
C3 production from CO2 reduction by concerted *CO trimerization on a single-atom alloy catalyst†

Abstract
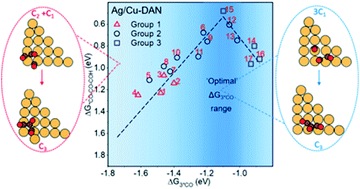
The direct electroreduction of carbon dioxide (CO2) and carbon monoxide (CO) to C3 products is challenging. The main reason is the competitive C2 production resulting from a traditional sequential C–C coupling mechanism. As a result, most catalysts could not facilitate C3 products since the carbon chain growth from C2 to C3 competes with C2 desorption. In this work, we carried out Density Functional Theory (DFT) calculations with implicit solvation effects on densely arrayed Cu nanopyramids (Cu-DANs). We demonstrate that the co-adsorption energy of three *CO intermediates (ΔG3*CO; from the CO2 or CO reactant) is a descriptor for C3 activity. An activity volcano plot was constructed based on this discovery, which can be used to predict the optimal range for ΔG3*CO adsorption strength. We demonstrate that by applying the single-atom alloy catalyst strategy, i.e. embedding Ag single metal onto Cu-DANs, we could successfully tune the ΔG3*CO strength toward the optimal range. In addition, the adsorbed *CO could form a long carbon chain on such a structure via a one-step concerted trimerization mechanism to form the key C3 reaction intermediate, avoiding the competitive C2 desorption pathway. Furthermore, Ag-doped Cu-DANs could effectively retain oxygen atoms in the hydroxyl group, which enabled a pathway towards direct electrosynthesis of a new C3 product (C3H8O2; 1,2-PDO) beyond the only available n-propanol. Our newly discovered concerted trimerization mechanism in combination with single-atom alloy catalysts paves the way for materials design toward more long-chain oxygenate generation.
- This article is part of the themed collection: Single-Atom Catalysis
 Please wait while we load your content...
Please wait while we load your content...

