
Promotion of the oxygen evolution reaction via the reconstructed active phase of perovskite oxide†

Abstract
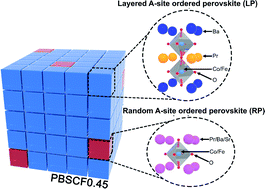
Developing a stable and highly efficient electrocatalyst for the oxygen evolution reaction (OER) is critical for renewable, safe, and emission-free energy technologies. Perovskite oxides with flexible and tunable electronic structures as functional electrocatalysts towards the OER have been investigated for decades. Various strategies for highly efficient perovskite catalysts have been proposed; however, precise insights are still lacking due to the diverse metal compositions and tunable structures. Among them, controlling the composition of the transition metals at the B-site of the double perovskite was considered as a dominant parameter to improve the electrocatalytic activity. It is known that several Co-based perovskites exhibit extraordinary OER activities and stabilities when the B-site is partially occupied by Fe. To elucidate the function of B-site tuning of perovskite oxides, PrBa0.5Sr0.5Co2−xFexO5+δ (PBSCF) catalysts with various x values were prepared. Interestingly, via X-ray diffraction (XRD) and transmission electron microscope (TEM) analysis we found that the PBSCF catalysts coexist with layered perovskite (LP) structures and randomly ordered perovskite (RP) structures. In parallel, density functional theory computational calculations were conducted to provide theoretical insights into our research results. Our findings show that doping Fe into Co-based perovskite oxides alters the intrinsic properties, yielding efficient OER activity and prolonged stability (@ 100 mA cm−2 over 2000 hours).
- This article is part of the themed collection: Special issue in honour of Prof. John Kilner’s 75th birthday
 Please wait while we load your content...
Please wait while we load your content...

