
Nitrogen-coordinated single-atom catalysts with manganese and cobalt sites for acidic oxygen reduction†
Guojie
Chao
a,
Yizhe
Zhang
a,
Longsheng
Zhang
*a,
Wei
Zong
b,
Nan
Zhang
a,
Tiantian
Xue
b,
Wei
Fan
 b,
Tianxi
Liu
a and
Yi
Xie
*c
b,
Tianxi
Liu
a and
Yi
Xie
*c
aKey Laboratory of Synthetic and Biological Colloids, Ministry of Education, School of Chemical and Material Engineering, International Joint Research Laboratory for Nano Energy Composites, Jiangnan University, Wuxi, China. E-mail: zhangls@jiangnan.edu.cn
bState Key Laboratory for Modification of Chemical Fibers and Polymer Materials College of Materials Science and Engineering, Donghua University, Shanghai, China
cHefei National Laboratory for Physical Sciences at Microscale, University of Science and Technology of China, Hefei, China. E-mail: yxie@ustc.edu.cn
First published on 23rd November 2021
Abstract
Developing a low-cost, highly active and durable catalyst for acidic oxygen reduction reaction (ORR) is of significant importance for proton-exchange electrolyte membrane fuel cell. In this work, we report an efficient catalyst based on Mn, Co and N co-doped carbon (MnCo–N–C) for acidic ORR. Electron microscopy and X-ray absorption spectroscopy indicate that atomic CoNx and MnNx sites are dispersed in the carbon matrices of MnCo–N–C. Raman spectroscopy verifies that the doping of Mn into carbon matrices can enable a higher graphitization degree, which can improve the corrosion resistance and catalytic durability of the MnCo–N–C catalyst towards ORR in challenging acidic media. As a result, the MnCo–N–C catalyst exhibits significantly improved ORR durability with a higher current retention of 81% after 50 h of test compared with that (52%) of the Co–N–C catalyst. Moreover, the half-wave potential of the MnCo–N–C catalyst increases by 100 mV compared with that of the Co–N–C catalyst.
Introduction
Challenges from the growing global energy demand provide a strong driving force for the rapid advancement of efficient conversion of renewable energy.1 Among others, proton-exchange electrolyte membrane fuel cells (PEMFCs) can convert renewable and carbon-free hydrogen energy into electricity, and have shown potential applications in portable electronic devices, electric vehicles and stationary power sources.2 One of the biggest issues in achieving the large-scale application of PEMFCs is to develop alternative catalysts to platinum group metal (PGM) catalysts for the sluggish oxygen reduction reaction (ORR) in acidic electrolytes.3–5Great efforts have been devoted to developing cost-effective, highly active and durable PGM-free catalysts for acidic ORR. Recently, a class of transition metals (Fe, Co, Mn, Cu, etc.) and N co-doped carbon (denoted as M–N–C) catalysts have received extensive attention due to their maximum atom-utilization efficiencies and encouraging performance towards acidic ORR.6–10 To date, compared to other M–N–C catalysts, the Fe–N–C catalyst has shown the highest catalytic activity, which is close to those of PGM catalysts towards acidic ORR.11–13 However, inevitable Fenton reactions between the generated H2O2 and dissolved Fe ions during the ORR process will produce hydroxyl and hydroperoxyl radical species, which would lead to fast degradation of ionomers and membranes in PEMFCs.14,15 Thus, the exploration of Fe-free alternative M–N–C catalysts that can barely induce Fenton reactions is desired for achieving durable and inexpensive PEMFCs.
Previous studies have illustrated that the catalytic activity of M–N–C catalysts follows the order of Fe > Co > Mn > Cu > Ni towards acidic ORR.16,17 Compared with Fe ions, Co ions are much less active in initiating Fenton reactions,18 making Co–N–C catalysts good candidates for Fe-free and PGM-free catalysts for acidic ORR. However, Co–N–C catalysts suffer from an undesired two-electron pathway during the acidic ORR process and generate substantial amounts of H2O2, which would induce H2O2 oxidative attacks on the catalysts and thereby lead to serious dissolution of active metal sites.19,20 Besides, carbon corrosion of catalysts in challenging acidic media would also promote the dissolution of active metal sites followed by catalyst degradation.21 Concomitantly, corroded carbon matrices with active oxygenated groups would induce a two-electron pathway and generate undesired H2O2 during the ORR process, which would accelerate the catalyst degradation.22 Recent studies have shown that introducing Mn into the carbon matrices of M–N–C catalysts can increase the degree of graphitization of carbon matrices, which is of great significance to enhance the corrosion resistance of carbon matrices.23 Meanwhile, it was reported that Mn ions are hardly involved in Fenton reactions due to the weak reaction activity between Mn ions and H2O2 species.24 We posit that doping Mn in carbon matrices of Co–N–C may catalyze the graphitic structures and improve the ORR stability of Co–N–C, so as to obtain ORR catalysts with both high activity and durability in acidic electrolytes.
Here, we report a Mn, Co and N co-doped carbon (MnCo–N–C) catalyst for acidic ORR, which can be synthesized via a facile method based on zeolitic imidazolate frameworks. Owing to their high specific surface area and abundant nitrogen sources, zeolitic imidazolate frameworks were thereby chosen as precursors for preparing the MnCo–N–C catalyst in this work. Our extensive characterization reveals that the configurations of atomic CoNx and MnNx sites are well embedded in the carbon matrices of the MnCo–N–C catalyst. Raman spectroscopy indicates that the doping of Mn into carbon matrices can induce a higher graphitization degree, thus improving the corrosion resistance of MnCo–N–C compared with that of Co–N–C. As a consequence, the MnCo–N–C catalyst exhibits greatly enhanced ORR durability with a current density retention of 81% over 50 h of continuous test, much higher than that (52%) of Co–N–C catalyst. Moreover, the MnCo–N–C catalyst shows a higher half-wave potential (E1/2) of 0.80 V (vs. the reversible hydrogen electrode, RHE), compared to that (0.70 V vs. RHE) of Co–N–C catalyst. Furthermore, a small negative shift of 25 mV in E1/2is achieved for MnCo–N–C catalyst after 5000 cycles of accelerated durability tests, in contrast to that (90 mV) of Co–N–C catalyst.
Results and discussion
As illustrated in Fig. S1,† the MnCo–N–C sample was obtained by carbonizing the Mn and Co co-doped zeolitic imidazolate framework (see synthesis details in the ESI†). As shown in the scanning electron microscopy (SEM) image (Fig. 1a), the MnCo–N–C sample shows a rhombododecahedral morphology with a size of 300–400 nm. Transmission electron microscopy (TEM) and energy-dispersive elemental mapping images reveal that Co, Mn, N and C elements are homogeneously distributed within the MnCo–N–C sample (Fig. 1b and c). High angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was further performed to obtain evidence of the Co and Mn distribution with atomic resolution (Fig. 1d). Small bright spots can be observed in the carbon matrices, indicating the configuration of single-metal sites in the MnCo–N–C sample. Most of the Mn and Co atoms exist separately during the formation of Co–N–C and Mn–N–C structures, and a few Mn and Co atoms exist as diatomic pairs. The contents of Mn and Co in the MnCo–N–C sample are 0.6 and 1.0 wt%, respectively (Table S1†). As shown in Fig. 2a, the X-ray diffraction (XRD) patterns of the MnCo–N–C sample and controls demonstrate that only two diffraction peaks of graphitic carbon are observed and no diffraction peaks of metals or oxides are found. | ||
| Fig. 1 Morphology characterization of MnCo–N–C sample. (a) SEM image. (b and c) TEM and corresponding element mapping images, respectively. (d) HAADF-STEM image. | ||
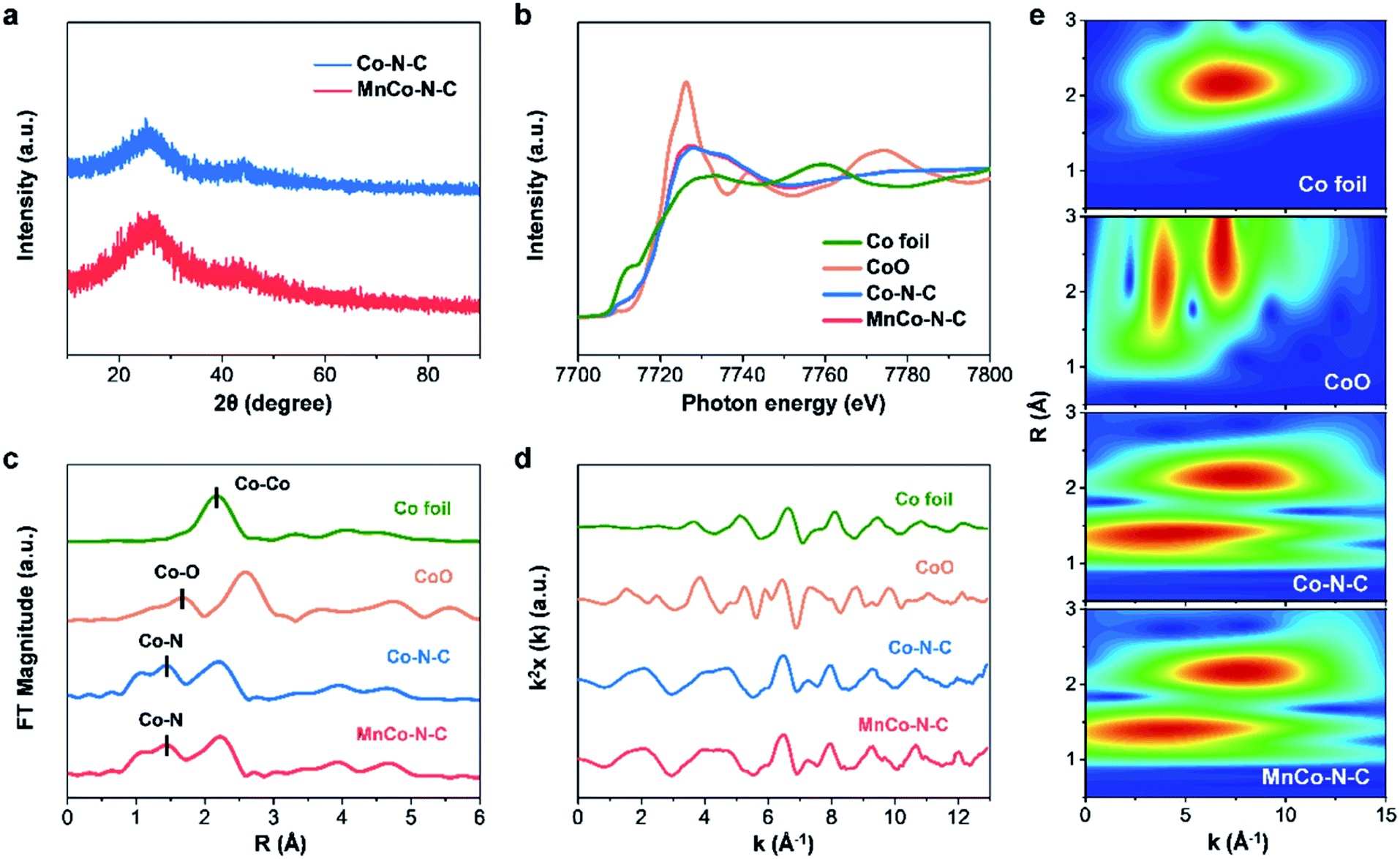 | ||
| Fig. 2 Structural characterization of MnCo–N–C and controls. (a) XRD patterns. (b and c) Co K-edge XANES and EXAFS spectra, respectively. (d and e) k2-weighted XAFS χ(k) and wavelet transform, respectively. | ||
To further get the coordination information of Co for MnCo–N–C and controls, we carried out X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) measurements. The Co K-edge XANES spectra show that the adsorption threshold positions of the two Co-doped M–N–C samples are located between Co foil and CoO (Fig. 2b), which illustrate that the valences of Co species in these two samples are situated between 0 and +2. The Fourier transform k2-weighted EXAFS spectra of Co–N–C and MnCo–N–C (Fig. 2c) show similar peaks at 1.43 Å, which are much shorter than the Co–O peak of the CoO reference at 1.67 Å and can be assigned to the Co–N contributions. In addition, the electron energy loss spectrum (Fig. S2†) illustrates that both N and Co elements exist in the MnCo–N–C sample, indicating that Co is coordinated with N at the atomic scale.6,26 Besides, the peaks observed at 2.22 Å for the Co–N–C and MnCo–N–C samples indicate the existence of Co clusters or nanoparticles. Since Co clusters or nanoparticles are inactive for the ORR,25–27 this study mainly focuses on nitrogen-coordinated single-atom Co sites (Co–N–C) with catalytic activity towards the ORR. The Fourier transform k2-weighted XAFS χ versus the reciprocal wave vector (Fig. 2d) indicates good qualities of the EXAFS data. Wavelet transform was further used to investigate the EXAFS oscillations of the samples. The wavelet transform analysis of both Co–N–C and MnCo–N–C catalysts shows intensity maxima at approximately 1.4 Å (k = 3 Å−1, R = 1.4 Å), which can be attributed to Co–N contributions (Fig. 2e), verifying the formation of CoNx sites.
X-ray photoelectron spectroscopy (XPS) measurements were further performed to reveal the chemical composition and states of MnCo–N–C and controls. As shown in the high-resolution N 1 s spectra of Co–N–C and MnCo–N–C (Fig. 3a and b), the fitted spectra have four types of N species, attributed to graphitic N, pyrrolic N, metal-N and pyridinic N, respectively. The metal-N species of MnCo–N–C can be associated with the atomically dispersed CoNx and MnNx moieties. Table S2† shows the contents of these four types of N species in samples, obtained from the fitting of XPS results. Note that the content of metal-N species in the MnCo–N–C catalyst (32.0%) is much higher than that of the Co–N–C catalyst (19.6%), which can be ascribed to MnNx and CoNx moieties in the MnCo–N–C catalyst. As shown in Fig. 3c and d, the high-resolution Co 2p spectra of MnCo–N–C and controls indicate the existence of CoNx and metallic Co species, in agreement with the EXAFS analysis. The high-resolution Mn 2p spectrum of the MnCo–N–C catalyst reveals that the Mn 2p1/2 peak (654.0 eV) and Mn 2p3/2 peak (642.0 eV) can be ascribed to the MnNx moieties (Fig. 3e), in accordance with those of previously-reported Mn–N–C catalysts.6,28 Compared with the Mn–N–C catalyst, the XPS peaks of Mn 2p of the MnCo–N–C catalyst show a positive shift of ∼0.9 eV (Fig. S3†), which may be ascribed to the interaction between Mn atoms and Co atoms. Raman measurements were conducted to further investigate the influence of Mn doping on the graphitization degree of carbon matrices. As shown in Fig. 3f and S4,† the Raman spectra of all the samples exhibit two peaks at 1354 and 1591 cm−1, which are respectively indexed to disordered sp3 carbon (D band) and graphitic sp2 carbon (G band). The D and G band intensity ratio of MnCo–N–C is lower than that of Co–N–C, verifying the higher graphitization degree of carbon matrices with Mn dopants.29,30 As reported, the doping of Mn into carbon matrices can induce the formation of ordered graphitic carbon with fewer defects and thus result in a higher graphitization degree.23 N2 adsorption–desorption measurements (Fig. S5†) show that both Co–N–C and MnCo–N–C samples show mesopores and the specific surface areas of Co–N–C and MnCo–N–C samples are 506 and 420 m2 g−1, respectively.
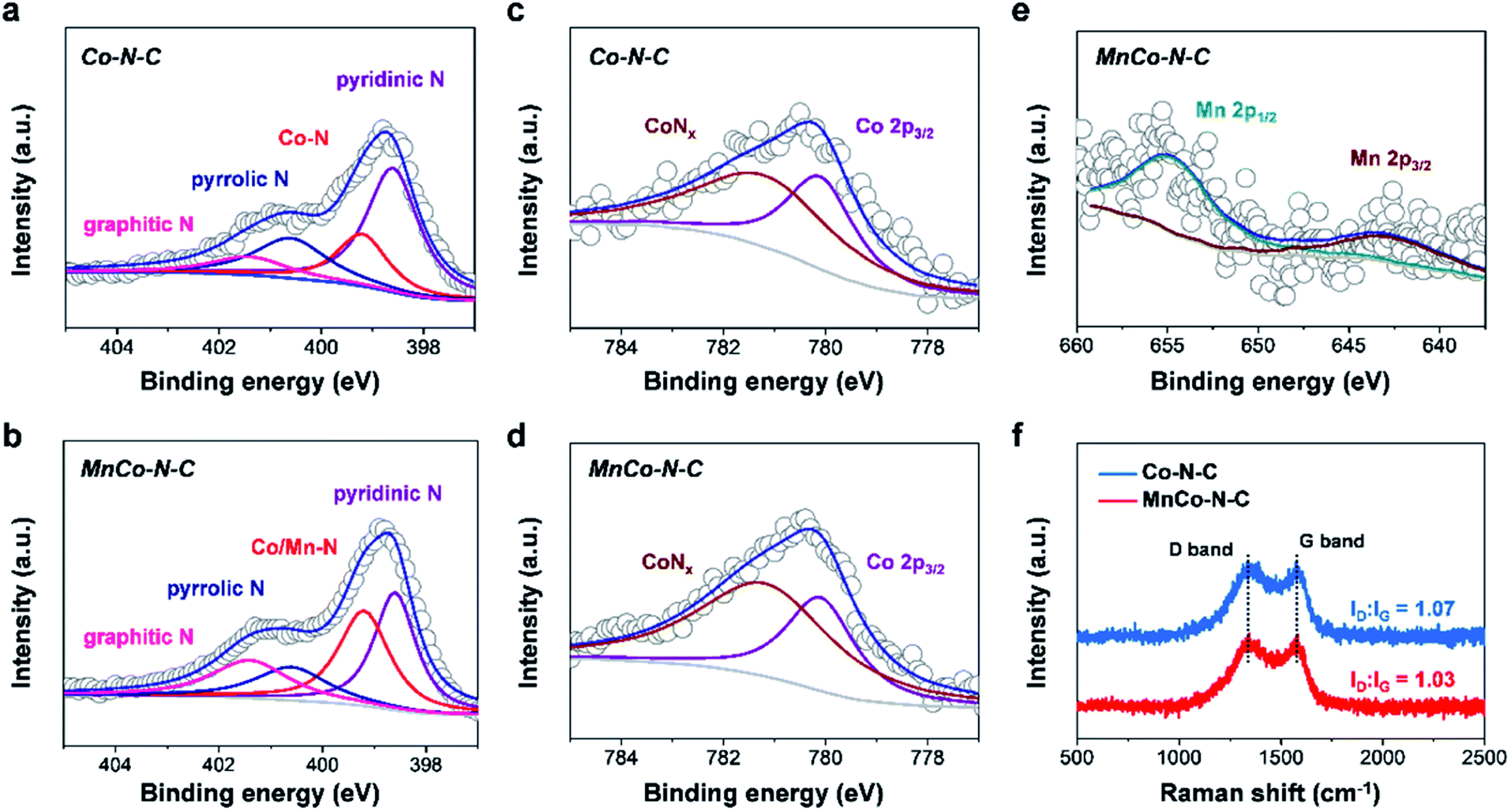 | ||
| Fig. 3 Structural characterization of MnCo–N–C and controls. High-resolution (a and b) N 1 s spectra, (c and d) Co 2p spectra and (e) Mn 2p spectrum, respectively. (f) Raman spectra. | ||
The electrochemical measurements of MnCo–N–C and controls were carried out to evaluate their ORR activities in acidic media. As shown in Fig. S6,† obvious reduction peaks are observed in the cyclic voltammetry (CV) curves of catalysts measured in O2-saturated media in contrast to those in N2-saturated media. Notably, compared with Co–N–C, the reduction peak of MnCo–N–C is more positive (Fig. 4a), suggesting its higher activity towards the ORR. Linear sweep voltammetry (LSV) was further performed to assess their ORR activities (Fig. 4b, S7 and S8†). The E1/2 (0.80 V vs. RHE) of MnCo–N–C is comparable to that (0.86 V vs. RHE) of Pt/C, which is higher than that (0.70 V vs. RHE) of Co–N–C and that (0.50 V vs. RHE) of Mn–N–C. As shown in Fig. S9,† by normalizing the obtained currents to the specific surface areas from the N2 adsorption–desorption analysis, the specific activities of the MnCo–N–C catalyst are found to be higher than those of the Co–N–C catalyst at various potentials. Besides, the MnCo–N–C sample has a smaller Tafel slope (Fig. S10†), indicating its faster kinetics towards the ORR. The electrochemical selectivity of the catalyst was further studied through rotating ring disk electrode (RRDE) polarization tests (Fig. 4c, S11 and S12†). Compared with the Co–N–C catalyst, the electron transfer number of the MnCo–N–C catalyst is close to 4 and the H2O2 yield of the MnCo–N–C catalyst is lower, indicating its higher selectivity over the four-electron reaction pathway towards the ORR.
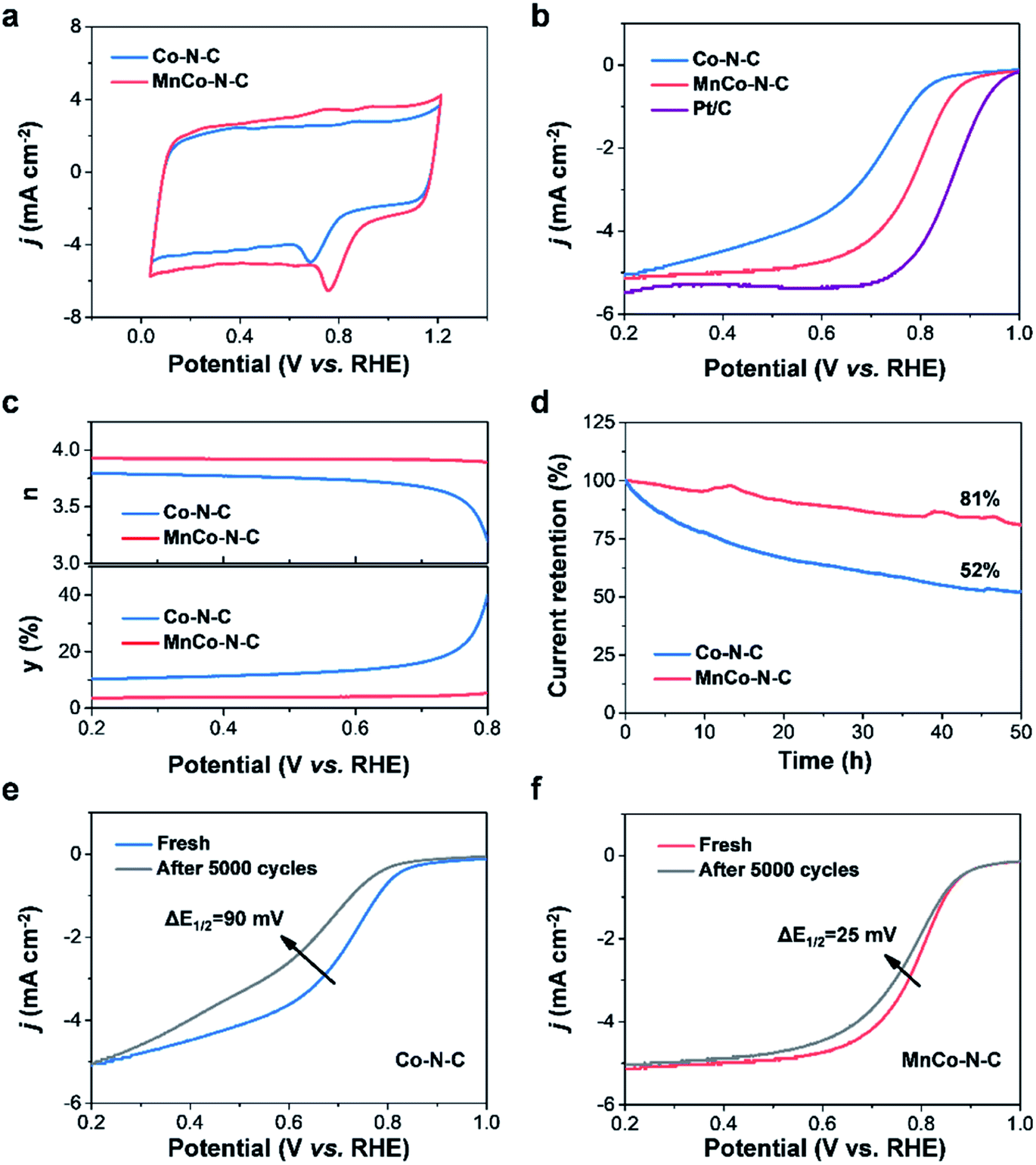 | ||
| Fig. 4 Electrochemical measurements of MnCo–N–C and controls. (a and b) CV and LSV curves obtained in O2-saturated electrolytes, respectively. (c) Electron transfer number (n) and H2O2 yield (y), and (d) the i–t experiments of catalysts, respectively. (e and f) LSV curves of catalysts before and after 5000 cycles of tests, respectively. | ||
The catalytic durability of MnCo–N–C and controls in acidic media was evaluated via current–time chronoamperometry (i–t) and accelerated durability tests (ADT). During the i–t experiment (Fig. 4d), MnCo–N–C shows a much higher current retention of 81% after continuous operation for 50 h, in comparison with that (52%) of Co–N–C. In challenging acidic electrolytes, corrosion of carbon matrices of the MnCo–N–C catalyst can be mitigated to some extent, which is still inevitable and can not be totally avoided. Besides, the undesired H2O2 that is generated during the ORR process would induce oxidative attacks on the MnCo–N–C catalyst and thereby cause dissolution of active metal sites.7,15 These observations would result in the performance decay of the MnCo–N–C catalyst with increase of time during the long-term ORR test. Moreover, MnCo–N–C exhibits good durability during the ADT experiment (Fig. 4e and f). After 5000 cycles, the E1/2 of MnCo–N–C exhibits a small negative shift of 25 mV, while the E1/2 of Co–N–C exhibits a rapid decay with a negative shift of 90 mV. These results, taken together, demonstrate that MnCo–N–C exhibits high catalytic activity, selectivity and durability towards the ORR in the challenging acidic media. Moreover, MnCo–N–C also exhibits superior methanol tolerance and excellent basic ORR performance (Fig. S13†). As shown in Fig. S14,† the LSV curves of MnCo–N–C and Pt/C catalysts obtained in O2-saturated 0.1 M KOH aqueous electrolytes reveal that the half-wave potential (0.89 V vs. RHE) of the MnCo–N–C catalyst is higher than that (0.84 V vs. RHE) of the commercial Pt/C catalyst, indicating its good ORR performance in alkaline electrolytes.
To further understand the ORR process on the surface of Co–N–C and MnCo–N–C, in situ ATR-SEIRAS measurements were further carried out (Fig. 5 and S15†). The ATR-SEIRAS spectra of these two catalysts show a vibration peak located at υ = 1020 cm−1 that becomes more pronounced as the experiments proceed, which can be ascribed to the *O2− intermediate.31–33 As clearly observed, the vibration peak of the *O2− intermediate becomes much stronger for Co–N–C than that of MnCo–N–C. Besides, compared with the Co–N–C catalyst, stronger vibration peaks of the *OOH intermediate at υ = 1212 cm−1 are observed in the spectra of the MnCo–N–C catalyst.33 As reported, during the ORR process, the selectivity for electroreduction of O2 to H2O2 (via the two-electron reaction pathway) or H2O (via the four-electron reaction pathway) is mainly determined by its propensity to break the O–O bond. The strong binding of *OOH intermediates on the catalyst surface will result in the predominance of the four-electron pathway over the two-electron pathway, which causes the *OOH intermediates to be reduced and dissociate into *O and *OH intermediates, thereby enabling electroreduction of O2 to H2O.19,34 By contrast, the strong binding of *O2− intermediates on the catalyst surface will shift the reaction mechanism to follow the two-electron reaction pathway to generate H2O2 (*O2− + 2H+ + e− → H2O2) during the ORR process.31,33 The faster kinetics of *OOH formation and adsorbed *O2− desorption of the MnCo–N–C catalyst can correlate well with its enhanced activity and selectivity towards the ORR. The lower yield of H2O2 of the MnCo–N–C catalyst during the ORR process would enable less H2O2 oxidative attacks on the catalyst and enhance the durability of the MnCo–N–C catalyst.
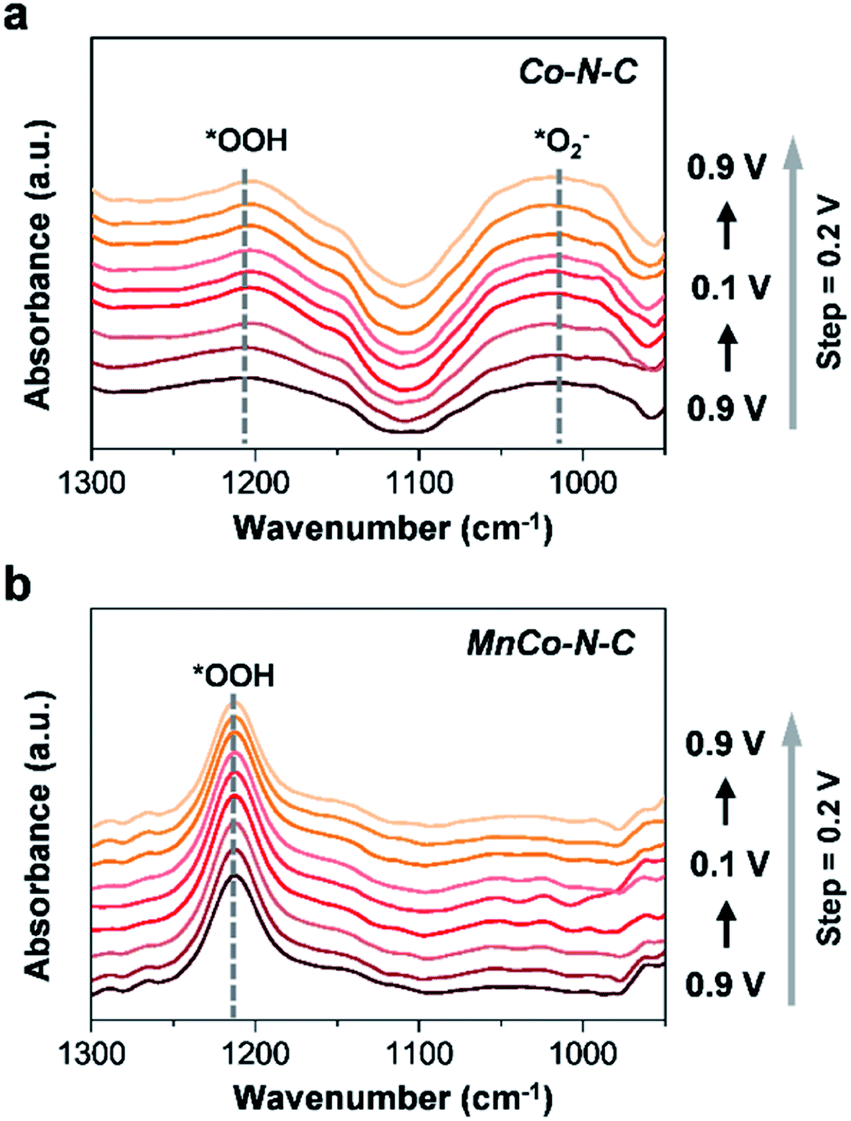 | ||
| Fig. 5 (a and b) In situ ATR-SEIRAS spectra of Co–N–C and MnCo–N–C, respectively. | ||
Conclusions
In summary, we demonstrate a novel design of acidic ORR catalyst by co-doping Mn, Co and N into carbon matrices, and the resultant MnCo–N–C catalyst shows greatly boosted activity, selectivity and durability compared with the Co–N–C catalyst. The HAADF-STEM, XAFS and XPS studies indicate that CoNx and MnNx atomic sites are dispersed in the carbon matrices of the MnCo–N–C catalyst. Besides, the Raman spectroscopy results verify that the doping of Mn can improve the graphitization of carbon matrices, thus enhancing the corrosion resistance and catalytic durability of the MnCo–N–C catalyst. The formation of ordered graphitic carbon with less active oxygenated groups can decrease the yield of H2O2 during the ORR process, which can decrease the undesired oxidative attacks on the catalyst and in turn promote the durability of the MnCo–N–C catalyst. This work provides insights for further advances in the development of high-performance acidic ORR catalysts for PEMFCs.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (2232019A3-03, and JUSRP121035), National Natural Science Foundation of China (21875033, and 52103260), China Postdoctoral Science Foundation (2021M690067), Natural Science Foundation of Jiangsu Province for Youths (BK20210482) and Jiangsu Province Postdoctoral Science Foundation (2021K053A).References
- X. X. Wang, M. T. Swihart and G. Wu, Nat. Catal., 2019, 2, 578–589 CrossRef CAS.
- L. Chong, J. G. Wen, J. Kubal, F. G. Sen, J. X. Zou, J. Greeley, M. Chan, H. Barkholtz, W. J. Ding and D. J. Liu, Science, 2018, 362, 1276–1281 CrossRef CAS.
- Y. X. Wang, X. Z. Cui, L. W. Peng, L. L. Li, J. L. Qiao, H. T. Huang and J. L. Shi, Adv. Mater., 2021, 33, 2100997 CrossRef CAS.
- H. Cheng, R. J. Gui, H. Yu, C. Wang, S. Liu, H. F. Liu, T. P. Zhou, N. Zhang, X. S. Zheng, W. S. Chu, Y. Lin, H. A. Wu, C. Z. Wu and Y. Xie, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104026118 CrossRef CAS.
- G. J. Chao, X. Y. An, L. S. Zhang, J. Tian, W. Fan and T. X. Liu, Compos. Commun., 2021, 24, 100603 CrossRef.
- X. H. Xie, C. He, B. Y. Li, Y. H. He, D. A. Cullen, E. C. Wegener, A. J. Kropf, U. Martinez, Y. W. Cheng, M. H. Engelhard, M. E. Bowden, M. Song, T. Lemmon, X. S. Li, Z. M. Nie, J. Liu, D. J. Myers, P. Zelenay, G. F. Wang, G. Wu, V. Ramani and Y. Y. Shao, Nat. Catal., 2020, 3, 1044–1054 CrossRef CAS.
- J. Z. Li, M. J. Chen, D. A. Cullen, S. Hwang, M. Y. Wang, B. Y. Li, K. X. Liu, S. Karakalos, M. Lucero, H. G. Zhang, C. Lei, H. Xu, G. E. Sterbinsky, Z. X. Feng, D. Su, K. L. More, G. F. Wang, Z. B. Wang and G. Wu, Nat. Catal., 2018, 1, 935–945 CrossRef CAS.
- F. Li, G. F. Han, H. J. Noh, S. J. Kim, Y. L. Lu, H. Y. Jeong, Z. P. Fu and J. B. Baek, Energy Environ. Sci., 2018, 11, 2263–2269 RSC.
- J. K. Li, M. T. Sougrati, A. Zitolo, J. M. Ablett, I. C. Oguz, T. Mineva, I. Matanovic, P. Atanassov, Y. Huang, I. Zenyuk, A. Di Cicco, K. Kumar, L. Dubau, F. Maillard, G. Drazic and F. Jaouen, Nat. Catal., 2021, 4, 10–19 CrossRef CAS.
- E. G. Luo, H. Zhang, X. Wang, L. Q. Gao, L. Y. Gong, T. Zhao, Z. Jin, J. J. Ge, Z. Jiang, C. P. Liu and W. Xing, Angew. Chem., Int. Ed., 2019, 58, 12469–12475 CrossRef CAS.
- Y. H. He, S. Hwang, D. A. Cullen, M. A. Uddin, L. Langhorst, B. Y. Li, S. Karakalos, A. J. Kropf, E. C. Wegener, J. Sokolowski, M. J. Chen, D. Myers, D. Su, K. L. More, G. F. Wang, S. Litster and G. Wu, Energy Environ. Sci., 2019, 12, 250–260 RSC.
- H. T. Chung, D. A. Cullen, D. Higgins, B. T. Sneed, E. F. Holby, K. L. More and P. Zelenay, Science, 2017, 357, 479–483 CrossRef CAS.
- H. G. Zhang, S. Hwang, M. Y. Wang, Z. X. Feng, S. Karakalos, L. L. Luo, Z. Qiao, X. H. Xie, C. M. Wang, D. Su, Y. Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS PubMed.
- C. H. Choi, H. K. Lim, M. W. Chung, G. Chon, N. R. Sahraie, A. Altin, M. T. Sougrati, L. Stievano, H. S. Oh, E. S. Park, F. Luo, P. Strasser, G. Drazic, K. J. J. Mayrhofer, H. Kim and F. Jaouen, Energy Environ. Sci., 2018, 11, 3176–3182 RSC.
- X. X. Wang, V. Prabhakaran, Y. H. He, Y. Y. Shao and G. Wu, Adv. Mater., 2019, 31, 1805126 CrossRef.
- H. L. Peng, F. F. Liu, X. J. Liu, S. J. Liao, C. H. You, X. L. Tian, H. X. Nan, F. Luo, H. Y. Song, Z. Y. Fu and P. Y. Huang, ACS Catal., 2014, 4, 3797–3805 CrossRef CAS.
- J. Masa, A. Q. Zhao, W. Xia, M. Muhler and W. Schuhmann, Electrochim. Acta, 2014, 128, 271–278 CrossRef CAS.
- S. T. Thompson, A. R. Wilson, P. Zelenay, D. J. Myers, K. L. More, K. C. Neyerlin and D. Papageorgopoulos, Solid State Ionics, 2018, 319, 68–76 CrossRef CAS.
- Y. Y. Sun, L. Silvioli, N. R. Sahraie, W. Ju, J. K. Li, A. Zitolo, S. Li, A. Bagger, L. Arnarson, X. L. Wang, T. Moeller, D. Bernsmeier, J. Rossmeisl, F. Jaouen and P. Strasser, J. Am. Chem. Soc., 2019, 141, 12372–12381 CrossRef CAS.
- D. Banham, S. Ye, K. Pei, J. Ozaki, T. Kishimoto and Y. Imashiro, J. Power Sources, 2015, 285, 334–348 CrossRef CAS.
- C. H. Choi, C. Baldizzone, J. P. Grote, A. K. Schuppert, F. Jaouen and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2015, 54, 12753–12757 CrossRef CAS.
- B. Q. Li, C. X. Zhao, J. N. Liu and Q. Zhang, Adv. Mater., 2019, 31, 1808173 CrossRef.
- S. Gupta, S. Zhao, X. X. Wang, S. Hwang, S. Karakalos, S. V. Devaguptapu, S. Mukherjee, D. Su, H. Xu and G. Wu, ACS Catal., 2017, 7, 8386–8393 CrossRef CAS.
- Y. H. Zhong, X. L. Liang, Z. S. He, W. Tan, J. X. Zhu, P. Yuan, R. L. Zhu and H. P. He, Appl. Catal., B, 2014, 150–151, 612–618 CrossRef CAS.
- G. G. Yang, J. W. Zhu, P. F. Yuan, Y. F. Hu, G. Qu, B. A. Lu, X. Y. Xue, H. B. Yin, W. Z. Cheng, J. Q. Cheng, W. J Xu, J. Li, J. S. Hu, S. C. Mu and J. N. Zhang, Nat. Commun., 2021, 12, 1734 CrossRef CAS.
- X. X. Wang, D. A. Cullen, Y. T. Pan, S. Hwang, M. Y. Wang, Z. X. Feng, J. Y. Wang, M. H. Engelhard, H. G. Zhang, Y. H. He, Y. Y. Shao, D. Su, K. L. More, J. S. Spendelow and G. Wu, Adv. Mater., 2018, 30, 1706758 CrossRef.
- B. You, N. Jiang, M. L. Sheng, W. S. Drisdell, J. Yano and Y. J Sun, ACS Catal., 2015, 5, 7068–7076 CrossRef CAS.
- S. C. Sun, G. Q. Shen, J. W. Jiang, W. B. Mi, X. L. Liu, L. Pan, X. W. Zhang and J. J. Zou, Adv. Energy Mater., 2019, 9, 1901505 CrossRef.
- G. J. Chao, L. S. Zhang, J. Tian, W. Fan and T. X. Liu, Compos. Commun., 2021, 25, 100703 CrossRef.
- T. Y. Zhu, Q. C. Feng, S. L. Liu and C. Zhang, Compos. Commun., 2020, 20, 100376 CrossRef.
- S. Nayak, P. U. Biedermann, M. Stratmann and A. Erbe, Electrochim. Acta, 2013, 106, 472–482 CrossRef CAS.
- S. Nayak, I. J. McPherson and K. A. Vincent, Angew. Chem., Int. Ed., 2018, 57, 12855–12858 CrossRef CAS.
- Y. Y. Wang, D. J. Chen, T. C. Allison and Y. Y. J. Tong, J. Chem. Phys., 2019, 150, 041728 CrossRef PubMed.
- S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens and J. Rossmeisl, Nat. Mater., 2013, 12, 1137–1143 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta08029f |
| This journal is © The Royal Society of Chemistry 2022 |