
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revealing the activity of Co3Mo3N and Co3Mo3N0.5 as electrocatalysts for the hydrogen evolution reaction†
Youyi
Sun
a,
Lewen
Wang
b,
Olga
Guselnikova
 cd,
Oleg
Semyonov
c,
James
Fraser
a,
Yecheng
Zhou
*b,
Núria
López
e and
Alexey Y.
Ganin
*a
cd,
Oleg
Semyonov
c,
James
Fraser
a,
Yecheng
Zhou
*b,
Núria
López
e and
Alexey Y.
Ganin
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: alexey.ganin@glasgow.ac.uk
bSchool of Materials Science & Engineering, Sun Yat-Sen University, Guangzhou 510006, Guangdong, P. R. China. E-mail: zhouych29@mail.sysu.edu.cn
cTomsk Polytechnic University, Lenina Av. 30, 634050 Tomsk, Russian Federation
dNational Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
eInstitute of Chemical Research of Catalonia, The Barcelona Institute of Science and Technology, 43007 Tarragona, Spain
First published on 13th December 2021
Abstract
The hydrogen evolution reaction (HER) from water is governed by the electrocatalysts used. Multiple factors such as crystal structure, composition and morphology dictate the final catalytic performance. However, as multicomponent materials are developed to replace noble metals in the HER, it has become increasingly difficult to identify intrinsically active materials. Hence, there is an imperative for phase-pure catalysts to be synthesized and tested without obscuring contributions from impurities or substrates. Herein, we demonstrate that phase-pure, unsupported Co3Mo3N achieves a competitively low overpotential (OVP) of 108 ± 8 mV at 10 mA cm−2 in 0.5 M H2SO4. Density functional theory (DFT) reveals weakly binding metal sites as the catalytic centres for the HER in the nitride. Remarkably, the N-deficient Co3Mo3N0.5 shows similar electrochemical properties but has limited chemical stability under cathodic bias. Thus, even though nitrogen sites play only a minor role in catalytic performance, their occupancy is crucial for the stability of nitride catalysts in the corrosive electrolyte. The composite of Co3Mo3N on Ni-foam sustains 10 ± 1 mA cm−2 at an applied potential of just 20 mV over extended time, highlighting the utility of nitrides for the future design of stable and active HER catalytic systems.
Introduction
Renewable energy sources are intermittent as seasonal changes limit their operation at full capacity.1,2 Hence, to mitigate the mismatch between supply and demand it is important to address the natural swings in energy output by storing the excess of renewable energy as fuels rather than halting the production.2–4 Hydrogen is an excellent fuel and can be produced sustainably by water electrolysis in an electrolyser.3 Proton Exchange Membrane (PEM) electrolysers are compact units for the production of green hydrogen from water at appreciable rates.5–7 The short start-up times and load flexibility make PEM electrolysers ideally suited for integration with renewable energy sources. However, platinum (which is the electrocatalyst of choice for PEM electrolysis) is scarce and expensive, making the search for electrocatalysts composed of earth-abundant elements important.8 Non-precious metal alloys are cheaper alternatives to Pt but their poor stability in acids makes them a suboptimal choice for PEM electrolysers.9–12 This limitation may be overcome by employing ternary nitrides instead of alloys, as a recent report demonstrated the superior stability of binary molybdenum nitrides over Mo metal in acidic conditions.13Remarkably, ternary nitrides have remained relatively unexplored as electrocatalysts in acidic media up to date (Table S1†) and those nitrides that had delivered OVPs in the vicinity of 100 mV at 10 mA cm−2 were generally synthesized with poorly defined crystal structure and compositions. Many investigated nitride catalysts contained impurity phases and/or were composite materials, with for example, high surface area metal-foam or carbons.14–18 This makes the explanation of their catalytic properties by theory a complex task because it is very challenging to build reliable computational models for multiphase systems.12,19–22 On the other hand, the electrochemical tests on materials that are free from substrates and secondary phases can provide a robust benchmark for comparison between existing and development of future nitride materials. However, despite being detrimental (especially when it comes to multicomponent systems), the studies of phase-pure, free-standing nitrides are rare23,24 and attempts to decouple the role of confounding factors (such as impurities and substrates) from the intrinsic catalyst's performance are urgently needed.
Here, we report that phase-pure Co3Mo3N and Co3Mo3N0.5 tested in free-standing form can deliver an OVP of just over 100 mV at 10 mA cm−2 in 0.5 M H2SO4. Hence, they outperform any free-standing metal nitrides previously reported in acidic electrolyte and thus, are intrinsically catalytic. Remarkably, the initial computational studies point out that the calculated free energy of hydrogen adsorption (ΔGH) on a single metal site is close to the optimal thermodynamic value of ΔGH = 0. However, the multiple adsorption sites model suggests that the HER proceeds over the weakly bonding metal sites with ΔGH > 0. Finally, we demonstrate that a composite material consisting of single-phase Co3Mo3N on Ni-foam can deliver a very strong performance by sustaining the benchmark current densities of 10 ± 0.7 and 50 ± 5 mA cm−2 at the applied potentials of only 20 mV and 80 mV respectively in 0.5 M H2SO4.
Experimental
Synthesis
Co3Mo3N was synthesised using an ammonia-free reaction route (ESI† for detailed procedures). An oxide precursor was converted into Co3Mo3N at 750 °C in the flow of H2/N2 gas for 5 hours. Heating Co3Mo3N at 750 °C for 2 hours in the flow of H2/Ar gas produced an isomorphic (morphology preserved), nitrogen-deficient Co3Mo3N0.5.25 Heating an oxide precursor on Ni-foam at 750 °C in the flow of H2/N2 gas for 5 hours produced the composite Co3Mo3N on Ni foam material.Materials characterisation
Powder X-ray diffraction (PXRD) was carried out on a Rigaku MiniFlex 6G equipped with a D/teX Ultra detector, a 6-position (ASC-6) sample changer and Cu sealed tube (Kα1 and Kα2 wavelengths – 1.5406 and 1.5444 Å respectively) operating in the Bragg–Brentano geometry. A sample was carefully spread over a silicon zero-background holder and flattened with a glass slide. In the case of Co3Mo3N on Ni-foam, the sample was attached with a scotch tape to the zero-background holder. Data collection was carried out using Rigaku SmartLab Studio II software (Rigaku Corporation, 2014).The stability studies were carried out by measuring PXRD directly on glassy carbon electrodes using the custom-built bracket26 on the Panalytical Xpert-Pro diffractometer with the Cu Kα (λ = 1.54184 Å) source operating in the Bragg–Brentano geometry.
Rietveld refinements27 of PXRD data were carried out using GSAS-II software.28 Refined parameters included unit cell parameters, sample displacement, strain, and crystallite broadening, as well as atomic position and Uiso for transition metals. Background was fitted using shifted Chebyshev polynomic shape. Pseudo-Voigt function was used for peak shapes.
Nitrogen adsorption–desorption isotherms were measured at −196 °C using a Quantachrome Autosorb Evo unit. The samples were degassed at 80 °C overnight under vacuum before the measurement.
Scanning Electron Microscopy (SEM) was performed using a Philips XL30 ESEM instrument equipped with an Oxford Instruments X-act spectrometer for Energy Dispersive X-ray Spectroscopy EDXS measurements. The INCA software was used for the data analysis. Cu foil was used as the calibration standard for the EDXS. The sample preparation involved dispersing ca. 1 mg of product in 1 ml of water on sonication for 5 minutes. 100 μl drop of resulting solution was casted on Cu foil producing a well-spread and homogenous coating on drying.
The X-ray photoelectron spectroscopy (XPS) was conducted using a Thermo Fisher Scientific XPS NEXSA spectrometer with a monochromated Al K alpha X-ray source operating at 1486.6 eV. The high-resolution spectra were recorded at the pass energy of 50 (eV) and energy resolution 0.1 (eV). The flood gun was used for the charge compensation. The analysed area was 200 μm2.
Electrochemical testing
All electrochemical measurements were performed using a Biologic SP-150 potentiostat with a three-electrode setup in a single cell compartment in 0.5 M H2SO4 (ESI† for a detailed procedure). The experimental errors were obtained by performing at least three independent measurements and by taking the deviations between these measurements as an error.DFT simulations
Surface structures were optimized in Vienna Ab initio Simulation Package (VASP 5.4.4), which implemented Projector Augment Wave (PAW) based on plane-wave basis.29,30 At least 10 Å of vacuum was implemented in our models to avoid layer interactions. Due to the large size of the unit cells, Γ-point was used to relax the structure. A Γ-point centred at 3 × 3 × 1 k-mesh was used for static energy calculations. The cut-off energy of plane wave basis was set at 550 eV. The exchange functional was the Perdew–Burke–Ernzerhof (PBE) of Generalized Gradient Approximation (GGA). In all calculations, the magnetic moments of the atoms were initially ferromagnetically aligned. Additional details are provided in the ESI.†Results and discussion
The phase purity of the product prepared in H2/N2 gas flow was assessed by powder X-ray diffraction (PXRD) with the resulting pattern matching the simulated pattern of Co3Mo3N. Therefore, the Rietveld refinement of the PXRD data (Fig. 1a and Table S2† for the refined parameters) was carried out using a previously reported Co3Mo3N structure (Fig. S1†) as a model.31 The results revealed that Co3Mo3N was the only product, and no other crystalline phases were present. The cubic unit cell parameter (a = 1101.39 ± 0.03 pm) is marginally smaller than literature data which is consistent with the results of elemental analysis for nitrogen (experiment N: 2.79 ± 0.04 wt%, theory N: 2.93 wt%) suggesting a minor nitrogen non-stoichiometry.25,31,32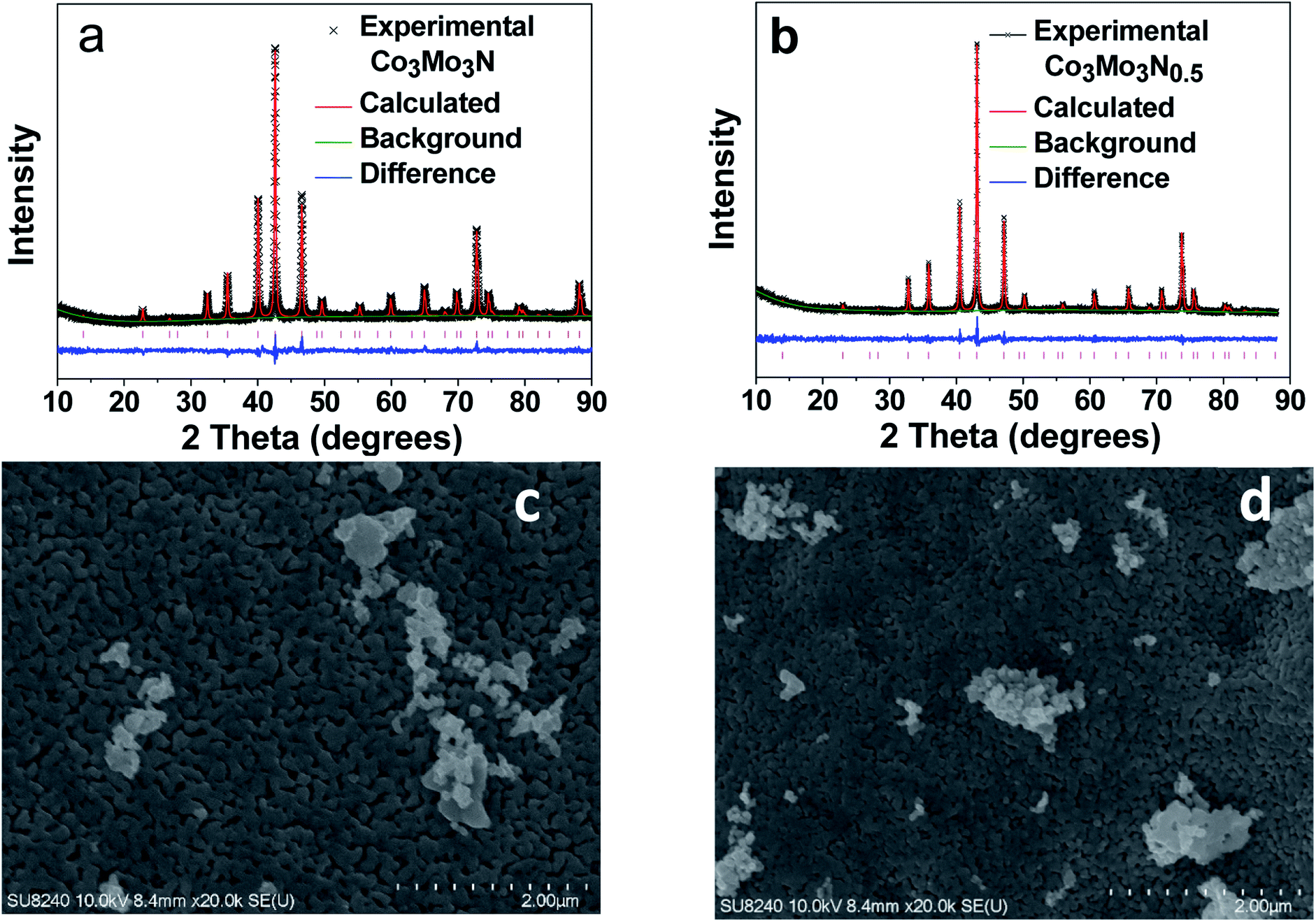 | ||
Fig. 1 Rietveld refinement of PXRD data (CuKα) for (a) Co3Mo3N and (b) Co3Mo3N0.5 using a structure model based on Co3Mo3N structure (space group: Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) mz). Measured data are shown as crosses; the calculated profile is shown by a solid red line. The difference profile is shown as blue line along the bottom of the plot. Vertical bars represent the reflection positions for the phase. High magnification SEM images for (c) Co3Mo3N and (d) Co3Mo3N0.5. mz). Measured data are shown as crosses; the calculated profile is shown by a solid red line. The difference profile is shown as blue line along the bottom of the plot. Vertical bars represent the reflection positions for the phase. High magnification SEM images for (c) Co3Mo3N and (d) Co3Mo3N0.5. | ||
Similarly, the PXRD diffraction pattern of Co3Mo3N annealed in H2/Ar was consistent with the simulated one for previously reported for Co3Mo3N0.5. The Rietveld refinement of the PXRD data (Fig. 1b and Table S3† for the refined parameters) further confirmed the single-phased nature of the sample with the unit cell parameter a = 1088.10 ± 0.01 pm. Hence, it is in an excellent agreement with the previously reported value of a = 1088.24 pm for a sample with composition determined as Co3Mo3N0.5 by neutron diffraction.25 Therefore, we concluded that the N-content in our sample must be close to the expected value of 0.5. This was further confirmed by with the results of elemental analysis on our sample which was consistent with the expected nitrogen content = 0.5 within the error of the measurement (Co3Mo3N0.5 – experiment: N: 1.43 ± 0.04 wt%; theory: N: 1.48 wt%).
Since the morphology of a catalyst may play an important role in the catalytic properties Scanning Electron Microscopy (SEM) was carried out to investigate the morphologies of Co3Mo3N and Co3Mo3N0.5. The images (Fig. 1c and d) demonstrate that both nitrides showed similar surfaces and thus, confirming that the conversion of Co3Mo3N into Co3Mo3N0.5 proceeds through a topotactic reaction. Topotactic reactions often lead to retention of the morphology between the precursor and the product. Therefore, the comparison between electrocatalytic properties of two materials is simplified due to similarity of their morphologies. Analysis with Energy Dispersive X-ray Spectroscopy (EDXS) showed that both nitrides had the same Co to Mo ratios (Table S4†). Furthermore, according to gas adsorption measurements both nitrides showed low surface areas of ca. 2 m2 g−1 (Fig. S2†). The similarity of the morphology and surface areas of the nitrides imply that any difference in catalytic performance between two nitrides could be attributed to the difference in their intrinsic crystal structure and stoichiometry. Overall, the SEM, EDXS and adsorption studies point out that the only difference between these two isomorphic nitrides is the occupancy of the N-sites.
Co3Mo3N is a metallic compound33 and thus, it is highly conductive making it a promising target for electrocatalytic studies. Although Co3Mo3N prepared through an ammonia route has been tested for the oxygen evolution reaction in alkaline media and showed relatively good performance;34 rather intriguingly, when it was tested for the HER in acidic media23 it was unable to achieve a benchmark current density of 10 mA cm−2. Therefore, it has been deemed as inactive and has not been investigated by any other groups since then.
Surprisingly, we found that Co3Mo3N and Co3Mo3N0.5 demonstrated overpotentials (OVPs) of 108 ± 8 mV and 117 ± 12 mV at 10 mA cm−2 respectively when tested by linear-sweep voltammetry (LSV) in 0.5 M H2SO4 (Fig. 2a). The standard deviations on the OVP values come from the numerous samples tested with all samples consistently showing OVPs close to 100 mV at 10 mA cm−2. Furthermore, both Co3Mo3N and Co3Mo3N0.5 catalysts achieved respectable cathodic current densities of more than 500 mA cm−2 (Fig. S3†). In addition, the evaluated turnover frequency (TOF = 14.1 s−1) at the benchmark current of 10 mA cm−2 (calculated following previously reported approach in the ESI†)35 suggests high catalytic performance. These results are comparable to some of the highest values previously reported for free-standing nitride materials (Table S1†). Therefore, we could not rationalize the reason for poor catalytic activity found in Co3Mo3N in previous research.23 A possible explanation could be the difference in synthetic methods, i.e. ammonia synthesis compared with H2/N2 route employed in our work. This could result in a different stoichiometry on the N-site. However, the nearly identical electrocatalytic performance of Co3Mo3N and Co3Mo3N0.5 suggests that nitrogen positions play a very limited role as active sites in the HER, otherwise both nitrides would have had a different catalytic performance due to the difference in N-stoichiometry. From this perspective, 5% H2/N2 gas mix (which is non-flammable at such low dilution levels) appears to be a viable substitution to ammonia. Ammonia has high vapour pressure and toxicity. This hinders its use even in well-equipped chemistry departments and prevents wider research into nitride materials.
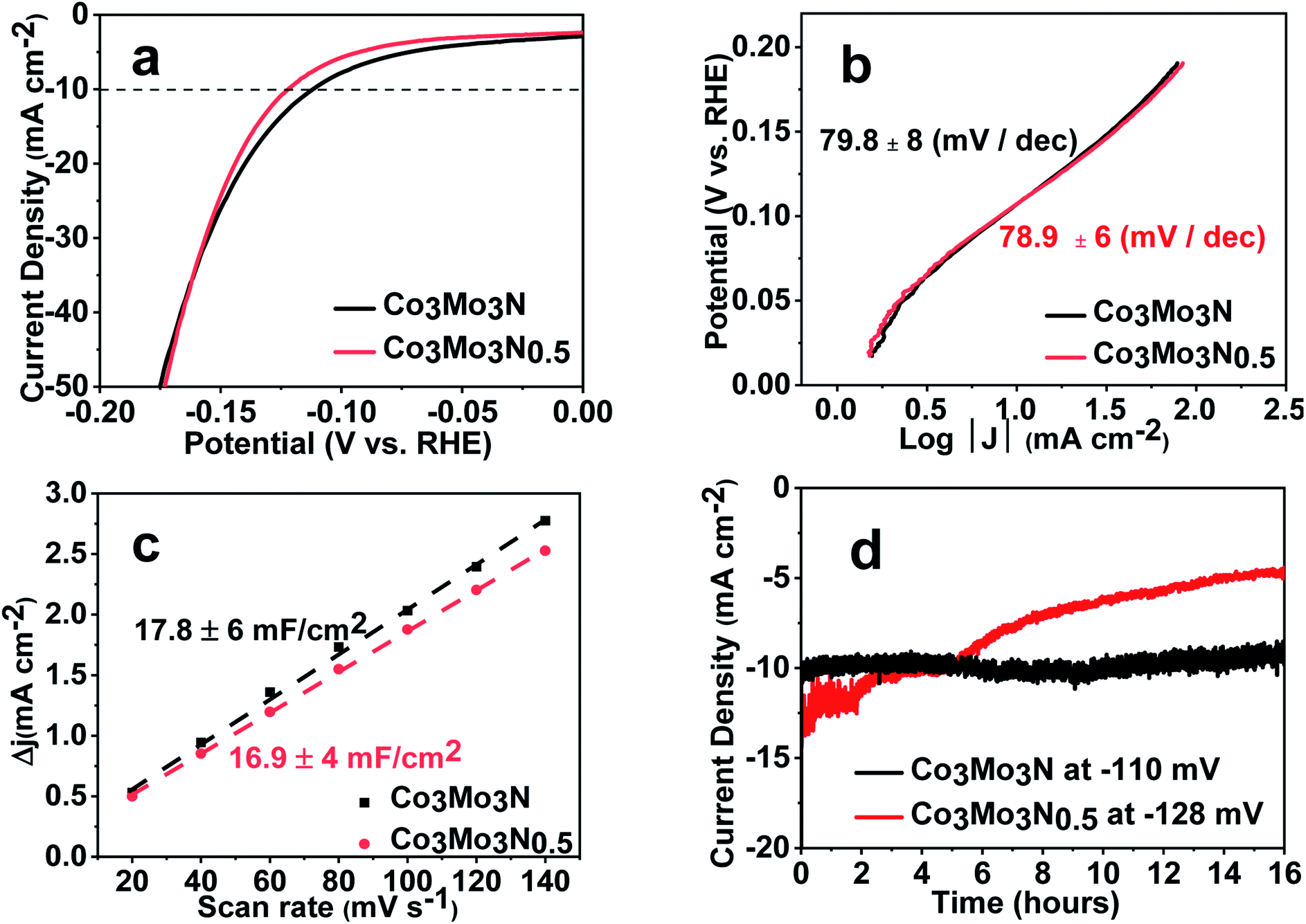 | ||
| Fig. 2 Electrochemical studies on Co3Mo3N and Co3Mo3N0.5 in 0.5 M H2SO4. (a) Comparison of the current densities obtained by linear sweep voltammetry at a scan rate of 5 mV s−1. Dashed line provides a guide to the eye at a current density of 10 mA cm−2. (b) Tafel plots and corresponding Tafel slopes. All current densities were corrected for resistance. (c) Current density differences of nitrides plotted against scan rates. The double layer capacitances (CDL) measured at 160 mV (vs. RHE). (d) Chronoamperometry profiles at an applied potential of −110 mV and −128 mV (vs. RHE). | ||
This striking similarity in electrochemical performance of two nitrides is further confirmed by the analysis of corresponding Tafel plots. Almost identical slopes of 80 ± 8 mV and 79 ± 6 mV dec−1 for Co3Mo3N and Co3Mo3N0.5 (Fig. 2b) suggest that the kinetics of the HER on the surface of either nitride must be the same. Although the attempts to select the current density range with a minimum transport limitation (for example, due to the bubble formation and diffusion) has been made, mass-transport correction are required to determine the rate determining step governed by electron transfer.36,37 These corrections are incredibly difficult to implement in practice on polycrystalline powders. Therefore, it is common to observe deviations from the “classical” slopes of 40 mV dec−1 (Heyrovsky mechanism) and 30 mV dec−1 (Tafel mechanism) (Table S1†). However, the similarity in Tafel slopes acts as an indicator that the nature of HER on both nitrides is the same and therefore, is likely independent of the occupancy of the nitrogen sites.
The similarity of the catalytic behaviour between Co3Mo3N and Co3Mo3N0.5 was further confirmed by the investigation of the capacitive currents by cycle voltammetry (CV) curves at various scan rates (Fig. S4 and S5†). The values of the double-layer capacitance (CDL) that are believed to be a good proxy of the catalyst' roughness factor,36 are identical in Co3Mo3N and Co3Mo3N0.5 and are 18 ± 6 and 17 ± 4 mF cm−2 respectively (Fig. 2c). This confirms that in both nitrides the catalytic behaviour happens on the same sites (or at least the same sites undergo the initial adsorption of hydronium ion at low overpotential).
We also carried out electrochemical impedance spectroscopy (EIS) experiments that showed (Fig. S6†) the same charge-transfer resistance (both iR compensated) values of Rct = 21 ± 7 Ω and Rct = 20 Ω ± 8 Ω for Co3Mo3N and Co3Mo3N0.5. This suggests that high charge-transfer properties and hydrogen evolution reaction kinetics are the same in both nitrides.
However, when tested at a constant applied potential of ca. 110 mV in 0.5 M H2SO4, Co3Mo3N retained the benchmark current density of 10 mA cm−2 over 16 hours, while for Co3Mo3N0.5 the current density decreased gradually under applied potential with time (Fig. 2d). This suggests that the lower nitrogen content in Co3Mo3N0.5 is responsible for the instability of the catalyst which is in line with the behaviour previously reported in the binary Mo–N system.13 LSV data also show that performance deteriorates after 1000 cycles in Co3Mo3N0.5 (Fig. S7†) while the activity of Co3Mo3N remains unchanged (Fig. S8†). The stability of Co3Mo3N after electrolysis was additionally confirmed by PXRD (Fig. S9†) and faradaic efficiency measurements by gas chromatography (Fig. S10†). In addition, the Rietveld refinement of PXRD data showed (Fig. S11†) that the unit cell parameters of Co3Mo3N before (a = 1100.4 ± 0.3 pm) and after (a = 1099.8 ± 0.4 pm) 1000 cycles are identical within the standard deviations. As the nitrogen deficiency or degradation of Co3Mo3N would result in the measurable shift of unit cell parameters we concluded that the sample remained unchanged.
In addition, X-ray photoelectron spectroscopy (XPS) measurements were carried out on Co3Mo3N before and after 1000 cycles in 0.5 M H2SO4. Only minor changes were observed in the high-resolution Mo 3d, Co 2p and Mo 3p spectra before and after electrochemical reaction (Fig. S12–S14†) suggesting that Co3Mo3N remains unchanged upon cycling. The measured XPS spectra are nearly identical to previously reported for Co3Mo3N as well.38–40 We deconvoluted the XPS data and found that the spectra displayed a range of binding energies corresponding to different oxidation states (Tables S5–S13†). The positions of the key peaks in the high resolution Co 2p3/2 at 778.1/778.4 ± 0.1 eV (before/after 1000 cycles) and Mo 3d5/2 at 227.7/227.9 ± 0.1 eV (before/after 1000 cycles) were in good agreement with the values of 778.5 eV and 228.2 eV reported by Hosono's group for Co3Mo3N prepared in ammonia at 785 °C.39 These values are consistent with the expected low valent state of metals due to the intermetallic nature of Co3Mo3N. We noted that although Mo–O and Co–O oxides were also present (inevitable as Co3Mo3N powders were handled in air), the peaks remained at the same position and intensity before and after electrolysis. Finally, the analysis of 380–430 eV region, where both signals from Mo 3p and N 1s are present, showed a striking similarity with the Mo 3p spectra previously reported in the literature.39 The deconvolution of the spectrum (Fig. S14†) allowed us to assign the position of N 1s peak at 397.0 ± 0.1 eV. This value is smaller than previously reported of 397.9 eV for Co3Mo3N nitride because previous investigators did not consider the presence of a small shoulder on Mo 3p1/2 peak which suggested there was a second component present. However, the overlapping of Mo 3p3/2 (and corresponding oxides) with N 1s signal complicated the precise analysis for N content which appeared to be strongly underestimated. However, as both samples analysed before and after 1000 cycles have the same (low N-content of 1.5%) it is unlikely that decomposition took place during the cycling.
Although our primary goal was to demonstrate that Co3Mo3N is a competent catalyst in free standing form; keeping in mind the potential application of Co3Mo3N in PEM electrolysers, we designed a method for synthesis of Co3Mo3N directly on Ni-foam. When heated at 750 °C in H2/N2 for 5 hours a metal oxide precursor immobilized on Ni foam converts into Co3Mo3N. According to PXRD analysis the resulting diffraction pattern of the composite material contains only peaks consistent with Ni and Co3Mo3N (Fig. S15†). When tested under applied potential of 20 mV the composite retained the current density of 10 ± 1 mA cm−2 for over 16 hours (Fig. S16†). In this respect, it outperforms 20% Pt on carbon which showed the current densities of 8 ± 2 mA cm−2 at an applied potential of 25 mV for over 16 hours. The catalyst remains stable under higher current densities as well, for example, when a potential of 80 mV was applied, it sustained the current density of 50 ± 2 mA cm−2 over 16 hours (Fig. S16†). The performance is similar with 20% Pt on carbon which demonstrated at the applied potential of 65 mV the current densities of 35 ± 1 mA cm−2 over 16 hours.
Overall, the experimental observations point out that the metal sites are likely responsible for catalytic behaviour while the nitrogen content is governing the stability of the nitrides. Therefore, to rationalise the catalytic behaviour of Co3Mo3N and Co3Mo3N0.5, we carried out DFT simulations of the electronic structure and investigated the most likely reaction sites. Guided by the previous theoretical research on activity of Co3Mo3N41 towards ammonia synthesis that found (111)-surface the most stable we built ten Co3Mo3N-(111) surfaces (Fig. S17–S20†). We then evaluated the surface energies of the various slabs (Table S14†) to assess the surface composition with the lowest energy surface, the resulting structure is shown in Fig. 3a. Firstly, there is a striking similarity of total density of states (DOS) between the surface with fully occupied nitrogen sites and the surface with a nitrogen vacancy (Fig. S20–S22†) showing that the electron rearrangement due to N-vacancies does not affect the overall electronic properties of the material. Additionally, the Mo-sites in proximity to the N-site show the same DOS independent of the occupancy on N-site (Fig. S21†). This explains the experimental findings that Co3Mo3N and Co3Mo3N0.5 show identical catalytic performance as the N-vacancy does not influence the DOS of metal sites in proximity to the vacancy. However, the N-vacancy disrupts a network of corner sharing [Mo6N] octahedra, which may explain the lower chemical stability of Co3Mo3N0.5 under prolonged electrolysis (Fig. 2d). The calculated energy release of ≈0.6 eV when protons are adsorbed on the vacancy sites indicates that a prolonged electrolysis could destabilise the structure when excess vacancies are present due to the occupation of these sites with H as the HER reaction proceeds.
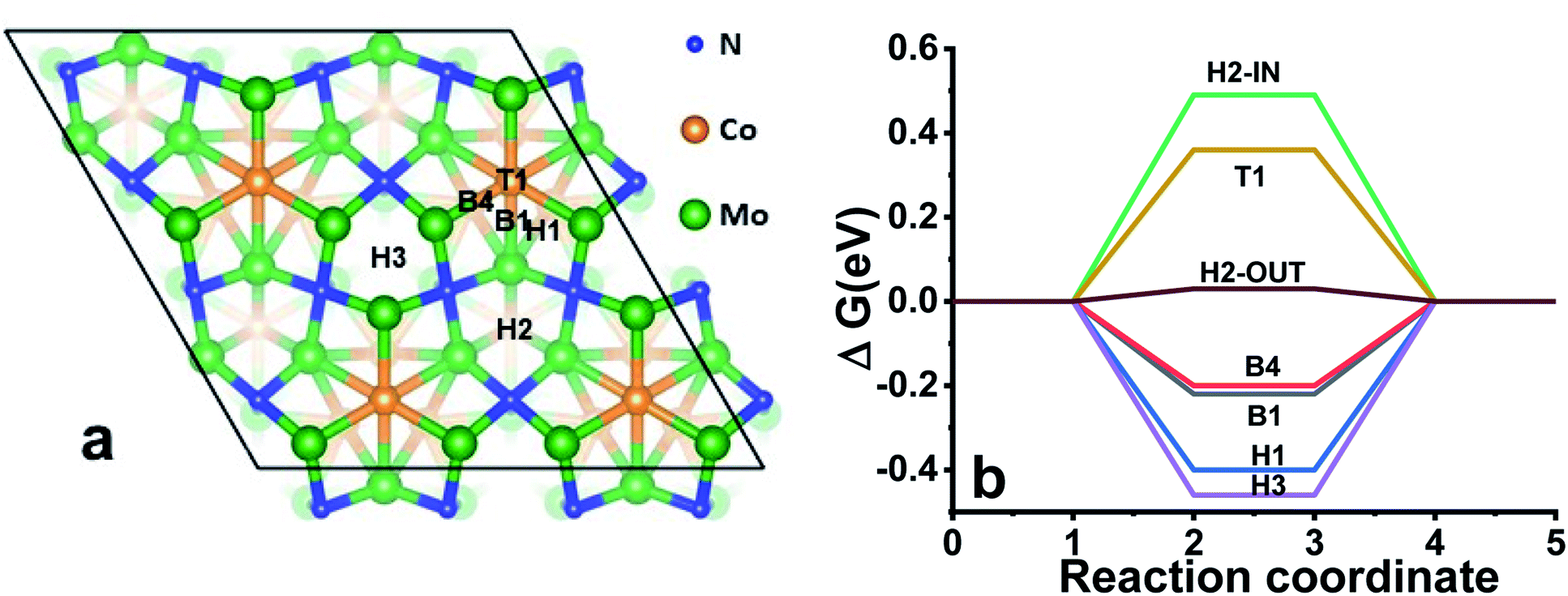 | ||
| Fig. 3 (a) The most energetically favourable surface of Co3Mo3N and the optimal reaction sites for the HER in a single site model. (b) The free energy diagram of the possible reaction sites on the Co3Mo3N-(111) surface at pH = 0. The labels for the reaction sites correspond to the sites shown in (a). | ||
Tables S15 and S16† give the calculated free energies of hydrogen adsorption ΔGH values for all the possible sites and Fig. 3b shows the optimal sites that could participate in the catalytic reaction. ΔGH can be taken as the simplest first order descriptor for predicting the HER activity in metals and alloys; and indeed, the closer this value is to zero, the closer the system is considered to the thermodynamic optimum.19,20,42 Fig. S23† shows the algorithm and how the model has been built based solely on the single adsorption site model (see Fig. S24–S32† for a better visualisation of the location of H on all sites in this model). In this simulation the hollow H2-OUT site has a ΔGH = 0.03 eV. This value places Co3Mo3N into the optimal range in comparison with transition metals when plotted against exchange current densities (Fig. S33†). Therefore, this model explains fairly well the relatively low OVP observed in Co3Mo3N.19,43,44 However, this is a simplified picture as the presented model is based on a single adsorption site. In practice, as the HER progresses, the protons could adsorb on multiple sites.
It has been proposed that the strongly adsorbing sites with negative ΔGH act as spectators rather than participating in the HER while the weakly bound hydrogens (enabled on sites with the ΔGH > 0) drive the HER forward.21,22,45 Therefore, we modelled the impact of the presence of strong adsorption sites (H3 > H1 > B1 > B4) on the reactive ability of the H2-OUT site (Tables S17 and S18† for the ΔGH of the sites and Fig. S34† for the algorithm how the model was built). The most stable configuration in the multiple site model is achieved when the reaction happens on the H2-IN site (see Fig. S35† for a better visualization). This site has ΔGH = 0.13 eV when two strongly adsorbing H1- and H3-sites are occupied. In addition, the adsorption is also possible at this point on the T2-site (unstable in the case of single site model). Thus, the overall trend, leading to the increase in ΔGH when multiple sites are occupied may explain that contrary to the simplified single site model (which gave a reaction site with ΔGH ∼ 0 eV) Co3Mo3N is still inferior to Pt. However, as in both models the adsorption of the hydrogen on the H2-IN site (Fig. S35†) which is in close proximity to the Co site suggests that the catalytic ability of Co3Mo3N may be tuned by substitution of Co with other transition metals such as Fe, Ni or Mn.
Conclusions
In summary, in Co3Mo3N it is the Co-site which appears to be catalytic even when a more complex model with adsorption on the neighbouring sites is considered. The intrinsic activity and stability of Co3Mo3N as a catalyst for the HER highlights an important feature of transition metal nitrides: while being inherently more stable than traditional metal alloys they could also have the metal sites that show ΔGH ∼ 0 eV. This suggests that corrosion-resistant transition metal nitrides, such as Co3Mo3N, may be re-designed into extrinsically superior catalytic systems by adopting the key principles from traditional transition metal alloy systems, such as nanostructuring and/or interaction with electroactive substrates. We examined the validity of this approach by testing a composite consisting of Co3Mo3N on Ni-foam. This was found to be highly active towards the HER suggesting the adaptability of ternary nitrides as viable alternatives to noble metals in PEM systems.Author contributions
A. Y. G. conceived and managed the experiments. Y. S. designed the experiments and carried them out. Y. S., A. Y. G. and Y. Z. analysed and plotted the data. A. Y. G. wrote the manuscript. Y. Z., L. W. and N. L. designed and carried DFT experiments. O. G. and O. S. carried out the XPS measurements and analysed/plotted the data. J. F. measured SEM data. All authors have contributed to writing the manuscript and have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. Y. G. acknowledges the University of Glasgow and EPSRC (EP/N509668/1) for supporting this work. Y. S. thanks China Scholarship Council for providing him with the scholarship. Y. Z. acknowledges the support of the Basic and Applied Research Project (No. 202102020495) of Guangzhou. L. W., Y. Z. and N. L. appreciate the National Super-Computer Center in Guangzhou (China) and the Barcelona Supercomputing Center BSC-RES (Spain) for providing computational resource. O. G. and O. S. acknowledge the support from the Tomsk Polytechnic University Enhancement programme.Notes and references
- S. Gangwar, D. Bhanja and A. Biswas, J. Renewable Sustainable Energy, 2015, 7, 013109 CrossRef.
- H. Stančin, H. Mikulčić, X. Wang and N. Duić, Renewable Sustainable Energy Rev., 2020, 128, 109927 CrossRef.
- Z. Chehade, C. Mansilla, P. Lucchese, S. Hilliard and J. Proost, Int. J. Hydrogen Energy, 2019, 44, 27637–27655 CrossRef CAS.
- F. Tong, M. Yuan, N. S. Lewis, S. J. Davis and K. Caldeira, iScience, 2020, 23, 101484 CrossRef PubMed.
- P. Aßmann, A. S. Gago, P. Gazdzicki, K. A. Friedrich and M. Wark, Curr. Opin. Electrochem., 2020, 21, 225–233 CrossRef.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- J. Koponen, A. Kosonen, V. Ruuskanen, K. Huoman, M. Niemelä and J. Ahola, Int. J. Hydrogen Energy, 2017, 42, 29648–29660 CrossRef CAS.
- H. Jin, B. Ruqia, Y. Park, H. J. Kim, H.-S. Oh, S.-I. Choi and K. Lee, Adv. Energy Mater., 2021, 11, 2003188 CrossRef CAS.
- J. N. Hansen, H. Prats, K. K. Toudahl, N. M. Secher, K. Chan, J. Kibsgaard and I. Chorkendorff, ACS Energy Lett., 2021, 6, 1175–1180 CrossRef CAS PubMed.
- L. Rößner and M. Armbrüster, ACS Catal., 2019, 9, 2018–2062 CrossRef.
- Y. Sun and A. Y. Ganin, Hydrogen, 2020, 1, 11–21 CrossRef.
- Z. Wang, Y.-R. Zheng, J. Montoya, D. Hochfilzer, A. Cao, J. Kibsgaard, I. Chorkendorff and J. K. Nørskov, ACS Energy Lett., 2021, 6, 2268–2274 CrossRef CAS.
- M. Kozejova, V. Latyshev, V. Kavecansky, H. You, S. Vorobiov, A. Kovalcikova and V. Komanicky, Electrochim. Acta, 2019, 315, 9–16 CrossRef CAS.
- H. Jin, Q. Gu, B. Chen, C. Tang, Y. Zheng, H. Zhang, M. Jaroniec and S. Z. Qiao, Chem, 2020, 6, 2382–2394 CAS.
- J. Zhao, Y. Zeng, J. Wang, Q. Xu, R. Chen, H. Ni and G. J. Cheng, Nanoscale, 2020, 12, 15393–15401 RSC.
- W. Wang, Y. Shao, Z. Wang, Z. Yang, Z. Zhen, Z. Zhang, C. Mao, X. Guo and G. Li, ChemElectroChem, 2020, 7, 1201–1206 CrossRef CAS.
- Y. Gu, A. Wu, Y. Jiao, H. Zheng, X. Wang, Y. Xie, L. Wang, C. Tian and H. Fu, Angew. Chem., Int. Ed., 2021, 60, 6673–6681 CrossRef CAS PubMed.
- J. Tan, Y. Mei, H. Shen, H. Liu, T. Azhagan, W. Song, T. Thomas, J. Liu, M. Yang and M. Gao, ChemCatChem, 2020, 12, 122–128 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Nørskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS PubMed.
- J. Kibsgaard, C. Tsai, K. Chan, J. D. Benck, J. K. Nørskov, F. Abild-Pedersen and T. F. Jaramillo, Energy Environ. Sci., 2015, 8, 3022–3029 RSC.
- P. Lindgren, G. Kastlunger and A. A. Peterson, ACS Catal., 2019, 10, 121–128 CrossRef.
- J. Ekspong, E. Gracia-Espino and T. Wagberg, J. Phys. Chem. C, 2020, 124, 20911–20921 CrossRef CAS.
- B. Cao, G. M. Veith, J. C. Neuefeind, R. R. Adzic and P. G. Khalifah, J. Am. Chem. Soc., 2013, 135, 19186–19192 CrossRef CAS PubMed.
- W. F. Chen, K. Sasaki, C. Ma, A. I. Frenkel, N. Marinkovic, J. T. Muckerman, Y. Zhu and R. R. Adzic, Angew. Chem., Int. Ed., 2012, 51, 6131–6135 CrossRef CAS PubMed.
- S. M. Hunter, D. Mckay, R. I. Smith, J. S. J. Hargreaves and D. H. Gregory, Chem. Mater., 2010, 22, 2898–2907 CrossRef CAS.
- J. C. McGlynn, M. Friskey and A. Y. Ganin, Sustainable Energy Fuels, 2020, 4, 4473–4477 RSC.
- H. M. Rietveld, A Profile Refinement Method for Nuclear and Magnetic Structures, J. Appl. Crystallogr., 1969, 2, 65–71 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. K. Jackson, R. C. Layland and H. C. Zur Loye, J. Alloys Compd., 1999, 291, 94–101 CrossRef CAS.
- J. O. Conway and T. J. Prior, J. Alloys Compd., 2019, 774, 69–74 CrossRef CAS.
- T. J. Prior and P. D. Battle, J. Mater. Chem., 2004, 14, 3001–3007 RSC.
- K. Zhang, L. Zhang, X. Chen, X. He, X. Wang, S. Dong, P. Han, C. Zhang, S. Wang, L. Gu and G. Cui, J. Phys. Chem. C, 2013, 117, 858–865 CrossRef CAS.
- L. Ma, L. R. L. Ting, V. Molinari, C. Giordano and B. S. Yeo, J. Mater. Chem. A, 2015, 3, 8361–8368 RSC.
- D. Li, C. Batchelor-McAuley and R. G. Compton, Appl. Mater. Today, 2020, 18, 100404 CrossRef.
- D. Li, C. Lin, C. Batchelor-McAuley, L. Chen and R. G. Compton, J. Electroanal. Chem., 2018, 826, 117–124 CrossRef CAS.
- K. Hada, M. Nagai and S. Omi, J. Phys. Chem. B, 2001, 105, 4084–4093 CrossRef CAS.
- Y. Tsuji, K. Ogasawara, M. Kitano, K. Kishida, H. Abe, Y. Niwa, T. Yokoyama, M. Hara and H. Hosono, J. Catal., 2018, 364, 31–39 CrossRef CAS.
- Y. Tsuji, M. Kitano, K. Kishida, M. Sasase, T. Yokoyama, M. Hara and H. Hosono, Chem. Commun., 2016, 52, 14369–14372 RSC.
- C. D. Zeinalipour-Yazdi, J. S. J. Hargreaves and C. R. A. Catlow, J. Phys. Chem. C, 2016, 120, 21390–21398 CrossRef CAS.
- R. Parsons, Trans. Faraday Soc., 1958, 54, 1053–1063 RSC.
- M. M. Jaksic, Int. J. Hydrogen Energy, 2001, 26, 559–578 CrossRef CAS.
- B. E. Conway and J. O'M Bockris, J. Chem. Phys., 1957, 26, 532–541 CrossRef CAS.
- A. Ge, G. Kastlunger, J. Meng, P. Lindgren, J. Song, Q. Liu, A. Zaslavsky, T. Lian and A. A. Peterson, J. Am. Chem. Soc., 2020, 142, 11829–11834 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08389a |
| This journal is © The Royal Society of Chemistry 2022 |