to enhance the conductivity. Moreover, the reported t+ value of 0.32 is marginally higher than that of PEO and implies that the majority of the conductivity can be attributed to anion mobility. When compared to these other systems, the conductivity of the crosslinked polyester reported in this work may be slightly lower at an equivalent temperature, however this material boasts a high transference number up to 0.84 and has the added advantage of comprising 90% bio-sourced content (by weight, for Xb samples, with 0.05 cross-linker equivalent, and excluding LiTSFI salt). This work is also comparable, in terms of both ionic conductivity and transference number, to poly(ethylene carbonate)-based SPEs reported by Tominaga and co-workers (e.g., t+ = 0.8, σ ≈ 10−6 S cm−1 at 40 °C),16 and not far from the performance of polycarbonate SPEs reported by Mecerreyes and co-workers, which in some instances incorporate large equivalents of ethylene oxide units in the polymer backbone.18,68–70Conclusions
A bio-derived polyester (poly(1)), derived from D-xylose and 10-undecenoic acid, has been rendered into SPE films via crosslinking of the unsaturated fatty acid-derived chains with 2,2-(ethylenedioxy)diethanethiol (EDDET) and incorporation of LiTFSI as a source of lithium ions. The importance of optimising salt molarity, polymer molar mass and crosslinking density has been demonstrated. As expected, increasing salt molarity results in an increased ionic conductivity, up to a maximum of 70 mol%. Low molar mass and crosslinking density have been shown to be the most effective for ionic conductivity, however if these are too low then the film properties and/or mechanical integrity of the materials are compromised. A maximum ionic conductivity of 1.0 × 10−5 S cm−1 has been achieved at 60 °C for an SPE sample with 70 mol% LiTFSI, 0.05 crosslinker equivalents and a molar mass of 24.7 kg mol−1Mn. A high lithium transference number of 0.84 has been measured and electrochemical stability window to +3.88 V (vs. Li/Li+). The SPE is thermally stable to >200 °C but can also achieve >50% hydrolytic degradation after 72 hours in 1.0 M NaOH at 50 °C, thereby demonstrating the degradability of the material under controlled mild conditions. The effect of revealing some of the OH groups of the xylose core enabled a larger peak conductivity of 3.5 × 10−5 S cm−1 to be achieved at 60 °C, although this could not be maintained due to the dynamic nature of the system.With the structural manifold offered by this platform of polymers, there is certainly scope to optimise the performance of this SPE system. Both the unsaturation in the fatty acid moiety and the OH groups of the sugar moiety that can be revealed offer possible routes of post-polymerisation modification. Moreover, there is also scope to explore the performance of the polyether analogue of this polyester which has also recently been reported.71
Experimental methods
Materials and methods
All reagents were purchased from commercial chemical suppliers without additional purification except from LiTFSI which was dried at 110 °C in a vacuum oven for 24 hours before being stored in an argon-filled glovebox. All solvents were supplied by VWR and used without purification or taken from a MBraun solvent purification system in the case of anhydrous solvents. All chemicals were stored at room temperature except from polymerisation catalysts and ethyl vinyl ether which were stored in a fridge at 3 °C. Lithium foil and EDDET were stored in an argon-filled glovebox. Thiol–ene reactions were performed in a PhotoRedox TC light box by HepatoChem with a 30 W UV lamp (λ = 365 nm). Silica gel was used as the stationary phase for column chromatography and plates were visualised with phosphomolybdic acid (10 wt% in ethanol) staining solution.Synthetic procedures
Polymerisation of 1 was performed according to the procedure previously reported by our group.47 In a typical procedure, monomer 1 (3.02 g, 5.78 mmol, 200 equiv.) was measured into a 100 mL two-neck round-bottom flask followed by addition of Grubbs 2nd generation catalyst (24.5 mg, 0.029 mmol, 1 equiv.) and methyl 10-undecenoate (26 μL, 0.12 mmol, 4 equiv.). The flask was attached to an overhead stirrer equipped with a PTFE-coated steel stirring rod, a PTFE stirring blade and a PTFE vacuum-tight stirrer bearing. The stirring was started at 200 rpm and a dynamic vacuum applied (ca. 1 mbar) as the flask was submerged into an oil bath at 90 °C. After ca. 10 minutes the stirring was reduced to 30 rpm and the reaction left stirring overnight. The vacuum was then stopped and the flask removed from the oil bath and allowed to cool to room temperature. The product was dissolved in THF (20 mL) and quenched with an excess of ethyl vinyl ether (5 mL). An aliquot was taken to confirm conversion of the monomer by 1H NMR before the polymer was filtered through cotton wool and precipitated from cold methanol (ca. 120 mL). The product was then isolated by centrifugation (4000 rpm, 10 minutes) before thorough drying in a vacuum oven (70 °C) for 24 hours to yield poly(1) as a viscous brown liquid (2.62 g, 100% conversion, 92% yield, Table 1 entry 5). 1H NMR (400 MHz, CDCl3): δ 5.93 (1H, d, J = 3.7 Hz), 5.40–5.32 (2H, m), 5.26 (2H, d, J = 3.1 Hz), 4.51–4.47 (2H, m), 4.28–4.16 (2H, m), 2.36–2.28 (4H, m), 2.02–1.91 (4H, m), 1.64–1.56 (4H, m), 1.52 (3H, s), 1.35–1.23 (23H, m); 13C NMR (101 MHz, CDCl3) δ 173.5, 172.6, 130.5, 112.4, 105.1, 83.6, 77.0, 76.0, 61.4, 34.2, 32.7, 29.8–29.1, 27.4, 26.9, 26.4, 25.0. NMR data is in agreement with literature values. Mn,SEC = 24.9 kg mol−1 (ĐM = 2.06). Tg = −20 °C.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 solution of trifluoroacetic acid and water (5 mL) was slowly added. At predetermined intervals aliquots were taken of the reaction mixture and deprotection was determined by 1H NMR spectroscopy (CDCl3). Once the desired deprotection was achieved (2 hours), the polymer was precipitated from cold methanol and centrifuged (3500 rpm, 5 minutes). The supernatant was removed, the polymer was collected and dried in a vacuum oven at 70 °C for 24 hours (0.672 g, 86% yield, 33% deprotection). Mn,SEC = 8.7 kg mol−1 (ĐM = 4.04). Tg = −21 °C, Tm = 21 °C.
:2 solution of trifluoroacetic acid and water (5 mL) was slowly added. At predetermined intervals aliquots were taken of the reaction mixture and deprotection was determined by 1H NMR spectroscopy (CDCl3). Once the desired deprotection was achieved (2 hours), the polymer was precipitated from cold methanol and centrifuged (3500 rpm, 5 minutes). The supernatant was removed, the polymer was collected and dried in a vacuum oven at 70 °C for 24 hours (0.672 g, 86% yield, 33% deprotection). Mn,SEC = 8.7 kg mol−1 (ĐM = 4.04). Tg = −21 °C, Tm = 21 °C.
Characterisation methods
Nuclear magnetic resonance (NMR) spectra were recorded on a 400 MHz Bruker NMR spectrometer and referenced to residual proton or 13C peaks of the CDCl3 solvent. Spectra were processed and analysed using Mnova software by Mestrelab.Size exclusion chromatography (SEC) was performed with a 1260 GPC/SEC MDS system from Agilent. Separation was achieved using two PLgel 5 μm MIXED-D 300 × 7.5 mm columns with a PLgel 5 μm MIXED 50 × 7.5 mm guard column. SEC-grade THF was used as the mobile phase and refractive index (RI) was used as a detection method. The columns and RI detector were all maintained at 35 °C. The system was calibrated using polystyrene standards in THF which allowed the determination of the number-average molar mass (Mn,SEC) and dispersities (ĐM) of polymer samples. All Mn values of SPEs refer to poly(1) prior to crosslinking.
Differential scanning calorimetry (DSC) was used to measure the glass transition temperature (Tg) of samples on a TA Instruments DSC Q20 employing the Q Series program. The experiment was performed under nitrogen gas (flow rate = 18 mL min−1) and samples were heated and cooled at a rate of 10 °C min−1 in a 10 μL Tzero aluminium pan with lid. The Tg was taken from the second heating cycle between −60 °C and +150 °C.
Fourier-transform infrared spectra (FTIR) were recorded on a Nicolet iS5 FT-IR spectrometer by Thermo Fisher Scientific or a Bruker Alpha II Platinum ATM spectrometer in the range of 400–4000 cm−1 and processed using Origin or OPUS software.
Thermogravimetric analysis (TGA) was performed using the Calisto program on a Setaram Setsys Evolution TGA 16/18. The analytical chamber was purged with argon (200 mL min−1) for 40 minutes prior to heating under an argon flow (20 mL min−1) from 30 °C to 600 °C with a ramp of 10 °C min−1. The reported degradation temperatures correspond to the temperature of peak of the mass loss derivative.
Electrochemical measurements were performed using a modified version of a TCS battery cell (RHD instruments) with blocking stainless steel current collectors connected to a Metrohm Autolab PGSTAT204 potentiostat with a FRA32M module. The sample and cell components were dried in a vacuum oven at 70 °C prior to cell assembly inside an argon-filled glovebox. Temperature control of the cell was achieved by submersion in a 1 litre beaker containing metallic thermal beads (Lab Armor) which was placed in a water bath. The cell temperature was monitored via a thermometer submerged into the thermal beads directly next to the cell. The cell was equilibrated at 65 °C overnight and then at each temperature for 1 hour before measurements were taken.
Ionic conductivity (σ) was determined by a two-electrode electrochemical impedance spectroscopy (EIS) measurement in the typical frequency range of 0.1 Hz to 0.5 MHz with an applied amplitude of 50 mV in a symmetrical SS|SPE|SS cell. NOVA 2.1 (Metrohm) software was used to analyse the results and apply a Randles equivalent circuit fitting to the obtained Nyquist plot. The bulk resistance (Rb) was identified as the resistor labelled ‘Rp’ (see ESI Fig. S6†) as per standard literature procedure,72 and also confirmed by changing the sample thickness and observing the resulting change in the magnitude of Rp.Rb was used to calculate σ using the equation:
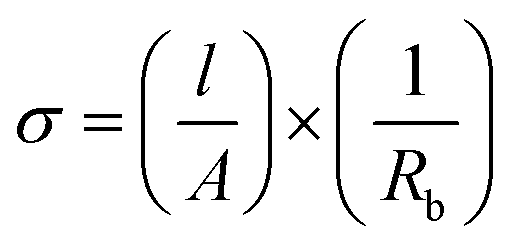
Lithium transference number (t
+
) was determined using a combined EIS and chronoamperometry method using a Li|SPE|Li symmetrical cell using lithium foil of 0.75 mm thickness and an applied voltage of 10 mV. The measurement was recorded at 70 °C and t+ calculated using the Bruce–Vincent equation:

Linear sweep voltammetry was used to determine the electrochemical stability in a Li|SPE|SS cell using a lithium counter/reference electrode at 25 °C with a scan rate of 1 mV s −1.
Wide angle X-ray scattering (WAXS) experiments were collected using a SAXSpoint 2.0 instrument by Anton Paar which employed a microfocus copper tube X-ray source (λ = 0.1542 nm) with point focus. The detector used was a hybrid photon counting detector which was at a distance of 109 mm from the sample holder. One frame with an acquisition time of 30 minutes was used for all samples.
Uniaxial tensile testing was performed on an Instron 3369 machine equipped with 50 N pneumatic grips. Samples were cut with dimensions of approximately 4 mm × 40 mm bars and gripped so that 20 mm of sample was exposed. Digital callipers were used to measure the thickness across the exposed 20 mm and an average was taken. A crosshead speed of 1 mm min−1 was used until the sample snapped. Where possible, three or more replicates of each film were measured and the results reported as an average (±standard error).
SPE degradation studies were performed after pre-soaking the SPE samples in water for 24 h (to remove LiTFSI and residual Irgacure 819) and dried at 100 °C in a vacuum oven. The SPEs were then degraded for 3 days with 0.1 M or 1.0 M HCl or NaOH at ambient temperature or 50 °C without stirring. After the degradation, the remaining solids were rinsed with 3 × 2 mL deionised water and then dried at 100 °C in vacuum oven.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Georgina Gregory for useful discussions. Analytical facilities were provided through the Material and Chemical Characterisation Facility (MC2) at the University of Bath. Research funding from the Engineering and Physical Sciences Research Council (DTP studentship to JRR, EP/L016354/1 CDT in Sustainable Chemical Technologies Studentship to MP), the University of Bath (studentship to MO), and the Royal Society (UF/160021 fellowship to AB) is also acknowledged.Notes and references
- International Energy Agency, Trends and Developments in Electric Vehicle Markets, Paris, p. 2021 Search PubMed.
- S&P Global Market Intelligence, https://www.spglobal.com/marketintelligence/en/news-insights/blog/top-electric-vehicle-markets-dominate-lithium-ion-battery-capacity-growth, accessed November 2021.
- S. Chen, K. Wen, J. Fan, Y. Bando and D. Golberg, J. Mater. Chem. A, 2018, 6, 11631–11663 RSC.
- B. Liu, Y. Jia, J. Li, S. Yin, C. Yuan, Z. Hu, L. Wang, Y. Li and J. Xu, J. Mater. Chem. A, 2018, 6, 21475–21484 RSC.
- K. Liu, Y. Liu, D. Lin, A. Pei and Y. Cui, Sci. Adv., 2018, 4, eaas9820 CrossRef PubMed.
- N. S. Choi, J. G. Han, S. Y. Ha, I. Park and C. K. Back, RSC Adv., 2015, 5, 2732–2748 RSC.
- C. Wang, K. Fu, S. P. Kammampata, D. W. McOwen, A. J. Samson, L. Zhang, G. T. Hitz, A. M. Nolan, E. D. Wachsman, Y. Mo, V. Thangadurai and L. Hu, Chem. Rev., 2020, 120, 4257–4300 CrossRef CAS PubMed.
- L. Long, S. Wang, M. Xiao and Y. Meng, J. Mater. Chem. A, 2016, 4, 10038–10069 RSC.
- Z. Xue, D. He and X. Xie, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- K. W. Gao, W. S. Loo, R. L. Snyder, B. A. Abel, Y. Choo, A. Lee, S. C. M. Teixeira, B. A. Garetz, G. W. Coates and N. P. Balsara, Macromolecules, 2020, 53, 5728–5739 CrossRef CAS.
- D. M. Halat, R. L. Snyder, S. Sundararaman, Y. Choo, K. W. Gao, Z. J. Hoffman, B. A. Abel, L. S. Grundy, M. D. Galluzzo, M. P. Gordon, H. Celik, J. J. Urban, D. Prendergast, G. W. Coates, N. P. Balsara and J. A. Reimer, Chem. Mater., 2021, 33, 4915–4926 CrossRef CAS.
- R. L. Snyder, Y. Choo, K. W. Gao, D. M. Halat, B. A. Abel, S. Sundararaman, D. Prendergast, J. A. Reimer, N. P. Balsara and G. W. Coates, ACS Energy Lett., 2021, 6, 1886–1891 CrossRef CAS.
- D. M. Pesko, M. A. Webb, Y. Jung, Q. Zheng, T. F. Miller, G. W. Coates and N. P. Balsara, Macromolecules, 2016, 49, 5244–5255 CrossRef CAS.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- M. A. Webb, Y. Jung, D. M. Pesko, B. M. Savoie, U. Yamamoto, G. W. Coates, N. P. Balsara, Z.-G. Wang and T. F. Miller, ACS Cent. Sci., 2015, 1, 198–205 CrossRef CAS PubMed.
- Y. Tominaga, Y. Kinno and K. Kimura, Electrochim. Acta, 2019, 302, 286–290 CrossRef CAS.
- M. Ebadi, T. Eriksson, P. Mandal, L. T. Costa, C. M. Araujo, J. Mindemark and D. Brandell, Macromolecules, 2020, 53, 764–774 CrossRef CAS PubMed.
- F. Ouhib, L. Meabe, A. Mahmoud, N. Eshraghi, B. Grignard, J.-M. Thomassin, A. Aqil, F. Boschini, C. Jérôme, D. Mecerreyes and C. Detrembleur, J. Mater. Chem. A, 2019, 7, 9844–9853 RSC.
- K. Saito, C. Jehanno, L. Meabe, J. L. Olmedo-Martínez, D. Mecerreyes, K. Fukushima and H. Sardon, J. Mater. Chem. A, 2020, 8, 13921–13926 RSC.
- F. P. Nkosi, M. Valvo, J. Mindemark, N. A. Dzulkurnain, G. Hernández, A. Mahun, S. Abbrent, J. Brus, L. Kobera and K. Edström, ACS Appl. Energy Mater., 2021, 4, 2531–2542 CrossRef CAS.
- Y. Li, Z. Sun, D. Liu, Y. Gao, Y. Wang, H. Bu, M. Li, Y. Zhang, G. Gao and S. Ding, J. Mater. Chem. A, 2020, 8, 2021–2032 RSC.
- P. Zhu, C. Yan, M. Dirican, J. Zhu, J. Zang, R. K. Selvan, C.-C. Chung, H. Jia, Y. Li, Y. Kiyak, N. Wu and X. Zhang, J. Mater. Chem. A, 2018, 6, 4279–4285 RSC.
- B. Kim, C.-G. Chae, Y. Satoh, T. Isono, M.-K. Ahn, C.-M. Min, J.-H. Hong, C. F. Ramirez, T. Satoh and J.-S. Lee, Macromolecules, 2018, 51, 2293–2301 CrossRef CAS.
- G. Lingua, P. Grysan, P. S. Vlasov, P. Verge, A. S. Shaplov and C. Gerbaldi, Macromolecules, 2021, 54, 6911–6924 CrossRef CAS PubMed.
- J. Ping, H. Pan, P.-P. Hou, M.-Y. Zhang, X. Wang, C. Wang, J. Chen, D. Wu, Z. Shen and X.-H. Fan, ACS Appl. Mater. Interfaces, 2017, 9, 6130–6137 CrossRef CAS PubMed.
- D. Devaux, D. Gle, T. N. T. Phan, D. Gigmes, E. Giroud, M. Deschamps, R. Denoyel and R. Bouchet, Chem. Mater., 2015, 27, 4682–4692 CrossRef CAS.
- D. T. Hallinan, S. A. Mullin, G. M. Stone and N. P. Balsara, J. Electrochem. Soc., 2013, 160, A464 CrossRef CAS.
- W.-S. Young, W.-F. Kuan and T. H. Epps III, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 1–16 CrossRef CAS.
- E. D. Gomez, A. Panday, E. H. Feng, V. Chen, G. M. Stone, A. M. Minor, C. Kisielowski, K. H. Downing, O. Borodin, G. D. Smith and N. P. Balsara, Nano Lett., 2009, 9, 1212–1216 CrossRef CAS PubMed.
- W. Huang, Q. Pan, H. Qi, X. Li, Y. Tu and C. Y. Li, Polymer, 2017, 128, 188–199 CrossRef CAS.
- K. Jeong, S. Park and S.-Y. Lee, J. Mater. Chem. A, 2019, 7, 1917–1935 RSC.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- F. Ahmed, D. Kim, J. Lei, T. Ryu, S. Yoon, W. Zhang, H. Lim, G. Jang, H. Jang and W. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 34102–34113 CrossRef CAS PubMed.
- E. A. Baroncini, D. M. Rousseau, C. A. Strekis and J. F. Stanzione, Solid State Ionics, 2020, 345, 115161 CrossRef CAS.
- S. Kalybekkyzy, A. F. Kopzhassar, M. V. Kahraman, A. Mentbayeva and Z. Bakenov, Polymers, 2021, 13, 15 CrossRef CAS.
- A. Thiam, C. Antonelli, C. Iojoiu, F. Alloin and J.-Y. Sanchez, Electrochim. Acta, 2017, 240, 307–315 CrossRef CAS.
- R. Singh, A. R. Polu, B. Bhattacharya, H. W. Rhee, C. Varlikli and P. K. Singh, Renewable Sustainable Energy Rev., 2016, 65, 1098–1117 CrossRef CAS.
- E. Lizundia and D. Kundu, Adv. Funct. Mater., 2021, 31, 2005646 CrossRef CAS.
- L. Hu, H. Wu, F. La Mantia, Y. Yang and Y. Cui, ACS Nano, 2010, 4, 5843–5848 CrossRef CAS.
- S. Ramesh, R. Shanti and E. Morris, Carbohydr. Polym., 2013, 91, 14–21 CrossRef CAS PubMed.
- Y. A. Samad, A. Asghar and R. Hashaikeh, Renewable Energy, 2013, 56, 90–95 CrossRef CAS.
- S. K. Kim, Y. Yoon, J. H. Ryu, J. H. Kim, S. Ji, W. Song, S. Myung, J. Lim, H.-K. Jung, S. S. Lee, J. Lee and K.-S. An, ChemSusChem, 2020, 13, 376–384 CrossRef CAS PubMed.
- I. Osada, S. M. Hosseini, S. Jeong and S. Passerini, ChemElectroChem, 2017, 4, 463–467 CrossRef CAS.
- H. Wang, L. Shu and S. Jiang, J. Appl. Polym. Sci., 2010, 117, 2790–2794 CAS.
- M. Zhang, A.-l. Zhang, Q. Li, F.-f. Li, S. Wang and S.-x. Li, J. Polym. Environ., 2019, 27, 2369–2379 CrossRef CAS.
- N. Zhang, A.-l. Zhang, Q.-f. Liu, M. Zhang, Q. Li and F.-f. Li, Ionics, 2018, 24, 3805–3813 CrossRef CAS.
- M. Piccini, D. J. Leak, C. J. Chuck and A. Buchard, Polym. Chem., 2020, 11, 2681–2691 RSC.
- K. Timachova, H. Watanabe and N. P. Balsara, Macromolecules, 2015, 48, 7882–7888 CrossRef CAS.
- Y. Tominaga, V. Nanthana and D. Tohyama, Polym. J., 2012, 44, 1155–1158 CrossRef CAS.
- S. Lascaud, M. Perrier, A. Vallée, S. Besner, J. Prud'homme and M. Armand, Macromolecules, 1994, 27, 7469–7477 CrossRef CAS.
- M. Watanabe, M. Togo, K. Sanui, N. Ogata, T. Kobayashi and Z. Ohtakilb, Macromolecules, 1984, 17, 2908–2912 CrossRef CAS.
- R. Dupon, B. L. Papke, M. A. Ratner and D. F. Shriver, J. Electrochem. Soc., 1984, 131, 586–589 CrossRef CAS.
- J. Motomatsu, H. Kodama, T. Furukawa and Y. Tominaga, Macromol. Chem. Phys., 2015, 216, 1660–1665 CrossRef CAS.
- I. D. Wu and F. C. Chang, Polymer, 2007, 48, 989–996 CrossRef CAS.
- R. Khurana, J. L. Schaefer, L. A. Archer and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7395–7402 CrossRef CAS PubMed.
- M. W. Schulze, L. D. McIntosh, M. A. Hillmyer and T. P. Lodge, Nano Lett., 2014, 14, 122–126 CrossRef CAS PubMed.
- K. M. Diederichsen, H. G. Buss and B. D. McCloskey, Macromolecules, 2017, 50, 3831–3840 CrossRef CAS.
- S. S. Zhang and G. X. Wan, J. Appl. Polym. Sci., 1993, 48, 405–409 CrossRef CAS.
- J. Evans, C. A. Vincent and P. G. Bruce, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, ACS Energy Lett., 2016, 1, 678–682 CrossRef CAS.
- S. Seki, Y. Kobayashi, H. Miyashiro, Y. Mita and T. Iwahori, Chem. Mater., 2005, 17, 2041–2045 CrossRef CAS.
- X. F. Yang, M. Jiang, X. J. Gao, D. Bao, Q. Sun, N. Holmes, H. Duan, S. Mukherjee, K. Adair, C. T. Zhao, J. W. Liang, W. H. Li, J. J. Li, Y. Liu, H. Huang, L. Zhang, S. G. Lu, Q. W. Lu, R. Y. Li, C. V. Singh and X. L. Sun, Energy Environ. Sci., 2020, 13, 1318–1325 RSC.
- J. Bidange, C. Fischmeister and C. Bruneau, Chem. –Eur. J., 2016, 22, 12226–12244 CrossRef CAS PubMed.
- T. M. McGuire, J. Bowles, E. Deane, E. H. E. Farrar, M. N. Grayson and A. Buchard, Angew. Chem., Int. Ed., 2021, 60, 4524–4528 CrossRef CAS PubMed.
- C. Fu, M. Iacob, Y. Sheima, C. Battaglia, L. Duchêne, L. Seidl, D. M. Opris and A. Remhof, J. Mater. Chem. A, 2021, 9, 11794–11801 RSC.
- I. L. Johansson, D. Brandell and J. Mindemark, Batteries Supercaps, 2020, 3, 527–533 CrossRef CAS.
- D. Zhang, L. Zhang, K. Yang, H. Wang, C. Yu, D. Xu, B. Xu and L.-M. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 36886–36896 CrossRef CAS PubMed.
- L. Meabe, N. Lago, L. Rubatat, C. Li, A. J. Müller, H. Sardon, M. Armand and D. Mecerreyes, Electrochim. Acta, 2017, 237, 259–266 CrossRef CAS.
- L. Meabe, T. V. Huynh, D. Mantione, L. Porcarelli, C. Li, L. A. O'Dell, H. Sardon, M. Armand, M. Forsyth and D. Mecerreyes, Electrochim. Acta, 2019, 302, 414–421 CrossRef CAS.
- L. Meabe, T. V. Huynh, N. Lago, H. Sardon, C. Li, L. A. O'Dell, M. Armand, M. Forsyth and D. Mecerreyes, Electrochim. Acta, 2018, 264, 367–375 CrossRef CAS.
- M. Piccini, J. Lightfoot, B. C. Dominguez and A. Buchard, ACS Appl. Polym. Mater., 2021, 3, 5870–5881 CrossRef CAS.
- S. B. Aziz, T. J. Woo, M. F. Z. Kadir and H. M. Ahmed, J. Sci.: Adv. Mater. Devices, 2018, 3, 1–17 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Mn,theo calculation, NMR spectra, example FTIR spectrum, SEC traces, Nyquist plots, crosslinking rate analysis, oligomeric SPE study, conductivity hysteresis monitoring, conductivity temperature normalisation and DSC plots. See DOI: 10.1039/d1ta10111k |
| ‡ Preliminary testing on SPEs prepared from oligomeric polymers indicated that 70 mol% was an optimal salt molarity (see ESI Table S4 and Fig. S10). |
| § Boiling point of EDDET (225 °C) is too high to be removed by evaporation. |
| ¶ In the industrial preparation of 10-undecenoic acid, methanolysis of castor oil yields the methyl ester of ricinoleic acid which is thermally cracked to obtain methyl 10-undecenoate. Saponification of methyl 10-undecenoate yields the 10-undecenoic acid. |
| This journal is © The Royal Society of Chemistry 2022 |