
Single Mo atoms paired with neighbouring Ti atoms catalytically decompose ammonium bisulfate formed in low-temperature SCR†
Junxiao
Chen
a,
Xue
Fang
a,
Zhouhong
Ren
b,
Weiye
Qu
a,
Xiaolei
Hu
a,
Zhen
Ma
 ac,
Liwei
Chen
b,
Xi
Liu
*b,
Yaxin
Chen
*a and
Xingfu
Tang
*acd
ac,
Liwei
Chen
b,
Xi
Liu
*b,
Yaxin
Chen
*a and
Xingfu
Tang
*acd
aDepartment of Environmental Science & Engineering, Fudan University, Shanghai, 200438, P. R. China. E-mail: tangxf@fudan.edu.cn; 16110740025@fudan.edu.cn
bSchool of Chemistry and Chemical, In-situ Center for Physical Science, Shanghai Jiao Tong University, Shanghai, 200240, P. R. China. E-mail: liuxi@sjtu.edu.cn
cShanghai Institute of Pollution Control and Ecological Security, Shanghai, 200092, P. R. China
dJiangsu Collaborative Innovation Centre of Atmospheric Environment & Equipment Technology, Nanjing University of Information Science & Technology, Nanjing, 210044, P. R. China
First published on 1st November 2021
Abstract
Selective catalytic reduction (SCR) of NOx with NH3 has been widely used for NOx emission control, but commercial catalysts inevitably suffer severe deactivation in SO2-containing stack gases at low temperatures because the ammonium bisulfate (NH4HSO4, ABS) formed in SCR blocks the surface active sites. We resolve this issue by developing a TiO2-supported single-atom Mo catalyst (Mo1/TiO2) that decomposes ABS at ∼225 °C, far lower than the dew point of ABS (∼260 °C). Single Mo atoms paired with the neighboring surface Ti atoms function as Mo–Ti acid–base dual sites, which respectively adsorb the NH4+ and HSO4− of ABS. After the oxidation of NH4+ by surface lattice oxygen on the Mo sites, electrons left behind on the dual sites are localized around the Fermi level, which allows them to transfer to the adsorbed HSO4− on the Ti sites, thus releasing SO2 at low temperatures. The Mo1/TiO2 catalyst with Mo–Ti acid–base dual sites enables the decomposition of ABS at low temperatures, and thus this work provides a way to effectively control NOx emission particularly from industrial boilers.
1 Introduction
Nitrogen oxides (NOx) emitted from mobile and stationary sources are not only major atmospheric pollutants, but also important precursors for the formation of ozone and secondary aerosols,1,2 and hence are severely harmful to human health and the environment. With the increasingly stringent environmental regulations, enormous efforts have been devoted to controlling NOx emissions.3,4 Selective catalytic reduction (SCR) of NOx with NH3 over V2O5-based catalysts is the state-of-the-art technology for abating NOx emission from high-temperature (300–400 °C) stack gases.5,6 Nevertheless, these catalysts will inevitably suffer severe deactivation when this technology is applied in low-temperature and SO2-containing stack gases typically from many industrial boilers, largely because the surface catalytic sites are covered by viscous ammonium bisulfate (NH4HSO4, ABS) formed in SCR.7–9 Hence, it is necessary to develop low-temperature ABS-resistant catalysts to ensure that SCR catalysts efficiently operate under such conditions.There are two main strategies to develop low-temperature ABS-resistant catalysts on the basis of the formation mechanism of ABS and its properties. One can be defined as a source-controlling strategy. ABS is formed mainly from two sequential reactions, i.e., SO2 + 1/2 O2 → SO3 and SO3 + H2O + NH3 → NH4HSO4,10 and thus the formation of ABS can be avoided by preventing the oxidation of SO2 to SO3. To slow down the reaction rate of SO2 oxidation, adjusting the redox properties of catalysts such as by lowering the loading of active components seems feasible.9,11 However, this strategy is not optimal because the lower reaction rate of SO2 oxidation is at the expense of the low SCR activity.12 Meanwhile, there is often a certain amount of SO3 in flue gases, which makes the formation of ABS inevitable by the reaction of SO3 with NH3 and H2O.
The other strategy is to develop SCR catalysts with a function of decomposing ABS at low temperatures. ABS deposited on active sites is reported to be usually decomposed in a relatively high temperature range owing to its viscosity and the electrostatic attraction between NH4+ and HSO4−.13–15 Two kinds of interaction are often important factors to determine the temperatures required for ABS decomposition: ABS-catalyst adsorption interactions and NH4+–HSO4− electrostatic interactions. Conventional methods are often focused on the interactions between ABS and catalysts and thus the lowest decomposition temperature for ABS was reported to be ∼340 °C.16–19 Nevertheless, an extremely strong electrostatic interaction between NH4+ and HSO4− exists in ABS,20 making the low-temperature decomposition of ABS to produce SO2 and N2 unfavorable.21 To address that challenge, our previous work successfully broke the electrostatic interaction by spatially separating NH4+ from HSO4− with the assistance of layered MoO3, which allows electron transfer between separated NH4+ and HSO4− mediated by catalysts, thus achieving a lower decomposition temperature of ABS at ∼275 °C.22 However, limited by the weak interaction between acidic MoO3 and HSO4−,23 this decomposition temperature is still high (higher than the reported lowest dew point of ABS, ∼260 °C).24 Therefore, dual sites for NH4+ and HSO4− adsorption and strong electron transfer ability are key requirements for catalysts to effectively decompose ABS at low temperatures.
Considering the adsorption model of ABS previously reported,25 a desired catalyst should possess acid–base dual sites to adsorb NH4+ and HSO4−, respectively. The electron transfer from NH4+ to HSO4− on that catalyst should be realized through the oxidation of NH4+ on the acid site together with the reduction of HSO4− on the base site by electrons left behind on the acid–base dual sites. It was reported that MoO3 has strong acidity to adsorb the NH4+ of ABS and the ability to oxidize NH4+,26,27 while the TiO2 support surfaces can provide the basic sites for adsorbing the acidic HSO4− of ABS.27,28 Hence, a catalyst with Mo–Ti acid–base dual sites should readily adsorb NH4+ and HSO4−, respectively. Moreover, owing to the unsaturated coordination configuration, the surface Mo ions often have the desired ability to facilitate the oxidation of NH4+,29 generating reduced Mo species. Due to the charge transfer between Mo and Ti atoms via Mo5+ + Ti4+ → Mo6+ + Ti3+,30 the produced Ti3+, which often has a higher electron state density near the Fermi level (Ef),31 favourably accelerates the reduction of HSO4−. What is more, it is feasible to fabricate the Mo–Ti acid–base dual sites because Mo species tend to disperse on the surface of TiO2 in the form of Mo single atoms,32 which can pair with neighboring Ti sites to form acid–base dual sites.
In this work, we developed a TiO2-supported single-atom Mo catalyst (Mo1/TiO2), which can decompose ABS at temperatures far lower than the dew point of ABS. Based on systematic characterization via density functional theory (DFT) calculation, aberration-corrected scanning transmission electron microscopy (AC-STEM), Raman spectroscopy, diffuse-reflectance infrared Fourier-transform (DRIFT) spectroscopy, Fourier transform infrared spectroscopy (FT-IR) and X-ray photoelectron spectroscopy (XPS), atomically dispersed Mo adatoms on the surface of TiO2 resulted in the formation of abundant Mo–Ti dual sites, which respectively adsorb the NH4+ and HSO4− of ABS. Further electron transfer from NH4+ to HSO4− mediated by these Mo–Ti dual sites was realized, resulting in Mo1/TiO2 decomposing ABS at a temperature as low as ∼225 °C. This work proposes a strategy for designing ABS-resistant catalysts to effectively control NOx emissions particularly from industrial boilers.
2 Experimental
2.1 Catalyst preparation
Titanium oxide (TiO2, 5–10 nm) was obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O) and ammonium bisulfate (NH4HSO4, ABS) were acquired from Sinopharm Chemical Reagent Co., Ltd (China).In a typical synthesis, a precursor solution was prepared by dissolving 0.147 g (NH4)6Mo7O24·4H2O into 20 mL deionized water under stirring. Then 2.0 g TiO2 was added and the slurry was evaporated to dryness in a water bath at 80 °C, accompanied by drying in an oven at 80 °C for 12 h. The powder was then calcined at 550 °C for 3 h to get 6 wt% MoO3/TiO2 (denoted as Mo1/TiO2 catalyst). Besides, 0.40 g TiO2 was further loaded on 1.0 g Mo1/TiO2 by impregnation and the obtained solid was calcined at 550 °C for 3 h to acquire the TiO2/Mo1/TiO2 catalyst.
As for MoO3/TiO2 catalysts with a series of MoO3 loadings on TiO2, they were synthesized by tuning the amount of (NH4)6Mo7O24·4H2O added to 0.073, 0.098, 0.123, 0.490, 0.736, or 0.981 g, respectively, following the same procedure used for the preparation of the Mo1/TiO2 catalyst. For 0.5 or 2 wt% ABS-loaded samples, 0.005 or 0.02 g ABS was dissolved in 20 mL deionized water and 1.0 g of the corresponding catalyst was added later. After evaporating to dryness at 80 °C in a water bath and drying at 80 °C for 12 h, ABS-deposited catalysts were obtained.
In order to better understand the electron transfer pathway, the Mo1/TiO2 catalyst was further treated by (i) NH4+ oxidation: the catalyst was pre-reduced under 50 mL min−1 3 vol% NH3/He for 3 h followed by a 50 mL min−1 Ar atmosphere for another 3 h at 230 °C; (ii) HSO4− reduction: 2 wt% ABS was loaded on the catalyst and the obtained sample was then treated at 300 °C for 3 h under a 500 mL min−1 N2 flow; (iii) re-oxidation: the catalyst obtained in step (ii) was oxidized at 300 °C for 3 h under a 500 mL min−1 mixed 3 vol% O2 + 97 vol% N2 flow.
2.2 Catalyst characterization
X-ray diffraction (XRD) patterns of the catalysts were collected via a Rigaku D/MAX2200V X-ray diffractometer at 40 kV and 40 mA using Ni-filtered Cu Kα radiation (λ = 0.15418 nm). Transmission electron microscopy (TEM, JEOL JEM-2100F) and high-resolution TEM (HRTEM) were recorded using a JEOL JEM-2100F transmission electron microscope. Besides, AC-STEM images and energy dispersive X-ray spectroscopy (EDX) measurements were performed on a probe-corrected scanning/transmission electron microscope (Thermo Fisher Thermis Z) equipped with a SuperEDX detector at an accelerating voltage of 300 kV. An XploRA confocal spectrometer (Jobin Yvon, Horiba Gr, France) was used to obtain Raman spectra. The Raman scattering was excited by an external-cavity diode (785 nm) and coupled with a 50× Olympus microscope objective (Olympus, 0.50 numerical aperture). The power of the laser was 9 mW. The Raman spectra were collected at a resolution of 3 cm−1 with two accumulations at a 10 s acquisition time using a 1200 lines per mm diffraction grating. To analyze the change in electron states after a series of treatments, XPS was performed on a Kratos Axis Ultra DLD system with a monochromatic Al-Kα X-ray gun (1486.6 eV). Spectra were calibrated by adjusting the C 1s peak to 284.6 eV. XPSPEAK 4.1 with a Shirley-type background was used to analyze and process XPS data. DRIFT spectroscopy of NH3 adsorption was conducted by accumulating 64 scans at 4 cm−1 resolution from 4000 to 1000 cm−1 on a Nicolet IS 50 Fourier transform infrared spectrometer equipped with a Harrick Scientific DRIFT cell and a mercury-cadmium-telluride MCT/A detector. After the catalyst was pretreated at 300 °C for 1 h to remove physically adsorbed water and trace residues in a flow of N2 (150 mL min−1) and then cooled to 50 °C, the background spectra were collected under a N2 flow. Then, the NH3 adsorption on the catalysts was achieved by exposing the catalysts to a flow of 500 ppm NH3/N2 (150 mL min−1) at 50 °C for 1 h. After removing physically adsorbed NH3 in a flow of N2 (150 mL min−1) for 1 h, the DRIFT spectra were collected with background correction. To observe the evolution of ABS on the surface of the catalyst, in situ DRIFTS was carried out. In order to remove the physically adsorbed water, pretreatment of the catalysts was undertaken in a flow of N2 (50 mL min−1) at 200 °C for 1 h. Then the temperature was raised to 260 °C and the N2 flow was changed to 25 mL min−1. The DRIFT spectra were collected with 4 cm−1 resolution and 64 scans between 4000 and 1000 cm−1 every two minutes. As for the FT-IR spectra of 0.5 wt% ABS-deposited samples, they were mixed with KBr at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :99, pressed, and dried under infrared light. The spectra were obtained with 4 cm−1 resolution and 64 scans between 4000 and 1000 cm−1.
:99, pressed, and dried under infrared light. The spectra were obtained with 4 cm−1 resolution and 64 scans between 4000 and 1000 cm−1.
2.3 Catalytic evaluation
All the above-synthesized catalysts were pressed, crushed, and sieved to 40–60 mesh for evaluation. Temperature-programmed decomposition (TPDC) experiments were performed in a fixed-bed quartz reactor (inner diameter of 6 mm) and detected using a Fourier-transform infrared spectrometer (Thermo Scientific Antaris IGS analyzer) under atmospheric pressure. Before TPDC experiments, 2 wt% ABS was loaded on the surface of each catalyst. During the test, 0.20 g sample was used and the N2 flow rate was 500 mL min−1 with a temperature ramp of 5 °C min−1. A temperature-programmed procedure was adopted to record the temperature data.NH3 oxidation experiments were performed and NH3 oxidation was detected using the same instrument as in TPDC experiments. In NH3 oxidation experiments, 0.50 g catalyst was used, and the gas flow rate was 500 mL min−1, containing 500 ppm NH3, 3 vol% O2, and balanced N2. The temperature ramp was 2 °C min−1.
The stability experiment of Mo1/TiO2 was evaluated at 260 °C in the same reactor used for the TPDC experiments, and the gas exiting the reactor was analyzed with an online chemiluminescence NO–NO2–NOx analyzer (42i-HL, Thermo Fisher Scientific, Waltham, MA). During the test, 0.50 g catalyst was used and the gas flow was 500 mL min−1, containing 3 vol% O2, 500 ppm NO, 500 ppm NH3, 500 ppm SO2, 5 vol% H2O, and balanced N2.
2.4 Density functional theory (DFT) calculations
All the spin polarized DFT + U calculations were performed using the VASP program with PW91 potentials, and the effective U for Ti 3d orbitals was set to be 4.2 eV. Self-consistence of the electronic energy is reached when the energy change is smaller than 10−5 eV. The force of each free atom converged to 0.03 eV Å−1 during geometric optimization. Besides, a 2 × 2 × 1 Monkhorst–Pack k-point grid was used for Brillouin zone sampling.3 Results and discussion
Anatase TiO2 (101) was selected as the exposed surface to anchor single Mo atoms because it is often the mainly exposed plane,33 and thus a Mo1/TiO2 model was constructed by using the DFT method to scheme the decomposition of ABS at low temperatures, as shown in Fig. 1a. A single Mo atom was fixed on TiO2 (101) since MoO3 tends to atomically disperse on the surface of TiO2.32,34 This acidic Mo atom (yellow ball in Fig. 1a) pairs with the neighboring surface basic Ti atom (blue ball in Fig. 1a) to form Mo–Ti acid–base dual sites, which respectively adsorb the NH4+ and HSO4− of ABS via acid–base interactions. Following the oxidation of NH4+ on Mo sites, electrons left on the Mo–Ti dual sites transfer to the Ti sites, where the reduction of HSO4− occurs and SO2 desorbs from the surface and is released. | ||
| Fig. 1 (a) Schematic diagram of the decomposition pathway of ABS over Mo1/TiO2. A single Mo atom (yellow ball) pairs with a Ti neighbour (blue ball) to form a Mo–Ti dual site, where the NH4+ of ABS is adsorbed and oxidized on Mo sites, while HSO4− is attached to and reduced on Ti sites. The wave-type arrow represents a possible electron transfer pathway between those two sites. (b) PDOS of the Mo site (4d) and Ti site (3d) in Mo1/TiO2–R. | ||
In order to shed light on the charge transfer ability of Mo–Ti dual sites, DFT calculations were performed to evaluate the electronic state of Mo and Ti in the pristine Mo1/TiO2 catalyst and the reduced one (Mo1/TiO2–R) by removing a linked oxygen in Mo–Ti dual sites to simulate the situation after NH4+ oxidation, and the projected density of states (PDOS) results are shown in Fig. 1b and S2.† For the pristine structure, both Ti 3d and Mo 4d showed peaks at ∼−0.9 eV (Fig. S2†), indicating the strong hybridization between Mo and Ti together with the bridging O atom, implying easy electron transfer between Mo and Ti.35 After the NH4+ oxidation by the Mo site, the finite density of states at the Fermi level emerged for Ti 3d due to higher occupancy of d orbitals.36 These electrons at the Fermi level possessed a greater electron-donating capacity which may be favorable for reducing HSO4− by transferring these high-energy charges to the anti-bonding orbitals of HSO4−. Therefore, such Mo–Ti dual sites can realize electron transport from NH4+ to HSO4− and thus the decomposition of ABS at low temperatures theoretically.
To verify the feasibility of this strategy, we synthesized a Mo1/TiO2 catalyst and explored the structure of Mo species on the surface of TiO2. No peak assigned to MoO3 appears on the XRD pattern of Mo1/TiO2, revealing a highly dispersed state of Mo ions on the surface of anatase TiO2 (Fig. S3†). A similar phenomenon was observed by TEM, i.e., no MoO3 particle was detected on TiO2 (Fig. S4†). Meanwhile, the EDX mapping image of Mo1/TiO2 displayed in Fig. 2b showed highly dispersed Mo ions. Furthermore, single Mo atoms dispersed on TiO2 (101) were clearly observed in the AC-STEM image of Mo1/TiO2 (Fig. 2c). As further analyzed by the two-dimensional simulated images of the selected area shown in Fig. 2d and the calculated structural model in Fig. S1,† single Mo atoms with a MoO5 motif were established. One Mo atom is bound to two twofold coordinated O atoms of TiO2 and two introduced hydroxy which link a Mo adatom and two fivefold coordinated Ti atoms, and one Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O dangling bond points away from the surface.32 These single Mo atoms coordinating with surrounding Ti atoms facilitate the formation of abundant Mo–Ti dual sites.
O dangling bond points away from the surface.32 These single Mo atoms coordinating with surrounding Ti atoms facilitate the formation of abundant Mo–Ti dual sites.
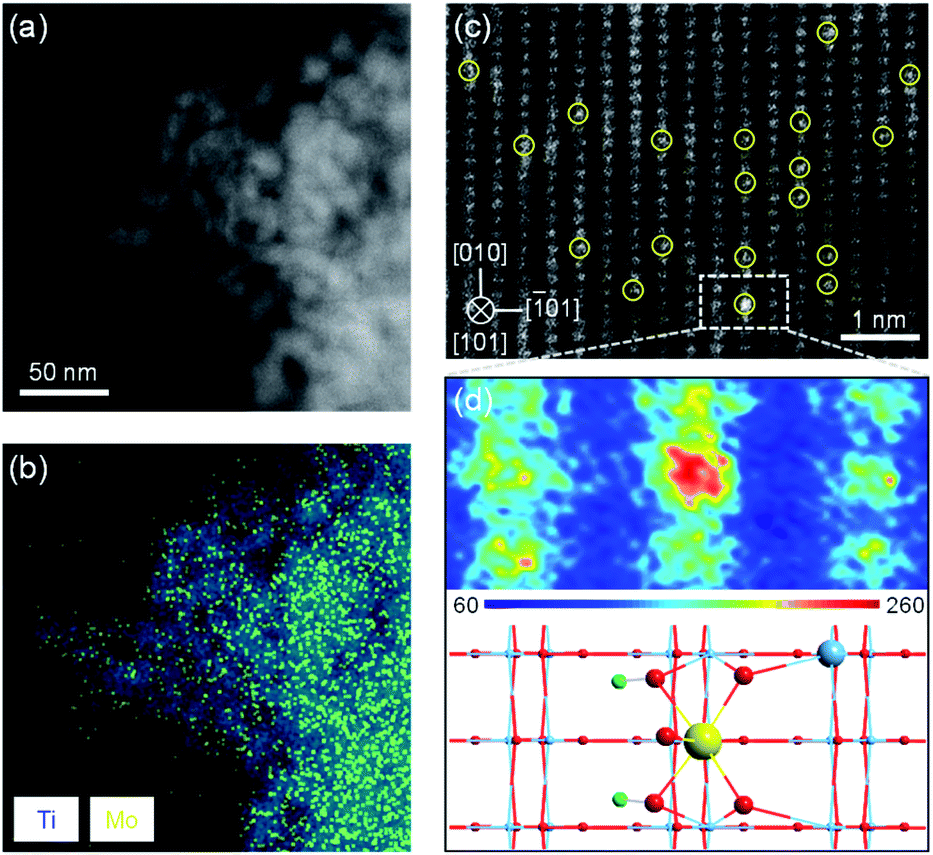 | ||
| Fig. 2 (a) AC-STEM and (b) EDX mapping images of Mo1/TiO2. (c) Atomic-resolution AC-STEM image of Mo1/TiO2. (d) Two-dimensional simulated image (above) and the corresponding structural model (below) of the selected area in (c). Light blue represents Ti atoms, red O atoms, green H atoms, and yellow Mo atoms. | ||
Furthermore, Raman spectra and NH3-DRIFT spectra were collected to determine the configuration of Mo atoms. In Fig. 3a, a small shoulder at ∼940 cm−1 appears in the Raman spectrum of Mo1/TiO2 compared with that of TiO2. As reported, surface molybdenum oxide species possess the terminal MoO Raman stretching in the range of 934–954 cm−1, while the peak shifted to higher frequency for bulk MoO3,37 indicating the existence of a dangling MoO bond. In the Raman spectrum of Mo1/TiO2, no specific signal can be observed at ∼875 cm−1 or ∼820 cm−1 assigned to the stretching mode of a Mo–O–Mo bond in surface or bulk molybdenum oxide species, respectively (Fig. S6†), evidencing the existence of single Mo atoms, in line with the results in Fig. 2. As for the NH3-DRIFT spectra shown in Fig. 3b, only a peak at ∼1600 cm−1 assigned to the adsorbed gas NH3 on Lewis sites appears in the spectrum of TiO2.29 However, when single Mo atoms were anchored on TiO2, significant signals at ∼1670 and ∼1455 cm−1 appeared, manifesting the increased NH3 coordination of Brønsted sites in the form of NH4+,29,38i.e., Mo–O–H structures were formed on the surface of Mo1/TiO2, which are totally different from that in bulk MoO3 where no relevant peak was observed. As a result, Mo single sites provide a particular Mo–O–H matrix for NH4+ adhesion.
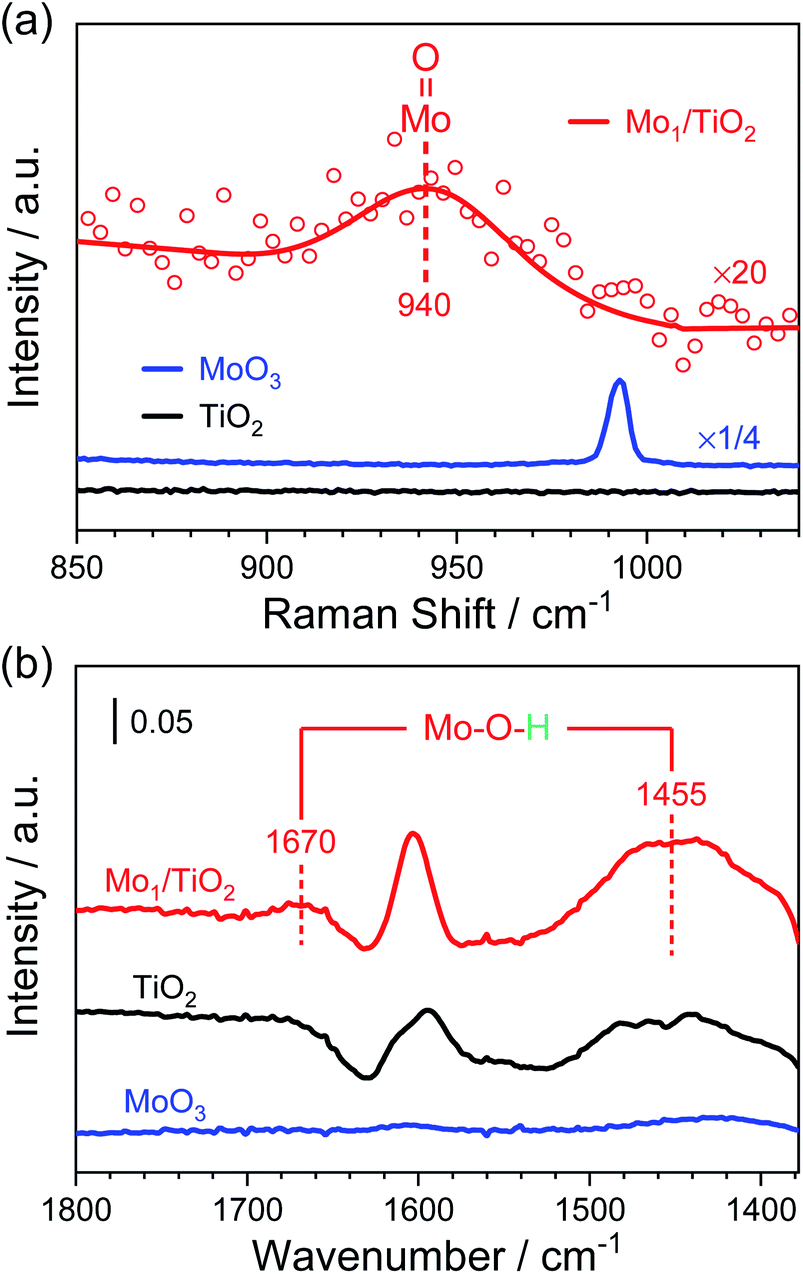 | ||
| Fig. 3 (a) Raman spectra and (b) NH3-DRIFT spectra of Mo1/TiO2, MoO3 and TiO2 catalysts. | ||
For the purpose of estimating the behaviour of adsorbed NH4+ on the Mo–Ti dual sites, NH3 oxidation experiments were implemented on Mo1/TiO2, MoO3 and TiO2. As shown in Fig. 4a, the onset of the NH3-oxidation curves of Mo1/TiO2 located at ∼200 °C, reflecting a strong ability of Mo1/TiO2 towards NH3 oxidation. Similarly, MoO3 started to catalyse NH3 oxidation at ∼200 °C, the same as Mo1/TiO2, while the onset temperature shifted to ∼300 °C for TiO2, showing that the Mo atoms of the dual sites are responsible for NH3 oxidation. Fig. 4b shows the Arrhenius plots for the rate (R) of NH3 oxidation together with activation energy (Ea) over Mo1/TiO2 and MoO3. The Ea of Mo1/TiO2 is 50 kJ mol−1, much lower than that of MoO3 (70 kJ mol−1), suggesting a strong interaction between Mo single atoms and neighboring Ti atoms which activates the surface lattice oxygen of Mo. Thus, combined with the results obtained from NH3-DRIFT spectra, the Mo atoms of Mo–Ti dual sites should be responsible for the adsorption and oxidation of NH4+ in ABS.
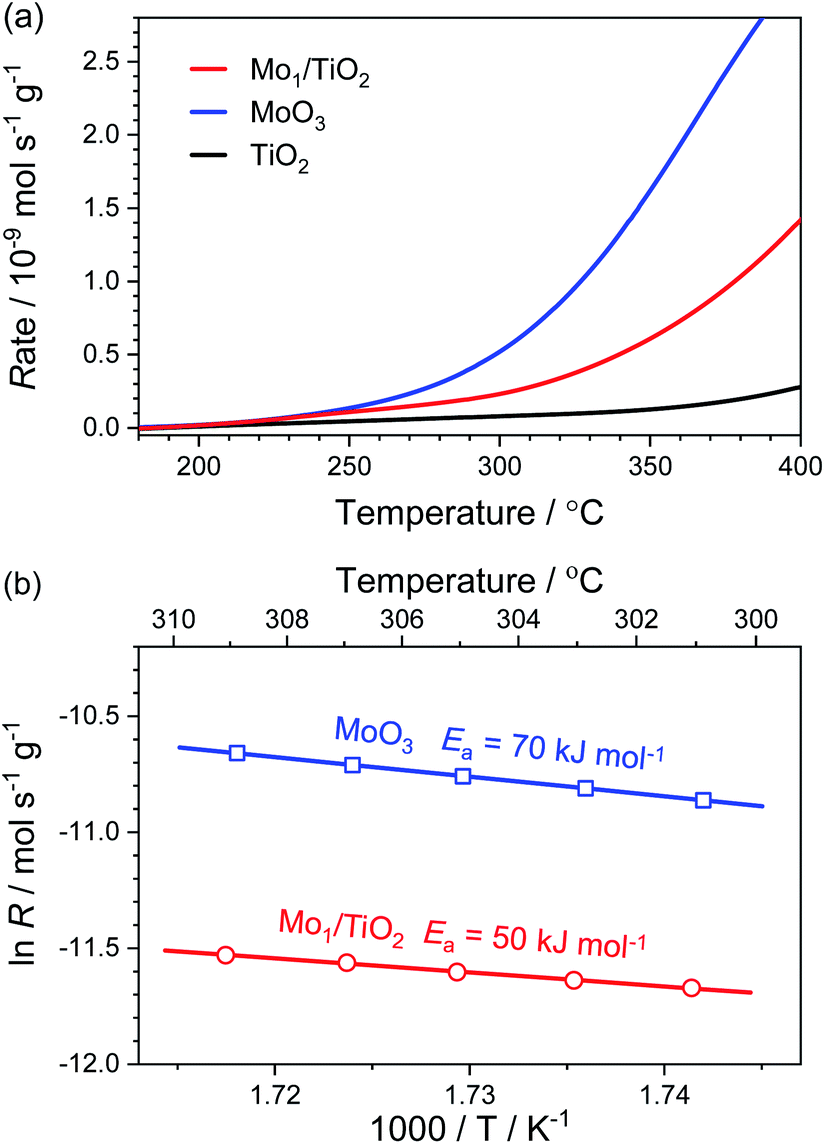 | ||
| Fig. 4 (a) Reaction rates of the NH3 oxidation as a function of temperature over Mo1/TiO2, MoO3 and TiO2. (b) Arrhenius plots for reaction rates with Ea over Mo1/TiO2 and MoO3. | ||
Based on the dual-site adsorption model of ABS,25 it is likely that HSO4− adsorbs on the neighboring Ti sites in consideration of NH4+ adsorption on Mo sites. FT-IR spectra were used to detect surface structures of catalysts after ABS deposition (Fig. 5a and S7†). For pure ABS, peaks at ∼1242, 1172, and 1072 cm−1, assigned to the bending vibrations of SO–H (δSO–H), symmetrical stretching vibrations of SO (vsSO), and asymmetrical stretching vibrations of S–O (vasS–O) in HSO4− emerged, respectively.28 After ABS was loaded on the surface of MoO3, only slight changes occur in the position of the peaks, demonstrating weak interactions between MoO3 and HSO4−. In contrast, the FT-IR spectra of ABS-deposited TiO2 and Mo1/TiO2 are both quite different from that of pure ABS. Both spectra have peaks at ∼1222 and 1134 cm−1 corresponding to the asymmetrical stretching vibrations of SO (vasSO) and vsSO, respectively, and a peak at ∼1042 cm−1 (vasS–O).28 The similar spectral features between the spectra of ABS-loaded TiO2 and Mo1/TiO2 illustrate that HSO4− is adsorbed on surface exposed Ti sites. Moreover, the disappearance of δSO–H and the appearance of vasSO in the spectra of Mo1/TiO2 compared to pure ABS indicates the transformation of HSO4− into SO42−,23via a strong interplay between TiO2 and HSO4−. The interaction resulted in obvious redshifts of vasS–O, indicating the increase of the S–O bond length and hence the reduction of its bonding energy,39 which is favorable for the cleavage of two S–O bonds of SO4− so as to release SO2. Hence, the co-adsorption of NH4+ and HSO4− of ABS on acid–base Mo–Ti dual sites was established, where Mo sites and Ti sites are responsible for the oxidation of NH4+ and the reduction of HSO4−, respectively.
To confirm whether such Mo–Ti dual sites realize low-temperature decomposition of ABS, the dynamic evolution of ABS on Mo1/TiO2 through time-resolved DRIFT spectra was observed to directly depict the decomposition process on the Mo–Ti dual sites (Fig. 5b). The strong peak at ∼1363 cm−1 could be assigned to SO42−,40 which gradually faded (here is the inversed peak becoming stronger and stronger with time), indicative of the decomposition of ABS. In contrast, the peak between ∼1500 and 1380 cm−1 characterized to be NH4+ adsorbed on Brønsted sites gradually increased.41 According to the decomposition equation of ABS: NH4HSO4 → 1/3 NH3 + 1/3 N2 + SO2 + 2 H2O,21 one third of the NH4+ species are eventually released in the form of NH3, and the formed NH3 will be adsorbed on the regenerated Brønsted sites, explaining the increase of signal between ∼1500 and 1380 cm−1 in Fig. 5b. Consequently, it is evident that the Mo–Ti dual sites indeed facilitate the decomposition of ABS at low temperatures.
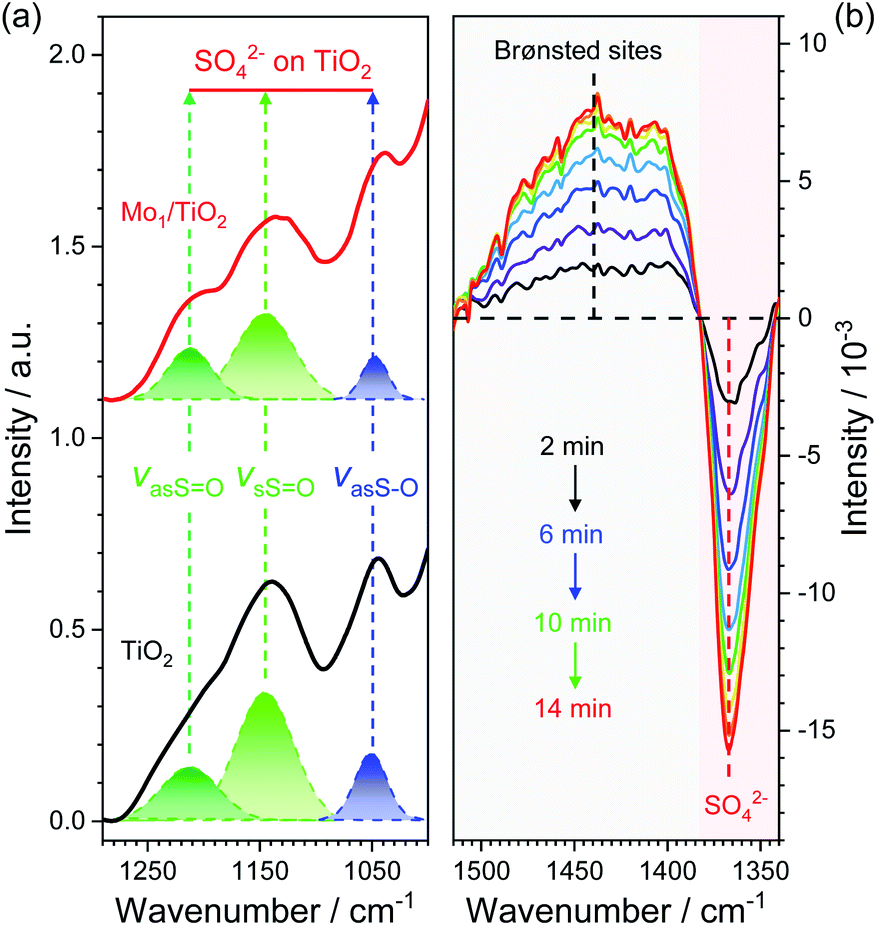 | ||
| Fig. 5 (a) FT-IR spectra of ABS-deposited TiO2 and Mo1/TiO2. (b) In situ DRIFT spectra of the transient reaction on the 0.5% ABS-deposited Mo1/TiO2 catalyst at 260 °C. The initial spectrum was deducted as the background. | ||
According to the above decomposition equation of ABS, we further determined the onset temperature for the decomposition of ABS by conducting TPDC experiments on catalysts pre-reduced in the NH3/N2 atmosphere (Fig. S8–S10†). The results from TPDC are shown in Fig. 6a. The onset temperature for pure ABS decomposition was measured to be ∼350 °C. Note that for Mo1/TiO2, the onset temperature greatly decreased down to a temperature as low as ∼225 °C, while the corresponding onset temperatures for TiO2 and MoO3 are ∼350 and ∼275 °C, respectively. These results indicate that the catalytic sites for the low-temperature decomposition of ABS on Mo1/TiO2 are the Mo–Ti dual sites rather than surface standalone Mo or Ti sites. For reference, we deposited ABS with the same loading on a conventional V2O5–WO3/TiO2 (VWTi) catalyst, and found that the onset temperature was ∼300 °C. This result is consistent with the fact that high decomposition temperature determined that conventional V2O5–WO3/TiO2 catalysts used at thermal plants often effectively operate only at temperatures higher than 300 °C. Otherwise, these catalysts will suffer deactivation.4 In contrast, Mo1/TiO2 can decompose ABS at temperatures as low as 225 °C, far lower than the reported lowest dew point of ABS.15 Because both Mo and Ti of Mo1/TiO2 are also important components of commercial V2O5–MoO3/TiO2 SCR catalysts, Mo1/TiO2 provides a crucial base for designing strong ABS-resistant SCR catalysts typically applied in many industrial boilers, as further confirmed by the high stability of the Mo1/TiO2 catalyst under simulation stack gas conditions of industrial boilers (Fig. S11†). Meanwhile, based on the results from TPDC experiments, the yield rates of SO2 were also calculated (Table S1†), and Mo1/TiO2 exhibited the highest yield rate, revealing a strong ability towards the decomposition of ABS.
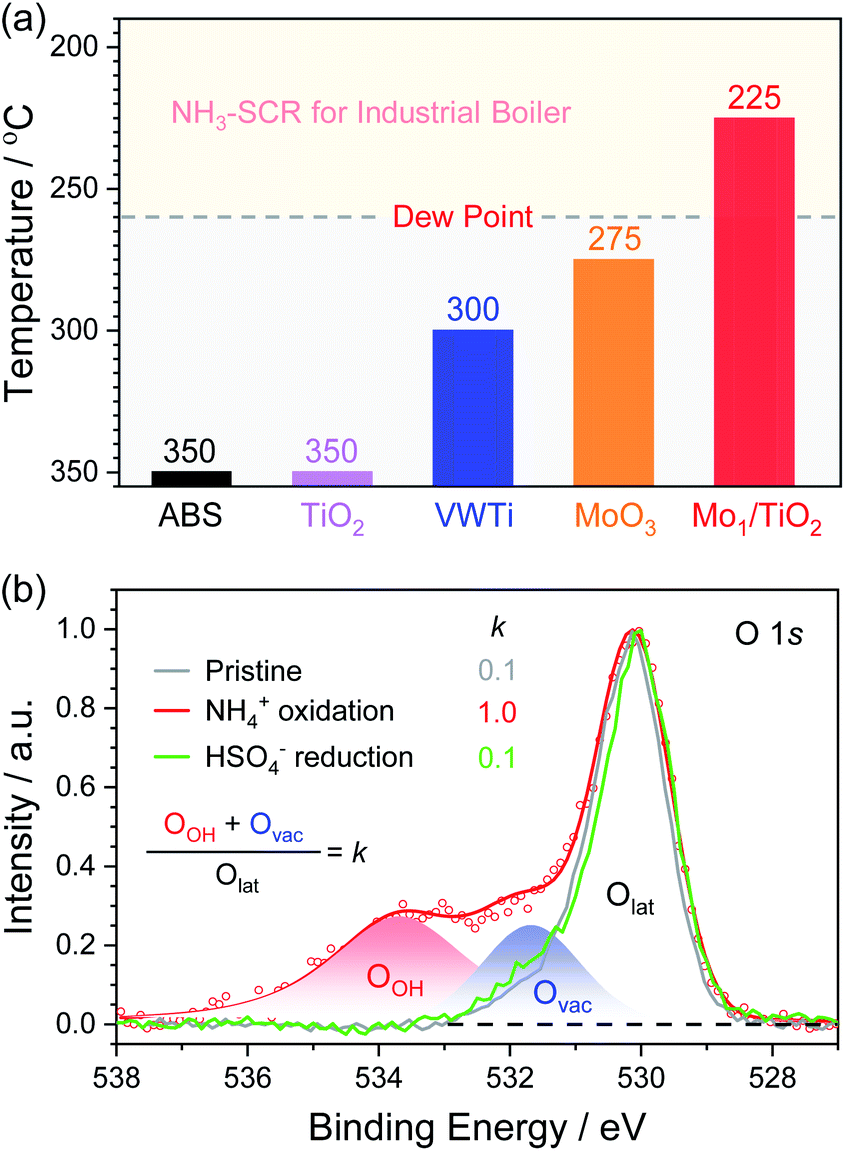 | ||
| Fig. 6 (a) Onset temperatures of the SO2 release over a series of ABS-deposited catalysts. (b) O 1s XPS of Mo1/TiO2 (pristine and after NH4+ oxidation and HSO4− reduction). | ||
The release of SO2 during TPDC reflects the reduction of HSO4− by the Mo–Ti dual sites. Hence, the decomposition process of ABS can be divided into three stages: NH4+ oxidation, HSO4− reduction, and re-oxidation of the catalyst by O2. XPS was adopted to probe the evolution of electron states of the pristine Mo1/TiO2 catalyst and those after each stage (Fig. 6b and S12†). After NH4+ oxidation, the ratio of Mo5+ increased from 41% to 64%, clearly confirming the electron transfer from NH4+ to single Mo atoms. Simultaneously, the ratio of oxygen vacancies (Ovac) to surface lattice oxygen (Olat) increased dramatically. Note that a new high-energy peak at 533.6 eV in the XPS of Fig. 6b related to oxygen in O–H (OOH) appeared after the NH4+ oxidation, consistent with the increase of the Brønsted sites in Fig. 5b. As illustrated in Fig. 1b, after the formation of oxygen vacancies via the oxidation of NH4+ on the Mo–Ti dual sites, electronic state densities of both Ti and Mo ions emerged at the Fermi level in Ti 3d and Mo 4d orbitals of the dual sites, which readily transfer to HSO4− adsorbed on the Ti site. Moreover, the protons of the Brønsted sites also play an important role in transferring charge between Mo and Ti ions of the dual sites,42 which is beneficial to the reduction of HSO4− and subsequent release of the produced SO2. Again, due to the desorption of SO2 concomitant with the H2O release resulting from the reduction of HSO4−, the remaining oxygen atoms filled the oxygen vacancies, accounting for the decrease of Ovac (Fig. 4b). Accompanied by the re-oxidation process, all the electronic states were eventually restored to the original states (Fig. S12†), completing the decomposition of ABS on the Mo1/TiO2 catalyst.
The above discussion demonstrates that single Mo atoms with neighboring Ti atoms function as Mo–Ti dual sites which provide Mo sites for the adsorption and oxidation of NH4+, and Ti sites for the adsorption and reduction of HSO4−. In order to further verify the effect of Mo–Ti dual sites in the decomposition process of ABS, we adjusted the surface structure of the catalyst by tuning the Mo loading or wrapping the exposed Mo single atoms with TiO2. The decomposition temperature remains basically stable (∼225 °C) at low Mo loadings, but increases to another constant (∼260 °C) at high loadings which is closer to that of bulk MoO3 (Fig. S13 and S14†). Meanwhile, further coverage of TiO2 also resulted in the delay of the temperature for the SO2 release (Fig. S15†). Therefore, the Mo–Ti acid–base dual sites, namely the pair of single Mo atom and neighboring Ti atom, are key to the decomposition of ABS over Mo1/TiO2 at low temperatures.
4 Conclusions
In summary, we designed a Mo1/TiO2 catalyst to solve the problem of ABS poisoning on low-temperature SCR catalysts. The Mo1/TiO2 catalyst successfully achieved the decomposition of ABS at ∼225 °C, which is superior to that of other catalysts reported previously. With various characterization techniques such as XRD, TEM, Raman and AC-STEM, single Mo atoms on TiO2 were defined, ensuring the formation of Mo–Ti acid–base dual sites by single Mo atoms pairing with the neighboring surface Ti atoms, which respectively adsorb the NH4+ and HSO4− of ABS, as confirmed by DRIFT and FT-IR spectra. After the oxidation of NH4+ by the surface lattice oxygen on the Mo sites, electrons left behind on the dual sites are localized around the Fermi level, which allows them to transfer to the adsorbed HSO4− on the Ti sites, releasing SO2 at low temperatures. Therefore, this work proposed a strategy for designing ABS-resistant catalysts for effectively controlling NOx emission particularly from industrial boilers.Author contributions
X. T., Y. C. and X. L. conceived and directed the research. J. C. conducted the experiments of catalyst preparation and evaluation, and performed the experimental characterization. X. F. conducted the DFT calculations. J. C., Z. R., W. Q., X. H., L. C. and Z. M. discussed the results and wrote the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21976037, 21777030) and the National Engineering Research Center for Synergetic Control of Air Pollutants and Greenhouse Gases (NEL-KF-201903).Notes and references
- J. H. Seinfeld, Science, 1989, 243, 745–752 CrossRef CAS PubMed.
- S. C. Anenberg, J. Miller, R. M. Injares, L. Du, D. K. Henze, F. Lacey, C. S. Malley, L. Emberson, V. Franco, Z. Klimont and C. Heyes, Nature, 2017, 545, 467–471 CrossRef CAS PubMed.
- S. Reis, P. Grennfelt, Z. Klimont, M. Amann, H. ApSimon, J. P. Hettelingh, M. Holland, A. C. LeGall, R. Maas, M. Posch, T. Spranger, M. A. Sutton and M. Williams, Science, 2012, 338, 1153–1154 CrossRef CAS.
- L. P. Han, S. X. Cai, M. Gao, J. Hasegawa, P. L. Wang, J. P. Zhang, L. Y. Shi and D. S. Zhang, Chem. Rev., 2019, 119, 10916–10976 CrossRef CAS.
- M. H. Zhu, J. K. Lai, U. Tumuluri, M. E. Ford, Z. L. Wu and I. E. Wachs, ACS Catal., 2017, 7, 8358–8361 CrossRef CAS.
- N. Y. Topsøe, Science, 1994, 265, 1217–1219 CrossRef.
- S. Matsuda, T. Kamo, A. Kato, F. Nakajima, T. Kumura and H. Kuroda, Ind. Eng. Chem. Prod. Res. Dev., 1982, 21, 48–52 CrossRef CAS.
- P. Forzatti and L. Lietti, Heterog. Chem. Rev., 1996, 3, 33–51 CrossRef CAS.
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS.
- C. Y. Zhou, L. N. Zhang, Y. Deng and S. C. Ma, Environ. Prog. Sustainable Energy, 2016, 35, 1664–1672 CrossRef CAS.
- H. Zhao, S. Bennici, J. Cai, J. Shen and A. Auroux, Catal. Today, 2010, 152, 70–77 CrossRef CAS.
- P. Forzatti, Catal. Today, 2000, 62, 51–65 CrossRef CAS.
- L. Y. Song, J. D. Chao, Y. J. Fang, H. He, J. Li, W. G. Qiu and G. Z. Zhang, Chem. Eng. J., 2016, 303, 275–281 CrossRef CAS.
- Y. Z. Xi, N. A. Ottinger and Z. G. Liu, Appl. Catal., B, 2014, 160, 1–9 CrossRef.
- I. Song, H. Lee, S. W. Jeon, I. A. M. Ibrahim, J. Kim, Y. Byun, D. J. Koh, J. W. Han and D. H. Kim, Nat. Commun., 2021, 12, 901 CrossRef CAS.
- R. Y. Qu, D. Ye, C. H. Zheng, X. Gao, Z. Y. Luo, M. J. Ni and K. F. Cen, RSC Adv., 2016, 6, 102436–102443 RSC.
- D. Ye, R. Y. Qu, H. Song, C. H. Zheng, X. Gao, Z. Y. Luo, M. J. Ni and K. F. Cen, RSC Adv., 2016, 6, 55584–55592 RSC.
- Z. Y. Fan, J. W. Shi, C. H. Niu, B. R. Wang, C. He and Y. H. Cheng, Chem. Eng. J., 2020, 398, 125572 CrossRef CAS.
- D. Ye, R. Y. Qu, C. H. Zheng, K. F. Cen and X. Gao, Appl. Catal. A, 2018, 549, 310–319 CrossRef CAS.
- J. W. DePalma, D. J. Doren and M. V. Johnston, J. Phys. Chem. A, 2014, 118, 5464–5473 CrossRef CAS PubMed.
- Y. Liske, S. Kapila, V. Flanigan, P. Nam and S. Lorbert, J. Hazard. Subst. Res., 1999, 2, 1–17 Search PubMed.
- Y. X. Chen, C. Li, J. X. Chen and X. F. Tang, Environ. Sci. Technol., 2018, 52, 11796–11802 CAS.
- J. Yu, E. Zhang, L. Wang, Z. Song, F. Kong, Y. Ma, H. Zhao and L. Sun, Energy Fuels, 2020, 34, 2107–2116 CrossRef CAS.
- J. R. Thøgersen, T. Slabiak and N. White, Ammonium bisulphate inhibition of SCR catalysts, Haldor Topsøe Inc, Frederikssund, 2007 Search PubMed.
- H. H. Phil, M. P. Reddy, P. A. Kumar, L. K. Ju and J. S. Hyo, Appl. Catal., B, 2008, 78, 301–308 CrossRef CAS.
- D. Liu, C. Wang, Y. Yu, B.-H. Zhao, W. Wang, Y. Du and B. Zhang, Chem, 2019, 5, 376–389 CAS.
- N. C. Jeong, J. S. Lee, E. L. Tae, Y. J. Lee and K. B. Yoon, Angew. Chem., Int. Ed., 2008, 47, 10128–10132 CrossRef CAS.
- D. Ye, R. Y. Qu, H. Song, X. Gao, Z. Y. Luo, M. J. Ni and K. F. Cen, Chem. Eng. J., 2016, 283, 846–854 CrossRef CAS.
- Z. W. Huang, Y. Y. Du, J. Zhang, X. M. Wu, H. Z. Shen and G. H. Jing, Environ. Sci. Technol., 2019, 53, 5309–5318 CrossRef CAS.
- F. Feng, W. Y. Yang, S. Gao, C. X. Sun and Q. Li, ACS Sustainable Chem. Eng., 2018, 6, 6166–6174 CrossRef CAS.
- S. Wendt, P. T. Sprunger, E. Lira, G. K. H. Madsen, Z. S. Li, J. Ø. Hansen, J. Matthiesen, A. Blekinge-Rasmussen, E. Lægsgaard, B. Hammer and F. Besenbacher, Science, 2008, 320, 1755–1759 CrossRef CAS PubMed.
- N. Doudin, G. Collinge, P. K. Gurunathan, M. S. Lee, V. A. Glezakou, R. Rousseau and Z. Dohnalek, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2017703118 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155409 CrossRef.
- S. F. Guo, Z. W. Huang, L. P. Wang, X. M. Wu, H. Z. Shen and G. H. Jing, J. Hazard. Mater., 2021, 418, 126289 CrossRef CAS.
- L. Dall'Acqua, I. Nova, L. Lietti, G. Ramis, G. Busca and E. Giamello, Phys. Chem. Chem. Phys., 2000, 2, 4991–4998 RSC.
- M. T. Greiner, M. G. Helander, W. M. Tang, Z. B. Wang, J. Qiu and Z. H. Lu, Nat. Mater., 2011, 11, 76–81 CrossRef PubMed.
- H. C. Hu, I. E. Wachs and S. R. Bare, J. Phys. Chem., 1995, 99, 10897–10910 CrossRef CAS.
- F. Giraud, C. Geantet, N. Guilhaume, S. Loridant, S. Gros, L. Porcheron, M. Kanniche and D. Bianchi, Catal. Today, 2021, 373, 69–79 CrossRef CAS.
- A. Goypiron, J. Devillepin and A. Novak, J. Raman Spectrosc., 1980, 9, 297–303 CrossRef CAS.
- G. Z. He, Z. H. Lian, Y. B. Yu, Y. Yang, K. Liu, X. Y. Shi, Z. D. Yan, W. P. Shan and H. He, Sci. Adv., 2018, 4, eaau4637 CrossRef CAS PubMed.
- I. Song, H. Lee, S. W. Jeon, T. Kim and D. H. Kim, Chem. Commun., 2020, 56, 15450–15453 RSC.
- E. C. Gentry and R. R. Knowles, Acc. Chem. Res., 2016, 49, 1546–1556 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08269h |
| This journal is © The Royal Society of Chemistry 2022 |