
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA review on dual-phase oxygen transport membranes: from fundamentals to commercial deployment
Ragnar
Kiebach
 *a,
Stéven
Pirou
a,
Lev
Martinez Aguilera
a,
Astri Bjørnetun
Haugen
a,
Andreas
Kaiser
a,
Peter Vang
Hendriksen
a,
María
Balaguer
b,
Julio
García-Fayos
b,
José Manuel
Serra
b,
Falk
Schulze-Küppers
c,
Max
Christie
d,
Liudmila
Fischer
ce,
Wilhelm Albert
Meulenberg
ce and
Stefan
Baumann
*c
*a,
Stéven
Pirou
a,
Lev
Martinez Aguilera
a,
Astri Bjørnetun
Haugen
a,
Andreas
Kaiser
a,
Peter Vang
Hendriksen
a,
María
Balaguer
b,
Julio
García-Fayos
b,
José Manuel
Serra
b,
Falk
Schulze-Küppers
c,
Max
Christie
d,
Liudmila
Fischer
ce,
Wilhelm Albert
Meulenberg
ce and
Stefan
Baumann
*c
aDepartment of Energy Conversion and Storage, Technical University of Denmark, Lyngby Campus, Anker Engelunds Vej, Building 301, DK-2800 Kgs. Lyngby, Denmark. E-mail: woki@dtu.dk; Fax: +45 46775688; Tel: +45 46775624
bInstituto de Tecnología Química (Universitat Politècnica de València – Consejo Superior de Investigaciones Científicas), Av. Naranjos s/n, E-46022 Valencia, Spain
cForschungszentrum Jülich GmbH, Institute of Energy and Climate Research, Materials Synthesis and Processing (IEK-1), 52425 Jülich, Germany. E-mail: s.baumann@fz-juelich.de; Fax: +49 2461612455; Tel: +49 2461618961
dLinde Inc., 175 East Park Drive, Tonawanda, NY 14150, USA
eUniversity of Twente, Faculty of Science and Technology, Inorganic Membranes, P. O. Box 217, Enschede 7500 AE, The Netherlands
First published on 17th January 2022
Abstract
Oxygen transport membranes (OTMs) are a promising alternative to cryogenic air separation (ASU) or pressure swing adsorption (PSA) for oxygen production. Using these ceramic membranes allows producing high purity oxygen on various scales in a continuous single-step process, at lower costs and power consumption, making it an advantageous technique for oxy-combustion in connection with carbon capture and delocalized oxygen production on a small scale. Moreover, their use in membrane reactors, directly utilizing the permeating oxygen in chemical reactions towards green chemistry, is an emerging research field. Especially dual-phase OTMs, where the membrane consists of a composite of a stable ionic conductor and a stable electronic conductor, are of high interest, because they can overcome the disadvantages of single-phase membranes like low chemical and mechanical stability at elevated temperatures and under harsh operation conditions. However, despite the progress in the development of dual-phase OTMs over the last years, and their potential applications in classic and emerging fields, challenges preventing their large-scale employment remain. This review aims to guide new studies that will promote the development and upscaling of dual-phase OTMs. Recent developments, current opportunities and challenges, and future directions of research are thoroughly discussed. Through this review paper, information about the basic working principle, properties, performance and current application in industry of dual-phase OTM membranes can be comprehended. Next to material properties, preparative methods and manufacturing are in focus, intending to accelerate development and upscaling of new materials and components. Furthermore, existing challenges and research strategies to overcome these are discussed, and focus areas and prospects of future application areas are suggested.
1 Introduction
Oxygen is a key product for numerous applications in different industrial sectors, and with more than 100 Mton produced annually1 it is an important chemical commodity accounting for ca. 25% of the world market of industrial gasses. Currently, the main markets for oxygen are steel production (55%), and the chemical industry (25%), but significant amounts are also used in power generation, hospitals or water treatment.2 In the future, the demand for oxygen is expected to grow, because large and medium scale clean energy technologies like oxy-fuel combustion and oxygen-blown biomass gasification, which will require oxygen as a feed, are expected to grow significantly within the next 30 years.The objective of this review paper is to focus on the recent advances of ceramic dual-phase composite membranes for the separation of oxygen from air. A comprehensive overview about the achieved results in terms of performance and stability is given, and existing issues and challenges with respect to separation performance, application and integration are addressed. Finally, different approaches to overcome these challenges and future development directions for dual-phase membranes for oxygen production and ways towards industrial realization are presented.
1.1 Advantages of oxygen transport membranes over state-of-the-art oxygen separation technologies
Oxygen is mainly produced from atmospheric gases separation, where fractional distillation of liquefied air, also known as cryogenic distillation, and pressure swing adsorption (PSA) are the two main used industrial separation technologies. Cryogenic distillation is a mature technology for oxygen production with capacities larger than 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Nm3 h−1 and purity of 99%. Since it works with sub-zero temperatures at −185 °C and 5–8 bar, it requires high investment and an energy demand of ca. 225 kW h ton−1. PSA is a semi-batch process usually designed for capacities up to 10000 Nm−3 h−1. It uses zeolites and pressurized systems to adsorb N2 and CO2 from the air and can generate oxygen with purity ca. 95%. Its energy demand varies from 245–525 kW h ton−1 depending on the use of vacuum (VPSA) to improve the absorption of the oxygen. Also other emergent technologies for oxygen production have been used in practical applications, like electrochemical water splitting, which generates high purity oxygen > 99.5% but at high capital cost and with a high energy demand, ca. 1320 kW h ton−1.1,3,4
000 Nm3 h−1 and purity of 99%. Since it works with sub-zero temperatures at −185 °C and 5–8 bar, it requires high investment and an energy demand of ca. 225 kW h ton−1. PSA is a semi-batch process usually designed for capacities up to 10000 Nm−3 h−1. It uses zeolites and pressurized systems to adsorb N2 and CO2 from the air and can generate oxygen with purity ca. 95%. Its energy demand varies from 245–525 kW h ton−1 depending on the use of vacuum (VPSA) to improve the absorption of the oxygen. Also other emergent technologies for oxygen production have been used in practical applications, like electrochemical water splitting, which generates high purity oxygen > 99.5% but at high capital cost and with a high energy demand, ca. 1320 kW h ton−1.1,3,4
Another promising air separation technology are dense ceramic membranes that separate oxygen from air at elevated temperatures, often referred to as “Oxygen Transport Membranes (OTMs)” or “Ion Transport Membrane (ITMs)”. Compared to state of the art technologies described above, this approach has significant advantages, and using thermally integrated separation modules based on ceramic OTMs can potentially reduce capital (CAPEX) and energy demand down to 147 kW h ton−1 (OPEX).1
The main advantages of dense permeation membranes include (i) infinite selectivity with respect to oxygen – resulting in a very pure product (>99.99% oxygen), (ii) the ability to thermally integrate oxygen separation into high temperature process like oxy-fuel combustion – reducing the energy needed for the separation process, (iii) the modular design of OTM reactors – which makes oxygen separation more versatile and economically viable also on small and medium scale, and (iv) a better process yield – e.g. as exclusively oxygen anions are allowed to diffuse through the membrane, this can cause considerable effects on yield and selectivity in chemical reactions,5e.g. by combining steam reforming and partial oxidation into one single step for the natural gas conversion.
1.2 Basic working principle of oxygen transport membranes
OTMs consist of gas-tight mixed ionic and electronic conductors (MIEC) that allow simultaneous transport of oxygen ions via oxygen vacancies and electrons by small polaron hopping in the crystal lattice. OTMs produce a flux of pure oxygen, based on a driving force created by the gradient between partial pressures of oxygen (pO2) on the two sides of the membrane. In general, such membranes work at high temperatures (>500 °C) because of the thermally activated diffusion process of oxygen ions, which is typically rate limiting compared to the electron transport. The oxygen flux through the membrane is for the case of fast surface exchange given by the Wagner eqn (1):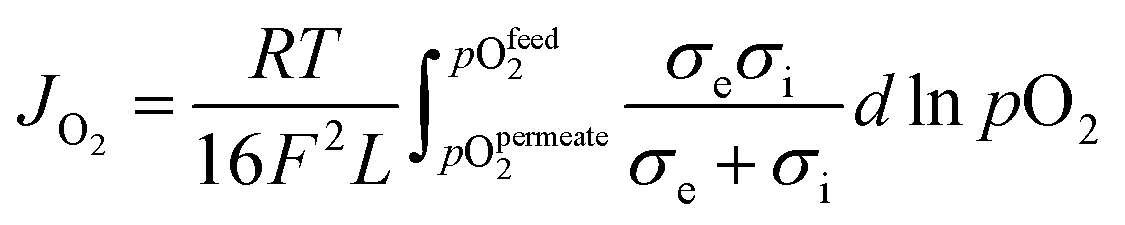 | (1) |
Assuming, the ambipolar conductivity 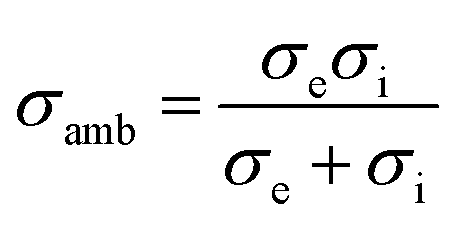 is independent of pO2, eqn (1) can be written as
is independent of pO2, eqn (1) can be written as
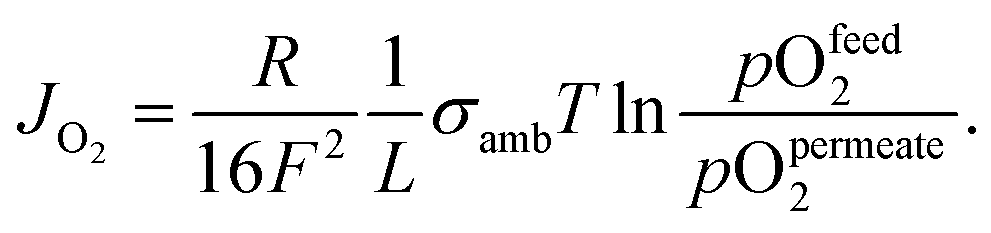 | (2) |
Eqn (2) shows that the permeation rate can be maximized by different measures, i.e. (i) operation conditions T and pO2-gradient (cf. Sub-section 2.4), (ii) materials development improving σamb (cf. Section 2), and (iii) membrane processing reducing its thickness L (cf. Sub-section 4.2).
1.3 Transport of oxygen through asymmetric membranes
According to eqn (2), the membrane thickness L should be as low as possible. Asymmetric membranes made of a thin selective membrane layer (5–30 μm) supported by a relatively thick porous support (200–1000 μm) are promising to minimize the actual membrane thickness while ensuring mechanical robustness.6Oxygen permeation through asymmetric MIEC membranes can be divided into six steps. Fig. 1 shows the assembly model of the steps represented as resistances in series. Zones I and VI represent the concentration polarization occurring in the gas phases (feed gas and sweep gas); zone V corresponds to the concentration polarization (sometimes referred to as mass transport resistance) in the pores of the porous support; zone II symbolizes the surface exchange including oxygen reduction, dissociation and incorporation into the oxide lattice at the high oxygen partial pressure side, while zone IV illustrates the reactions in the opposite order at the low-oxygen partial pressure side in order to reconstitute the oxygen molecule. Finally, zone III represents the bulk transport of the oxygen ions into the dense selective membrane layer. The dominating rate-limiting process (largest resistance) governs the overall performance. The rate-limiting process depends on several parameters such as the membrane material, the membrane geometry or the operating conditions.
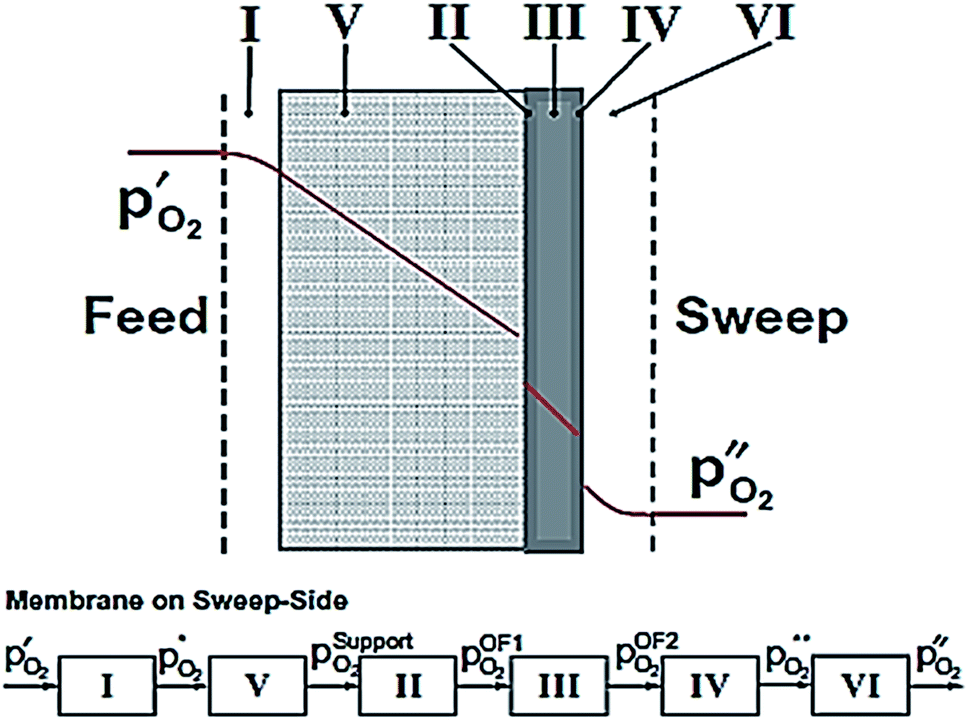 | ||
| Fig. 1 Model of resistances representing the steps of the oxygen permeation through asymmetric membranes. Reproduced with permission.7 Copyright 2014, Elsevier. | ||
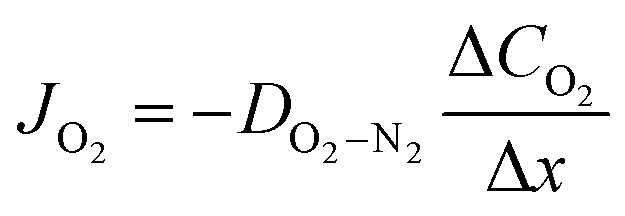 | (3) |
In lab scale testing, due to the relatively fast diffusion of the oxygen in air (large values of DO2−N2) the loss of oxygen activity is commonly neglected. However, the effect is highly dependent on the fluid dynamics and, thus, the design of the module/reactor must also consider the feed/sweep gas flow rates.
In contrast, the diffusion of the gas through the porous support (zone V, Fig. 1) usually induces a more significant loss of the driving force, which in case of high fluxes can become fully rate determining.8 The associated resistance is highly dependent on the microstructure of the support material (porosity, pore size, pore connectivity (opening diameter), tortuosity, etc.) as well as the gas mixture.9,10
In a porous support, molecular and Knudsen diffusion, surface diffusion as well as convective flow contribute to the overall transport, whereby the microstructural features of the porous structure determine the dominating processes. For the description of the overall gas transport through a porous medium, two main models are discussed in the literature, the Dusty Gas Model (DGM)11 and the Binary Friction Model (BFM), which was developed by Kerkhof12 who identified an error in the DGM. Nevertheless, both models are applicable to asymmetric OTMs.13,14 Due to the high complexity of these models, also simplifications are suggested.14
One example is a convection-diffusion approach considering a total diffusive oxygen flux (Fick) and an additional convective flux driven by an absolute pressure difference resulting in ref. 8
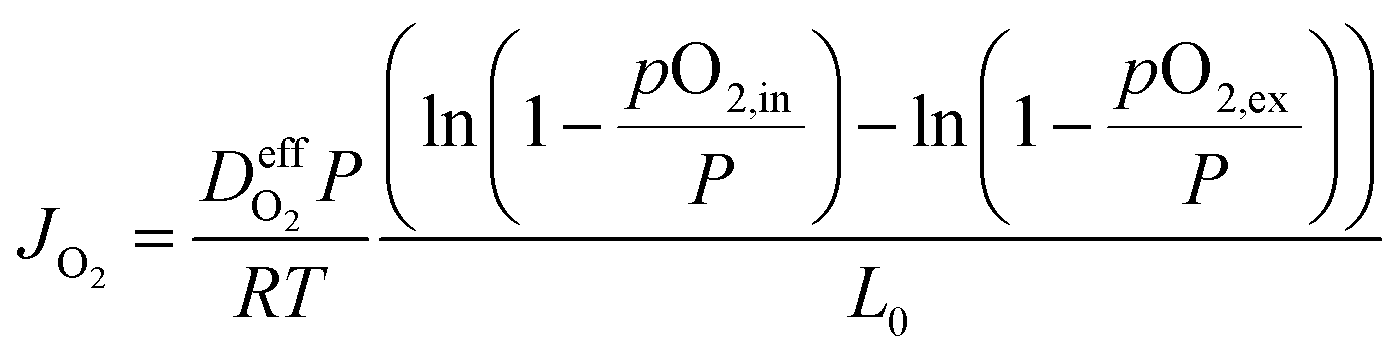 | (4) |
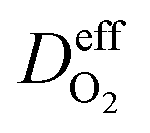 is the effective gas diffusivity, accounting for the effects of the porous medium and gas–solid friction (Knudsen diffusion).
is the effective gas diffusivity, accounting for the effects of the porous medium and gas–solid friction (Knudsen diffusion).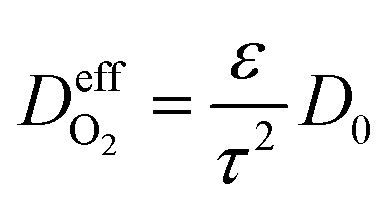 | (5) |
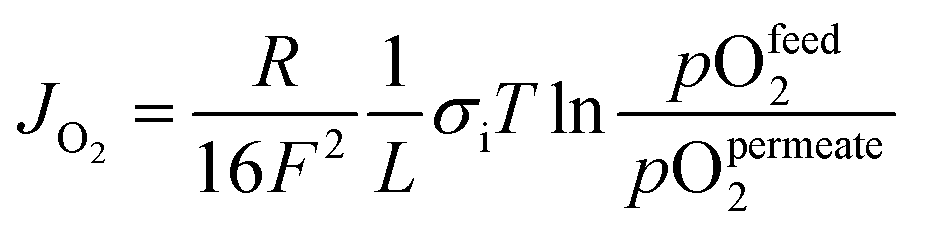 | (6) |
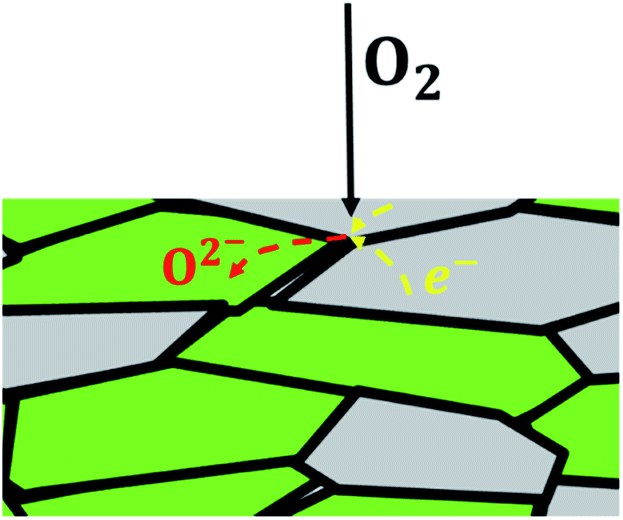
In dual-phase composites, the ionic and electronic conductance is realized in separate phases. Therefore, it is important to ensure an interconnected network of both phases providing sufficient pathways for both charge carriers, Fig. 2. Since in most cases the electronic conductivity is still higher compared to the ionic one, the fraction of ion conductor should be as high as possible whereas the fraction of electron conductor should be as high as necessary to sustain a percolating network.16
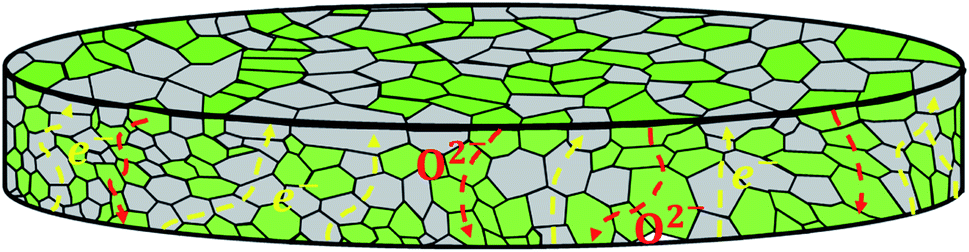 | ||
| Fig. 2 Schematic overview of a dual-phase composite containing one ionic conducting (green) and one electronic conducting (grey) phase. | ||
| O2 + e− → O2− | (7) |
| O2− + e− → O22− | (8) |
| O22− → 2O− | (9) |
| 2O− + 2e− → 2O2− | (10) |
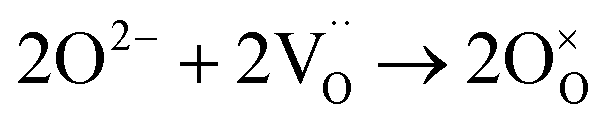 | (11) |
At the low-oxygen partial pressure side (zone IV, Fig. 1), the reactions occur in the opposite order, representing thus the oxidation, association and desorption of oxygen molecules. Each of these reactions can be the limiting rate step for the overall surface exchange reaction.
In the steady state, the oxygen flux across the membrane is assumed to be proportional to the chemical potential drop over the interface (linear kinetics):
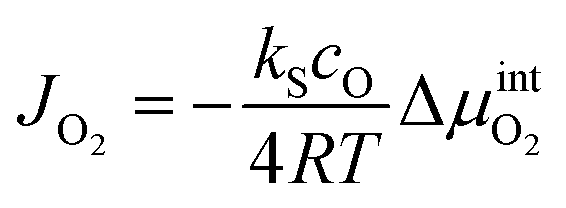 | (12) |
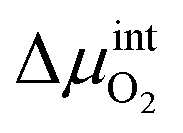 is the chemical potential drop across the interface, kS is the surface exchange coefficient, and cO denotes the oxide ion concentration at the surface.
is the chemical potential drop across the interface, kS is the surface exchange coefficient, and cO denotes the oxide ion concentration at the surface.
Bouwmeester and Burggraaf20,21 introduced the characteristic thickness Lc to define the membrane thickness corresponding to transition from predominant bulk diffusion limitation to the state when the oxygen permeation is governed by the surface exchange. Lc is defined by the ratio between the self-diffusion coefficient of oxygen (DS) and the surface exchange coefficient (kS):
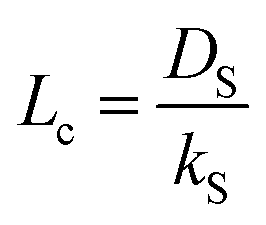 | (13) |
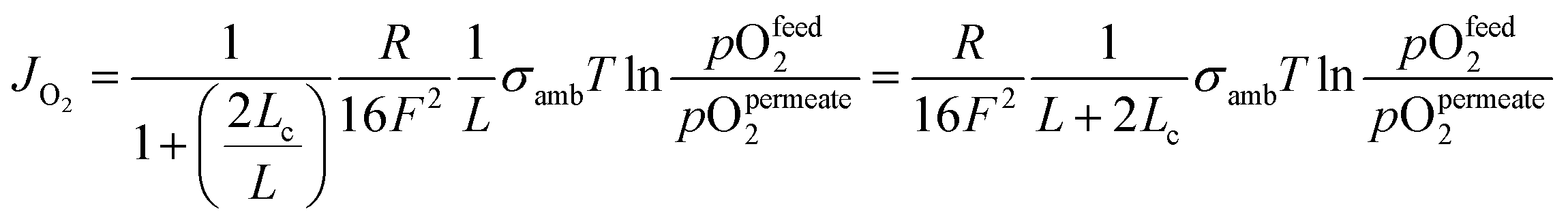 | (14) |
Fig. 3 displays the thickness dependence of perovskite single-phase membranes made of BSCF.17 The asymmetric membranes were placed with the support to the feed side. Pure oxygen was used as feed gas in order to minimize support effects. The characteristic thickness achieved by fitting the experimental data to eqn (14) (Lc = 43 μm) is in very well agreement with other literature sources. Applying a catalytically active porous BSCF layer, i.e. increasing surface area at the permeate side, significantly increases the oxygen permeation rate, revealing severe surface limitation.
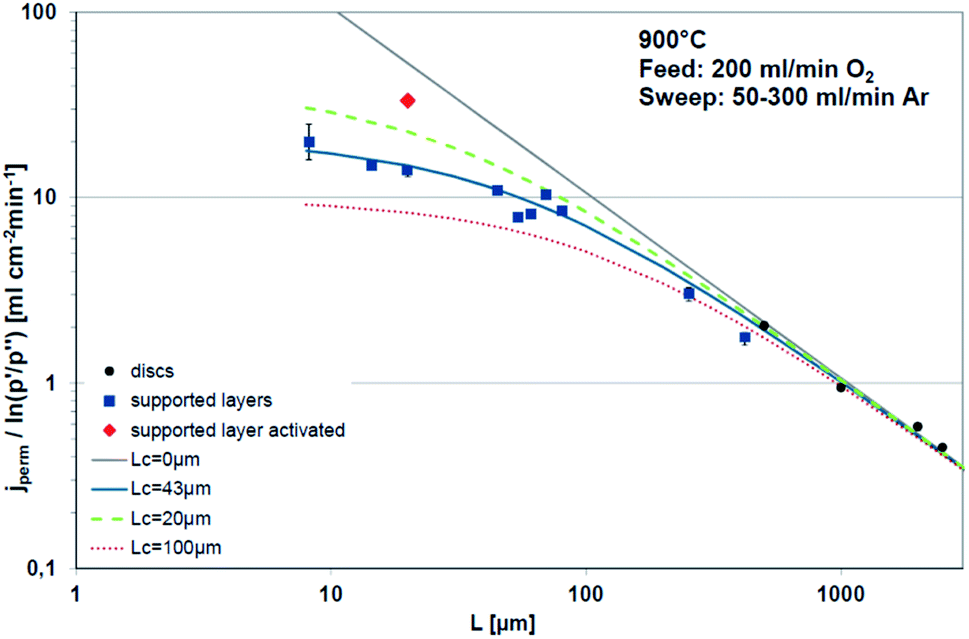 | ||
| Fig. 3 Thickness dependence of BSCF membranes fitted using eqn (14). Reproduced with permission.17 Copyright 2015, IOP Science. | ||
In case of dual-phase membranes, the surface exchange is of high importance already at high membrane thicknesses. The surface exchange reactions (7)–(11) can only occur at the triple phase boundaries (TPB) of air (providing molecular oxygen O2), electron conductor (providing electrons e) and ion conductor (providing oxygen vacancies 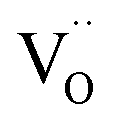 ) as depicted in Fig. 4. This effect is well known from cathode research in Solid Oxide Fuel Cells (SOFCs) and must be overcome by coating of porous catalyst layers providing electronic or mixed ionic electronic conductivity.
) as depicted in Fig. 4. This effect is well known from cathode research in Solid Oxide Fuel Cells (SOFCs) and must be overcome by coating of porous catalyst layers providing electronic or mixed ionic electronic conductivity.
 | ||
| Fig. 4 Schematic overview of the oxygen uptake in a dual-phase membrane material. | ||
2 Materials for dual-phase oxygen transport membranes
2.1 Material challenges in state-of the-art OTMs based on single phase MIEC
OTMs based on single phase MIEC materials have been studied for decades, showing high oxygen permeation flux.26–35 The oxygen conduction phenomenon exhibited by MIEC materials is attributed to their ability to support oxygen vacancies and lattice disorder, which allows the relatively rapid and sustainable transport of oxygen ions under the appropriate conditions.36–40 Most MIEC materials used in OTMs display a perovskite or Ruddlesden–Popper crystal structure.Perovskite materials are defined by the general formula ABO3, corresponding in general to A2+B4+O3 (A1+B5+O3 or A3+B3+O3 are also possible). In this formula, A and B correspond to two cations of very different sizes, the A atoms being larger than the B atoms.
The ionic conductivities in these materials can be enhanced by substituting lower valence cations for both A and B sites, because the deficiency from the substitution results in an increase of oxide ion vacancies. The electronic conductivity can also be increased by the addition of aliovalent cations. The B cation is oxidized and thus an electron hole is formed. After doping with other metal cations, the perovskite can be symbolized by the formula 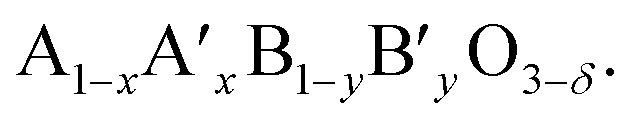 Usually, A ions are alkaline-earth metals such as Ca2+, Sr2+ and Ba2+, and B ions are transition metals such as Co3+ and Fe3+. Among the various combination of chemical compounds, Ba1−xSrxCo1−yFeyO3−δ (BSCF) and La1−xSrxCo1−yFeyO3−δ (LSCF) appear to be the materials with the highest reported oxygen permeation. Despite its good performance, BSCF has several drawbacks that limit its use as a membrane material. One of the most critical is its instability under CO2 and SO2 containing atmospheres.41–43 BSCF also has high chemical and thermal expansion.44 The lattice expansion arising from the phase transition (cubic to hexagonal) occurs in the 850–900 °C temperature range in which OTMs are usually operated. This lattice expansion can result in chemical instability and mechanical failure.45 LSCF has been intensively investigated as a membrane material for oxygen separation from air46,47 and cathode material for SOFCs.48–51 This material has a high electronic conductivity (310 S cm−1 for La0.2Sr0.8Co0.8Fe0.2O3−δ at 900 °C) and a good ionic conductivity (0.87 S cm−1 for La0.2Sr0.8Co0.8Fe0.2O3−δ at 900 °C).49 While studies showed its relative stability in CO2 if the Sr-content is limited,46,47 it is not stable in SOx-containing atmospheres due to the formation of SrSO4.52,53 This makes LSCF an inappropriate candidate material for OTMs developed for applications in which stability in low pO2 and/or SOx is required.
Usually, A ions are alkaline-earth metals such as Ca2+, Sr2+ and Ba2+, and B ions are transition metals such as Co3+ and Fe3+. Among the various combination of chemical compounds, Ba1−xSrxCo1−yFeyO3−δ (BSCF) and La1−xSrxCo1−yFeyO3−δ (LSCF) appear to be the materials with the highest reported oxygen permeation. Despite its good performance, BSCF has several drawbacks that limit its use as a membrane material. One of the most critical is its instability under CO2 and SO2 containing atmospheres.41–43 BSCF also has high chemical and thermal expansion.44 The lattice expansion arising from the phase transition (cubic to hexagonal) occurs in the 850–900 °C temperature range in which OTMs are usually operated. This lattice expansion can result in chemical instability and mechanical failure.45 LSCF has been intensively investigated as a membrane material for oxygen separation from air46,47 and cathode material for SOFCs.48–51 This material has a high electronic conductivity (310 S cm−1 for La0.2Sr0.8Co0.8Fe0.2O3−δ at 900 °C) and a good ionic conductivity (0.87 S cm−1 for La0.2Sr0.8Co0.8Fe0.2O3−δ at 900 °C).49 While studies showed its relative stability in CO2 if the Sr-content is limited,46,47 it is not stable in SOx-containing atmospheres due to the formation of SrSO4.52,53 This makes LSCF an inappropriate candidate material for OTMs developed for applications in which stability in low pO2 and/or SOx is required.
Ruddlesden–Popper phases can be described with the general formula An+1MnO3n+1 (with n = 1, 2, 3, …∞), where A is a cation of large ionic radius (lanthanide or alkaline earth) and M a transition metal (M = Co, Ni, Cu, etc.). La2NiO4+δ and its derivative materials La2−xSrxNi1−yMyO4+δ (M = Fe, Cu, Co) are the Ruddlesden–Popper materials that have been investigated the most as OTM materials.54–64 These materials exhibit high oxygen diffusion and surface exchange coefficients at intermediate temperatures together with moderate thermal expansion coefficients (TECs).59 Several studies attest that the substitution of strontium for lanthanum (0 ≤ x ≤ 0.75) results in an increase of the electrical conductivity.55,63,64 Aguadero et al. demonstrated that La1.25Sr0.75NiO4+δ exhibits a conductivity of 235 S cm−1 in air at 850 °C, while La2NiO4+δ displays only 60 S cm−1 under the same operating conditions.55 Although pure La2NiO4+δ shows good stability in CO2 due to the absence of any alkaline earth elements, its performance drops to zero instantaneously when adding low amounts of SOx.65 As possible reason for the performance drop the formation of a dense layer of sulphur-containing reaction products on the surface of the sample, was mentioned by the authors. Unfortunately, it was not possible to identify the phase composition of the formed phase.
2.2 Dual-phase composite systems for OTMs
As outlined above, the performance wise promising single-phase MIEC materials are chemically not stable in CO2 or SO2 containing atmospheres. Conversely, materials stable under the targeted harsh operation conditions do not present sufficiently high oxygen permeation fluxes to be of commercial interest. Dual-phase systems, where the membrane consists of a composite of a stable ionic conductor and a stable electronic conductor, can overcome the disadvantages of single-phase membranes. Ideally, they combine the best characteristics of the used compounds to achieve both a large oxygen permeability and good chemical and mechanical stability, at elevated temperatures and under harsh conditions (i.e. atmospheres containing H2O, CO2, SOx, etc.). Fig. 5 presents a schematic illustration of an asymmetric dual-phase membrane. As unique feature compared to single-phase membranes microstructural aspects are of utmost importance in dual-phase membranes apart from the material selection. First, a good percolation of both ionic and electronic conducting phases is fundamental for dual-phase composites enabling oxygen permeation. Therefore, most of the publications work with mixtures of 40–60% of ionic and 60–40% of electronic conducting phases, respectively. Typically the conductivity of the electronic conductor is much higher compared to the conductivity of the ion conductor. Therefore, maximum performance can be expected at maximum content of the ionic conductor provided that the second phase ensures sufficient electronic conductance (cf. Section 7.1). In consequence, the performance of a selected material combination even with identical volume fractions can be highly dependent on the processing.66,67 Moreover, the microstructure of the phase mixture determines the triple phase boundaries being crucial for oxygen surface exchange reactions (cf. Section 1.3.3).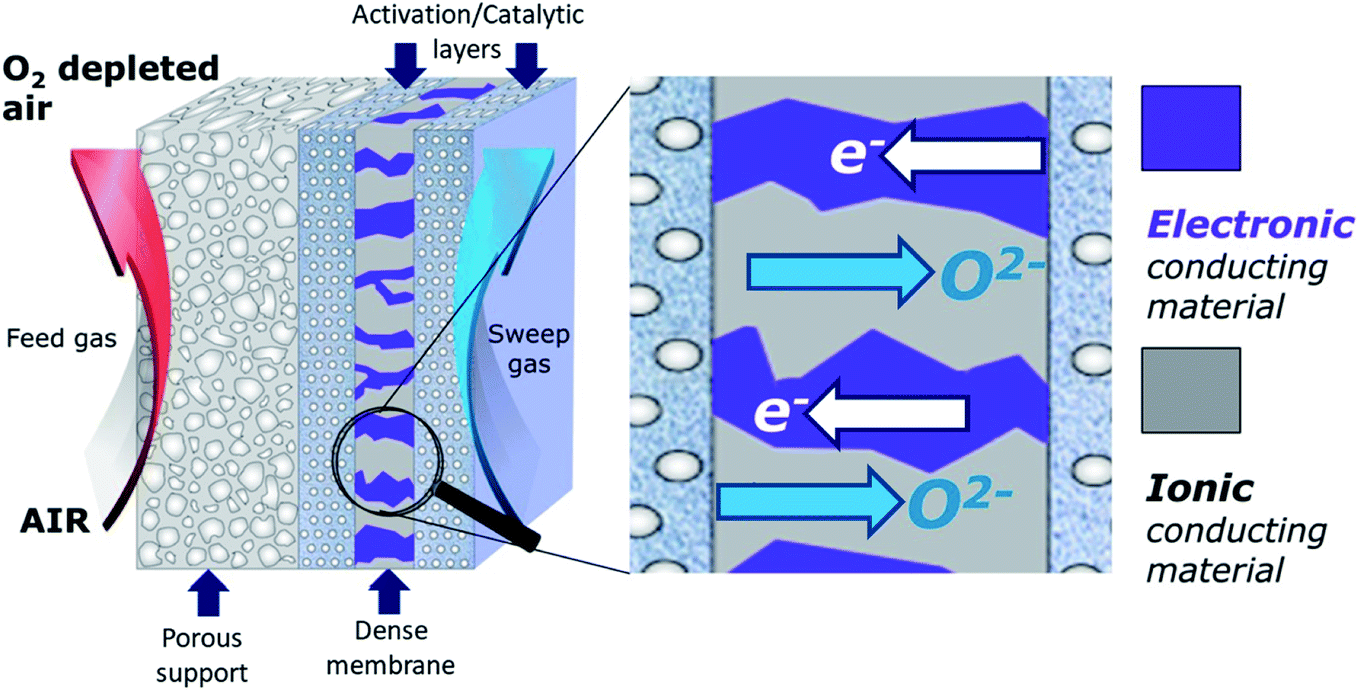 | ||
| Fig. 5 Schematic draw of an asymmetric dual-phase membrane. | ||
Already in this early work Chen et al. highlighted the importance of obtaining continuous percolative pathways for both electrons and ions by comparing results from 40% and 30% of Pd metallic phase in a composite with yttrium stabilized zirconia (YSZ).5,74 This made the difference between forming a percolative or non-percolative network through the membrane, respectively. The total conductivity of the YSZ–30%Pd composite was about one order of magnitude smaller than that of the YSZ, while the conductivities of the YSZ–40%Pd composites were up to three orders of magnitude higher than that of YSZ ceramic at 900 °C.5 The lack of continuous phase blocks the electrons when they find the ionic YSZ phase, and they are restricted to move via electron–hole hopping through the lattice. In terms of oxygen permeability, the percolative composite showed a JO2 value two orders of magnitude larger than that of the non-percolative composite YSZ–30%Pd.
The ionic conductivity of 25 mol% Er2O3-stabilized bismuth oxide (BE25) is more than two orders of magnitude higher than the one of conventional YSZ. Hence, BE25 has been tested as the ionic phase for composites in combination with metallic silver and gold.75 The increase in oxygen permeability also is two orders of magnitude over that of YSZ composite, while all three mentioned composites are bulk controlled. On the other hand, the nature of the noble metal influences the oxygen permeability. Silver is a very good electrode material promoting the oxygen exchange process at the surface of the composite, whereas gold is inert to the surface process and it becomes the rate limiting step. Pd and Pt are also good catalytic metals that can enhance the surface reactions,76 but their cost may be prohibitive in industrial applications.
Similarly, Kim et al. made dual-phase membranes based on Bi1.5Y0.3Sm0.2O3 (BYS)–Ag.73 They added samarium oxide in order to improve the chemical stability with regard to yttria stabilized bismuth oxide (BY). The electrical conductivity of the BYS–Ag 40% membrane was 104 to 105 times higher than the one of BYS–30%Ag membrane and showed the typical metallic behavior, i.e. the electrical conductivity decreased with increasing temperature, when the electron mobility is reduced. However, both BYS–Ag showed improved oxygen permeation fluxes of 10 and more than 50 times compared to that of the BYS membrane. This indicates that not only an improvement arising from the existence of a continuous electronic network is achieved, but also the non percolative spread silver can catalyze the oxygen exchange process at the surface of BYS.73
By doping the bismuth oxide with alkaline earth oxides, good oxide ion conductors can be achieved. BaO reacts with Bi2O3 to form rhombohedral layered structures BaBi8O13. At room temperature, this rhombohedral phase has relatively low ionic conductivity. On heating, BaBi8O13 undergoes a phase transition (568 °C), which make it a very good oxide ion conductor. 25 vol% of Ag was used to form a cermet composite membrane, which showed high oxygen permeation fluxes in the temperature range of 570–700 °C and are limited by surface reaction rates.77 SrO stabilized bismuth oxide possesses high ionic conductivity at temperatures of 600–800 °C, but the absence of electronic conductivity and the low rate of oxygen exchange across the interface between the gas and solid phase prevents good oxygen permeation through this material. The dense dual-phase composite membrane made from strontium-stabilized bismuth oxide and silver, (Bi2O3)0.74(SrO)0.26–(BSO)/40%Ag improved the oxygen permeability, being rate-limited by oxygen-ion conduction through the oxide phase of the composite instead. As it could be expected from bismuth oxide, a dramatic change of oxygen flux occurs in the range of 680–700 °C, which corresponds to the phase transformation of the bismuth oxide, limiting the operation condition of this composite.69
More recently, membranes combining metals with doped-ceria fluorite-structured ceramics have been reported, e.g. CGO–14%Ag–CuO composite. This composite showed similar oxygen diffusivity and thermal expansion to CGO combined with much higher surface exchange coefficient, pointing out that it could be a suitable material for oxygen permeation membranes although the maximum operation temperature has to be considered.78
To summarize, cermet membranes have been studied and have demonstrated that with a continuous pathway for ionic and electronic conduction and large catalytic activity towards oxygen exchange, pure oxygen can be produced. However, (i) the high costs arising from the use of noble metals, (ii) the common mismatch of the TECs between the ceramic and the metallic materials, and (iii) the relatively poor oxygen permeability limit the application of cermet membranes at industrial level.
2.2.2.1 Potential ionic conductors for dual-phases OTMs. Materials for which the electrical conduction is predominately by ions, rather than by electrons and holes (negligible electronic conductivity) are considered ionic conductors. Reports the ionic conductivity of some of the most studied ceramic materials for oxygen transport. Fluorite-type oxides represent the most common ionic conducting materials. The fluorite structure is represented by AO2, where A is a large tetravalent cation such as Zr4+ or Ce4+. Oxides based on Zr or Ce have been particularly investigated for use as ionic conductors in dual-phase OTM materials.
At room temperature, ZrO2 has a monoclinic crystal structure. Nevertheless, when the temperature increases, the crystal structure of ZrO2 transforms to the tetragonal (>1000 °C) and cubic structures (>2300 °C).80 The cubic ZrO2 presents the advantage of having a higher ionic conductivity than the monoclinic crystal structure. Divalent or trivalent cation oxides can be added to pure ZrO2 in order to stabilize the cubic phase at room temperature.80–82 In addition, lower valent substituents lead to the formation of oxygen vacancies for charge compensation leading to high oxygen ion conductivity.
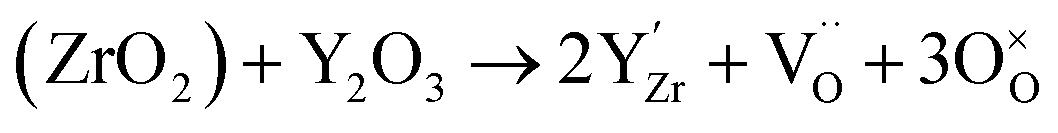 | (15) |
Yttria-doped zirconia has been particularly investigated as an OTM material and as an electrolyte material for SOFCs due to its high ionic conductivity, its thermodynamic stability in oxidizing and reducing atmospheres and its good mechanical properties.83 The highest ionic conductivity among (ZrO2)1−x(Y2O3)x materials is obtained for x = 0.08 (8YSZ), with 0.03 S cm−1 at 850 °C.84 Further addition of yttria will decrease the ionic conductivity due to enhanced association of the oxygen vacancies and dopant cations, which results in defective complexes with low mobility.85 During the past decades, other zirconia-based oxide ion conductors consisting of aliovalent dopants substituting zirconia such as (ZrO2)1−x–(M2O3)x (M = Sc,86–97 Yb,87,89,91,92,94,95,97 Gd,89,91,92,94,97 Dy,89,91,92,94,97 Eu,89,91,92,94 Er,87,91,92,94,97 Nd,87,94 La,87,94 Sm,87,94 Ce,86,91 Ho,94 Pr,94 Tb,94 Lu94) and ternary systems of two oxides co-doping zirconia like (ZrO2)1−(x+y)–(M2O 3)x–(M′2O3)y have been studied. Artemov et al. reported that the ionic conductivity of (ZrO2)0.89(Sc2O3)0.10(Y2O3)0.01 corresponds to 0.12 S cm−1 at 850 °C,98 which makes it a significantly better ionic conductor than 8YSZ (Fig. 6).
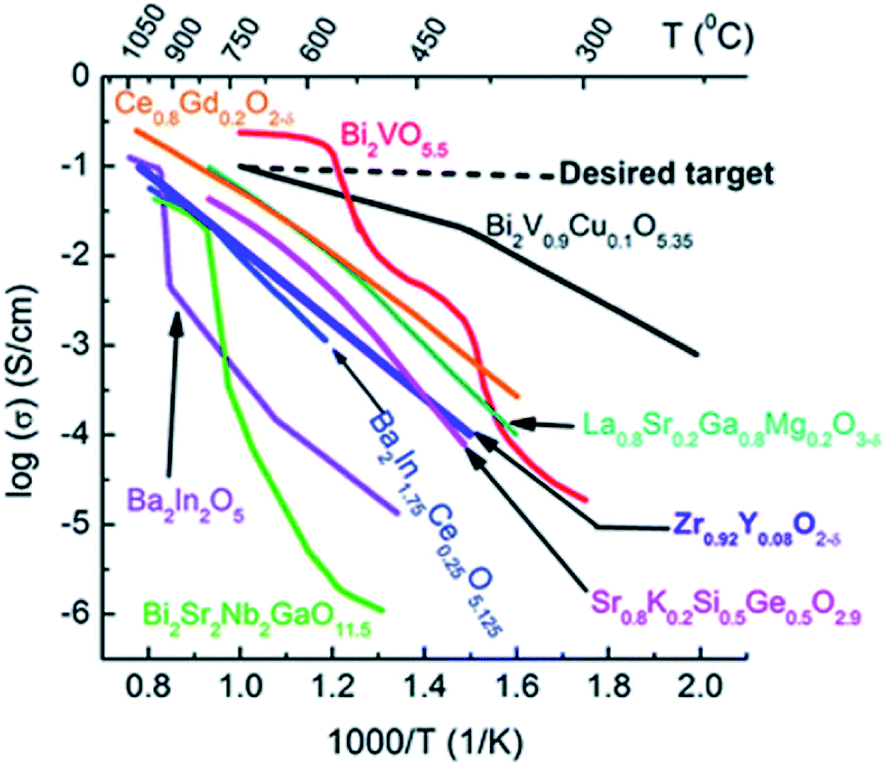 | ||
| Fig. 6 Oxide ion conductivities of some materials as a function of the temperature.99 Copyright 2016 Royal Society of Chemistry (RSC) Publishing, Open access. | ||
Unlike ZrO2, CeO2 has a stable cubic fluorite structure at room temperature and, therefore, the structure does not need to be stabilized. However, the partial substitution of Ce4+ by divalent or trivalent ions is desired because it creates oxygen vacancies in the structure, due to the lower valence of the doping ions compared with Ce4+.100–102 Over the past decades, many substitutions of ceria with alkaline earth or rare earth oxides have been researched in order to increase its ionic conductivity. Systems based on CeO2–M2O3 (M = Gd,87,89–91,102–105 Sm,89,90,102–104,106 Y,89,90,102,104 Dy,104 Nd,104 Eu,104 Yb,104 La,90,102,104 Sc102) and CeO2–M′O (M′ = Mg,102,104 Sr,102,104 Ba,102,104 Ca89) were reported. Gadolinia and Samaria-doped ceria show the highest conductivities among the doped ceria materials. The high performances were attributed to the good match in ionic radii.107Fig. 7 shows the dependence of the ionic conductivity on ionic radius of M3+ for (CeO2)0.8(M2O3)0.2 systems at 800 °C. The oxygen vacancies make CGO one of the fastest oxide ion conductors, in spite of being an electronic conductor at high temperature in reducing atmosphere. Ionic conductivities of 0.06 S cm−1 and 0.078 S cm−1 were found at 800 °C and 850 °C for a dopant level of 20 mol% Gd (Ce0.8Gd0.2O2−δ), respectively.108–110 At intermediate temperatures, e.g. 500 °C, doped ceria shows superior conductivity as compared to zirconia materials.111
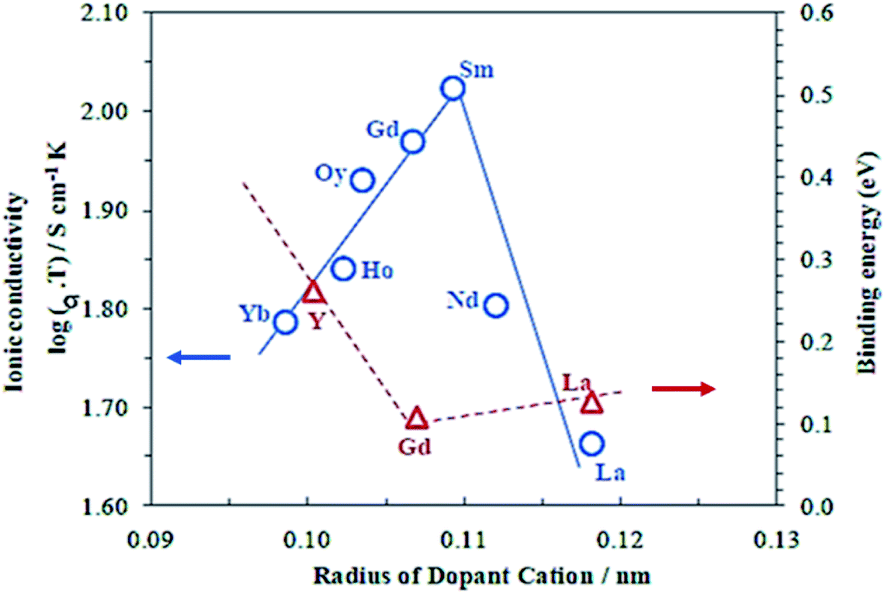 | ||
| Fig. 7 Dependence of the ionic conductivity on ionic radius of M3+ for (CeO2)0.8(M2O3)0.2 systems at 800 °C. Reproduced with permission.89 Copyright 2003, Elsevier. | ||
Bi2O3-doped metal oxides are another class of ionic conductors with high conductivity in comparison to doped ZrO2 and CeO2.112 Nevertheless, most bismuth oxide materials have extremely poor strength and tend to reduce in low partial pressure atmospheres, making them unsuitable for the intended industrial applications.97,113 The stability can be improved by the addition of vanadium, forming an aurivillius structural phase, which also facilitates metal doping to increase the ionic conductivity.
2.2.2.2 Possible electronic conductors for dual-phases OTMs. Electronic conducting oxides have been investigated for dual-phase membranes as replacements for the expensive noble metals originally used. These oxides can mainly be grouped in two categories of materials: spinel oxides type and perovskites.
2.2.2.2.1 Electronic conducting oxides with spinel structure. Oxide spinels are described by the general formula AB2O4, where in most of the cases A and B are tetrahedral and octahedral cation sites in a cubic close packing of oxygen. The conductivity of spinels originates from “hopping” of charges between octahedral sites.114 Consequently, the presence of aliovalent octahedral cations is beneficial to conduction. Fe3O4 has a conductivity over 100 S cm−1 at room temperature, among the highest for this group of material.115 On this basis, ferrite, manganite and cobaltite families of spinels are the best-suited as high-temperature conductors because of their multiple valence states. Petric et al. showed that among a selection of twenty-six spinels, MnCo2O4 and Mn1.7Cu1.3O4 present the two highest electrical conductivities with 60 S cm−1 at 800 °C and 225 S cm−1 at 750 °C, respectively.115 On the other hand, aluminium and chromium spinels are not good conductors because of their strong octahedral sites preferring valence states of Al3+ and Cr3+. A general drawback for this class of materials is their instability in low pO2 environments, which could limit the possible application areas (see Chapter 5).
2.2.2.2.2 Electronic conducting oxides with perovskite structure. Over the past decades, perovskites consisting of La as A-site cation and transition metals (Cr, Mn, Fe, Co and Ni) as B-site cation have been extensively investigated.116–121 Manganites, ferrites, nickelates and cobaltites present higher electrical conductivities than chromates but also a lower stability in reducing atmospheres, which is one of the key requirement for some OTM applications (e.g. partial oxidation of methane into syngas).122 Due to their excellent stability also in low pO2 atmospheres, lanthanum chromite-based materials are also considered as interconnect materials for SOFCs.123–129 Moreover, the electrical conductivity of LaCrO3 (0.6–1.0 S cm−1 at 1000 °C)122 can be improved by using suitable dopants. Dopants may also help to densify the lanthanum chromite-based materials, which are usually difficult to sinter. A-site and B-site dopants can be introduced to LaCrO3, forming the derivative chromates with the general formula La1−xAxCr1−yMyO3−δ, where A is typically an alkaline earth metal (A = Sr, Ca) and M is a transition metal (M = Mn, Fe, Co, Ni, Ti, Cu and Al). A-site dopants are generally acceptor type, and they enhance densification and improve the electrical conductivity.130–134 Previous studies showed that calcium and strontium doping on the A-site enhance the LaCrO3 sintering by the formation of liquid phase (CaCrO4/SrCrO4) which dissolves back into the lattice with increase in temperature.130–134 Accordingly, LaCrO3 has a relative low density of 50% at 1450 °C, while (La0.6Ca0.4)1.02CrO3 is 95% dense already at 1350 °C.135 B-site dopants also enhance the conductivity but also maintain the thermal and crystal structure stability.122 For example, the substitution of Cr with aliovalent transition metals like Cu and Ni was used to increase the electronic conductivity of the lanthanum chromite while keeping the thermal and chemical stability of the material.120
 are in contact.
are in contact.
There are several strategies for increasing the number of TPB to overcome the surface exchange limitations. On the one hand, enlarging the surface specific area by the roughness of the dense membrane increases the number of active sites for the incorporation or release of O2. This can be done, for example, by chemical etching136,137 or by the deposition of a porous layer.26 On the other hand, the distribution of a catalyst for the ORR along the membrane surface boosts the surface exchange rate. The most effective and therefore most common method of increasing the TPB number and the surface exchange coefficient (kex) is to combine both strategies in such way that the porous layer is made of MIEC catalytic material with further particle catalytic load.
There are several known elements and compounds that promote the surface exchange reactions (adsorption, dissociation, recombination, desorption). Noble metals have been used for the ORR reactions, like Pt and Pd.138,139 However, the high price, limited accessibility and proneness to poisoning prevent their commercial use and they are limited to laboratory measurements.
To decrease the cost of the catalytic layers, other metal oxides and compounds have been tested following similar strategies as those for SOFC cathodes. Perovskites made of LaMO3 (where M is a transition metal Fe, Co, Ni, Cr or Mn) are high p-type electronic conductors. These La perovskites are the state-of-the-art materials for the promotion of oxygen surface reactions and the selection of the dopants will also depend on the chemical compatibility with the membrane material, the stability in the operation atmosphere for a determined application and on the mechanical similarities in terms of chemical expansion coefficient.140,141 La1−xSrxMnO3 (LSM) is still the material of choice in applications at high temperature. In order to improve the performance of ORR, ion conducting materials are commonly added to form dual-phase composites as for the bulk membranes, preferably with the materials composing the membrane to ensure the compatibility.142,143 Regarding the activation of OTMs with dual-phase materials, formulations considering perovskite/fluorite and spinel/fluorite composites are amongst the most considered. With respect to the first, LSM-based structures such as LSM–CGO and LSM–YSZ have been used for the activation of OTMs in several works.144–146 Other examples of perovskite/fluorite catalyst activation are Ce0.8Sm0.2O1.9–La0.6Sr0.4Co0.2Fe0.8O3−δ (SDC–LSCF)147 and Ce0.8Gd0.2O2−x–LaCo0.2Ni0.4Fe0.4O3−x (CGO–LCNF).148 A similar strategy has been undertaken by combining fluorites and spinels, resulting in MIEC porous structures such as (Y2O3)0.01(Sc2O3)0.10(ZrO2)0.89–MnCo2O4 (10Sc1YSZ–MCO),149 (Y2O3)0.08(ZrO2)0.92–MnCo2O4 (8YSZ–MCO)149 and Fe2NiO4–Ce0.8Tb0.2O2−δ (NFO–CTO).150 Moreover, oxygen permeation rate was enhanced by further activating the porous NFO–CTO layers with Pr6O11 on both sides of the membrane.150
The catalysts are traditionally distributed throughout the porous layer by several approaches, e.g. particle deposition over membrane surface,151 or by means of infiltration in porous backbones.152 For example, asymmetric CGO membranes with 2% mol. of Co and surfaces activated with Pd nano-particles for oxy-fuel and chemical production applications.153 Atomic layer deposition (ALD) was used to deposit Pt and (Mn0.8Co0.2)3O4 layer onto the surface of porous LSM–YSZ backbone, thus extending the active zone of triple-phase boundary to the entire internal surface of the LSM–YSZ backbone.154 Other literature reports on a cobalt-free multi-phase nanocomposite in which tailored decomposition of the nominal compound could improve the surface reactions rate of a membrane. Such a nanocomposite combines Sr0.9Ce0.1Fe0.8Ni0.2O3−δ as a single perovskite main phase (77.2 wt%) and a second layered Ruddlesden–Popper phase (13.3 wt%), and minor phases surface-decorating with NiO (5.8 wt%) and CeO2 (3.7 wt%) minor phases.155
Lately, in situ nanocatalyst exsolution has emerged as a method for catalyst distribution in OTMs, especially when they are used in fuel cells and in catalytic membrane reactors.156–159 The main feature of this technique is that the metallic nanoparticle originates from the oxide lattice via the reduction of the oxide. The exsolved metallic nanocatalyst remains anchored to the bulk and homogeneously distributed over the surface of the grains. By controlling the redox process, it is possible to tune the amount and size of the nanoparticles. It has several advantages over the traditional impregnation or infiltration methods.160,161 Since the nano-catalyst particles are attached to the bulk oxide particles, agglomeration through cycling is avoided.
| Materials | Geom. | L (μm) | Flux (μmol cm−2 s−1) | T (°C) | Atm. pOfeed2/pOpermeate2 | Ref. | |
|---|---|---|---|---|---|---|---|
| 1 | BaBi8O13–Ag | Planar | 1500 | 0.078 | 650 | Air/0.000015 | 77 |
| 2 | Bi1.5Er0.5O3–Ag | Planar | 1600 | 0.119 | 800 | Air/0.026 | 76 |
| Planar | 230 | 0.107 | 700 | Air/He | 163 | ||
| Planar | 230 | 0.159 | 750 | Air/He | |||
| Planar | 230 | 0.209 | 800 | Air/He | |||
| Planar | 230 | 0.283 | 830 | Air/He | |||
| Planar | 230 | 0.309 | 852 | Air/He | |||
| 3 | Bi1.5Er0.5O3–Au | Planar | 1030 | 0.034 | 800 | Air/0.0015 | 76 |
| 4 | Bi1.48Sr0.52O3–Ag | Planar | 1000 | 0.050 | 700 | Air/0.0024 | 69 |
| 5 | Bi1.5Y0.3Sm0.2O3–Ag | Planar | 1300 | 0.58 | 850 | Air/0.009 | 73 |
| 6 | Bi1.5Y0.3Sm0.2O3–La0.8Sr0.2MnO3−δ | Hollow fiber | 290 | 0.39 | 850 | Air/He | 164 |
| Hollow fiber | 290 | 0.013 | 650 | Air/He | |||
| 7 | CeO2–La0.2Sr0.8CoO3 | Tubular | 10 | 0.007 | 850 | Air/He | 165 |
| 8 | Ce0.8Gd0.2O1.9–Ba0.95La0.05FeO3−δ | Planar | 1000 | 0.224 | 925 | Air/He | 166 |
| Planar | 400 | 0.313 | 850 | Air/He | |||
| Planar | 400 | 0.508 | 925 | Air/He | |||
| 9 | Ce0.8Gd0.2O1.9–Ba0.95La0.05Fe0.9Nb0.1O3−δ | Planar | 1000 | 0.146 | 925 | Air/CO2 | |
| 10 | Ce0.8Gd0.2O1.9–CoFe2O4 | Planar | 1000 | 0.006 | 700 | 0.21/0.0001 | 167 |
| Planar | 1000 | 0.135 | 950 | Air/He | 110 | ||
| 11 | Ce0.8Gd0.2O2−δ–Cu0.6Ni0.4Mn2O4 | Planar | 800 | 0.076 | 900 | Air/N2 | 168 |
| 12 | Ce0.8Gd0.2O2−δ–FeCo2O4 | Planar | 1000 | 0.082 | 850 | Air/Ar | 16 |
| 13 | Ce0.8Gd0.2O2−δ–GdBaCo2O5+δ | Planar | 620 | 0.21 | 950 | Air/He | 169 |
| 14 | Ce0.8Gd0.2O2−δ–LaCo0.2Ni0.4Fe0.4O3−δ | Planar | 630 | 0.552 | 1000 | Air/Ar | 148 |
| 15 | Ce0.8Gd0.2O2−δ–La0.5Sr0.5Fe0.8Cu0.2O3−δ | Planar | 600 | 0.427 | 950 | Air/He | 170 |
| Planar | 600 | 0.261 | 950 | Air/CO2 | |||
| 16 | Ce0.8Gd0.2O1.9–La0.7Sr0.3MnO3 | Planar | 600 | 0.08 | 950 | Air/He | 171 |
| Planar | 1000 | 0.04 | 950 | Air/He | 172 | ||
| 17 | Ce0.8Gd0.2O1.9–La0.8Sr0.2Fe0.8Co0.2O3 | Planar | 1000 | 0.063 | 950 | Air/He (0.2/0.01) | |
| 18 | Ce0.8Gd0.2O2−δ–MnCo1.9Fe0.1O4 | Planar | 800 | 0.104 | 900 | Air/N2 | 168 |
| 19 | Ce0.8Gd0.2O2−δ–MnFe2O4 | Planar | 1000 | 2.68 | 1000 | Air/Ar+5% H2 | 173 |
| Planar | 250 | 13.4 | 1000 | Air/Ar+10% CH4 | |||
| 20 | Ce0.8Gd0.2O2−δ–PrBaCo2O3−δ | Planar | 1000 | 0.169 | 925 | Air/CO2 | 174 |
| 21 | Ce0.8Gd0.2O2−δ–PrBaCo1.5Fe0.5O3−δ | Planar | 1000 | 0.172 | 925 | Air/CO2 | |
| 22 | Ce0.8Gd0.2O2−δ–PrBaCoFeO3−δ | Planar | 1000 | 0.187 | 925 | Air/CO2 | |
| 23 | Ce0.8Gd0.2O2−δ–PrBaCo0.5Fe1.5O3−δ | Planar | 1000 | 0.271 | 925 | Air/CO2 | |
| Planar | 1000 | 0.342 | 925 | Air/He | |||
| Planar | 600 | 0.420 | 925 | Air/He | |||
| 24 | Ce0.8Gd0.2O2−δ–Pr0.6Sr0.4Co0.5Fe0.5O3−δ | Planar | 500 | 0.463 | 900 | Air/He | 175 |
| Planar | 500 | 0.351 | 900 | Air/CO2 | |||
| Planar | 700 | 0.373 | 900 | Air/He | 176 | ||
| Planar | 700 | 0.284 | 900 | Air/CO2 | |||
| Planar | 1000 | 0.216 | 900 | Air/He | 175 | ||
| Planar | 1000 | 0.149 | 900 | Air/CO2 | |||
| 25 | Ce0.8Gd0.2O2−δ–Pr0.6Sr0.4Co0.5Fe0.4Nb0.1O3−δ | Planar | 500 | 0.336 | 900 | Air/He | |
| Planar | 500 | 0.254 | 900 | Air/CO2 | |||
| Planar | 700 | 0.269 | 900 | Air/He | 176 | ||
| Planar | 700 | 0.224 | 900 | Air/CO2 | |||
| Planar | 1000 | 0.134 | 900 | Air/He | 175 | ||
| Planar | 1000 | 0.112 | 900 | Air/CO2 | |||
| 26 | Ce0.8Gd0.2O1.9–Sr0.8Gd0.2FeO3 | Planar | 500 | 0.55 | 950 | 0.21/0.005 | 177 |
| Planar | 500 | 0.25 | 850 | 0.21/0.005 | |||
| Planar | 1000 | 0.3 | 950 | 0.21/0.005 | |||
| Planar | 1000 | 0.14 | 850 | 0.21/0.005 | |||
| Planar | 500 | 3.41 | 950 | Air/syngas | |||
| 27 | Ce0.9Gd0.1O2−δ–Ag | Planar | 1000 | 0.011 | 700 | Air/Ar | 178 |
| Planar | 1000 | 0.13 | 700 | Air/CH4 | |||
| 28 | Ce0.9Gd0.1O1.95–Ag–CuO | Planar | 1000 | 0.103 | 800 | Air/N2 | 179 |
| 29 | Ce0.9Gd0.1O2−δ–Ba0.5Sr0.5Co0.8Fe0.2O3−δ | Planar | 500 | 0.812 | 875 | Air/He | 180 |
| Planar | 500 | 1.338 | 950 | Air/He | |||
| Planar | 500 | 0.5 | 950 | Air/CO2 | |||
| 30 | Ce0.9Gd0.1O2−δ–Fe2O3 | Planar | 500 | 0.2 | 1000 | Air/CO2 | 181 |
| 31 | Ce0.9Gd0.1O1.95–LaCoO3 | Planar | 1000 | 0.0742 | 800 | Air/N2 | 179 |
| 32 | Ce0.9Gd0.1O2−δ–La0.8Ca0.2FeO3−δ | Planar | 110 | 0.0866 | 900 | Air/He | 182 |
| Planar | 110 | 0.0791 | 900 | Air/CO2 | |||
| 33 | Ce0.9Gd0.1O1.95–La0.6Sr0.4CoO3−δ | Planar | 1000 | 0.153 | 800 | Air/N2 | 179 |
| 34 | Ce0.9Gd0.1O2−δ–La0.6Sr0.4Co0.2Fe0.8O3−δ | Planar | 21 | 3.88 | 800 | Air/He | 162 |
| Planar | 21 | 6.57 | 900 | Air/He | |||
| Planar | 21 | 10.45 | 1000 | Air/He | |||
| 35 | Ce0.9Gd0.1O1.95–(La0.6Sr0.4)0.99Co0.2Fe0.8O3−δ | Planar | 1000 | 0.136 | 800 | Air/N2 | 179 |
| 36 | Ce0.9Gd0.1O1.95–La0.75Sr0.25Cr0.97V0.03O3−δ | Planar | 1000 | 0.0448 | 800 | Air/N2 | |
| 37 | Ce0.9Gd0.1O1.95–La0.6Sr0.4FeO3−δ | Planar | 1000 | 0.0926 | 800 | Air/N2 | |
| Planar | 100 | 1.052 | 900 | Air/He | 183 | ||
| 38 | Ce0.9Gd0.1O2−δ–(La0.6Sr0.4)0.98FeO3−δ | Tubular | 15 | 0.75 | 900 | 0.21/0.01 | 184 |
| Tubular | 10 | 1.567 | 850 | Air/N2 | 185 | ||
| Tubular | 10 | 11.12 | 850 | Air/H2 | |||
| 39 | Ce0.9Gd0.1O2−δ–La0.7Sr0.3MnO3−δ | Planar | 30 | 1.64 | 850 | Air/He | 186 |
| 40 | Ce0.9Gd0.1O2−δ–NiFe2O4 | Planar | 500 | 0.19 | 950 | Air/He | 187 |
| Planar | 500 | 0.16 | 950 | Air/CO2 | |||
| 41 | Ce0.9Gd0.1O2−δ–SrCo0.8Fe0.1Nb0.1O3−δ | Planar | 1000 | 0.36 | 900 | Air/He | 188 |
| Planar | 600 | 0.6 | 900 | Air/He | |||
| 42 | Ce0.9Gd0.1O1.95–Zn0.96Al0.02Ga0.02O1.02 | Planar | 1100 | 0.16 | 860 | Air/N2 | 189 |
| Planar | 1100 | 0.4 | 940 | Air/N2 | |||
| 43 | Ce0.85Gd0.1Cu0.05O2−δ–La0.6Ca0.4FeO3−δ | Planar | 500 | 0.52 | 950 | Air/CO2 | 190 |
| Planar | 500 | 0.65 | 950 | Air/He | |||
| 44 | Ce0.8Gd0.15Cu0.05O2−δ–SrFeO3−δ | Planar | 500 | 0.63 | 900 | 0.9/CO2 | 191 |
| Planar | 500 | 0.42 | 900 | Air/He | |||
| 45 | Ce0.8Gd0.1Pr0.1O2−δ–CoFe2O4 | Hollow fiber | 200 | 0.209 | 900 | Air/He | 192 |
| Hollow fiber | 200 | 0.44 | 950 | Air/He | |||
| Hollow fiber | 200 | 0.657 | 1000 | Air/He | |||
| Hollow fiber | 200 | 0.299 | 950 | Air/CO2 | |||
| 46 | Ce0.8La0.2O2−δ–La0.5Sr0.5Fe0.8Cu0.2O3−δ | Planar | 600 | 0.149 | 950 | Air/He | 170 |
| Planar | 600 | 0.067 | 950 | Air/CO2 | |||
| 47 | Ce0.75Nd0.25O1.875–Nd1.8Ce0.2CuO4 | Planar | 600 | 0.2 | 900 | 0.1/0.003 | 193 |
| Planar | 1030 | 0.12 | 900 | 0.1/0.003 | |||
| Planar | 1030 | 0.07 | 900 | 0.1/0.01 | |||
| Planar | 1030 | 0.04 | 850 | 0.1/0.01 | |||
| Planar | 1030 | 0.02 | 800 | 0.1/0.01 | |||
| 48 | Ce0.8Nd0.2O2−δ–La0.5Sr0.5Fe0.8Cu0.2O3−δ | Planar | 600 | 0.261 | 950 | Air/He | 170 |
| Planar | 600 | 0.09 | 950 | Air/CO2 | |||
| 49 | Ce0.8Nd0.2O2−δ–Nd0.5Sr0.5Fe0.8Al0.2O3−δ | Planar | 600 | 0.117 | 800 | Air/He | 194 |
| Planar | 600 | 0.337 | 900 | Air/He | |||
| Planar | 600 | 0.743 | 1000 | Air/He | |||
| Planar | 600 | 0.022 | 800 | Air/CO2 | |||
| Planar | 600 | 0.177 | 900 | Air/CO2 | |||
| Planar | 600 | 0.606 | 1000 | Air/CO2 | |||
| 50 | Ce0.9Nd0.1O2−δ–Nd0.6Sr0.4CoO3−δ | Planar | 400 | 0.418 | 900 | Air/He | 195 |
| Planar | 400 | 0.672 | 1000 | Air/He | |||
| Planar | 600 | 0.41 | 950 | Air/CO2 | |||
| 51 | Ce0.9Nd0.1O2−δ–Nd0.6Sr0.4FeO3−δ | Planar | 600 | 0.358 | 950 | Air/CO2 | 196 |
| 52 | Ce0.9Nd0.1O2−δ–Nd0.6Sr0.4Fe0.8Al0.2O3−δ | Planar | 600 | 0.058 | 800 | Air/He | 194 |
| Planar | 600 | 0.199 | 900 | Air/He | |||
| Planar | 600 | 0.455 | 1000 | Air/He | |||
| Planar | 600 | 0.015 | 800 | Air/CO2 | |||
| Planar | 600 | 0.116 | 900 | Air/CO2 | |||
| Planar | 600 | 0.389 | 1000 | Air/CO2 | |||
| 53 | Ce0.8Pr0.2O2−δ–La0.5Sr0.5Fe0.8Cu0.2O3−δ | Planar | 600 | 0.418 | 950 | Air/He | 170 |
| Planar | 600 | 0.112 | 950 | Air/CO2 | |||
| 54 | Ce0.85Pr0.1Cu0.05O2−δ–Pr0.4Sr0.6Fe0.8Cu0.2O3−δ | Planar | 600 | 1.06 | 1000 | Air/He | 197 |
| Planar | 600 | 0.687 | 1000 | Air/CO2 | |||
| 55 | Ce0.85Pr0.1Cu0.05O2−δ–Pr0.4Sr0.6Fe0.9Cu0.1O3−δ | Planar | 600 | 1.187 | 1000 | Air/He | |
| Planar | 600 | 0.709 | 1000 | Air/CO2 | |||
| 56 | Ce0.85Pr0.1Cu0.05O2−δ–Pr0.4Sr0.6Fe0.95Cu0.05O3−δ | Planar | 600 | 1.194 | 1000 | Air/He | |
| Planar | 600 | 0.731 | 1000 | Air/CO2 | |||
| 57 | Ce0.85Pr0.1Cu0.05O2−δ–Pr0.6Sr0.4Fe0.8Cu0.2O3−δ | Planar | 600 | 0.799 | 1000 | Air/He | |
| Planar | 600 | 0.351 | 1000 | Air/CO2 | |||
| 58 | Ce0.85Pr0.1Cu0.05O2−δ–Pr0.6Sr0.4Fe0.9Cu0.1O3−δ | Planar | 600 | 0.724 | 1000 | Air/He | |
| Planar | 600 | 0.463 | 1000 | Air/CO2 | |||
| 59 | Ce0.85Pr0.1Cu0.05O2−δ–Pr0.6Sr0.4Fe0.95Cu0.05O3−δ | Planar | 600 | 0.716 | 1000 | Air/He | |
| Planar | 600 | 0.604 | 1000 | Air/CO2 | |||
| 60 | Ce0.9Pr0.1O2−δ–La0.5Sr0.5Fe0.9Cu0.1O3−δ | Planar | 500 | 0.694 | 900 | Air/He | 198 |
| Planar | 500 | 0.53 | 900 | Air/CO2 | |||
| 61 | Ce0.9Pr0.1O2−δ–Mn1.5Co1.5O4−δ | Planar | 300 | 0.358 | 1000 | Air/CO2 | 199 |
| Planar | 300 | 0.276 | 950 | Air/CO2 | |||
| Planar | 500 | 0.164 | 1000 | Air/He | |||
| Planar | 500 | 0.149 | 1000 | Air/CO2 | |||
| 62 | Ce0.9Pr0.1O2−δ–Nd0.5Sr0.5Fe0.9Cu0.1O3−δ | Planar | 650 | 0.761 | 950 | Air/He | 200 |
| Planar | 650 | 0.470 | 950 | Air/CO2 | |||
| 63 | Ce0.9Pr0.1O2−δ–Pr0.6Ca0.4FeO3−δ | Planar | 300 | 0.433 | 900 | Air/He | 201 |
| Planar | 300 | 0.746 | 1000 | Air/He | |||
| Planar | 300 | 0.179 | 900 | Air/CO2 | |||
| Planar | 300 | 0.463 | 1000 | Air/CO2 | |||
| 64 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4FeO3−δ | Planar | 600 | 0.168 | 900 | Air/He | 202 |
| Planar | 600 | 0.343 | 1000 | Air/CO2 | |||
| Planar | 600 | 0.108 | 900 | Air/He | |||
| Planar | 600 | 0.211 | 1000 | Air/CO2 | |||
| 65 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.8Al0.2O3−δ | Planar | 330 | 0.769 | 1000 | Air/He | 203 |
| Planar | 600 | 0.485 | 950 | Air/He | |||
| Planar | 600 | 0.582 | 1000 | Air/He | |||
| Planar | 600 | 0.254 | 950 | Air/CO2 | |||
| Planar | 600 | 0.343 | 1000 | Air/CO2 | |||
| 66 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.6Al0.4O3−δ | Planar | 400 | 0.836 | 1000 | Air/He | 204 |
| 67 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.8Bi0.2O3−δ | Planar | 600 | 0.04 | 800 | Air/He | 205 |
| Planar | 600 | 0.075 | 900 | Air/He | |||
| Planar | 600 | 0.286 | 1000 | Air/He | |||
| Planar | 600 | 0 | 800 | Air/CO2 | |||
| Planar | 600 | 0.02 | 900 | Air/CO2 | |||
| Planar | 600 | 0.243 | 1000 | Air/CO2 | |||
| 68 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.85Bi0.15O3−δ | Planar | 600 | 0.031 | 800 | Air/He | |
| Planar | 600 | 0.11 | 900 | Air/He | |||
| Planar | 600 | 0.293 | 1000 | Air/He | |||
| Planar | 600 | 0 | 800 | Air/CO2 | |||
| Planar | 600 | 0.035 | 900 | Air/CO2 | |||
| Planar | 600 | 0.249 | 1000 | Air/CO2 | |||
| 69 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.9Bi0.1O3−δ | Planar | 600 | 0.046 | 800 | Air/He | |
| Planar | 600 | 0.13 | 900 | Air/He | |||
| Planar | 600 | 0.337 | 1000 | Air/He | |||
| Planar | 600 | 0.01 | 800 | Air/CO2 | |||
| Planar | 600 | 0.043 | 900 | Air/CO2 | |||
| Planar | 600 | 0.274 | 1000 | Air/CO2 | |||
| 70 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.95Bi0.05O3−δ | Planar | 600 | 0.051 | 800 | Air/He | |
| Planar | 600 | 0.143 | 900 | Air/He | |||
| Planar | 600 | 0.344 | 1000 | Air/He | |||
| Planar | 600 | 0.025 | 800 | Air/CO2 | |||
| Planar | 600 | 0.043 | 900 | Air/CO2 | |||
| Planar | 600 | 0.302 | 1000 | Air/CO2 | |||
| 71 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.975Bi0.025O3−δ | Planar | 600 | 0.062 | 800 | Air/He | |
| Planar | 600 | 0.16 | 900 | Air/He | |||
| Planar | 600 | 0.387 | 1000 | Air/He | |||
| Planar | 600 | 0.034 | 800 | Air/CO2 | |||
| Planar | 600 | 0.061 | 900 | Air/CO2 | |||
| Planar | 600 | 0.336 | 1000 | Air/CO2 | |||
| 72 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.99Bi0.01O3−δ | Planar | 600 | 0.072 | 800 | Air/He | |
| Planar | 600 | 0.191 | 900 | Air/He | |||
| Planar | 600 | 0.527 | 1000 | Air/He | |||
| Planar | 600 | 0.025 | 800 | Air/CO2 | |||
| Planar | 600 | 0.067 | 900 | Air/CO2 | |||
| Planar | 600 | 0.463 | 1000 | Air/CO2 | |||
| 73 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.5Co0.5O3−δ | Planar | 500 | 0.179 | 800 | Air/He | 206 |
| Planar | 500 | 0.44 | 900 | Air/He | |||
| Planar | 500 | 0.806 | 1000 | Air/He | |||
| Planar | 500 | 0.082 | 800 | Air/CO2 | |||
| Planar | 500 | 0.34 | 900 | Air/CO2 | |||
| Planar | 500 | 0.754 | 1000 | Air/CO2 | |||
| 74 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.9In0.1O3−δ | Planar | 600 | 0.275 | 900 | Air/He | 202 |
| Planar | 600 | 0.556 | 1000 | Air/He | |||
| Planar | 600 | 0.166 | 900 | Air/CO2 | |||
| Planar | 600 | 0.423 | 1000 | Air/CO2 | |||
| 75 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.925In0.075O3−δ | Planar | 600 | 0.221 | 900 | Air/He | |
| Planar | 600 | 0.536 | 1000 | Air/He | |||
| Planar | 600 | 0.108 | 900 | Air/CO2 | |||
| Planar | 600 | 0.358 | 1000 | Air/CO2 | |||
| 76 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.95In0.05O3−δ | Planar | 600 | 0.168 | 900 | Air/He | |
| Planar | 600 | 0.518 | 1000 | Air/He | |||
| Planar | 600 | 0.108 | 900 | Air/CO2 | |||
| Planar | 600 | 0.246 | 1000 | Air/CO2 | |||
| 77 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.975In0.025O3−δ | Planar | 600 | 0.128 | 900 | Air/He | |
| Planar | 600 | 0.536 | 1000 | Air/He | |||
| Planar | 600 | 0.108 | 900 | Air/CO2 | |||
| Planar | 600 | 0.408 | 1000 | Air/CO2 | |||
| 78 | Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.99In0.01O3−δ | Planar | 600 | 0.36 | 900 | Air/He | |
| Planar | 600 | 0.799 | 1000 | Air/He | |||
| Planar | 600 | 0.166 | 900 | Air/CO2 | |||
| Planar | 600 | 0.597 | 1000 | Air/CO2 | |||
| 79 | Ce0.8Sm0.2O2−δ–Ba0.95La0.05Zr0.1Fe0.5Co0.4O3−δ | Planar | 1000 | 0.313 | 925 | Air/He | 207 |
| 80 | Ce0.8Sm0.2O1.9–LaBaCo2O5 | Planar | 600 | 0.46 | 950 | 0.21/0.005 | 208 |
| 81 | Ce0.8Sm0.2O2−δ–La0.7Ca0.3CrO3−δ | Planar | 1000 | 0.11 | 950 | Air/He | 209 |
| 82 | Ce0.8Sm0.2O2−δ–La0.5Sr0.5Fe0.8Cu0.2O3−δ | Planar | 600 | 0.251 | 950 | Air/He | 170 |
| Planar | 600 | 0.149 | 950 | Air/CO2 | |||
| 83 | Ce0.8Sm0.2O1.9–La0.8Sr0.2CrO3 | Planar | 300 | 0.14 | 950 | 0.21/0.0092 | 210 |
| Tubular | 1100 | 0.86 | 950 | Air/CO (0.21/10−15) | 211 | ||
| 84 | Ce0.8Sm0.2O2−δ–La0.9Sr0.1FeO3−δ | Planar | 1100 | 0.642 | 900 | Air/CO | 212 |
| Planar | 1100 | 0.159 | 950 | Air/CO2 | |||
| Planar | 1100 | 0.159 | 950 | Air/He | |||
| 85 | Ce0.8Sm0.2O1.9–La0.8Sr0.2Cr0.5Fe0.5O3−δ | Hollow fiber | 240 | 0.005 | 750 | Air/He | 213 |
| Hollow fiber | 240 | 0.084 | 850 | Air/He | |||
| Hollow fiber | 240 | 0.362 | 950 | Air/He | |||
| Hollow fiber | 240 | 0.003 | 750 | Air/CO2 | |||
| Hollow fiber | 240 | 0.047 | 850 | Air/CO2 | |||
| Hollow fiber | 240 | 0.362 | 950 | Air/CO2 | |||
| Hollow fiber | 240 | 0.773 | 750 | Air/CO | |||
| Hollow fiber | 240 | 1.905 | 850 | Air/CO | |||
| Hollow fiber | 240 | 3.539 | 950 | Air/CO | |||
| 86 | Ce0.8Sm0.2O1.9–La0.8Sr0.2MnO3 | Hollow fiber | 300 | 0.32 | 950 | Air/He | 214 |
| Hollow fiber | 300 | 0.3 | 950 | Air/CO2 | |||
| 87 | Ce0.8Sm0.2O2−δ–PrBaCo2O5+δ | Planar | 600 | 0.238 | 925 | Air/He | 215 |
| 88 | Ce0.8Sm0.2O1.9–Sm0.8Ca0.2CoO3 | Planar | 500 | 0.172 | 950 | Air/He | 216 |
| Planar | 500 | 0.119 | 950 | Air/CO2 | |||
| 89 | Ce0.8Sm0.2O1.9–Sm0.6Ca0.4CoO3 | Planar | 500 | 0.41 | 950 | 0.21/0.0066 | 217 |
| 90 | Ce0.8Sm0.2O1.9–Sm0.8Ca0.2Co0.5Mn0.5O3 | Planar | 500 | 0.187 | 850 | 0.21/0.005 | 218 |
| Planar | 500 | 0.254 | 900 | 0.21/0.005 | |||
| Planar | 500 | 0.463 | 940 | 0.21/0.005 | |||
| 91 | Ce0.8Sm0.2O1.9–Sm0.6Ca0.4FeO3 | Planar | 500 | 0.336 | 950 | 0.21/0.006 | 217 |
| 92 | Ce0.8Sm0.2O1.9–Sm0.6Sr0.4FeO3−δ | Planar | 1000 | 0.224 | 950 | Air/He | 219 |
| Planar | 600 | 0.425 | 950 | Air/He | |||
| Planar | 420 | 0.507 | 950 | Air/He | |||
| Planar | 180 | 0.709 | 950 | Air/He | |||
| 93 | Ce0.8Sm0.2O2−δ–Sm0.6Sr0.4Fe0.7Al0.3O3−δ | Planar | 500 | 0.455 | 900 | Air/He | 220 |
| 94 | Ce0.8Sm0.2O3−δ–Sm0.3Sr0.7Fe0.8Cu0.2O3−δ | Planar | 600 | 0.776 | 950 | Air/He | 221 |
| Planar | 600 | 1.01 | 1000 | Air/He | |||
| Planar | 600 | 0.858 | 1000 | Air/CO2 | |||
| 95 | Ce0.8Sm0.2O3−δ–Sm0.5Sr0.5Fe0.8Cu0.2O3−δ | Planar | 600 | 0.754 | 950 | Air/He | 221 |
| Planar | 600 | 0.948 | 1000 | Air/He | |||
| Planar | 600 | 0.836 | 1000 | Air/CO2 | |||
| 96 | Ce0.8Sm0.2O2−δ–Sm0.6Sr0.4Fe0.8Cu0.2O3−δ | Planar | 500 | 0.575 | 900 | Air/He | 220 |
| 97 | Ce0.8Sm0.2O2−δ–SrCO3–Co3O4 | Planar | 500 | 0.694 | 900 | Air/He | 222 |
| 98 | Ce0.8Sm0.2O2−δ–SrCo0.9Nb0.1O3−δ | Planar | 800 | 1.15 | 950 | Air/He | 223 |
| Planar | 800 | 0.388 | 950 | Air/CO2 | |||
| 99 | Ce0.8Sm0.2O2−δ–Sr2Fe1.5Mo0.5O5+δ | Planar | 600 | 0.019 | 750 | Air/He | 224 |
| Planar | 600 | 0.077 | 850 | Air/He | |||
| Planar | 600 | 0.149 | 925 | Air/He | |||
| Planar | 600 | 0.011 | 750 | Air/CO2 | |||
| Planar | 600 | 0.043 | 850 | Air/CO2 | |||
| Planar | 600 | 0.116 | 925 | Air/CO2 | |||
| 100 | Ce0.8Sm0.2O1.9–Y0.8Ca0.2Cr0.8Co0.2O3 | Planar | 1300 | 0.23 | 950 | Air/N2 | 225 |
| 101 | Ce0.85Sm0.15O1.925–Sm0.6Sr0.4FeO3 | Planar | 500 | 0.34 | 950 | 0.21/0.005 | 226 |
| Planar | 500 | 2.7 | 950 | Air/Syngas | |||
| Planar | 160 | 0.746 | 950 | Air/He | 227 | ||
| 102 | Ce0.85Sm0.15O1.925–Sm0.6Sr0.4Al0.3Fe0.7O3 | Planar | 40 | 2.91 | 950 | Air/He | 228 |
| 103 | Ce0.9Sm0.1O1.95–MnCo1.5Ni0.5O4 | Planar | 300 | 1.1 | 1000 | Air/He | 229 |
| Planar | 300 | 7 | 1000 | Air/Ar,CH4 | |||
| 104 | Ce0.9Sm0.1O1.95–MnFe2O4 | Planar | 300 | 6 | 1000 | Air/Ar,CH4 | |
| Planar | 133 | 10 | 1000 | Air/Ar,CH4 | |||
| 105 | Ce0.8Sm0.15Bi0.05O2−δ–Sm0.6Sr0.4Fe0.7Al0.3O3−δ | Planar | 500 | 0.313 | 900 | Air/He | 220 |
| 106 | Ce0.8Sm0.15Bi0.05O2−δ–Sm0.6Sr0.4Fe0.8Cu0.2O3−δ | Planar | 500 | 0.522 | 900 | Air/He | |
| 107 | Ce0.8Sm0.1Bi0.1O2−δ–Sm0.6Sr0.4Fe0.8Cu0.2O3−δ | Planar | 500 | 0.597 | 900 | Air/He | |
| 108 | Ce0.8Sm0.05Bi0.15O2−δ–Sm0.6Sr0.4Fe0.8Cu0.2O3−δ | Planar | 500 | 0.619 | 900 | Air/He | |
| 109 | Ce0.8Tb0.2O2−δ–Fe2NiO4 | Planar | 600–700 | 0.104 | 850 | Air/Ar | 150 |
| 110 | Ce0.8Tb0.2O2−δ–NiFe2O4 | Planar | 680 | 0.15 | 1000 | Air/CO2 | 145 |
| 111 | Ce0.8Tb0.2O2−δ–NiFe2O4 + La0.6Sr0.4Co0.2Fe0.8O3−δ | Planar | 8 + 10 | 3.582 | 1000 | Air/Ar | 230 |
| Planar | 8 + 10 | 4.179 | 1000 | Air/CO2 | |||
| 112 | La0.15Sr0.85Ga0.3Fe0.7O3−δ–Ba0.5Sr0.5Fe0.2Co0.8O3−δ | Planar | 1990 | 0.352 | 915 | Air/He | 231 |
| 113 | (La0.9Sr0.1)0.98Ga0.8Mg0.2O3−δ–La2Ni0.8Cu0.2O4+δ | Planar | 650 | 0.027 | 900 | 0.21/0.013 | 232 |
| Planar | 1000 | 0.017 | 900 | 0.21/0.013 | |||
| 114 | (ZrO2)0.92(Y2O3)0.08–Boron doped MgLaCrOλ | Planar | 800 | 0.4 | 1100 | Air/H2 | 68 |
| 115 | (ZrO2)0.92(Y2O3)0.08–In0.9Pr0.1 | Planar | 800 | 2.30 | 1100 | Air/H2 | |
| Planar | 800 | 1.71 | 1100 | Air/CH4 | |||
| Planar | 300 | 5.50 | 1100 | Air/H2 | |||
| Planar | 300 | 4.09 | 1100 | Air/CH4 | |||
| Planar | 250 | 6.10 | 1100 | Air/H2 | |||
| 116 | (ZrO2)0.92(Y2O3)0.08–In0.95Pr0.025Zr0.025 | Planar | 300 | 5.80 | 1100 | Air/CH4 | |
| 117 | (ZrO2)0.92(Y2O3)0.08–La0.8Sr0.2CrO3−δ | Planar | 115 | 0.91 | 750 | Air/CO | 233 |
| Planar | 115 | 1.12 | 850 | Air/CO | |||
| 118 | (ZrO2)0.92(Y2O3)0.08–La0.7Sr0.3MnO3−δ | Planar | 100 | 0.194 | 850 | Air/He | 234 |
| Planar | 100 | 0.336 | 900 | Air/He | |||
| Planar | 100 | 0.403 | 950 | Air/He | |||
| Planar | 50 | 0.285 | 800 | Air/He | 235 | ||
| Planar | 50 | 0.535 | 850 | Air/He | |||
| Planar | 50 | 0.781 | 900 | Air/He | |||
| 119 | (ZrO2)0.92(Y2O3)0.08–Pd | Planar | 800 | 2.1 | 1100 | Air/H2 | |
| Planar | 800 | 1.56 | 1100 | Air/CH4 | |||
| 120 | (ZrO2)0.92(Y2O3)0.08–Pt | Planar | 800 | 1.8 | 1100 | Air/H2 | |
| Planar | 800 | 1.34 | 1100 | Air/CH4 | |||
| 121 | (ZrO2)0.92(Y2O3)0.08–SrCo0.4Fe0.6O3−δ | Planar | 1200 | 0.597 | 750 | 0.21/0.001 | 236 |
| Planar | 2000 | 0.269 | 850 | 0.21/0.001 | |||
| 122 | Zr0.8Y0.2O1.9–La0.8Sr0.2CrO3−δ | Tubular | 1230 | 0.0092 | 950 | Air/He | 237 |
| Tubular | 1230 | 0.032 | 930 | Air/He–CO (80–20%) | |||
| 123 | Zr0.8Y0.2O2−δ–La0.8Sr0.2Cr0.5Fe0.5O3−δ | Planar | 120 | 0.045 | 900 | Air/Ar | 238 |
| Planar | 120 | 0.131 | 900 | Air/H2 | |||
| Planar | 120 | 0.896 | 900 | Air/CO | |||
| 124 | Zr0.84Y0.16O1.92–La0.8Sr0.2Cr0.5Fe0.5O3−δ | Hollow fiber | 270 | 0.247 | 950 | Air/He | 239 |
| Hollow fiber | 270 | 3.37 | 950 | Air/CO | |||
| Planar | 30 | 1.791 | 900 | Air/CO | 240 | ||
| Planar | 20 | 1.65 | 950 | Air/CO | 241 | ||
| Planar | 5 | 1.455 | 900 | Air/CO | 242 | ||
| 125 | Zr0.84Y0.16O1.92–La0.8Sr0.2MnO3−δ | Planar | 150 | 0.19 | 900 | 0.21/0.002 | 243 |
| Hollow fiber | 160 | 0.21 | 950 | Air/He | 244 | ||
| 126 | Zr0.789Sc0.198Ce0.012O1.90–(La0.8Sr0.2)0.95Cr0.5Fe0.5O3−δ | Planar | 20 | 2.64 | 900 | Air/H2 | 245 |
| Planar | 200 | 0.552 | 900 | Air/H2 | |||
| Planar | 300 | 0.396 | 900 | Air/H2 | |||
| 127 | Zr0.79Sc0.2Ce0.01O2−δ–La0.7Sr0.3MnO3−δ | Planar | 40 | 1.231 | 900 | Air/He | 235 |
| Planar | 42.7 | 1.194 | 900 | Air/He | 246 | ||
| 128 | Zr0.802Sc0.18Y0.018O1.901–(La0.825Sr0.175)0.94Cr0.72Mn0.26V0.02O3−δ | Tubular | 20–30 | 0.664 | 900 | Air/H2–CO | 247 |
| 129 | (ZrO2)0.89(Sc2O3)0.10(Y2O3)0.01–LaCrO3 | Planar | 1000 | 0.05 | 900 | Air/N2 | 121 |
| 130 | (ZrO2)0.89(Y2O3)0.01(Sc2O3)0.10–LaCr0.85Cu0.10Ni0.05O3−δ | Planar | 1000 | 0.198 | 950 | Air/N2 | 120 |
| Planar | 1000 | 0.183 | 950 | Air/CO2 | |||
| Planar | 110 | 0.762 | 950 | Air/N2 | |||
| Planar | 110 | 0.743 | 950 | Air/CO2 | |||
| 131 | (ZrO2)0.89(Y2O3)0.01(Sc2O3)0.10–MnCo2O4 | Planar | 7 | 0.216 | 750 | Air/N2 | 248 |
| Planar | 7 | 0.366 | 800 | Air/N2 | |||
| Planar | 7 | 0.619 | 850 | Air/N2 | |||
| Planar | 7 | 0.94 | 900 | Air/N2 | |||
| Planar | 7 | 1.052 | 940 | Air/N2 | |||
| Planar | 7 | 0.291 | 750 | Air/CO2 | |||
| Planar | 7 | 0.321 | 800 | Air/CO2 | |||
| Planar | 7 | 0.388 | 850 | Air/CO2 | |||
| Planar | 7 | 0.493 | 900 | Air/CO2 | |||
| Planar | 7 | 0.604 | 940 | Air/CO2 | |||
| 132 | (ZrO2)0.89(Y2O3)0.01(Sc2O3)0.10–Zn0.98Al0.02O1.01 | Planar | 1000 | 0.246 | 925 | Air/N2 | 249 |
| Planar | 8 | 0.119 | 925 | Air/N2 |
Fig. 8 and 9 were plotted from data reported in literature and summarize the performance of planar and tubular/hollow fiber dual-phase membranes to transport oxygen. It is important to mention that comparing the performance of membranes manufactured and tested in different conditions is very difficult. Indeed, as described in the Section 1.3, multiple processes can be limiting the performance of the membranes. Consequently, many parameters such as: (i) the thickness of the dense and selective membrane layer, (ii) test set-up geometrical design as well as gas flow rates affecting the driving force applied across the actual membrane, (iii) the presence of catalytic layers to facilitate the oxidation and reduction of oxygen, etc., can greatly influence the performance of the membranes. In order to compare as fairly as possible the performances of dual-phase membranes, Fig. 8 and 9 summarize all dual-phase OTMs composed of a dense separation layer of 300 μm or thinner which were tested using air as a feed gas and an inert gas as a sweep gas (He, Ar, N2 or CO2) leading to a nominal pO2-gradient of approx. 0.21/10−5.
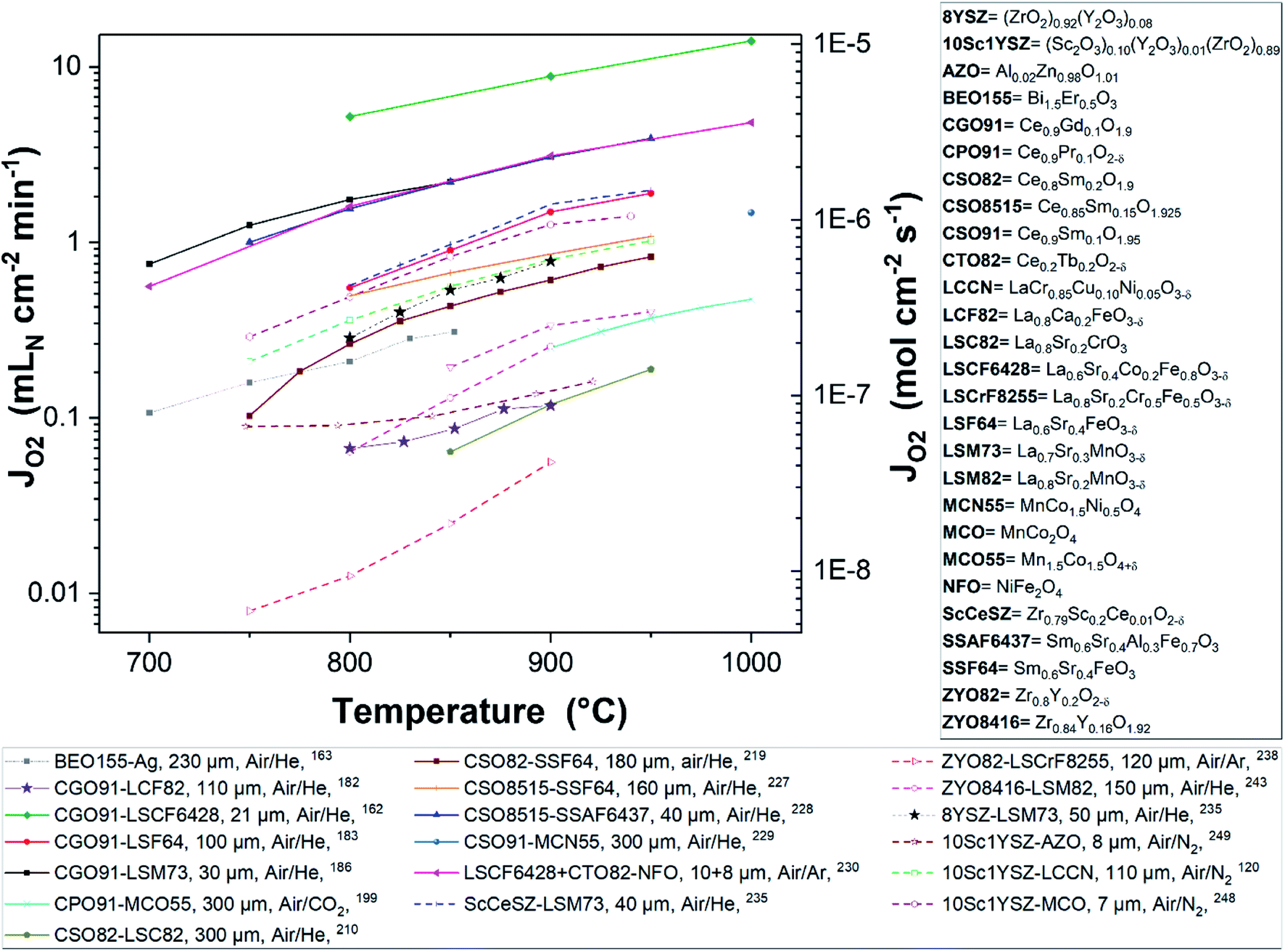 | ||
| Fig. 8 Oxygen permeation flux of various thin (L ≤ 300 μm) planar dual-phase OTMs as a function of the temperature. | ||
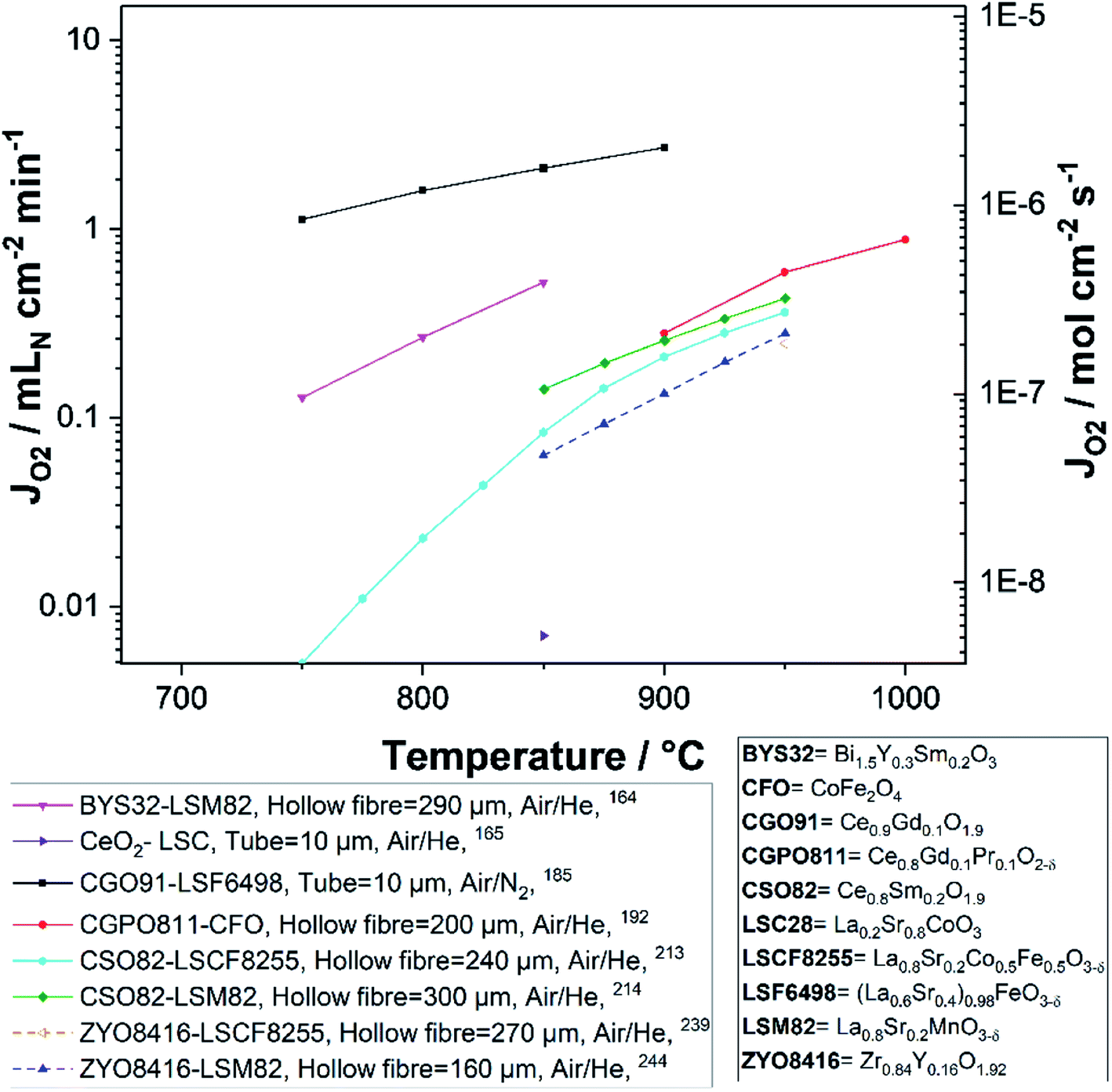 | ||
| Fig. 9 Oxygen permeation flux of various thin (L ≤ 300 μm) tubular and hollow fiber dual-phase OTMs as a function of the temperature. | ||
As shown in Fig. 8 and 9, most of the thin (L ≤ 300 μm) dual-phase OTMs reported in literature and tested using air as a feed gas and an inert gas as a sweep gas are planar (19 planar membranes, 7 tubular/hollow fiber membranes). In general, ceria-based membranes (solid lines) display higher oxygen fluxes than zirconia-based membranes (dashed lines). This is due to the fact that oxygen transport is typically limited by the ionic conductivity, hence the best ion conductors lead to the highest performance. It is a 21 μm thick Ce0.9Gd0.1O2−δ–La0.6Sr0.4Co0.2Fe0.8O3−δ dual-phase membrane (coated with Ba0.5Sr0.5Co0.8Fe0.2O3−δ porous catalytic layers) recently developed by Nam et al. that shows the highest reported oxygen permeation flux with 10.45 μmol cm−2 s−1 at 1000 °C in air/He.162 Among the zirconia-based membranes, the Zr0.79Sc0.2Ce0.01O2−δ–La0.7Sr0.3MnO3−δ, 10Sc1YSZ–MCO and 10Sc1YSZ–LaCr0.85Cu0.10Ni0.05O3−δ (LCCN) composites display the three highest oxygen permeation rates.
In literature, a few studies present long-term permeation tests of dual-phase OTMs developed for CO2 capture via oxy-fuel combustion. Pirou et al. manufactured and tested 7 μm thick 10Sc1YSZ–MnCo2O4 asymmetric membranes over 1730 hours in pure CO2.248 The study showed an initial degradation of 21% during the first 1100 hours, due to catalytic degradation, after which stable performance was achieved. The 10Sc1YSZ–MnCo2O4 (70/30 vol%) asymmetric membrane itself was considered stable in CO2 atmosphere and thus it could be a good candidate for use in industrial applications where contact with CO2 is required. The same research group manufactured 10Sc1YSZ–Al0.02Zn0.98O1.01 asymmetric membranes and performed a 900 h long-term electrical conductivity measurement under pure CO2. The test demonstrated the instability of the Al0.02Zn0.98O1.01 phase in very mildly reducing atmosphere leading to low permeation performances, which compromises possible industrial applications.249 Dual-phase membranes made of 60 wt% Ce0.9Pr0.1O2−δ–40 wt% Pr0.6Sr0.4Fe0.5Co0.5O3−δ were tested at 950–1000 °C for a total duration of about 500 h using pure He (for ≈40 h) and pure CO2 (for ≈460 h) as sweep gases. The study underlines the excellent stability of the membrane in CO2 and conclude that Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4Fe0.5Co0.5O3−δ is a promising composite membrane material for industrial applications dealing with oxy-fuel process for CO2 capture.250 Similarly, 75 wt% Ce0.8Sm0.2O1.9–25 wt% SmMn0.5Co0.5O3 and 75 wt% Ce0.8Sm0.2O1.9–25 wt% Sm0.8Ca0.2Mn0.5Co0.5O3 OTMs were tested for a total duration of 500 h (150 h in pure He + 350 h in pure CO2), showing limited oxygen permeation fluxes up to 0.40 ml min−1 cm−2 but good stability in CO2.218
Long-term operations for partial oxidation of methane (POM) were investigated on 75 wt% Ce0.85Sm0.15O1.925–25 wt% Sm0.6Sr0.4Fe0.7Al0.3O3−δ (SDC–SSFA) and 75 wt% Ce0.85Sm0.15O1.925–25 wt% Sm0.6Sr0.4FeO3−δ (SDC–SSF) dual-phase composite membranes. SDC–SSFA membranes were tested at 950 °C for 1100 h. Pure CH4 was used as feed gas. Throughout the operation, CH4 conversion and CO selectivity were greater than 98%.251 Similarly, SDC–SSF membranes were tested at 940 °C and for 500 h achieving CH4 conversion and CO selectivity > 98% for pure CH4 used as feed gas.226 At industrial level, Praxair, Inc. developed OTMs for carbon capture power systems and fuel synthesis applications. Fig. 10 presents the oxygen flux degradation of their ScYSZ–LSCF OTMs over about 7000 h in syngas reforming and non-reforming modes.252
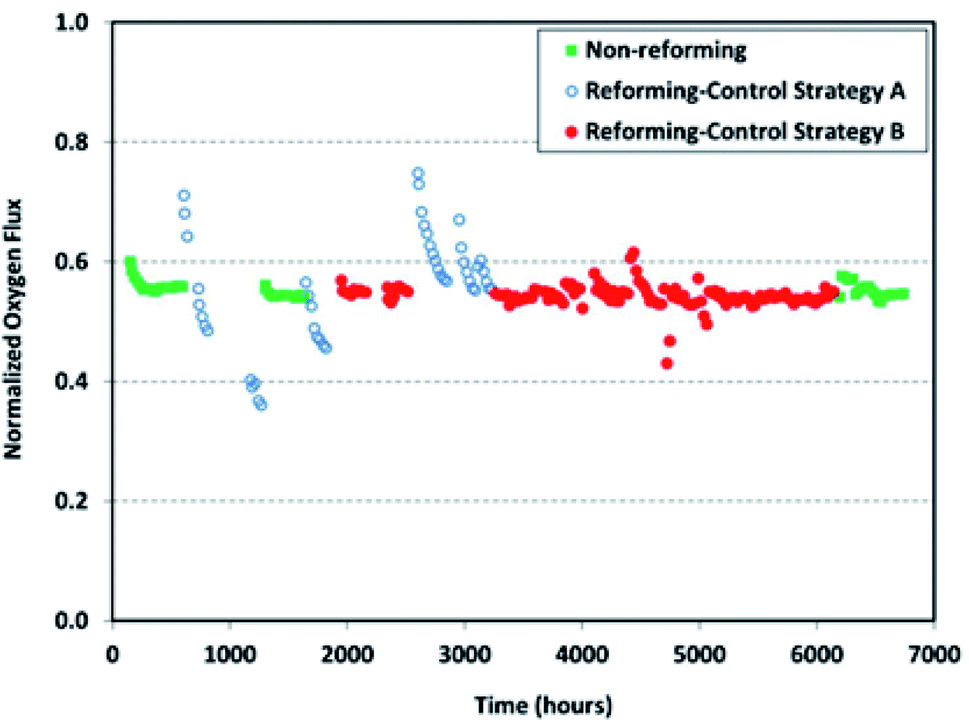 | ||
| Fig. 10 Long-term stability test of Praxair, Inc. OTMs in syngas reforming and non-reforming modes.252 | ||
Several other studies published short stability tests lasting between 200 h to 500 h.120,225,234,253–255 However, the small number of studies reporting long term stability tests over 500 h underlines that over the recent years research have mainly been focused on material selection and initial performance rather than stability. Consequently, there is a lack of knowledge in this domain for dual-phase OTMs.
3 Membrane geometry and architectures
Several different configurations, shapes and targeted operation conditions for membrane modules exist. Three architectures of OTM-membranes can be differentiated: single-hole tubes (“tubular”), plates (“planar”) and multi-channel monoliths (e.g. “honey-comb”). There are also crossovers of the aforementioned architectures, like the plate-and tube illustrated in Fig. 11 below.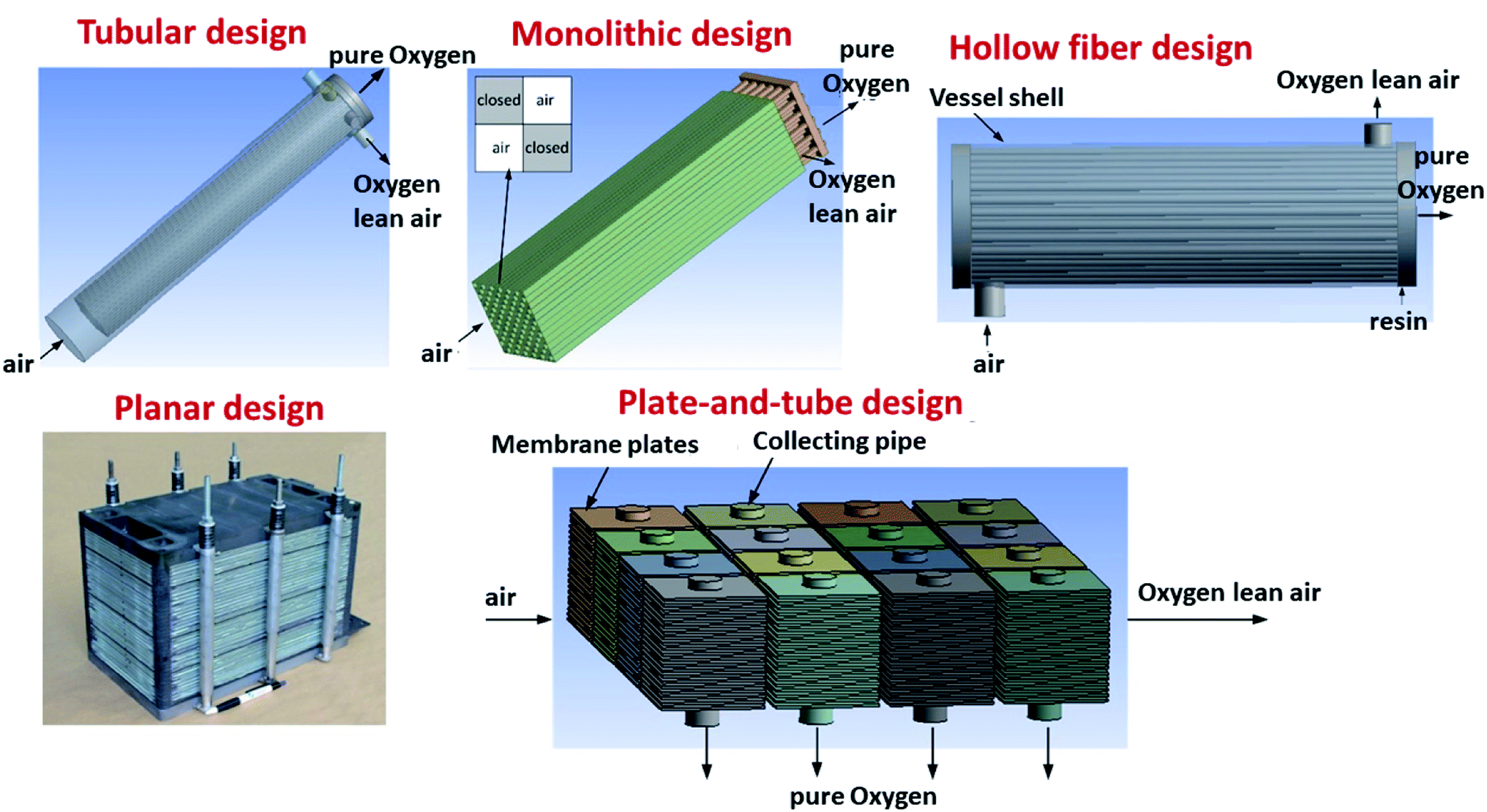 | ||
| Fig. 11 Different design configurations for OTM modules. | ||
The most common geometric shapes of OTMs are planar and tubular. The advantages and disadvantages of the two different designs are discussed in the Sub-sections below.
3.1 Planar membrane geometry
The most common membrane configuration used for oxygen permeation testing at small lab-scale is a simple flat dense disk, typically prepared by pressing powders followed by sintering at high temperature. These disks can be easily integrated into test setups with two chambers at different pO2, which provide the required driving force. It is well established that reducing the thickness of a membrane enhances the oxygen permeation rate due to the shorter distance of oxide ion diffusion. However, the thickness of self-supporting membranes is limited to about 100 μm because of their low mechanical strength. Most of the values provided in Table 1 are obtained by these types of samples and measurements.A strategy to further reduce the thickness of the active membrane down to 5–50 μm without jeopardizing the mechanical strength are supported membranes (often called asymmetric membranes). Basically, these membrane architectures can be divided into the following functional layers (Fig. 5):
(i) A porous support ensuring the mechanical stability of the assembly while at the same time providing the high porosity and low tortuosity required for diffusion of gases through the support.
(ii) Porous interlayer(s) facilitating the transition from the macroporous support to the dense membrane.
(iii) A thin dense membrane layer.
This architecture allows the thickness of the dense permeating layer to be notably reduced and the oxygen permeation rate is maximized, up to a point where ion diffusion through the bulk oxide is no longer the rate limiting step. Indeed, the overall supported asymmetric membrane is typically governed by more than one transport process depending on the total flux achieved. In the case of very high-performing asymmetric single-phase membranes, besides surface exchange a significant porous support limitation can be observed. In dual-phase composites the total flux is expected to be lower and, thus, the support influence is smaller and surface exchange becomes the dominating rate limiting step in particular at lower temperatures. At high temperatures and utilizing surface activation, bulk diffusion limitation might become significant again, but it is expected that surface exchange will remain as the critical process.
For successful demonstration of this concept, several requirements have to be fulfilled, starting with a small thermal and chemical expansion mismatch and good chemical compatibility between the different layers, combined with good interfacial anchoring. Strategies on how such structures can be realized are outlined in Chapter 4. A way for ensuring material compatibility is by considering the use of porous supports fabricated from the same membrane material itself. From the economic point of view, this solution may not be feasible since membrane materials are normally expensive and considerable amounts of powder are needed for production of the supports. Other alternatives that are usually explored are the use of porous substrates consisting of cheaper materials such as YSZ,238,248,256 Al2O3,257,258 MgO259–262 and metallic alloys.263 Several attempts to obtain such robust and high performing asymmetric membranes have been reported.
Fig. 12 shows an example of a planar dual-phase membrane made of Ce0.8Gd0.2O2−δ–FeCo2O4 developed by Forschungszentrum Jülich GmbH and the Technical University of Denmark for oxy-fuel combustion.
 | ||
| Fig. 12 Post-sintered picture (a) and SEM image of a fractured cross-section (b) of a planar dual-phase membrane component made from Ce0.8Gd0.2O2−δ–FeCo2O4. The post-sintered picture of the membrane was taken on the 3YSZ porous support side. | ||
Another example is asymmetric membranes based on SrCo0.4Fe0.5Zr0.1O3−δ (SCFZ) and MgO. In this case, the thermal expansion behavior between the dense membrane layer and the support could be matched by using composites of the materials in both layers (60 wt% SCFZ/40 wt% MgO in the dense membrane layer, 40 wt.%SCFZ/60 wt% MgO in the support).264 It should be noted that MgO is not a conductor, therefore the term “dual-phase” is partly misleading here. A different strategy to obtain asymmetric OTMs was reported for Ce0.85Sm0.15O1.925 (SDC)–Sm0.6Sr0.4FeO3−δ (SSF). In a first step, a monolithic composite membrane (flat disc) was prepared by conventional solid-state sintering. Subsequently, a dense thin layer upon a porous support was fabricated by selectively dissolving the perovskite phase using HCl. By using this method, it was possible to reduce TEC mismatch, and oxygen fluxes over 1 ml cm−2 s−1 (ref. 227) were reported (cf.Table 1, Chapter 2). The importance of optimizing the microstructure of the porous support layer in asymmetric membranes was demonstrated by Kovalevsky et al.265 In his work asymmetric membranes consisting of a 70% SrFeO3−δ/30% SrAl2O4 composite were prepared by using pore forming additives, dry pressing and sintering. The oxygen permeation measurements showed that the performance was limited by the gas diffusion through the porous support, and only slightly higher permeation flux values as compared to the monolithic membrane made of the same composition were measured. Recent developments aiming to increase gas diffusion in the porous support are pore orientation perpendicular to the dense layer, often in connection with nanosized catalytic particles inside the pores to enhance the reaction rate.265
In conclusion, a trade-off between porosity, layer thickness and mechanical strength is needed for an asymmetric membrane arrangement. Even though the performance of planar OTMs has been improved significantly in the last years, one can expect challenges in up-scaling and module construction, as typically the sealing needs to be placed in the high temperature zone of the stack/module. Such challenges are well-known from the field of SOFCs, and several promising solutions exist to solve them. These options are expected to be (partly) adoptable for planar OTM modules. In the case of dual-phase membranes, the chemical and mechanical compatibility with the support is an additional challenge.
3.2 Tubular membranes
An alternative to the planar is the tubular design. The advantages of this geometry lie in the reduced sealing area and the higher mechanical robustness and thermal shock resistance. These two advantages make tubular geometry to be currently the preferred solution for operation in pressurized environments, as described for OTM modules used in industrial applications (see Chapter 6). The main disadvantages of the tubular design often mentioned are the increased materials and fabrication costs due to the thick tube walls and the significant lower packing density (active area per volume) in modules. Here one must consider that a low packing density is not always a disadvantage, e.g. in OTM applications that involve heat transfer. In these application, a stacked planar design may not be an efficient way of facilitating heat transfer, and tubes would be a better design for that purpose. While the lower packing density is an intrinsic design problem which is difficult to optimize, the materials and fabrication costs can be partly reduced by the use of asymmetric tubes, in which the active membrane is applied on a porous support (see asymmetric membranes in 3.1).Examples for the optimization of this type of membrane are reported by X. Yin and co-workers. The authors describe the manufacturing of an asymmetric CeO2 based porous supported tubular membrane, coated with a CeO2–La0.2Sr0.8CoO3−δ composite. By using this composite, a dense and gas tight layer was obtained as the CeO2 presence buffered the thermal stress appearing from the different CTEs of the used materials.165 Other work on the production and testing of tubular dual-phase membranes considered formulations such as CeO2–La0.2Sr0.8CoO3,165 Ce0.8Sm0.2O1.9–La0.8Sr0.2CrO3,211 SrFeO3−δ–SrAl2O4,266 Ce0.9Gd0.1O1.95–La0.6Sr0.4FeO3−δ,184,185 and Sc0.2Y0.02Zr0.89O2−δ–LaCr0.85Cu0.10Ni0.05O3−δ.267 The most advanced developments to date in the field of tubular OTMs are related to the R&D activities of Praxair, Inc. and are described in detail in Chapter 6.
3.3 Hollow fiber membranes
Hollow fibers (also referred to as capillary or microtubular membranes) provide a high membrane surface area per unit volume of reactor compared to the conventional disc and tubular forms typically used. Hollow fibers typically have diameters in the range of 1–2 mm, with wall thicknesses of about 0.1–0.3 mm, which is significantly thinner compared to state-of-the-art disk and tube shaped membranes with thicknesses of 1–2 mm. On the other hand, the sealing and upscaling of hollow fiber membranes is challenging. In recent years, interest in hollow fibers has been growing, and a number of groups are now fabricating and characterizing the performance of different dual-phase hollow fiber membranes.164,192,213,214,268–270 For example, Zhang et al. developed a Ce0.85Sm0.15O1.925–Sm0.6Sr0.4Al0.3Fe0.7O3−δ (SDC–SSAF) hollow fiber membrane aimed for oxy-fuel related applications.268 Their membranes reached a maximum O2 flux of 2.6 ml min−1 cm−2 at 950 °C under an oxygen partial pressure difference of 1/0.02 atm (feed/permeate). Other studies dedicated to oxy-fuel applications used the same ionic conductor (SDC) but combined it with other electron-conducting materials such as La0.8Sr0.2MnO3−δ,214 La0.8Sr0.2Cr0.5Fe0.5O3−δ (ref. 213) and La0.7Ca0.3CrO3.269 Bi1.5Y0.3Sm0.2O3–La0.8Sr0.2MnO3−δ dual-phase composite hollow fiber membranes were also fabricated for oxygen separation. An oxygen permeation flux of 0.52 ml min−1 cm−2 was obtained at 850 °C under a gradient of air/helium.164 Bi et al. manufactured and characterized Pr0.1Gd0.1Ce0.8O2−δ–CoFe2O4 membranes having wall thicknesses of about 200 μm.192 A maximum oxygen permeation flux of 0.88 ml min−1 cm−2 was achieved using He as sweep gas and air as a feed gas at 1000 °C. The same study also demonstrates successful operation at 950 °C in a pure CO2 atmosphere for more than 200 h without any noticeable performance degradation or membrane deterioration. Recently, Chen et al. fabricated Ce0.8Sm0.2O2−δ–carbonate dual-phase hollow fiber membranes for CO2 separation.270 At 700 °C, their membranes produced maximum CO2 permeation fluxes of 4.78 ml min−1 cm−2 and 5.46 ml min−1 cm−2 under the feed condition of 50%CO2–50%N2 and 5% H2–47.5%CO2–47.5%N2, respectively, showing a great potential as commercially applied membrane for separation of CO2 from different gas-mixture situations.4 Manufacturing of dual-phase membranes
In this chapter the manufacturing techniques for obtaining dual-phase membrane in the geometries discussed in the previous chapter are reviewed, as illustrated in Fig. 13 below. | ||
| Fig. 13 Typical core steps in processing of planar and tubular multilayer dual-phase OTMs. | ||
Dual-phase OTMs are still in an earlier stage of development, especially when it comes to fabrication and up-scaling to pre-pilot scale. As a consequence, only a limited number of publications directly report on the manufacturing of dual-phase oxygen membranes. Nevertheless, the authors strongly believe that for accelerating the progress in the R&D of OTMs, more information on important fabrication methods and challenges in the preparation of multi-layer single phase membranes should be available. For this reason and to give the reader a comprehensive overview on membrane manufacturing, a review in the following sub-chapters on existing studies and potential technologies is given.
4.1 Preparation of dual-phase membrane powders
The fabrication of the raw powders plays a crucial role for the performance of dual-phase membranes. Powder properties, such as surface area, particle size distribution, purity and homogeneity of the two powder phases, combined with the sintering profile strongly affect the final microstructure of the two phases in the dual-phase membrane. Furthermore, the microstructure, e.g. the porosity, tortuosity of the ionic- or electronic-pathways, percolation or contact area between both phases determine the membrane performance. For example, a fine particle size of the powder of the two phases is required for fabrication of a thin membrane layer (few tens of microns) and its complete densification. Here, we introduce the most relevant powder manufacturing methods for cercer composite powders for dual-phase OTMs.Powder manufacturing techniques can be categorized into either solid state (mixed powder) or wet chemical methods (e.g. co-precipitation, sol–gel, hydrothermal and spray techniques).271 The most conventional and direct method for dual-phase material preparation is the mixed powder method, in which a mixture of metal oxides, salts, or carbonates is treated by mixing/grinding211,272 and subsequent high temperature calcination.273 During the calcination, the crystalline phases form by metal and oxygen ion diffusion at the surface of the mixed metal oxides/salts/carbonates.274–277 Advantages of the powder-mixing route are the use of already available precursors, industrial equipment and the low cost, which makes this route suitable for industrial scale. Drawbacks of the method are the high calcination temperature, leading to large grain size, low surface area, poor chemical homogeneity, the formation of undesired phases or non-stoichiometry due to partial decomposition of products, as reported for barium-containing perovskites.278,279 An additional milling step is required to improve sinterability, which can introduce more impurities.273
As an alternative to the simultaneous, direct synthesis of the two powder phases by the mixed powder method, the dual-phase powder can be prepared by mixing one fine, pure oxide phase and precursors of the other phase, or by mixing the phase-pure, fine oxide powders of both phases. If reactive sintering is envisaged to form one (or both) of the phases, the phase transition increases reactivity and potentially reduces the sintering temperature.110,280
Wet synthesis is an alternative to using only solid precursors. For example, a precursor solution or suspension of one of the phases can be used to coat stoichiometric amounts of the other phase. One variety of this is the packing method, where the less abundant powder phase is the dispersed component, such that the grains of this minority phase will be embedded into the continuous network of the majority phase grains after the mixture is calcined.281 A risk, especially with a low phase volume, is that the minor phase might not percolate, which will compromise the ambipolar conductivity and reduce the oxygen flux. The inverse of the packing method is the loading method,281 where the major powder phase is mixed into a solution of the components of the minor phase. The minor phase grains will then deposit on the main phase. Such routes have been proposed by Zhu et al.281
An attractive wet chemical route for synthesis of dual-phase powders is the one pot method,187,227,281 which allows the two compositions to be synthesized by a single step following typical solution fabrication methods for ceramics e.g. sol–gel,144,221,282 co-precipitation,283,284 Pechini method,16,201,285 hydrothermal synthesis,121,286–288 or spray pyrolysis.289–292 All of these processes utilize a precursor solution, but the crystallites or powders are produced in different ways. The main advantage relative to the mixed powder method are significantly lower calcination temperatures for forming the desired oxide phases, resulting in powders with smaller size, higher purity and homogeneity.
The sol–gel process for oxide ceramics usually uses a colloidal dispersion of metal alkoxides. The sol (i.e. solution) is transformed to a gel by hydrolysis and polymerization of the precursors at relatively low temperatures, immobilizing all homogeneously distributed components.271,273Co-precipitation is based on an aqueous solution of the metal cations, mixed with a solution containing the precipitant. The precipitated product can be separated from the liquid by filtration.271,273 Both processes use thermal decomposition of the precursors at higher temperature to form the structural phases, resulting in homogeneous, nano-sized grains in a well distributed mixture of the two phases.
Spray pyrolysis is also based on a solution or suspension of the metal cations or precursors, which are atomized into a heated chamber.292,293 During atomization, the solvent is evaporated forming spherical agglomerates of sub-micron primary particles. Those usually have to be calcined in order to obtain the target phases. Alternatively, in flame spray pyrolysis the precursors are directly converted to the respective oxides during the spray process by feeding the precursor solution through a capillary into a hot flame.294 Spray drying is a related, lower-temperature process used to mix and granulate nanoparticles of fine oxide powders into micron-sized agglomerates of well-defined shape, particle size distribution and surface area.293 This allows for easy handling and direct use for the following shaping steps, which can be advantageous for example for homogenous mixing and handling of a mixture of oxides and for extrusion or pressing of membrane support structures.
In hydrothermal synthesis, an aqueous solution of metal cations is heated above the boiling point of water inside an autoclave. By reaching the vapour saturation pressure, the targeted product crystallizes out from the fluid in the targeted phase composition, making a subsequent calcination superfluous.271,294
Comparing the wet chemical methods discussed in this chapter, one can generally conclude that powder uniformity and distribution decreases as follows: one pot methods (i.e. sol gel, Pechini, co-precipitation) > loading method > powder mixing method > packing method.271,281
4.2 Shaping of dual-phase membranes
A variety of shaping methods are used for dual-phase OTMs. The choice of shaping method depends on the membrane geometry (planar, tubular or capillary) and configuration (symmetric or asymmetric). An asymmetric membrane configuration with porous support, catalytic layers and a thin membrane layer usually requires several shaping steps. The most used techniques include pressing, extrusion, tape casting and spinning to produce support structures and dip coating, screen printing, spraying and lamination of tapes to apply thin film porous catalytic and dense membrane layers. Other shaping techniques, such as thermal spraying, PVD (Physical Vapour Deposition), CVD (Chemical Vapour Deposition) and sol–gel have even been rarely reported for the fabrication of mixed-conducting asymmetric OTM architectures or dual-phase membranes. Reports on the fabrication of real-size dual-phase membranes for practical applications are scarce, due to challenges in combination of materials (Chapter 2) and in the sintering of multilayer architectures with such materials (Chapter 4.3).4.2.1.1 Pressing. In dry pressing, powders or granules are generally compacted in the axial direction between two stamps in hardened metal molds. As can be seen from Table 1, the majority of scientific studies on dual-phase OTMs are actually based on millimeter-thick, dry pressed and sintered self-standing planar dual-phase membranes. The reason for this approach is the simplicity of the fabrication process and the membrane architecture, which allows the fast preparation of small samples with a very simple geometry and robustness. Most of these studies aim to characterize the performance (oxygen fluxes, stability) of new dual-phase membrane materials for scientific studies or fast material screening, with a minimum of required equipment and fabrication effort. For the measurement of reactive membrane materials, surface passivation layers formed during high temperature sintering can be removed from the pressed and sintered membrane layers to allow the application of catalytic layers and testing.295
4.2.1.2 Tape casting. Tape casting is a proven, industrial-scale process for the cost-effective production of planar ceramic components, such as SOFC,296 capacitors,297 and solid state batteries.298 A practical overview on the topic of tape casting was published by Mistler and Twiname.299 A ceramic powder is dispersed in a slurry, consisting of solvent, surfactants, binder and plasticisers to form a flexible, mechanically stable, non-sintered sheet, called “green” tape. The slurry is cast by a doctor blade process onto a flat surface, usually a coated polymer foil, steel band, paper or glass, followed by slow drying inside a drying chamber with well controlled atmosphere.300–303
Asymmetric membranes can be manufactured by sequential tape casting (co-casting)296,302 or lamination of separately casted green tapes.149,304 In lamination, the membrane and substrate layers are first separately casted and afterwards combined by lamination, which is the joining of the green tapes by a hot pressing process between two roles or in a hot press.305 The slurry formulation of the tapes needs to be adjusted to ensure sufficient interface adhesion to avoid delamination during debinding (or sintering). Sequential tape casting (or co-casting) eliminates the need for hot pressing. First, a slurry of the thin membrane layer is cast on the carrier foil. The slurry of the thicker support is then cast onto the dried or semi-dried membrane layer, dissolving binder at the surface of the membrane layer, leading to better cross-linking of the binder chains of both layers. Laminated or co-cast structures are usually cut to the desired green size before debinding and sintering.
For introduction of porosity in support or catalytic layers, pore forming agents can be added to the slurry, potentially in combination with other techniques, such as freeze casting,230,241,306 leaching307 or phase inversion183,240,243 to further increase porosity and pore size in the membrane support layer.
4.2.1.3 Screen printing. In screen printing, a cost-effective, industrial scale thick-film coating technology, a mesh is used to transfer the ink onto a substrate. A squeegee is moving a ceramic ink across a screen to press and fill it into the open mesh apertures. During the reverse stroke of the squeegee, the screen touches the substrate temporarily to transfer the ink on the substrate surface. A good understanding of the complex relation between ink composition and rheology of highly loaded inks (or pastes) and the screen-printing parameters is of crucial importance.308 The inks should preferably have high solid loading (resulting in high packing and green density) to help densification of the dual-phase membrane layer and reduce shrinkage of the catalytic layers upon sintering on a (pre-)sintered membrane. Several studies are related to the screen printing of dual-phase membrane and catalytic layers in asymmetric OTMs. For example, Gaudillere et al.230 screen printed a NFO/CTO gas-tight ceramic top layer on the top of a LSCF porous support made by ice templating. Garcia-Fayos309 and Lobera310 used screen printing to evaluate catalyst layers for oxidative coupling of methane and oxy-fuel/syngas production on ceria based OTM membrane reactors, respectively. The same group screen printed porous LSM–CGO catalytic inks, based on an ethylcellulose and terpineol formulation, on pressed NFO–CTO dual-phase membranes for investigation of the stability of the membranes in CO2 and SO2 atmospheres.144 Ramasamy et al.253 applied LSCF activation layers by screen printing on Ce0.8Gd0.2O2−δ–FeCo2O4 dual-phase OTMs.
4.2.1.4 RF sputtering. RF or Radio Frequency Sputtering is a versatile method to produce thin films in the range of 50 nm to 5 μm over a large area with high uniformity.311,312 It consists of applying an energetic wave through an inert gas (typically argon) in a vacuum chamber which becomes ionized.313 The generated high energy ions (Ar+) produce a plasma by bombarding a target material, thus resulting in the release of atoms (positive ions) which are accumulated on the surface of a target face (substrate) forming a thin layer. Faster deposition rates are achieved with RF magnetron sputtering by using magnets behind the target material, thus trapping electrons and preventing them to bombard the substrate.
Deposition of thin films by RF sputtering is mainly considered for applications such as electronics, improvement of optical properties, layer protection and photocatalysis.312,314,315 Nevertheless, applicability in OTMs has also been prospected in the past recent years,316 especially by depositing MIEC thin layers by means RF magnetron co-sputtering.317 The work conducted by Solís et al. on the deposition of 150 nm-thick NiFe2O4–Ce0.8Gd0.2O2−δ nanocomposite thin films on BSCF asymmetric planar membranes demonstrated RF magnetron sputtering as a potential route for OTM manufacturing to overcome bulk diffusion limitations. In this work, the deposited nanocomposite layer presented suitable MIEC features for permitting oxygen permeation as well as providing protective features against CO2 exposure as can be seen in Fig. 14.
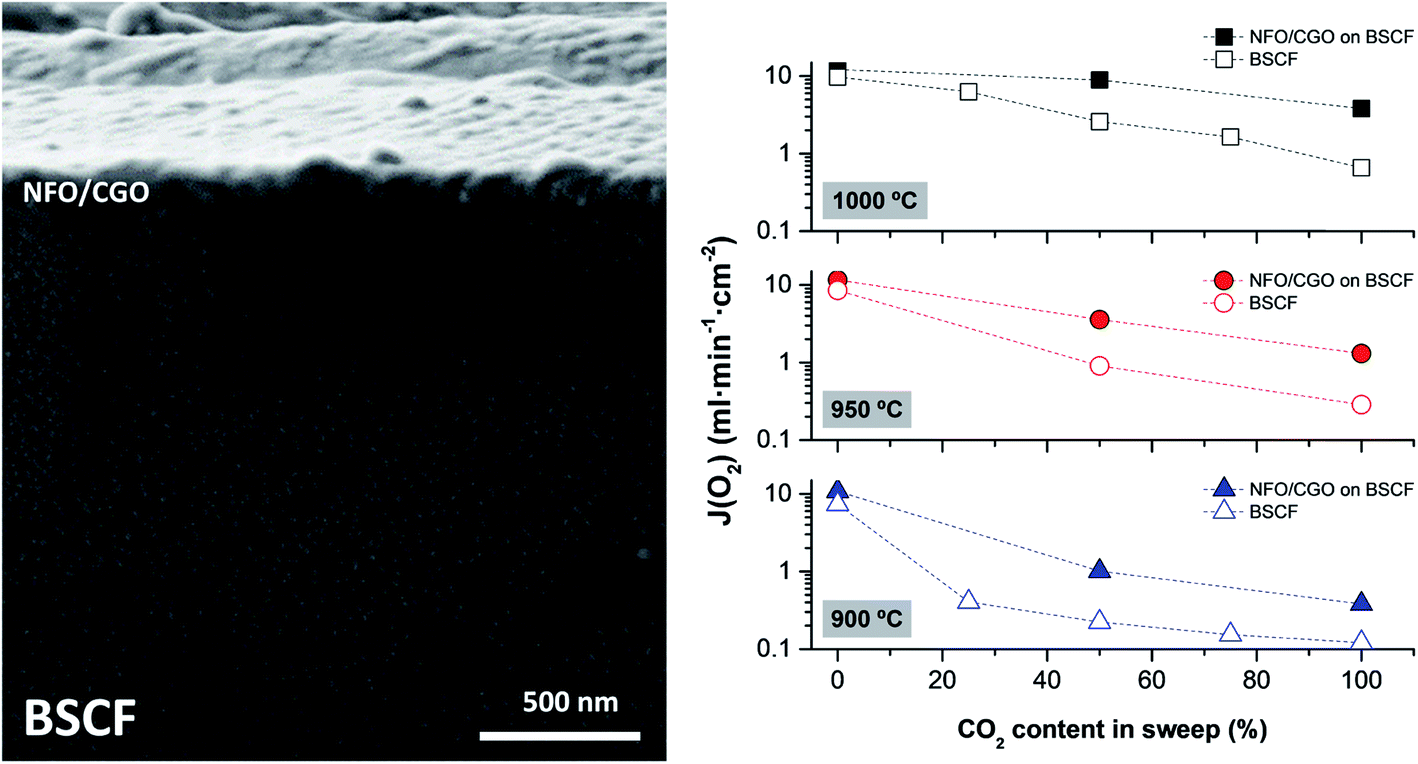 | ||
| Fig. 14 (Left) Cross-section FESEM image of NFO–CGO layer deposited on a supported all-BSCF membrane. (Right) Permeation results of the bare and co-sputtered membrane as a function of testing temperature and CO2 concentration in the sweep gas (experimental conditions: 300 ml min−1 air feeding, 300 ml min−1 Ar/CO2 mixtures sweeping). Reproduced with permission.317 Copyright 2018, Wiley-VCH. | ||
4.2.2.1 Extrusion. Porous tubular supports for OTMs have been produced by two main types of extrusion processes: water based and thermoplastic. With respect to dual-phase membranes, these tubular structures have mostly been extruded as porous support structures to enable the subsequent application of catalytic and membrane layers (10–50 μm thick) by spray- or dip-coating. The requirements on the extruded porous, tubular supports are challenging: a sufficiently high gas permeability, mechanical strength, chemical stability and suitable sintering activity for co-firing with the functional layers. Due to the high solid loading and packing in extrusion, it is difficult to design and fabricate tubes with sufficiently high gas permeability, which do not compromise other properties. The challenges with design of extrusion feedstocks are discussed at the end of this sub-chapter, whereas co-firing is presented in Section 4.3.2.
4.2.2.2.1 Water based extrusion. For the fabrication of thin-walled tubes (diameter of 5 to 20 mm and a wall thickness of 0.5 to 2 mm) the most common high-volume fabrication technique is water-based extrusion, in which a plastic ceramic feed of fine powder material318 is continuously fed through a charging hopper, evacuated and transported by a screw (auger) to the die. The length of the tube is determined by cutting. OTM tubes up to a length of 1 meter have been produced by water based extrusion.319,320
Asymmetric, tubular oxygen membranes have been fabricated based on aqueous extrusion of support tubes, for example BSCF membranes by Hoffmann and Pipphardt et al.321,322 and La2NiO4+δ membranes by Dahl et al.323 Nevertheless, these support materials exhibited poor mechanical properties and chemical stability. Therefore, the extrusion of aqueous ceramic pastes of partially stabilized zirconia (3YSZ) with addition of pore formers has been successfully optimized by different groups324,325 or companies (see Section 5) to obtain tubular support structures with enhanced mechanical stability, gas permeability and sinterability for use in asymmetric OTMs. The aqueous extrusion of tubular supports for use in dual-phase membrane systems of La0.2Sr0.8CoO3−δ/Ce0.8Gd0.2O2−δ on a tubular CeO2 support326 and a YSZ–Ag composite on a porous YSZ–Ni composite tube327 have recently been reported.
4.2.2.2.2 Thermoplastic extrusion. Thermoplastic extrusion allows the fabrication of tubes with wall thickness below 1 mm, where a mixture of fine, sinterable powders with molten polymers (a feedstock) is forced through a hot auger and die to shape continuous components of a fixed cross-sectional area by the fast cooling of the thermoplastic feedstock immediately after exiting the die. The obtained tube requires the removal of the thermoplastic binder system via thermal debinding,262,267 solvent extraction or catalytic methods before densification during sintering.328
4.2.2.2.3 Compositional and microstructural design of extrusion for tubular OTM supports. Porous ceramic supports for asymmetric dual-phase membranes must exhibit a good balance between sufficient mechanical strength and high permeability. This is required to reduce concentration polarization or pressure gradients due to gas transport limitations, which would negatively affect the performance of the membrane.6 For use in OTMs or SOFCs, water-based and thermoplastic extrusion have been used for the fabrication of relatively thick (0.5–1 mm) tubular support structures. Achieving the following set of requirements is still challenging in the development of supports for asymmetric dual-phase membranes: (1) a high pore volume (>50%), (2) pore channels of width much above 1 micron, without bottle necks, (3) a feedstock with fine raw powders, which allows co-firing with additional catalytic and membrane layers and (4) sufficient mechanical strength.
Pippardt et al.322 fabricated BSCF OTM tubes with a diameter of 12.25 mm and wall thickness of 1.25 mm (before firing) by water based extrusion of polymethyl methacrylate (PMMA) spheres, micron-sized BSCF powder (2.7 micron) and hydroxypropyl methylcellulose as binder that allowed a one-step co-firing with a Ba0.5Sr0.5(Co0.8Fe0.2)0.97Zr0.03O3-δ (BSCF3Zr) membrane. However, matching the shrinkage of membrane, catalyst and support layers for more complex multilayer systems, utilizing different type of micron-sized raw powders with high melting points and sintering temperatures (e.g. LCCN based dual-phase composites) and different chemical composition is very challenging. CaTi0.9Fe0.1O3−δ (CTF) support tubes for asymmetric CTF membranes were fabricated from a mixture of sub-micron CTF powder, a mixture of charcoal and starch as pore-formers and a binder system with a ram extruder.329 Gas flow limitations in the support structure were suspected to contribute to limitations in oxygen permeation even at moderate fluxes of 0.16 ml min−1 cm−2 at 1000 °C in air/argon atmosphere.
Ramachandran et al.330,331 optimized thermoplastic feedstocks for extrusion of MgO tubes for use in asymmetric Ce0.9Gd0.1O1.95−δ (CGO10) membranes262 and for CGO10–La0.6Sr0.4FeO3−δ dual-phase OTMs.332 Different types of graphite and PMMA were used as pore formers, but gas permeabilities could not be increased above 10−15 m2. By replacing MgO with 3YSZ as a structural support material in this thermoplastic feedstock system, Haugen et al. could increase the mechanical strength of the membrane tubes significantly and reached gas permeability values of 10−14 m2.267 The permeabilities of these extruded support tubes are still significantly lower than those for tubular structures with micron-sized, directional pores produced by a freeze casting technique,333 see the next section on slip casting.
4.2.2.2 Slip casting. Slip casting is a process to form a solid cast of a ceramic green body by adsorbing water from an aqueous suspension (slip or slurry) on the surface of a porous mould (gypsum or porous polymer molds) by capillary forces. In drain casting, the cast copies the inner contour of the mould surface whereas in solid casting the slip is refilled until the entire mould is filled up. For both techniques, the (wall) thickness of the cast is limited because with increasing wall thickness, the formed cast will lower the speed of further cast formation due to reduced water extraction.
Saint-Gobain has demonstrated the fabrication of highly permeable tubular YSZ tubes with large pore size and radially aligned porosity by a combination of a slip casting process and rotational freezing for use as supports in OTMs.334 The method is based on a conventional ice-templating process in a rotatory mould.335 The pore volume could be adjusted by the solid loading, the pore size by the freezing temperature and the overall tube thickness by the volume of slurry initially poured into the mould. Significantly higher gas permeabilities of up to 2.96 × 10−13 m2 have been demonstrated for these YSZ tubes with a wall thickness of 2 mm in comparison to ceramic membrane support tubes prepared by extrusion, utilizing sacrificial pore-formers.331,336
4.2.2.3 Dip coating. Dip-coating consists of depositing a liquid film with a thickness between 1 to 100 μm by immersion of a substrate in a precursor solution or a slurry.337,338 After withdrawal of the substrate with constant speed, the precursor film is dried through evaporation of the liquid, and the film consolidates on the substrate. According to the nature of the deposited material, a further post-treatment, such as curing or sintering, might be performed.339,340
Reports on the fabrication of tubular multilayer OTMs by dip coating (and co-firing) for dual-phase membranes are scarce. Usually, two to four dip coating steps onto the porous support tube are required: the composite membrane layer, and one or more activation layers (on each side of the membrane). Due to the very limited number of studies on the fabrication of tubular asymmetric dual phase membranes until now, we will include and explain in the following section one of the main challenges, the co-firing of the multilayers, on studies with single phase membranes.
4.2.2.3.1 Dip coating of dual-phase membrane layers. For asymmetric, single phase tubular OTMs of La0.2Sr0.8Fe0.8Ta0.2O3−δ (LSFT), Gurauskis et al.27,341 suggested different pre-calcination and sintering steps for the co-firing of the different layers to achieve the desired densification of the porous support and the gas-tight LSFT membrane layer. Similarly, Dahl et al.323 fabricated asymmetric LaNiO4−δ membranes by dip coating on porous support tubes of the same material. Liu et al.239 enhanced the oxygen permeation rate of Zr0.84Y0.16O1.92–La0.8Sr0.2Cr0.5Fe0.5O3−δ dual-phase hollow fiber membrane by applying a type of dip coating process to infiltrate Ce0.8Sm0.2O1.9 nanoparticles. Ovtar et al.185 described the dip coating of four layers on a MgO support tube, a porous CGO active layer, a dense dual-phase membrane layer, a porous CGO active layer on the inner and outer side of membrane and an impregnation layer to improve the oxygen transport properties of tubular Ce0.9Gd0.1O1.95–La0.6Sr0.4FeO3−δ composite, asymmetric oxygen permeation membranes.
Haugen et al.267 have reported the fabrication of asymmetric membranes composed of different materials. The dip coating of a porous (Y2O3)0.03(ZrO2)0.97 (3YSZ) tube with a porous 10Sc1YSZ activation layer and a thin composite oxygen membrane layer of LCCN–10Sc1YSZ led to severe challenges with the subsequent co-sintering. The high co-sintering temperatures of about 1450 °C, required for full densification of the LCCN composite membrane layer, resulted in Cr evaporation, loss of LCCN phase, formation of insulating LaZr2O7 phase, and crack formation in the membrane.
4.2.2.4 Slurry spraying. Tubular OTMs can be coated by slurry spraying, in which a suspension of powders is sprayed on a support tube by using a spray gun. The coating usually has a thickness of 10 to 50 μm after one coating pass and is subsequently dried and sintered.
For example Yin et al.165 sprayed a mixed conducting La1−xSrxCoO3−δ (LSCO) membrane on a ceria support tube. Ritchie342 prepared a coating of La0.5Sr0.5Fe0.8Ga0.2O3−δ membrane on a high-purity porous α-alumina tube for a syngas membrane reactor by spray deposition. Z. Liu343 produced an asymmetric membrane of SrCo0.4Fe0.5Zr0.1O3−δ by a spin-spraying process.
4.3 Membrane sintering
The sintering of dual-phase membranes can result in several challenges related to (i) the homogeneous densification of the dual-phase materials, (ii) reactions or evaporation of the dual-phase materials during the high temperature treatment and (iii) the formation of thermo-mechanical stresses and defects in asymmetric membrane architectures during co-firing due to an inaccurate debinding process, mismatches in shrinkage rates or thermal expansion. These challenges are discussed here comprehensively for the first time after a brief general introduction into debinding and sintering processes.4.3.2.1 Densification of dual-phase membrane materials. Once both starting composite phases are formed, they need to be dense and have intimate contact between grains. However, it is often very difficult to sinter such compacts sufficiently because compositions of each phase generally require different optimal sintering temperatures.248,267,354 Conventional sintering processes comprise the uniaxial or isostatic pressing of green disks and later sintering at high temperature during several hours. The high temperature densification always causes grain growth and consequent grain boundary and interfacial issues. Thus, various techniques have been developed to lower the sintering temperature. One way is to introduce sintering aids, e.g. metal oxides. MnO2, Mn2O3, Ga2O3, Co3O4, Fe2O3, or CoO have been proposed due to their lower melting point with respect to the dual-phase membrane materials.355 This promotes diffusion of material at the particle surface, which lowers the sintering temperature and reduces the final grain size. Occasionally, the addition of these sintering aids may also introduce electrical conductivity.356,357
Spark Plasma Sintering (SPS) is an advanced sintering method that permits separate grain growth and densification processes. In this method, high current amplitude pulse (>1000 A) is applied through a graphite die at a low voltage (5 V), internally heating the sample via the “Joule heating mechanism” instead of using an external heating source.358 Since the sample is self-heated from both inside and outside, the heating rate and mass transfer speed are both rapid and localized so that the sintering process generally is very fast (within a few minutes). Thus, compared with the conventional sintering methods explained above, the SPS process can produce dense ceramics in a very short sintering time and at relatively low temperatures, which retains the nano-size and nano-structure and avoids coarsening and decomposition of the composite phases.354,359
4.3.2.2 Co-sintering of asymmetric dual-phase membrane architectures. Asymmetric dual-phase OTMs usually consist of a multi-layer structure with a thin-film OTM layer on a porous support, often with additional catalytic layers. Such asymmetric, dual-phase membrane structures offer improved performance due to reduced thickness of the membranes layer according to the Wagner equation, improved mechanical and dimensional stability, and can be fabricated by cost effective shaping processes, such as extrusion and dip coating or tape casting and lamination, as described above for tubular and planar devices, respectively. For the manufacturing of these asymmetric membranes, the simultaneous sintering of the different layers (co-sintering) is one of the most critical steps.360–362 The different layers often consist of different materials, the membrane layer need to be sintered close to 100% of theoretical density, whereas the porous supporting layer should not reach much above 50% of theoretical density. The challenge with the high-temperature co-sintering of dual-phase membranes is to avoid the formation of cracks, delaminations, the peeling-off of coatings, interface reactions or material evaporation. Apart from compositional related challenges (e.g. membrane material instabilities or mismatch in thermal expansion), defects occur mainly due to residual stress development inside the structure in response to the differential shrinkage in the constituent layers. i.e. the layer with faster shrinkage rate is constrained by the layer that shrinks slower, resulting in constrained sintering. In order to produce defect-free asymmetric structures, it is therefore important to understand, and control stress generation by ensuring shrinkage match during the entire sintering process.
Experimental work and modelling of the co-sintering of different types of tubular and planar asymmetric OTMs have recently been reported. For co-firing of planar asymmetric membranes with porous/dense multi-layer systems and similar shrinkage rates, in situ studies with optical dilatometry363 have shown that significant warpage can occur without destruction of the membrane. Thin dense CGO membranes on a porous support with Co3O4 as sintering additive resulted in warping in a narrow temperature range of less than 100 °C. This lead to a concave shape at temperatures just below the optimum firing temperature of 1030 °C due to a higher densification rate in the dense membrane layer than in the porous support, whereas a convex shape was observed above 1030 °C, when the densification rate of the support was dominant. Multi-scale modelling of shape distortion of such planar, porous/dense bi-layers has been performed by Molla et al.362 Transient stress development during constrained sintering of such bi-layered structures has also been modelled for tubular configurations by Molla et al. for a dense Ce0.9Gd0.1O1.95−δ membrane layer on porous MgO support tubes.360
5 Potential applications for dual-phase membranes
Possible application areas can be divided into chemical conversion/membrane reactor applications (Section 5.1), in which the permeated oxygen can be used to create higher value chemicals, and in “oxy-fuel processes” (Section 5.2), in which the produced oxygen is used to enhance combustion processes.5.1 Membrane reactors and chemical conversion processes based on OTMs
In the near future a transition towards a circular economy364 is required. Besides methane conversion from (in the short term) natural gas and in particular biogas, the utilization of renewable energy together with abundant resources, i.e. H2O, CO2, is the key to success. Membrane reactors have the potential to be part of the solution; as outlined by Plazaola et al.,365 “the integration of oxygen membranes in conventional chemical processes has shown promising results, which improves, in all the cases, the efficiency of the processes because of the in situ air separation. Moreover, in some of these processes, an increase in the overall performance of the process has been achieved. On the other side, the oxygen flux and its stability in long-term experiments needs to be further improved to make these membrane reactor processes economically viable”. Membrane reactors based on OTMs can offer an increased energy efficiency due to the process intensification, i.e. combining reaction and separation tasks typically realized in series.366 For instance, membrane reactors can integrate oxygen separation, steam reforming and partial oxidation into a single step for the conversion of methane from natural gas or biogas. In the past, the majority of research was targeted towards the conversion of natural gas into syngas (a mixture of H2 and CO) in order to produce valuable chemical products, i.e. polymers and fuels (Gas-to-Liquid (GTL)).Typically, the conditions in membrane reactors are considered “hard” for the OTMs, and often corrosive and reducing atmosphere as well as high pressure at high temperatures are found. Another challenge is the integration of specific catalysts facilitating the targeted reactions, which must be compatible, i.e. both membrane and catalyst must maintain their performance when brought in direct contact. In this context, dual-phase membranes composed of two inherently stable phases offer high potential compared to the less stable single phase MIEC membranes. Nevertheless, the systematic trade-off and finding an optimum balance between performance and stability is still the main driver for materials development.
In the following Sub-sections, a few potential reaction schemes for OTM based reactors are described.
| CH4 + 0.5O2 → CO + 2H2, ΔHR =−36 kJ mol−1 | (16) |
The use of OTMs to perform the POM reaction is claimed to have the advantage of obtaining higher CO selectivity because oxygen is directly provided to the methane in its activated, i.e. ionized, form. Therefore, the oxygen partial pressure in the gas mixture remains low minimizing CO2 formation.
Several investigations for the POM reaction reveal feasibility obtaining high CO selectivity and methane conversions close to 100%.365 Besides typical single phase membranes also few dual-phase membranes were reported, e.g. YSZ–(LaSr) (CrFe)O3.367 A detailed status of the industrial activities in this application can be found in Chapter 6.
| 2CH4 + O2 → C2H4 + 2H2O, ΔHR =−88.3 kJ mol−1 | (17) |
The typical reaction temperature is in the range of 750–950 °C, required for the C–H bond activation. Major drawbacks occruing at these temperatures are (i) competition of the desired coupling reaction with oxidation reactions of both methane and ethylene, as well as (ii) consecutive reactions leading to selectivity-conversion problems.
Studies on the OCM (without using membranes) were first performed in the 1980–90s, due to the attractiveness of obtaining ethylene and other light alkenes by means of such direct way. A very wide variety of catalysts were tested aiming to achieve high selectivity and yields to C2 hydrocarbons; mainly consisting of alkali, alkaline earth, rare earth and transition metal oxides.368 However, the poor results obtained led to a decreased interest in the mid of 1990's, mainly due to the difficulty of reaching the economically viable minimum yield for ethylene (16–30%).
More promising recent OCM studies have focused on the utilization of MIEC membranes distributing oxygen in the ionic form O2−. This decreases considerably the oxygen partial pressure in the gas bulk phase and, thus, the formation of COx. In consequence, the reaction towards ethylene production is favored.
Several in particular single phase materials have been tested in the past, mainly perovskites and fluorites,365 but also a few dual-phase membranes as reported by Yaremchenko et al. in 2008.266 Besides stability issues of membrane materials, the complexity of the OCM reaction leads to chemical engineering issues in reaction control, e.g. residence time, choice of catalyst etc. Therefore, the performance in this early-stage development is still low, but expected to increase when an interdisciplinary approach is applied.
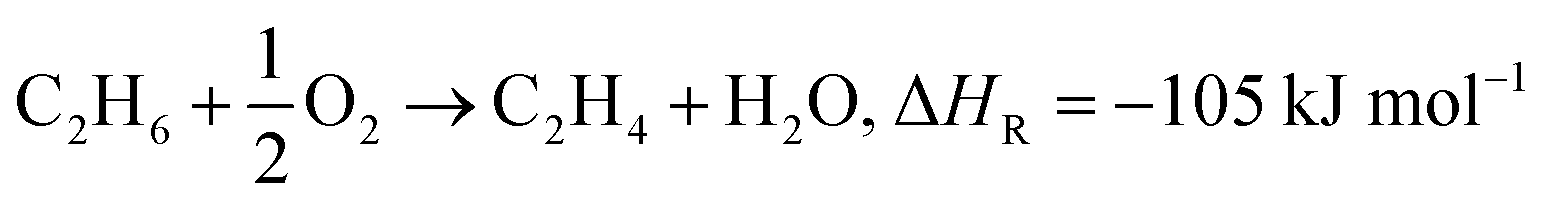 | (18) |
The drawback of this reaction concept is the need of molecular oxygen (or enriched air) to perform the oxidative de-hydrogenation at high conversion rates. Here undesired combustion reactions, in particular highly exothermic total oxidation of ethane and ethylene (ΔHR = −1428 and ΔHR = −1323 kJ mol−1, respectively), can decrease ethylene selectivity and yield (i.e. selectivity-conversion problem). Therefore, the use of OTMs in ODHE reactions is of great interest, since ionic oxygen supply could minimize undesired side-reactions by reducing oxygen partial pressure, enabling higher ethylene yields.
In recent years, some groups have been performing ODHE tests over MIEC membranes, mainly using perovskites as BSCF and BCFZ,369 as well as rare earth-doped ceria.370 High ethylene yields have been obtained at 850 °C for surface activated ceramic membranes, up to 81% for BSCF,371 and 84% in the case of CTO.371 Despite the promising results obtained on single phase MIEC membranes, there are no published works focused on ODHE reaction on composite membranes. However, dual-phase membranes are a very promising in particular because of their typically better stability and performance at intermediate temperatures, and should be tested in ODHE reactions in the near future.
Water dissociation into hydrogen and oxygen has been demonstrated to be possible by means of the utilization of single phase,372 and also of dual phase membranes.373 However, to perform the reaction coupling the water splitting reaction with other reactions on the permeate side (e.g. POM, OCM or ODHE) is required, generating a high oxygen partial pressure gradient, and displacing the water splitting equilibrium reaction to the formation of H2 and O2 due to continuous oxygen extraction by means of permeation. Classical single-phase perovskitic MIEC membranes, i.e. cobaltites or ferrites, are rather prone towards deep reduction and, thus, unstable. Only few materials are specifically developed for stable operation in these and only these conditions, i.e. reducing atmosphere at both sides of the membrane.374 Therefore, dual-phase membranes are promising due to their applicability to both oxidizing and reducing atmospheres.
In early works, water thermolysis studies conducted on composite membranes considered as materials the use of Gd0.2Ce0.8O1.9−δ–Gd0.08Sr0.88Ti0.95Al0.05O3±δ,373 Ni–Ce0.8Gd0.2O1.9−δ or Cu–Ce0.8Gd0.2O1.9−δ cermet membranes,375 40% vol. belonging to the metallic phase. For the Ni–CGO cermet membrane a H2 production rate of 6 ml min−1·cm−2 at 900 °C was obtained from the decomposed steam at the feed side. Recently, Liang et al. considered a dual-phase membrane consisting of Ce0.8Sm0.2O2−δ–Sr2Fe1.5Mo0.5O5+δ for coupling POM and water splitting reactions.376 With this approach, they obtained a CO selectivity of 98%, a CH4 conversion of 97% on the POM side and a H2 production of 1.5 cm3 (STP) min−1 cm−2 on the H2O splitting side. It is expected that in near future the number of studies including thermolysis of CO2 will significantly increase because of the importance of mitigating climate change as well as the progress made in materials development of dual-phase membranes. One example of this is the work conducted on Ce0.9Pr0.1O2−δ–Pr0.6Sr0.4FeO3−δ dual-phase membranes for the one-step thermochemical conversion of CO2 and H2O to synthesis gas coupled with POM on the other membrane side,377 obtaining a syngas production rate of 1.3 ml min−1 cm−2 at 930 °C for a H2O/CO2 feed ratio of 5:1 with H2O and CO2 conversions of 1.7% and 4.2%, respectively.
Another emerging field is the use of plasma-assisted decomposition of CO2.378,379 CO2-plasmas are generated, e.g. by microwaves, forming CO and O in significant conversions at reasonable temperatures of 1000–1500 °C. The oxygen needs to be extracted immediately to avoid recombination to CO2. The process can also be applied to other plasmas, such as H2O plasmas for hydrogen generation. The technology is in early stage and requires interdisciplinary R&D in all aspects, in particular oxygen separation. This could be a promising case for membranes due high, but reasonable temperatures, and high oxygen content not necessarily requiring reducing atmospheres at the permeate side.
5.2 Oxy-fuel combustion
Oxy-fuel combustion is considered to be an energy-efficient process for Carbon Capture and Storage (CCS) in power plants or other combustion processes. It consists in the combustion of coal (or another fuel) under an oxygen-rich atmosphere. Operating under such conditions presents the following advantages: (i) an increase in the combustion efficiency, permitting a higher flame temperature in the furnace, (ii) a flue gas stream containing mainly CO2 (80–88%) and H2O, but neither CO nor NOx, and (iii) the ability to capture easily generated CO2 by means of CCS systems. The process requires high purity oxygen (>95%) in large quantities, which is typically produced by Air Separation Unit (ASU) or by PSA. OTMs are a promising alternative for oxygen production at the high temperature and the integration of OTM into high temperature processes as coal oxy-combustion could save energy in the oxygen production because the operation temperature of the OTM reactor and the combustion temperature (850–1000 °C) are in the same range. Indeed, the theoretical energy demand for oxygen production via OTMs would be significantly lower compared to cryogenic air separation. Several studies reported the effective energy demand values of 147 kW per h per tO2 (ref. 380) and 185 kW per h per tO2 (ref. 381) for plants equipped with the OTM technology, while the energy consumption for cryogenic distillation has been put at 220–245 kW per h per tO2 for large scale production,382–384 and 280–340 kW per h per tO2 for small-to medium-scale plants.381The integration of OTMs in the oxy-fuel combustion process can be applied in two ways called the 4-end mode and the 3-end mode. Fig. 15a and b illustrate the two options, respectively. The two processes can be distinguished according to two parameters: the membrane integration (direct/indirect) and the operation mode (sweep gas/vacuum). The 3-end mode membrane module is indirectly integrated to oxy-fuel combustion power plants. In this configuration, the membrane module generates pure oxygen, which is subsequently diluted with recirculated flue gas to control the combustion process in the boiler. The membrane module is, therefore, not in direct contact with the flue gas. Vacuum pumps are required to remove the oxygen from the membrane. Conversely, the 4-end mode integrates the membrane module in direct contact with the flue gas. The recirculated flue gas is used as a sweep gas on the permeate side of the membrane and is thus directly diluting the oxygen and ready for combustion.385 Consequently, the 4-end mode membrane module does not require additional turbomachinery and consumes less energy than the 3-end mode. Up to 60% reduction in capture energy demand compared to cryogenic air separation can be achieved by using thermally integrated separation modules (4-end mode) based on ceramic OTMs.380,384
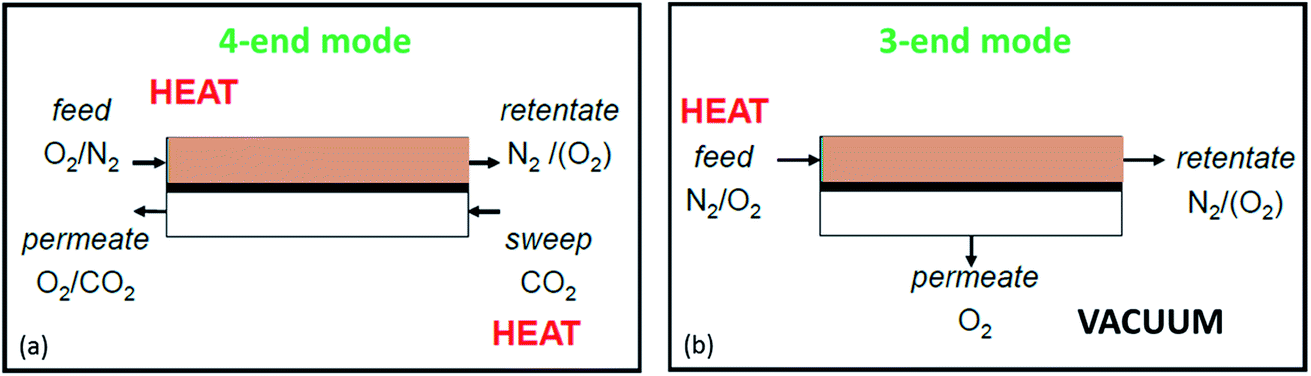 | ||
| Fig. 15 Illustrations of (a) 4-end mode (direct) and (b) 3-end mode (indirect) integration of OTMs into oxy-fuel combustion power plants. | ||
The main drawback for OTMs to be used in oxy-fuel process are the harsh operation environments to which the membranes are exposed when in direct contact with the fuel or flue gas. The composition of the flue gas of oxy-fuel power plants is influenced by several parameters: oxygen purity, fuel composition and air intrusion. Therefore, the composition of the flue gas varies from case to case. Nevertheless, it is commonly composed mainly of CO2 (80–90 mol%) and contains a limited amount of N2 (8–10 mol%), H2O (2–3 mol%), O2 (2–3 mol%) and SO2 (200–500 ppm).386 No OTM exhibited sufficiently high performances under such conditions to be commercialized yet. Therefore, the main effort required for the integration of the OTM technology in oxy-fuel combustion power plants is to develop high performance and stable membranes under realistic power plant conditions. Dual-phase membranes have proven to be stable in carbon dioxide containing environments and have showed acceptable oxygen permeation values, being in the range 0.2–0.52 ml min−1 cm−2 at a temperature about 1000 °C. It should be noticed that these results were obtained in non-reducing atmospheres (no H2, CH4 or CO present); in the presence of reducing gases higher fluxes can be expected.
In terms of performance and stability in CO2, also recent results from Luo and co-workers197,202,205 should be high-lighted, in which a new group of cobalt-free Cu-based dual-phase oxygen permeation membranes made up of Ce0.9Pr0.1O2 and PrxSr1−xFe1−yMyO3 (M = Cu, Bi, In) is reported. The composition with Pr0.4Sr0.6Fe0.95Cu0.05O3 yielded in an oxygen permeation flux of 0.98 ml min−1 cm−2 when using CO2 as sweep gas, showing potential for application in CO2 capture based on the oxy-fuel combustion.
Dual-phase OTMs were also tested under SO2 atmosphere showing lowered oxygen permeation fluxes due to SO2 adsorption in competition to oxygen blocking active sites for the surface exchange reactions. Nevertheless, no structural degradation/chemical reactions such as sulfate-formation were found after SO2 exposure and, thus, performances recover in clean atmosphere.144,145,149 These results, despite being promising are still too low for considering composite materials as ready-to-use in oxy-fuel installations, for what it is needed further investigation on the matter. In this context, coating high-performance LSCF or BSCF MIEC membranes with thin, protective dual-phase layer (e.g. Fe2NiO4–Ce0.8Tb0.2O2) as recently reported by Gaudillere et al.230 seems to be an interesting concept which should be further explored.
6 Existing industrial applications for oxygen transport membranes
6.1 Praxair, Inc. integrated gasification combined cycle
Significant efforts have been made to develop OTMs for integration in the diverse industrial applications described above. The state-of-the-art OTM in industry is a CH4-reforming pilot-scale system developed by Praxair, Inc., which can be attached to, for example, a coal-gasification train (Fig. 16) in the so-called integrated gasification combined cycle (IGCC). The OTM system is used to adjust the H2/CO ratio in the syngas stream from the coal gasification process. Syngas mixtures with the proper H2/CO ratio can be used subsequently in catalytic reactions to generate liquid fuels, as well as oxygenated organic compounds, such as methanol, acetic acid, and dimethyl ether (DME).1,24,387–390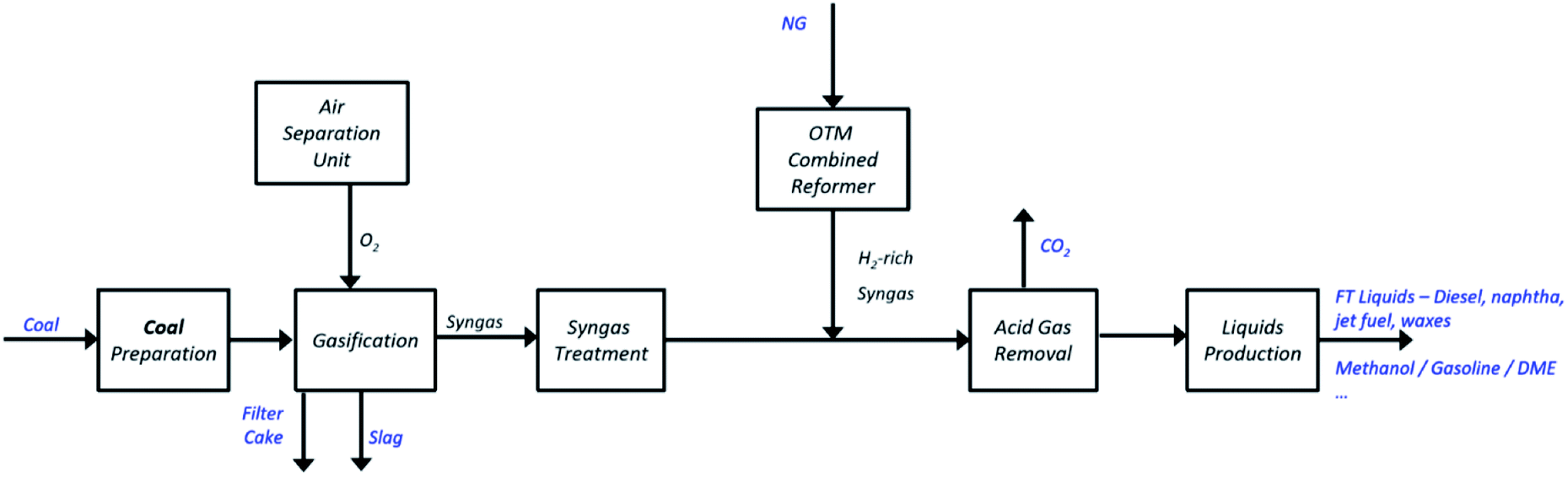 | ||
| Fig. 16 Diagram of the integrated gasification combined cycle (IGCC). The syngas is enhanced by an OTM reforming system (NG = natural gas, FT = Fischer Tropsch, DME = dimethyl ether).389 | ||
The concept for an OTM system is shown in Fig. 17 and is based on the integration of steam methane reforming (SMR), auto-thermal reforming (ATR) and air separation unit (ASU) processes in a single reactor working between 900 °C and 1050 °C at 27.5 bar.252 The ATR occurs in the OTM tube, where the OTM also acts as the ASU. The combination of the units increases the thermal efficiency by combining endothermic (SMR) and exothermic (ATR) processes, as well as increasing the yield of H2 and CO in the syngas product. The steam reforming step partially converts the CH4 into synthesis gas, as well as converting heavier hydrocarbons into CH4, H2 and carbon oxides. When supplied with CH4 conversion rates of >99% and 70 vol% H2 content in the syngas with a H2/CO ratio of 3.4 can be obtained.389
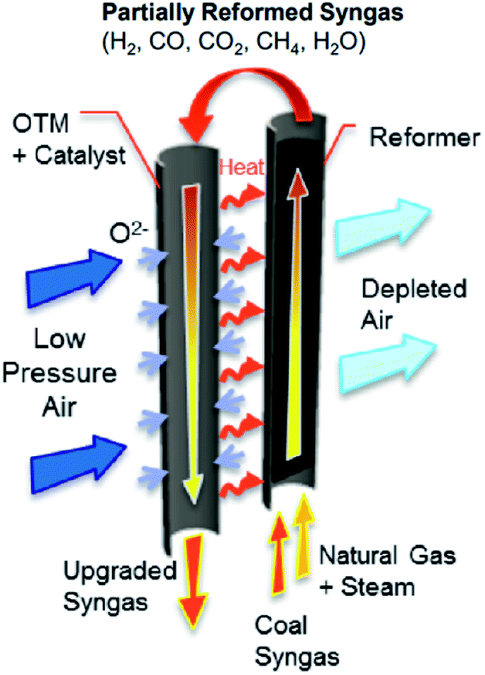 | ||
| Fig. 17 OTM combined reformer for IGCC power systems concept developed by Praxair, Inc.252 | ||
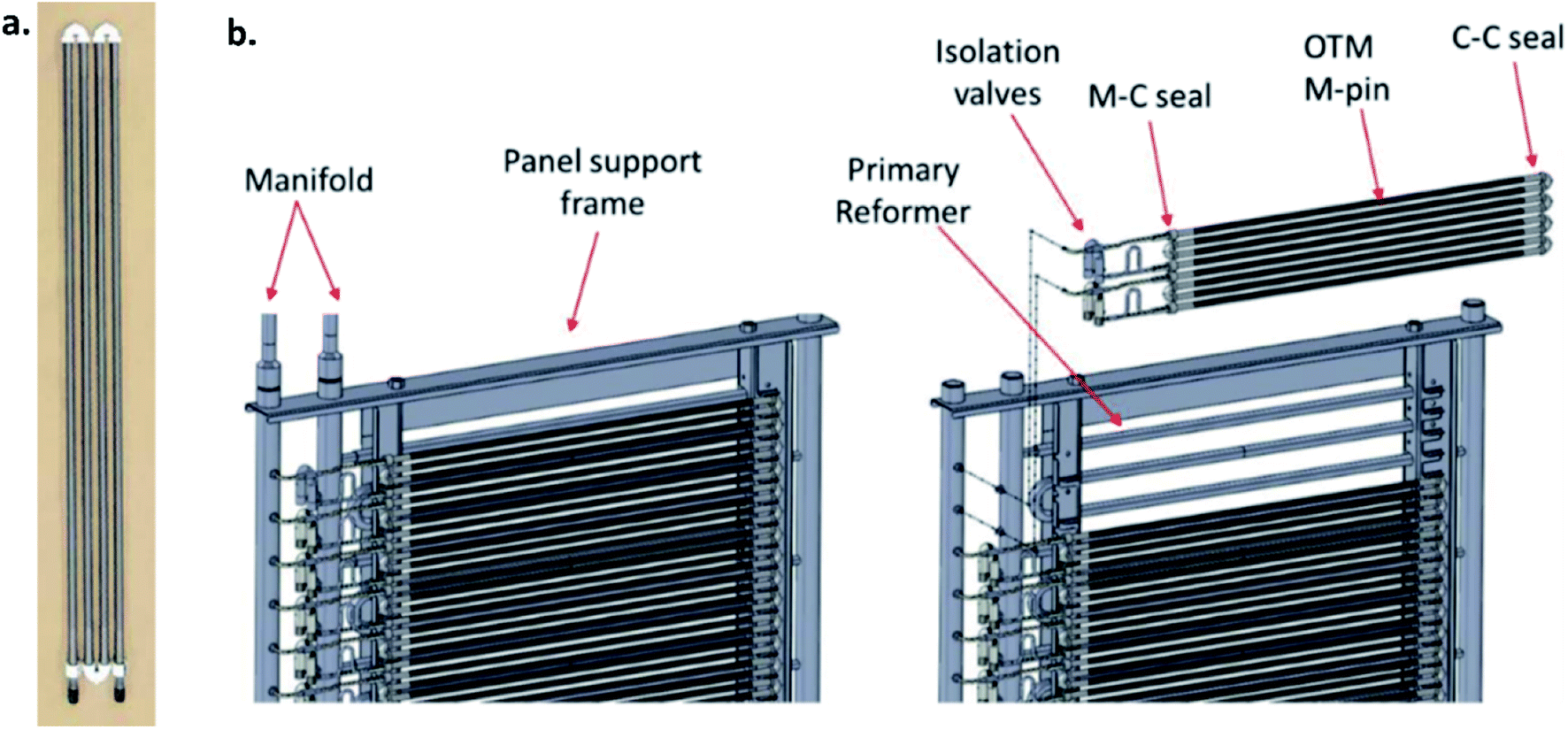 | ||
| Fig. 18 Arrangement of the (a) M-pin assembly and (b) OTM unit panel arrays.389 | ||
 | ||
| Fig. 19 Picture of (a) a single OTM unit panel array and (b) scaling panel size concept for large-scale applications.389 | ||
Preliminary techno-economic analysis has shown that carbon capture increases from 83% to 92% by using the OTM combined reformer in an IGCC plant and a HHV net plant efficiency could increase from 32% to 35% in comparison to the coal gasification plant. The expected cost of the plant using OTM is estimated as $3840 USD kW−1.252,389
The primary reformer is a tube made of a metal alloy stable at high temperature, such as Inconel 625 or 800HT, with an inner diameter of 1.25 cm. A catalyst-coated spiral metal monolith made of 800HT alloy (Fig. 20) is inserted inside the metal alloy tubes. The reforming catalyst is a Praxair, Inc. formulation based on Ni, Rh, Al2O3, CeO2 and YSZ with high coking resistance. The SMR unit operates between 13.8 and 29.3 bar at temperatures between 800 °C and 900 °C with a steam-to-carbon ratio of 1.5.389,392
 | ||
| Fig. 20 Picture of (a) the components of the primary reformer (tube and catalysts support) and (b) catalyst-coated spiral metal monolith.389 | ||
The secondary reformer technology, in its current form, has been in development since 2010. It consists of a tubular OTM with a diameter of approximately 10 mm. The OTM operates at temperatures between 900 °C and 1000 °C, thus, materials with exceptional redox resistance are required. The OTM is a multilayer tubular architecture consisting of thin dense and porous functional layers supported on a mechanically robust yttria-doped zirconia porous support fabricated by extrusion. The sintered tubes have thickness of approximately 1 mm and are designed to work at 29 bar and 1000 °C; however, they are have demonstrated the ability to withstand burst pressures in excess of 100 bar.389,393 A schematic of the architecture of the membrane is shown in Fig. 21.
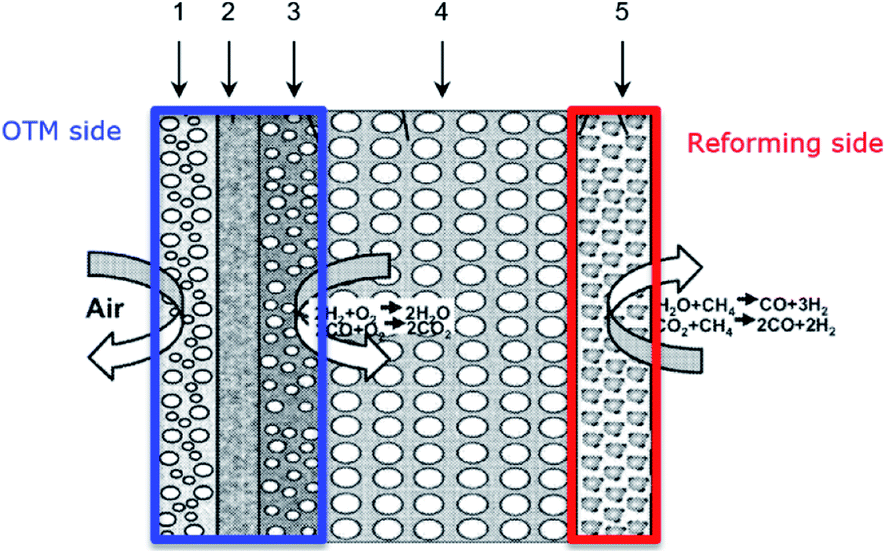 | ||
| Fig. 21 Schematic of the OTM developed by Praxair®. (1) Surface exchange layer; (2) active membrane; (3) fuel oxidation layer; (4) porous support; (5) reforming catalyst layer.393 | ||
The inner side of the tube (layer 5 in Fig. 21) is coated with a reforming porous catalyst layer based on Ni, Rh, Al2O3 and YSZ ca. 15 μm thick deposited by wash-coating. The active oxygen separation membrane (layer 2 in Fig. 21) is based on a dual-phase composite of (La0.8Sr0.2)0.98Cr0.3Fe0.7O3−δ (LSCrF) and Zr0.802Sc0.18Y0.018O2−δ (ScYSZ) with an approximate 40:60 volumetric ratio. On the opposing surfaces of the separation membrane are two porous catalytic layers (surface exchange layer and fuel oxidation layer) of the same dual-phase composite. The thicknesses of the surface exchange, membrane and fuel oxidation layers are approximately 10, 15 and 15 μm respectively. Fig. 22 shows the OTM tubes and the microstructure of the OTM and reforming side, as well as a picture of the OTMs after sintering.389
 | ||
| Fig. 22 Microstructure of the OTM showing the functional layers of the OTM (left), a picture of the sintered membranes with the different coatings (middle) and the reforming catalyst layer (right).389 | ||
The multi-step fabrication process of an OTM tube is summarised in Fig. 23. The process starts with debinding of the extruded tubes at 1050 °C for 4 h in air. After cooling, the fuel oxidation and the membrane layers are deposited. The coated layers and the support are sintered at 1350–1400 °C for 6 hours in an inert atmosphere (e.g. N2). Finally, the surface exchange and the reforming catalyst layers are deposited and pre-sintered at 1250 °C for 30 minutes in air atmosphere. A yield of 96% is reported following this method.389
 | ||
| Fig. 23 Fabrication process of the oxygen membranes.393 | ||
Fig. 24a shows the normalized oxygen flux, where ‘1’ is the oxygen flux targeted by Praxair, Inc.389 As observed, the operation of the membrane is very stable during the test. Similar stability tests were performed using 24 bar for 1000 h showing high stability with normalized oxygen flux around 1.0. Thermal treatment in reducing conditions at 1400 °C showed that the dual-phase components and catalysts are unaffected and no traces of La2Zr2O7 and SrZrO3 were identified.
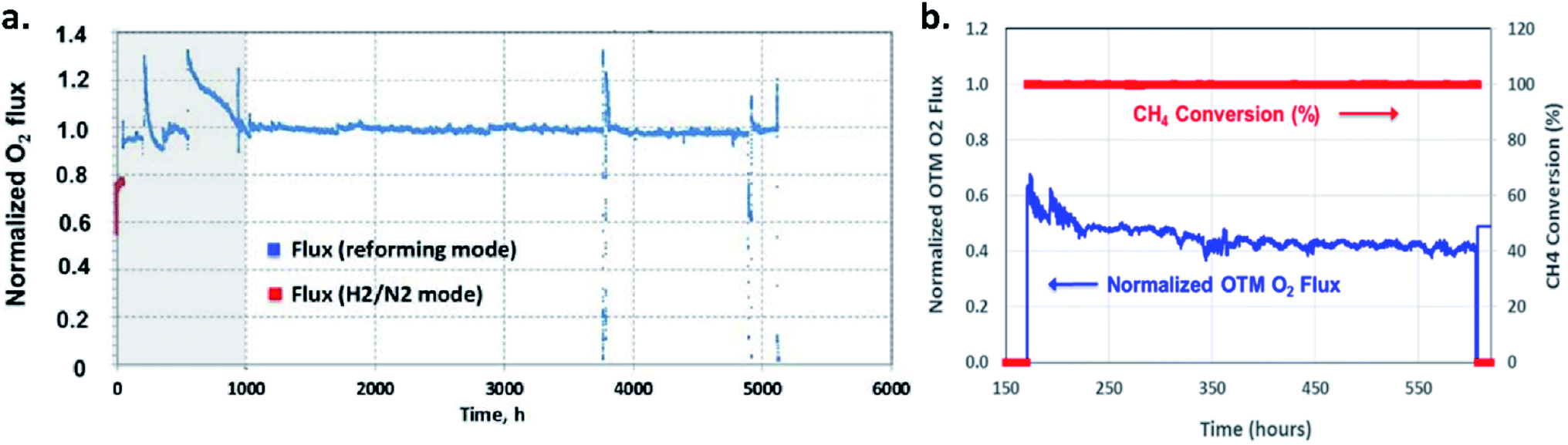 | ||
| Fig. 24 (a) Long term test of the OTM combined reformer single tube using simulated syngas at 950 °C and 13.8 bars. (b) OTM single panel oxygen flux tests using synthetic coal syngas/NG/steam at 10.3 bar and average temperature of 970 °C.389 | ||
Fig. 24b shows the tests of an OTM single panel consisting of six primary reformers and nine OTM secondary reformers using a mixture of simulated coal syngas/natural gas/steam for more than 500 hours at a pressure of 10.3 bar.389 The natural gas accounted for 30% of the high heating value (HHV) of the fuel feed. After an initial equilibrium period, a stable oxygen flux is obtained. The overall methane conversion after both reformers was 99.8%.
Similar methane conversion (99.5%) was obtained in an IGCC pilot plant, using a 72 tube-multi panel unit working at 13.8 bar for 800 h.389 As in the panel tests, a simulated coal syngas/natural gas/steam stream reacted with the oxygen provided by the OTM. The composition of the feed gas was 12 vol% CH4, 37% H2O, 31 vol% H2 and 20 vol% N2. After the IGCC, syngas with composition 0.04 vol% CH4, 61.4 vol%, H2, 8.2 vol% CO, 4.4 vol% CO2 and 25.8 vol% N2 was obtained.389
Although additional work is necessary to improve the instrumentation and control system of the pilot plant, Praxair has demonstrated important progress towards the commercialization and application of OTMs to produce high-quality syngas and enhance the syngas produced by coal-gasification, reducing the amount of cryogenically-produced oxygen required.
6.2 OTM for blue hydrogen production
Linde, formally Praxair R&D has developed world-class ceramic membrane technology which has been demonstrated at pilot-scale for conversion of natural gas to syngas. The existing pilot-scale test infrastructure in Tonawanda, NY is currently being re-purposed to demonstrate an advanced OTM-SMR blue hydrogen technology. Tubular ceramic membranes are integrated as heating elements into a Steam Methane Reformer (SMR) furnace, replacing conventional air-fired burners. Combustion of hydrocarbon fuel is facilitated by oxygen that has been transported from low-pressure furnace air, through the ceramic membrane to pressured fuel contained in arrays of manifolded, ceramic membrane burner elements. On fueling, the elements ‘light-up’ and radiate heat to conventional SMR tubes. CO2 that would normally leave in the SMR flue gas as a combustion product from conventional air-fired burners is concentrated as CO2 in the OTM ceramic burner element outlet manifold and remains isolated from the large volume of air that flows through the furnace interior. As such, the OTM burner elements facilitate air-separation, oxy-combustion, and CO2 capture in a single component. In addition to these features, the ceramic burners require no external oxygen feed, require no solvents or sorbents, produce no NOx emissions from combustion, and in addition to CO2 also recover the water in the combustion products, resulting in significantly less water consumption than a conventional SMR producing hydrogen.The results of preliminary economic assessments at a scale of 50000 Nm3 hH2−1, a natural gas price of $3/MMBtu, and a power price of $50/MW h, suggest that OTM-SMR technology has the potential to reduce the cost of CO2 capture by $30–40/tonne as compared to the best-known post-combustion capture technologies today.
The basic process for the OTM-SMR is shown in Fig. 25. A mixture of high-pressure, preheated, and desulfurized natural gas and steam enters conventional SMR tubes containing catalyst where endothermic steam-methane reforming reactions convert the fuel into a syngas mixture of H2, CO, CO2, H2O and residual CH4. The mixture proceeds to cooling and a water-gas shift to further convert residual CO to hydrogen, followed by hydrogen recovery within a pressure swing adsorption (PSA) unit. A purified, high pressure hydrogen product exits the PSA as primary product, while residual CO, CO2, CH4, and unrecovered hydrogen exit as a low-pressure tail gas stream. In a conventional SMR, this tail gas stream would be combusted with air and some additional natural gas in the SMR furnace providing heat to the primary reforming process and additional process heat recovery and steam generation from the flue gas. In a conventional SMR, flue gas discharged to atmosphere contains all of the carbon from the natural gas provided to the system in the form of CO2. In the OTM-SMR process, the PSA tail gas is compressed, heated, and fed to the ceramic OTM burner elements which can be thought of as ‘oxy-fuel gas heating elements’, in that they perform the function of combusting the PSA tail gas fuel with pure oxygen. The oxygen for combustion is generated via in situ electrochemical separation from preheated low-pressure air circulated through the furnace interior. The tubular OTM burner elements ‘light-up’ and glow as the fuel contained inside the elements is combusted with oxygen and the released heat is radiated to the SMR tubes. The carbon that would normally leave in the SMR flue gas, is concentrated as CO2 in the OTM burner element outlet pipe. Once cooled and dried, the concentrated CO2 stream may be sent to a liquefier, or further purified, and compressed for pipeline transport.
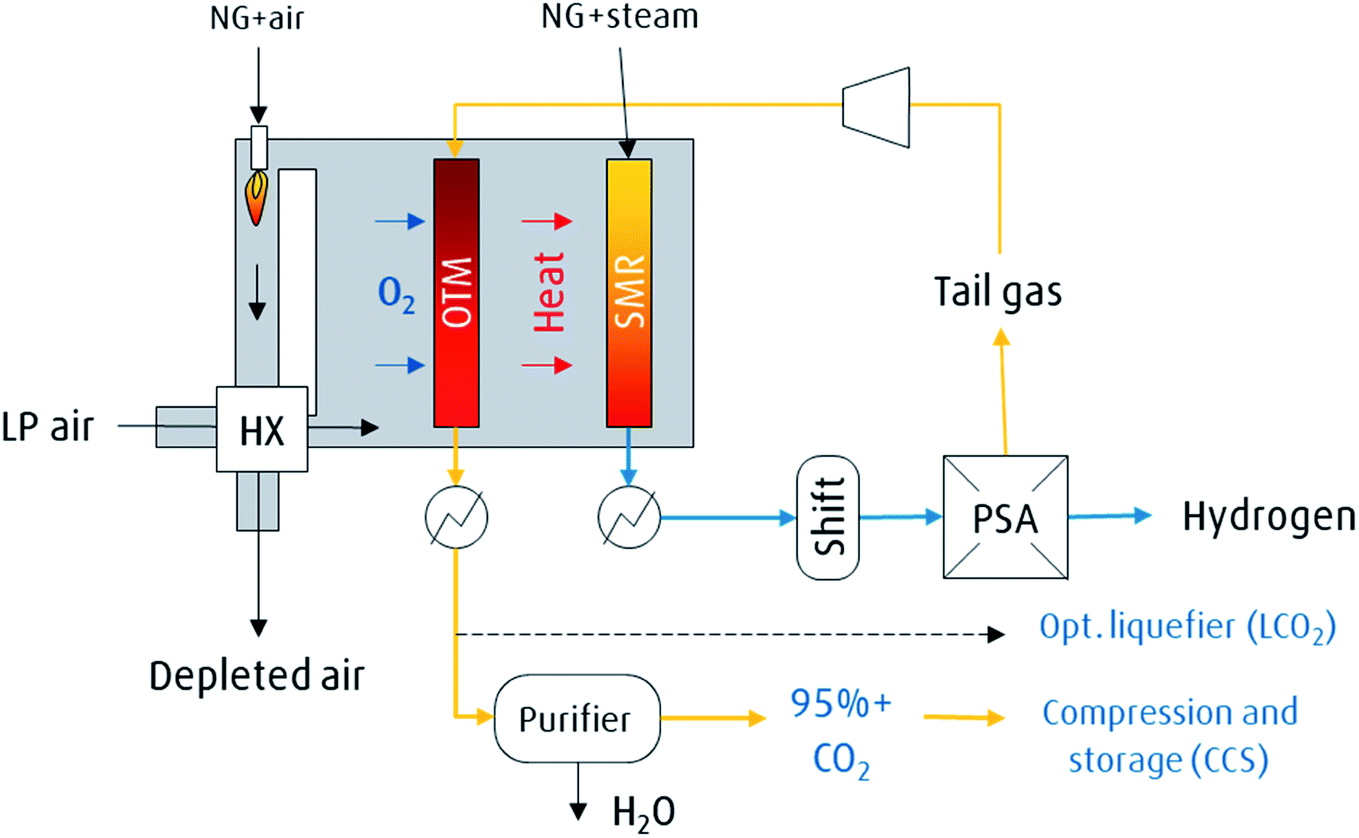 | ||
| Fig. 25 OTM- SMR high-level process schematic. | ||
7 Potential and required development steps for future industrial applications – an outlook
7.1 Potential performance of dual-phase membranes
As a first step to investigate the applicability and potential performance of a dual-phase composite system, a “best-case estimation” of the oxygen flux should be made. Even if the “best-case scenario” cannot technically be achieved, this approach has the benefit to assess if the selected dual-phase composite can in theory reach the performance required by the targeted application.An example of such an estimation for a dual-phase OTM is described below. As “best case situation” it is assumed that the Wagner equation (eqn (2)) is valid in the entire parameter range i.e. neglecting surface exchange limitation as well as any impact of porous supports required for thin membranes.
Dual-phase OTMs typically show permeation rate limitations by the ionic conductivity. Therefore, a maximum portion of the ionic conductor is beneficial. In consequence, a best-case situation exists if the (hypothetical) dual-phase membrane consists only of the ion conducting phase, and assuming an infinite electronic conductivity. In this case the ambipolar conductivity equals the ionic one.
As ion conducting phase 20 mol% Gd-doped ceria (CGO20) is chosen as an example due to its high ionic conductivity at intermediate temperatures, i.e. 500–600 °C.394 The targeted permeation rate value (benchmark) considered the technical relevant minimum flux requirement by the OTM community varies from 1 to 10 ml cm−2 min−1.395,396
Using reported data for CGO20 (σ410 °Cionic = 8,15 × 10−4 S cm−1; Ea = 70 kJ mol−1),397 the upper bound of the oxygen permeation rate can be calculated according to eqn (2) considering the thickness L, the absolute temperature T, and the driving force 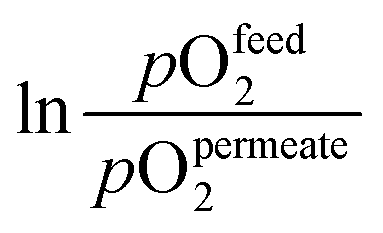 as parameters. Here, p′ (corresponding to pOfeed2) is fixed to air at ambient pressure, i.e. p′ = 0.21 bar. For p′′ (corresponding to pOpermeate2) two concentrations are chosen arbitrarily, i.e. p′′ = 0.005 bar mimicking typical oxygen permeation tests using air and inert gas (Ar or He) as feed and sweep gases, respectively and p′′ = 10−15 bar exemplarily for membrane reactor applications.
as parameters. Here, p′ (corresponding to pOfeed2) is fixed to air at ambient pressure, i.e. p′ = 0.21 bar. For p′′ (corresponding to pOpermeate2) two concentrations are chosen arbitrarily, i.e. p′′ = 0.005 bar mimicking typical oxygen permeation tests using air and inert gas (Ar or He) as feed and sweep gases, respectively and p′′ = 10−15 bar exemplarily for membrane reactor applications.
In Fig. 26, the dependency of the oxygen flux on pressure gradient, thickness and operating temperature is illustrated in an Arrhenius plot. The orange line represents the hypothetical oxygen flux across a 1 mm thick membrane lab operated in a 0.21/0.005 partial pressure gradient. It should be noticed that both thickness and pressure gradient are the most common test conditions reported, as shown in Table 1.
The reduction of the membrane thickness to 50 μm leads to significant improvement of the hypothetical oxygen flux as illustrated by a shift along the Y-direction (green line). The same tendency occurs by reduction of the sweep side partial pressure from 0.005 bar to 10−15 bar (brown line). As expected, the highest fluxes can be achieved with a thin membrane operated at high driving force (violet line). In these hypothetical cases the “best-case estimation” equals the benchmark performance of 1 ml cm−2 min−1 already at temperatures above 470 °C.
For comparison, Fig. 26 also shows experimental data of 1 mm thick membrane pellets (single phase perovskite La0.6Sr0.4Co0.2Fe0.8O3−δ (LSCF) and CGO20-based surface activated dual-phase composite). The experimental data were driving force normalized to p′′ = 0.005 bar (an average value of O2 concentration measured by mass spectrometry during the permeation test). At 1000 °C, LSCF comes close to the benchmark (coincidently equal to the upper bound of CGO) and utilizing thin supported membranes it is reported that it can easily exceed the benchmark value at temperatures above 800 °C.9,398 However, due to a relatively high activation energy, LSCF reaches only 2.5% of the upper bound of CGO at 650 °C. The second material (black squares, Fig. 26), a CGO-based composite using FeCo2O4 spinel as second phase (CGO–FCO),16,253 shows significant lower activation energy. Below 800 °C a better performance compared to LSCF, reaching approx. 30% of the upper bound at 650 °C, can be observed. Please note, that this specific membrane is given here as an example for illustration, and the authors consider it as one out of many applicable composites. However, the benefit of CGO20-based dual-phase membranes in comparison to single-phase perovskites at lower temperature is considered to be systematic.
 | ||
Fig. 26 The modelling of best-case performance of CGO20 for L = 0.05 and 1 mm, 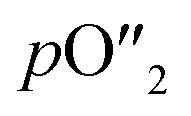 = 0.005 and 10–15 bar, dashed line represents oxygen flux of 1 ml cm−2 min−1 as well as experimental data of LSCF and CGO–FCO 1 mm thick pellets. = 0.005 and 10–15 bar, dashed line represents oxygen flux of 1 ml cm−2 min−1 as well as experimental data of LSCF and CGO–FCO 1 mm thick pellets. | ||
While for thicker membranes, i.e. 1 mm, the experimentally measured performance and the “best case estimations” are in reasonable agreement, large discrepancies can be found for thinner membranes. As example, the experimental data for an 11 μm thin supported CGO–FCO composite membrane (prepared by tape casting) is shown in Fig. 27. While the measured performance exceeds the benchmark at higher temperatures, the gap between the respective upper bound (orange coloured line) and experimental values increases significantly. Moreover, the activation energy drastically increases below 900 °C. This is strongly indicating surface exchange limitations, as discussed in Sub-section 1.3.3. In particular the surface activation of the membrane support interface, a place which is not easily accessible for catalyst integration, seems performance limiting. In consequence, an advanced activation using a Ce/Pr-based catalyst infiltrated into the support as well as a porous composite top-coating was carried out. A clear performance improvement at intermediate temperatures (Fig. 27, green square symbols) was achieved, but nevertheless there is obviously still dramatic need for optimization.
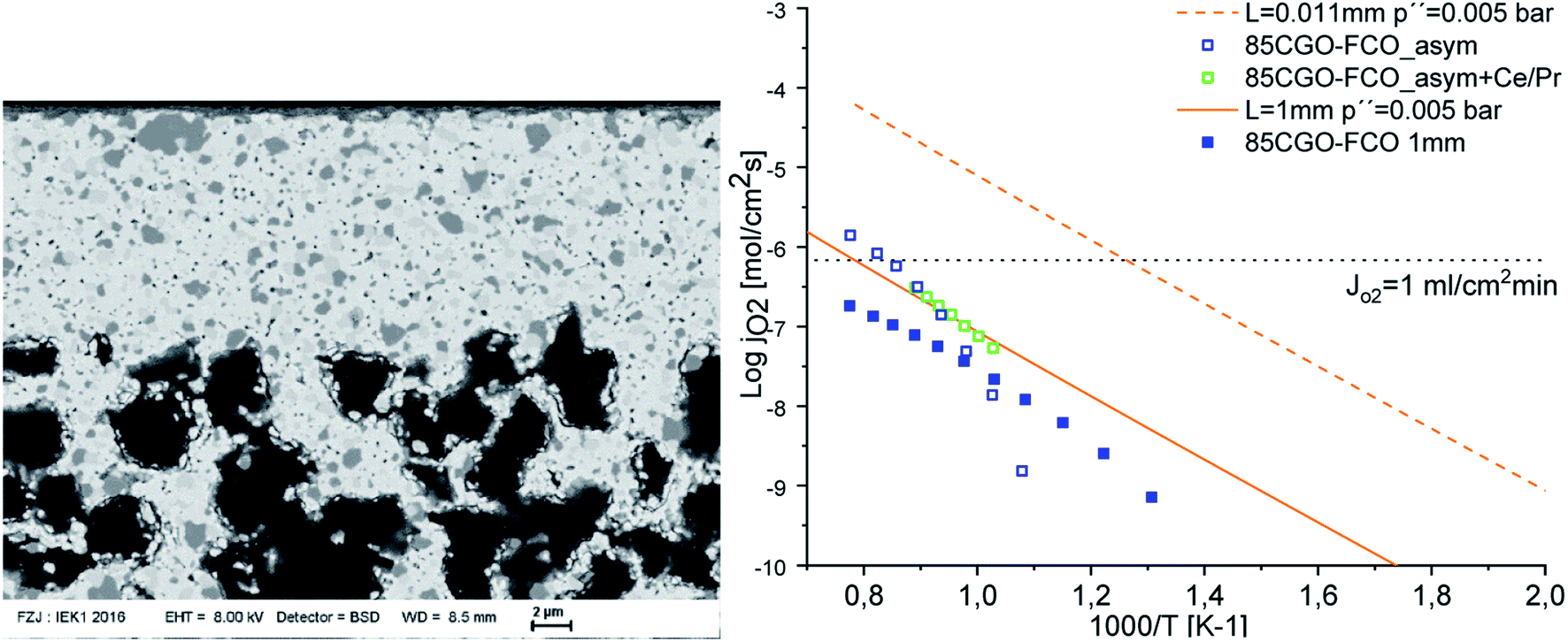 | ||
| Fig. 27 Left: 11 μm thin asymmetric membrane made of Ce0.8Gd0.2O2−δ–FeCo2O4 composite (CGO–FCO) right: performance of asymmetric CGO–FCO with porous LSCF top-coating as well as advanced surface activation using (Ce,Tb)O2–NiFe2O4 composite porous top coating and a Ce/Pr-based catalyst infiltrated into both top coating and porous CGO–FCO support. | ||
Another example of theoretical estimation of dual-phase OTMs performances was described in the literature.120 The authors calculated the theoretical oxygen permeation fluxes of 1000 μm thick and 110 μm thick 10Sc1YSZ–LCCN OTMs and compared them to the experimental values. Theoretical oxygen permeation fluxes were calculated considering different scenario: (i) the permeation flux is mainly limited by bulk diffusion limitation, the oxygen flux can be characterized by the Wagner equation (eqn (2)), (ii) the permeation flux is also limited by surface exchange kinetics, the contribution of the catalyst layers (8YSZ–LSM in this study) to the overall resistance of the membrane must also be taken into account, and (iii) the oxygen permeation flux was calculated considering the contribution of the catalyst layers and a tortuosity factor corresponding to the fact that the ionic path is “blocked” by the electronic conducting phase in the case of dual-phase OTMs. For these calculations the 8YSZ–LSM resistances were taken from studies published by Kim et al.399 and Barfod et al.400 The tortuosity factor was estimated at 2 from the literature.179 The ambipolar conductivity of the membrane was approximated as σamb ≃ σionic ≃ χσionic,10Sc1YSZ, where χ is the volume percentage of 10Sc1YSZ in the composite membrane. The ionic conductivities of 10Sc1YSZ from 750 °C to 950 °C were selected in a study of Irvine et al.401 The experimental (symbols) and theoretical (lines) oxygen permeation fluxes through 10Sc1YSZ–LCCN OTMs published in this study are presented Fig. 28. The figure shows that the theoretical oxygen permeation fluxes become fairly close the experimental ones once the Wagner equation and the contribution of the catalyst layers and the tortuosity factor are considered. More details about these theoretical calculations can be found in the ESI material of the study.120
 | ||
| Fig. 28 Experimental (symbols) and theoretical (lines) oxygen permeation fluxes through 10Sc1YSZ–LCCN (70–30 vol%) membranes coated with a 8YSZ–LSM (50–50 vol%) catalyst layer on both sides as a function of the temperature. The experiment was performed in air/N2 atmosphere. | ||
The principal trends of this specific examples are expected to be general for dual-phase OTM. Therefore, the approach described here will help to analyse future developments and to identify the bottlenecks requiring optimizations.
Required future R&D directions includes microstructuring of dual phase membranes in order to utilize the potential of the chosen ion conductor as much as possible, i.e. maximizing the ambipolar conductivity. A real breakthrough, however, can only be expected once a novel material with superior ionic conductivity is developed, which is currently not in sight. The material/microstructure optimization should be accompanied with developing thin membranes, e.g. asymmetric membranes or capillaries/hollow fibres. In this context special attention must be laid on catalysts and their application facilitating oxygen surface exchange.
7.2 Required development steps for future industrial applications
Besides recent progress in OTM research, more R&D efforts are necessary to bring OTM technology to market. The ones considered most relevant are summarized below:Attention has to be paid that typical oxygen reduction catalysts used in OTMs, e.g. cobaltites and ferrites, might have negative impact on the targeted chemical reactions. Therefore, cross-cutting activities with the catalysis community are required in order to find suitable catalysts and ways to integrate these into the membrane assembly.
A vision for future application areas and a market entry vision are illustrated in Fig. 29. OTM technology is expected to enter the market in special niche applications, in which pure oxygen is required “on demand” on a relatively small scale. Such application could be chemistry laboratories (for combustion analyzers, calorimeters, etc.) or medical application (here one must consider long and costly validation steps). Most likely single-phase MIEC membranes are the preferred option over dual-phase membranes in these niche applications, as no exposure to reducing atmospheres or high impurity concentrations are expected. In parallel, more process intensified schemes, i.e. membrane reactors, need to be pursued where dual-phase membranes are very promising due to corrosive and reducing atmospheres.
 | ||
| Fig. 29 Application areas and potential market entering vision for dual-phase OTMs. | ||
The next market segment is the on-site production of oxygen on a medium scale, e.g. to cover the supply of hospitals, in the specialty ceramic or glass industry for various small scale oxy-fuel processes or in the food industry. These ‘stepping-stones’ are considered very important in the roll out strategy of the OTM technology. The reliability and performance stability demonstrated in these early markets are expected to create references for the technology and help to find investors for larger demonstration projects anticipated in the future.
The future energy sector is expected to develop in a direction of more decentralized power generation with less use of fossil fuels. Nevertheless, carbon capture and utilization (CCU) technologies are needed to match the trend in the circular economy relying on “renewable” fuels and polymers. Here OTM technology for use in oxy-fuel schemes as well as gas-to-X (e.g. biogas upgrading) is a promising solution. Due to the modular design of membrane technology, it is well suited for small and medium scale whereas mature technology like cryogenic air separation needs large-scale centralized facilities with additional transport expenditure and PSA requires too much energy. Once the technology is established on medium scale, large scale application such as steel industry, cement production, or bulk chemical industry can be targeted for large scale technology demonstration. Lower TRL research, of course, is required already in earlier stages.
Author contributions
Ragnar Kiebach performed conceptualization, writing – original draft, writing – review & editing, visualization and supervision; Stéven Pirou carried out investigations, writing – original draft, writing – review & editing and visualization; Lev Martinez Aguilera carried writing – original draft; Astri Bjørnetun Haugen performed writing-original draft and writing-review and editing; Andreas Kaiser performed writing-original draft and writing-review and editing, Peter Vang Hendriksen participated in writing-review and editing; María Balaguer participated in conceptualization, investigation, writing-original draft, writing-review and editing; Julio García-Fayos performed investigation, writing-original draft, writing-review and editing; José Manuel Serra carried out conceptualization, writing-original draft, writing-review and editing; Falk Schulze-Küppers performed writing- original draft; Liudmila Fischer carried out investigations; Wilhelm Albert Meulenberg carried out writing – review & editing; Stefan Baumann writing – conceptualization, original draft & writing – review & editing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This project was supported in part by the FLEXSNG project. This project has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 101022432, and form the Danish Council for Independent Research (DFF) for funding within the H2Now project (Grant No. 9041-00334B). This work was also funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – 387282673.References
- S. Smart, J. C. Diniz Da Costa, S. Baumann and W. A. Meulenberg, Adv. Membr. Sci. Technol. Sustain. Energy Environ. Appl., 2011, 255–292 CAS.
- J. Emsley, Nature's Building Blocks: an A-Z Guide to the Elements, Oxford University Press, 2001 Search PubMed.
- T. Banaszkiewicz, M. Chorowski and W. Gizicki, AIP Conf. Proc., 2014, 1573, 1373–1378 CrossRef CAS.
- S. Alavandi, J. Seaba and G. Subbaraman, Emerging and Existing Oxygen Production Technology Scan and Evaluation, 2018 Search PubMed.
- C. S. Chen, B. A. Boukamp, H. J. M. Bouwmeester, G. Z. Cao, H. Kruidhof, A. J. A. Winnubst and A. J. Burggraaf, Solid State Ionics, 1995, 76, 23–28 CrossRef CAS.
- S. Baumann, W. A. Meulenberg and H. P. Buchkremer, J. Eur. Ceram. Soc., 2013, 33, 1251–1261 CrossRef CAS.
- P. Niehoff, S. Baumann, F. Schulze-Küppers, R. S. Bradley, I. Shapiro, W. A. Meulenberg, P. J. Withers and R. Vaßen, Sep. Purif. Technol., 2014, 121, 60–67 CrossRef CAS.
- F. Schulze-Küppers, S. Baumann, W. A. Meulenberg and H. J. M. Bouwmeester, J. Memb. Sci., 2020, 596, 117704 CrossRef.
- J. M. Serra, J. Garcia-Fayos, S. Baumann, F. Schulze-Küppers and W. a. Meulenberg, J. Memb. Sci., 2013, 447, 297–305 CrossRef CAS.
- C. Gaudillere, J. Garcia-Fayos and J. M. Serra, J. Mater. Chem. A, 2014, 2, 3828–3833 RSC.
- E. A. Mason and A. P. Malinauskas, Gas Transport in Porous Media: the Dusty-Gas Model, Chemical e., Amsterdam; New York, 1983 Search PubMed.
- P. J. A. M. Kerkhof, Chem. Eng. J. Biochem. Eng. J., 1996, 64, 319–343 CrossRef CAS.
- A. Häffelin, C. Niedrig, S. F. Wagner, S. Baumann, W. A. Meulenberg and E. Ivers-Tiffée, J. Electrochem. Soc., 2014, 161, F1409–F1415 CrossRef.
- U. Unije, R. Mücke, P. Niehoff, S. Baumann, R. Vaßen and O. Guillon, J. Memb. Sci., 2017, 524, 334–343 CrossRef CAS.
- H. J. M. Bouwmeester, Catal. Today, 2003, 82, 141–150 CrossRef CAS.
- M. Ramasamy, S. Baumann, J. Palisaitis, F. Schulze-Küppers, M. Balaguer, D. Kim, W. A. Meulenberg, J. Mayer, R. Bhave, O. Guillon and M. Bram, J. Am. Ceram. Soc., 2016, 99, 349–355 CrossRef CAS.
- S. Baumann, P. Niehoff, F. Schulze-Kuppers, M. Ramasamy, W. A. Meulenberg and O. Guillon, ECS Trans., 2015, 66, 21–33 CrossRef CAS.
- S. J. Xu and W. J. Thomson, Chem. Eng. Sci., 1999, 54, 3839–3850 CrossRef CAS.
- P. J. Gellings and H. J. M. Bouwmeester, Catal. Today, 1992, 12, 1–105 CrossRef CAS.
- H. J. M. Bouwmeester and A. J. Burggraaf, in The CRC Handbook of Solid State Electrochemistry, CRC Press, 1997 Search PubMed.
- H. J. M. Bouwmeester, H. Kruidhof and A. J. Burggraaf, Solid State Ionics, 1994, 72, 185–194 CrossRef CAS.
- J. A. Lane, S. J. Benson, D. Waller and J. A. Kilner, Solid State Ionics, 1999, 121, 201–208 CrossRef CAS.
- Energy Information Administration, Annu. Rev. Energy, 2011, 2011, 117 Search PubMed.
- M. Puig-Arnavat, S. Soprani, M. Søgaard, K. Engelbrecht, J. Ahrenfeldt, U. B. Henriksen and P. V. Hendriksen, RSC Adv., 2013, 3, 20843–20854 RSC.
- J. H. Joo, G. S. Park, C. Y. Yoo and J. H. Yu, Solid State Ionics, 2013, 253, 64–69 CrossRef CAS.
- S. Baumann, J. M. Serra, M. P. Lobera, S. Escolástico, F. Schulze-Küppers and W. A. Meulenberg, J. Memb. Sci., 2011, 377, 198–205 CrossRef CAS.
- J. Gurauskis, Ø. F. Lohne, D. S. Lagergren, E. T. Wefring and K. Wiik, J. Eur. Ceram. Soc., 2016, 36, 1427–1434 CrossRef CAS.
- Z. Shao, W. Yang, Y. Cong, H. Dong, J. Tong and G. Xiong, J. Memb. Sci., 2000, 172, 177–188 CrossRef CAS.
- Y. Teraoka, T. Nobunaga and N. Yamazoe, Chem. Lett., 1988, 503–506 CrossRef CAS.
- Z. Shao, G. Xiong, J. Tong, H. Dong and W. Yang, Sep. Purif. Technol., 2001, 25, 419–429 CrossRef CAS.
- J. Tong, W. Yang, R. Cai, B. Zhu and L. Lin, Catal. Lett., 2002, 78, 129–137 CrossRef CAS.
- S. Liu and G. R. Gavalas, J. Memb. Sci., 2005, 246, 103–108 CrossRef CAS.
- T. Nagai, W. Ito and T. Sakon, Solid State Ionics, 2007, 177, 3433–3444 CrossRef CAS.
- P. Zeng, Z. Shao, S. Liu and P. Z. Xu, Sep. Purif. Technol., 2009, 67, 304–311 CrossRef CAS.
- H. Luo, B. Tian, Y. Wei and H. Wang, AIChE J., 2010, 56(3), 604–610 CAS.
- S. Gopalan, J. Miner. Met. Mater. Soc., 2002, 54, 26–29 CrossRef CAS.
- A. J. Jacobson, S. Kim, A. Medina, Y. L. Yang and A. Abeles, Mater. Res. Soc., 1998, 497, 29–34 CrossRef CAS.
- A. Leo, S. Liu and J. C. D. da Costa, Int. J. Greenh. Gas Control, 2009, 3, 357–367 CrossRef CAS.
- W. Zhou, R. Ran and Z. Shao, J. Power Sources, 2009, 192, 231–246 CrossRef CAS.
- W. Yang, H. Wang, X. Zhu and L. Lin, Top. Catal., 2005, 35, 155–167 CrossRef CAS.
- M. Arnold, H. Wang and A. Feldhoff, J. Memb. Sci., 2007, 293, 44–52 CrossRef CAS.
- E. Bucher, A. Egger, G. B. Caraman and W. Sitte, J. Electrochem. Soc., 2008, 155, B1218 CrossRef CAS.
- M. Pilar Lobera, S. Escolastico, J. Garcia-Fayos and J. M. Serra, ChemSusChem, 2012, 5, 1587–1596 CrossRef PubMed.
- R. Kriegel, R. Kircheisen and J. Töpfer, Solid State Ionics, 2010, 181, 64–70 CrossRef CAS.
- S. Švarcová, K. Wiik, J. Tolchard, H. J. M. Bouwmeester and T. Grande, Solid State Ionics, 2008, 178, 1787–1791 CrossRef.
- A. S. Möbius, Charakterisierung perowskitischer Hochtemperaturmembranen zur Sauerstoffbereitstellung für fossil gefeuerte Kraftwerksprozesse, RWTH Aachen University, 2010 Search PubMed.
- O. Ravkina, T. Klande and A. Feldhoff, J. Memb. Sci., 2015, 480, 31–38 CrossRef CAS.
- L.-W. Tai, M. M. Nasrallah, H. U. Anderson, D. M. Sparlin and S. R. Sehlin, Solid State Ionics, 1995, 76, 259–271 CrossRef CAS.
- J. W. Stevenson, T. R. Armstrong, R. D. Carneim, L. R. Pederson and W. J. Weber, J. Electrochem. Soc., 1996, 143, 2722–2729 CrossRef CAS.
- A. Petric, P. Huang and F. Tietz, Solid State Ionics, 2000, 135, 719–725 CrossRef CAS.
- H. Ullmann, N. Trofimenko, F. Tietz, D. Stöver and A. Ahmad-Khanlou, Solid State Ionics, 2000, 138, 79–90 CrossRef CAS.
- R.-R. Liu, S. Taniguchi, Y. Shiratori, K. Ito and K. Sasaki, ECS Trans., 2011, 35, 2255–2260 CrossRef CAS.
- D. Wang and J. Leng, Chem. Res. Chinese Univ., 2012, 28, 866–868 CAS.
- M. Greenblatt, Solid State Mater. Sci., 1997, 2, 174–183 CrossRef CAS.
- A. Aguadero, M. J. Escudero, M. Pérez, J. A. Alonso, V. Pomjakushin and L. Daza, Dalt. Trans., 2006, 4377–4383 RSC.
- V. V. Kharton, A. A. Yaremchenko, A. L. Shaula, M. V. Patrakeev, E. N. Naumovich, D. I. Logvinovich, J. R. Frade and F. M. B. Marques, J. Solid State Chem., 2004, 177, 26–37 CrossRef CAS.
- E. N. Naumovich and V. V. Kharton, J. Mol. Struct. THEOCHEM, 2010, 946, 57–64 CrossRef CAS.
- A. L. Shaula, E. N. Naumovich, A. P. Viskup, V. V. Pankov, A. V. Kovalevsky and V. V. Kharton, Solid State Ionics, 2009, 180, 812–816 CrossRef CAS.
- V. V Kharton, A. P. Viskup, E. N. Naumovich and F. M. B. Marques, J. Mater. Chem., 1999, 9, 2623–2629 RSC.
- D. ping Huang, Q. Xu, F. Zhang, W. Chen, H. xing Liu and J. Zhou, Mater. Lett., 2006, 60, 1892–1895 CrossRef.
- S.-Y. Jeon, M.-B. Choi, H.-N. Im, J.-H. Hwang and S.-J. Song, J. Phys. Chem. Solids, 2012, 73, 656–660 CrossRef CAS.
- J. B. Smith and T. Norby, J. Electrochem. Soc., 2006, 153, A233–A238 CrossRef CAS.
- Y. Takeda, R. Kanno, M. Sakano, O. Yamamoto, M. Takeda, Y. Bando, H. Akinaga, K. Takita and J. B. Goodenough, Mat. Res. Bull., 1990, 25, 293–306 CrossRef CAS.
- V. V. Vashook, S. P. Tolochko, I. I. Yushkevich, L. V. Makhnach, I. F. Kononyuk, H. Altenburg, J. Hauck and H. Ullmann, Solid State Ionics, 1998, 110, 245–253 CrossRef CAS.
- S. Engels, T. Markus, M. Modigell and L. Singheiser, J. Memb. Sci., 2011, 370, 58–69 CrossRef CAS.
- F. Zeng, S. Baumann, J. Malzbender, A. Nijmeijer, L. Winnubst, O. Guillon, R. Schwaiger and W. A. Meulenberg, J. Memb. Sci., 2021, 628, 119248 CrossRef CAS.
- L. Fischer, K. Neuhaus, C. Schmidt, K. Ran, P. Behr, S. Baumann, J. Mayer and W. A. Meulenberg, J. Mater. Chem. A, 2021 10.1039/d1ta05695f.
- T. J. Mazanec, T. L. Cable and J. G. Frye, Solid State Ionics, 1992, 53, 111–118 CrossRef.
- K. Wu, S. Xie, G. S. Jiang, W. Liu and C. S. Chen, J. Memb. Sci., 2001, 188, 189–193 CrossRef CAS.
- J. W. ten Elshof, N. Q. Nguyen, M. W. den Otter and H. J. M. Bouwmeester, J. Electrochem. Soc., 1997, 144, 4361–4366 CrossRef CAS.
- K. Kobayashi and T. Tsunoda, Solid State Ionics, 2004, 175, 405–408 CrossRef CAS.
- F. T. Akin and J. Y. S. Lin, J. Memb. Sci., 2004, 231, 133–146 CrossRef CAS.
- J. Kim and Y. S. Lin, J. Memb. Sci., 2000, 167, 123–133 CrossRef CAS.
- C. S. Chen, Fine Grained Zirconia-Metal Dual Phase Composites (ref 74 Chen thesis 1994), University of Twente, 1994 Search PubMed.
- C. S. Chen and a J. Burggraaf, J. Appl. Electrochem., 1999, 29, 355–360 CrossRef CAS.
- C. S. Chen, H. Kruidhof, H. J. M. Bouwmeester, H. Verweij and A. J. Burggraaf, Solid State Ionics, 1996, 86–88, 569–572 CrossRef CAS.
- T. H. Lee, Y. L. Yang and A. J. Jacobson, Solid State Ionics, 2000, 134, 331–339 CrossRef CAS.
- P. Seeharaj and A. Atkinson, Solid State Ionics, 2011, 204–205, 46–52 CrossRef CAS.
- W. Bai, J. Feng, C. Luo, P. Zhang, H. Wang, Y. Yang, Y. Zhao and H. Fan, Int. J. Hydrogen Energy, 2021, 46, 36257–36290 CrossRef CAS.
- D. Wang, Y. Guo, K. Liang and K. Tao, Sci. China, Ser. A, 1999, 42, 80–86 CAS.
- H. Arashi and H. Naito, Solid State Ionics, 1992, 53–56, 431–435 CrossRef CAS.
- Y. Nigara, Y. Kosaka, K. Kawamura, J. Mizusaki and M. Ishigame, Solid State Ionics, 1996, 86–88, 739–744 CrossRef CAS.
- H. Yanagida, K. Koumoto, K. Komoto and M. Miyayama, The Chemistry of Ceramics, 1996, 173 Search PubMed.
- X. J. Chen, K. A. Khor, S. H. Chan and L. G. Yu, Mater. Sci. Eng. A, 2002, 335, 246–252 CrossRef.
- V. V. Kharton, E. N. Naumovich and A. a. Vecher, J. Solid State Electrochem., 1999, 3, 61–81 CrossRef CAS.
- J. W. Fergus, J. Power Sources, 2006, 162, 30–40 CrossRef CAS.
- J. B. Goodenough, Annu. Rev. Mater. Res., 2003, 33, 91–128 CrossRef CAS.
- S. J. Skinner and J. A. Kilner, Mater. Today, 2003, 30–37 CrossRef CAS.
- T. Ishihara, N. M. Sammes and O. Yamamoto, in High-temperature Solid Oxide Fuel Cells: Fundamentals, Design and Applications, 2003, pp. 83–117 Search PubMed.
- J. W. Fergus, J. Miner. Met. Mater., 2007, 56–62 CrossRef CAS.
- O. Yamamoto, Electrochim. Acta, 2000, 45, 2423–2435 CrossRef CAS.
- Y. Arachi, H. Sakai, O. Yamamoto, Y. Takeda and N. Imanishai, Solid State Ionics, 1999, 121, 133–139 CrossRef CAS.
- S. P. S. Badwal, F. T. Ciacchi and D. Milosevic, Solid State Ionics, 2000, 136–137, 91–99 CrossRef CAS.
- T. H. Estell and S. N. Flengas, Chem. Rev., 1970, 70, 339–376 CrossRef.
- O. Yamamoto, Y. Arati, Y. Takeda, N. Imanishi, Y. Mizutani, M. Kawai and Y. Nakamura, Solid State Ionics, 1995, 79, 137–142 CrossRef CAS.
- A. Weber and E. Ivers-Tiffée, J. Power Sources, 2004, 127, 273–283 CrossRef CAS.
- V. V. Kharton, F. M. B. Marques and A. Atkinson, Solid State Ionics, 2004, 174, 135–149 CrossRef CAS.
- V. G. Artemov, I. E. Kuritsyna, S. P. Lebedev, G. a Komandin, P. O. Kapralov, I. E. Spektor, V. V Kharton, S. I. Bredikhin and a a Volkov, Russ. J. Electrochem., 2014, 50, 690–693 CrossRef CAS.
- W. H. Kan, A. J. Samson and V. Thangadurai, J. Mater. Chem. A, 2016, 4, 17913–17932 RSC.
- S. Wang, T. Kobayashi, M. Dokiya and T. Hashimoto, J. Electrochem. Soc., 2000, 147, 3606–3609 CrossRef CAS.
- B. C. H. Steele, Solid State Ionics, 2000, 129, 95–110 CrossRef CAS.
- M. Mogensen, N. M. Sammes and G. A. Tompsett, Solid State Ionics, 2000, 129, 63–94 CrossRef CAS.
- J. M. Ralph, A. C. Schoeler and M. Krumpelt, J. Mater. Sci., 2001, 36, 1161–1172 CrossRef CAS.
- H. Inaba and H. Tagawa, Solid State Ionics, 1996, 83, 1–16 CrossRef CAS.
- E. Ivers-Tiffée, A. Weber and D. Herbstritt, J. Eur. Ceram. Soc., 2001, 21, 1805–1811 CrossRef.
- X. Sha, Z. Lü, X. Huang, J. Miao, L. Jia, X. Xin and W. Su, J. Alloys Compd., 2006, 424, 315–321 CrossRef CAS.
- H. Yahiro, K. Eguchi and H. Arai, Solid State Ionics, 1989, 36, 71–75 CrossRef CAS.
- M. Balaguer, C. Solís and J. M. Serra, J. Phys. Chem., 2012, 116, 7975–7982 CAS.
- M. Mogensen, T. Lindegaard, U. R. Hansen and G. Mogensen, J. Electrochem. Soc., 1994, 141, 2122–2128 CrossRef CAS.
- Y. Lin, S. Fang, D. Su, K. S. Brinkman and F. Chen, Nat. Commun., 2015, 6, 6824 CrossRef CAS PubMed.
- J. Zhang, C. Lenser, N. H. Menzler and O. Guillon, Solid State Ionics, 2020, 344, 115138 CrossRef CAS.
- H. J. B. P. J. Gellings, The CRC Handbook of Solid State Electrochemistry, 1997 Search PubMed.
- S. P. S. Sukhvinder, P. S. Badwal and F. T. Ciacchi, Adv. Mater., 2001, 13, 993–996 CrossRef.
- T. Horita, Y. Xiong, H. Kishimoto, K. Yamaji, N. Sakai, M. E. Brito and H. Yokakawa, Solid Oxide Fuel Cells IX, 2001, 603–610 Search PubMed.
- A. Petric and H. Ling, J. Am. Ceram. Soc., 2007, 90, 1515–1520 CrossRef CAS.
- H. U. Anderson, Solid State Ionics, 1992, 52, 33–41 CrossRef CAS.
- J. Mizusaki, Solid State Ionics, 1992, 52, 79–91 CrossRef CAS.
- H. Yokokawa, N. Sakai, T. Kawada and M. Dokiya, Solid State Ionics, 1992, 52, 43–56 CrossRef CAS.
- R. E. Williford and P. Singh, J. Power Sources, 2004, 128, 45–53 CrossRef CAS.
- S. Pirou, J. M. Bermudez, B. T. Na, S. Ovtar, J. H. Yu, P. V. Hendriksen, A. Kaiser, T. R. Reina, M. Millan and R. Kiebach, J. Memb. Sci., 2018, 552, 115–123 CrossRef CAS.
- Y. Xu, S. Pirou, P. Zielke, S. B. Simonsen, P. Norby, P. V. Hendriksen and R. Kiebach, Ind. Eng. Chem. Res., 2018, 57(7), 2123–2130 CrossRef CAS.
- S. Gupta, M. K. Mahapatra and P. Singh, Mater. Sci. Eng. R Reports, 2015, 90, 1–36 CrossRef.
- N. Sakai, H. Yokokawa, T. Horita and K. Yamaji, Int. J. Appl. Ceram. Technol., 2004, 1, 23–30 CrossRef CAS.
- J. G. M. Furtado and R. N. Oliveira, Rev. Matéria, 2008, 13, 147–153 CAS.
- P. Singh and N. Q. Minh, Int. J. Appl. Ceram. Technol., 2004, 1, 5–15 CrossRef CAS.
- N. Xu, H. Zhao, W. Wei, X. Lu, W. Ding and F. Li, Int. J. Hydrogen Energy, 2010, 35, 7295–7301 CrossRef CAS.
- Z. G. Yang, J. W. Stevenson and P. Singh, Adv. Mater. Process., 2003, 161, 34–37 CAS.
- J. W. Fergus, Solid State Ionics, 2004, 171, 1–15 CrossRef CAS.
- T. Horita, in Perovskite Oxide for Solid Oxide Fuel Cells, 2009, pp. 285–296 Search PubMed.
- M. Mori, Y. Hiei and N. M. Sammes, Solid State Ionics, 1999, 123, 103–111 CrossRef CAS.
- M. Mori, Y. Hiei and N. M. Sammes, Solid State Ionics, 2000, 135, 743–748 CrossRef CAS.
- L. A. Chick, J. Liu, J. W. Stevenson, T. R. Armstrong, D. E. McCready, G. D. Maupin, G. W. Coffey and C. A. Coyle, J. Am. Ceram. Soc., 1997, 80, 2109–2120 CrossRef CAS.
- S. Simner, J. Hardy, J. Stevenson and T. Armstrong, J. Mater. Sci. Lett., 2000, 19, 863–865 CrossRef CAS.
- S. Simner, J. S. Hardy, J. W. Stevenson and T. R. Armstrong, J. Mater. Sci., 1999, 34, 5721–5732 CrossRef CAS.
- J. D. Carter, M. M. Nasrallah and H. U. Anderson, J. Mater. Sci., 1996, 31, 157–163 CrossRef CAS.
- Z. Wang, H. Liu, X. Tan, Y. Jin and S. Liu, J. Memb. Sci., 2009, 345, 65–73 CrossRef CAS.
- Y. Wei, Q. Liao, Z. Li and H. Wang, Sep. Purif. Technol., 2013, 110, 74–80 CrossRef CAS.
- J. Kim and Y. S. Lin, AIChE J., 2000, 46, 1521–1529 CrossRef CAS.
- D. Han, J. Sunarso, X. Tan, Z. Yan, L. Liu and S. Liu, Energy and Fuels, 2012, 26, 4728–4734 CrossRef CAS.
- S. Y. Istomin and E. V Antipov, Russ. Chem. Rev., 2013, 82, 686–700 CrossRef.
- S. Dwivedi, Int. J. Hydrogen Energy, 2020, 45(44), 23988–24013 CrossRef CAS.
- L. Navarrete, M. Balaguer, V. B. Vert and J. M. Serra, Fuel Cells, 2017, 17, 100–107 CrossRef CAS.
- M. Balaguer, V. B. Vert, L. Navarrete and J. M. Serra, J. Power Sources, 2013, 223, 214–220 CrossRef CAS.
- J. Garcia-Fayos, M. Balaguer and J. M. Serra, ChemSusChem, 2015, 8, 4242–4249 CrossRef CAS PubMed.
- M. Balaguer, J. García-Fayos, C. Solís and J. M. Serra, Chem. Mater., 2013, 25, 4986–4993 CrossRef CAS.
- K. Zhang, G. Zhang, Z. Liu, J. Zhu, N. Zhu and W. Jin, J. Memb. Sci., 2014, 471, 9–15 CrossRef CAS.
- T. Liu, Y. Chen, S. Fang, L. Lei, Y. Wang, C. Ren and F. Chen, J. Memb. Sci., 2016, 520, 354–363 CrossRef CAS.
- J. Garcia-Fayos, M. Balaguer, S. Baumann and J. M. Serra, J. Memb. Sci., 2018, 548, 117–124 CrossRef CAS.
- S. Pirou, J. García-Fayos, M. Balaguer, R. Kiebach and J. M. Serra, J. Memb. Sci., 2019, 580, 307–315 CrossRef CAS.
- J. García-Fayos, R. Ruhl, L. Navarrete, H. J. M. Bouwmeester and J. M. Serra, J. Mater. Chem. A, 2018, 6, 1201–1209 RSC.
- A. Leo, S. Liu and J. C. Diniz da Costa, J. Memb. Sci., 2009, 340, 148–153 CrossRef CAS.
- L. Navarrete, C. Solís and J. M. Serra, J. Mater. Chem. A, 2015, 3, 16440–16444 RSC.
- J. García-Fayos, M. Søgaard, A. Kaiser and J. M. Serra, Sep. Purif. Technol., 2019, 216, 58–64 CrossRef.
- Y. Chen, K. Gerdes, S. A. Paredes Navia, L. Liang, A. Hinerman and X. Song, Nano Lett., 2019, 19, 8767–8773 CrossRef CAS PubMed.
- Y. Song, Y. Chen, M. Xu, W. Wang, Y. Zhang, G. Yang, R. Ran, W. Zhou and Z. Shao, Adv. Mater., 2020, 32, 1–9 Search PubMed.
- D. Papargyriou and J. T. S. Irvine, Solid State Ionics, 2016, 288, 120–123 CrossRef CAS.
- J. Carneiro and E. Nikolla, Nano Res., 2019, 12, 2081–2092 CrossRef CAS.
- J. H. Kim and J. W. Yun, J. Electrochem. Sci. Technol., 2019, 10, 335–343 CAS.
- S. Vecino-Mantilla, P. Gauthier-Maradei, M. Huvé, J. M. Serra, P. Roussel and G. H. Gauthier, ChemCatChem, 2019, 11, 4631–4641 CrossRef CAS.
- H. Han, J. Park, S. Y. Nam, K. J. Kim, G. M. Choi, S. S. P. Parkin, H. M. Jang and J. T. S. Irvine, Nat. Commun., 2019, 10, 1–8 CrossRef PubMed.
- O. Kwon, S. Sengodan, K. Kim, G. Kim, H. Y. Jeong, J. Shin, Y. W. Ju, J. W. Han and G. Kim, Nat. Commun., 2017, 8, 1–7 CrossRef PubMed.
- G. D. Nam, G. Lee, S. Choi, J. Lee, S. J. Song and J. H. Joo, J. Mater. Chem. A, 2020, 8, 23884–23893 RSC.
- C. S. Chen, H. Kruidhof, H. J. M. Bouwmeester, H. Verweij and A. J. Burggraaf, Solid State Ionics, 1997, 99, 215–219 CrossRef CAS.
- C. Yang, Q. Xu, C. Liu, J. Liu, C. Chen and W. Liu, Mater. Lett., 2011, 65, 3365–3367 CrossRef CAS.
- X. Yin, L. Hong and Z. L. Liu, J. Memb. Sci., 2006, 268, 2–12 CrossRef CAS.
- H. Cheng, N. Zhang, X. Xiong, X. Lu, H. Zhao, S. Li and Z. Zhou, ACS Sustain. Chem. Eng., 2015, 3, 1982–1992 CrossRef CAS.
- I. Kagomiya, T. Iijima and H. Takamura, J. Memb. Sci., 2006, 286, 180–184 CrossRef CAS.
- U. Pippardt, J. Böer, C. Bollert, A. Hoffmann, M. Heidenreich, R. Kriegel, M. Schulz and A. Simon, J. Ceram. Sci. Technol., 2014, 5, 309–316 Search PubMed.
- M. B. Choi, S. Y. Jeon, H. J. Hwang, J. Y. Park and S. J. Song, Solid State Ionics, 2010, 181, 1680–1684 CrossRef CAS.
- G. Chen, Z. Zhao, M. Widenmeyer, T. Frömling, T. Hellmann, R. Yan, F. Qu, G. Homm, J. P. Hofmann, A. Feldhoff and A. Weidenkaff, J. Memb. Sci., 2021, 639, 2–13 CrossRef.
- V. V. Kharton, A. V. Kovalevsky, A. P. Viskup, F. M. Figueiredo, A. A. Yaremchenko, E. N. Naumovich and F. M. B. Marques, J. Electrochem. Soc., 2000, 147, 2814–2821 CrossRef CAS.
- V. V. Kharton, A. V. Kovalevsky, A. P. Viskup, A. L. Shaula, F. M. Figueiredo, E. N. Naumovich and F. M. B. Marques, Solid State Ionics, 2003, 160, 247–258 CrossRef CAS.
- H. Takamura, K. Okumura, Y. Koshino, A. Kamegawa and M. Okada, Journal of Electroceramics, 2004, 13, 613–618 CrossRef CAS.
- B. Jiang, H. Cheng, L. Luo, X. Lu and Z. Zhou, J. Mater. Sci. Technol., 2014, 30, 1174–1180 CrossRef CAS.
- H. Cheng, P. Wang, H. Zhao, K. Li, X. Lu and Q. Xu, Ceram. Int., 2017, 43, 6477–6486 CrossRef CAS.
- C. Liu, H. Cheng, G. Yan, H. Zhao, X. Lu and P. Wang, J. Mater. Sci., 2018, 53, 11962–11976 CrossRef CAS.
- X. Zhu and W. Yang, AIChE J., 2008, 54, 665–672 CrossRef CAS.
- E. Ruiz-Trejo, P. Boldrin, J. L. Medley-Hallam, J. Darr, A. Atkinson and N. P. Brandon, Chem. Eng. Sci., 2015, 127, 269–275 CrossRef CAS.
- A. J. Samson, M. Søgaard and P. Vang Hendriksen, J. Memb. Sci., 2014, 470, 178–188 CrossRef CAS.
- J. Xue, Q. Liao, Y. Wei, Z. Li and H. Wang, J. Memb. Sci., 2013, 443, 124–130 CrossRef CAS.
- H. Luo, H. Jiang, K. Efimov, F. Liang, H. Wang and J. Caro, Ind. Eng. Chem. Res., 2011, 50, 13508–13517 CrossRef CAS.
- J. H. Park, Y. Il Kwon, G. D. Nam and J. H. Joo, J. Mater. Chem. A, 2018, 6, 14246–14254 RSC.
- S. Cheng, H. Huang, S. Ovtar, S. B. Simonsen, M. Chen, W. Zhang, M. Søgaard, A. Kaiser, P. V. Hendriksen and C. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 4548–4560 CrossRef CAS PubMed.
- J. Gurauskis, S. Ovtar, A. Kaiser, M. Søgaard and P. V. Hendriksen, ECS Trans., 2014, 64, 251–258 CrossRef CAS.
- S. Ovtar, J. Gurauskis, A. Bjørnetun Haugen, C. Chatzichristodoulou, A. Kaiser and P. V. Hendriksen, J. Memb. Sci., 2017, 523, 576–587 CrossRef CAS.
- J. H. Joo, K. S. Yun, J. H. Kim, Y. Lee, C. Y. Yoo and J. H. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 14699–14707 CrossRef CAS PubMed.
- H. Luo, K. Efimov, H. Jiang, A. Feldhoff, H. Wang and J. Caro, Angew. Chem., Int. Ed., 2011, 50, 759–763 CrossRef CAS PubMed.
- J. Zhou, X. Tang, D. He, C. Wu, Y. Zhang, W. Ding, Y. Jin and C. Sun, J. Alloys Compd., 2015, 646, 204–210 CrossRef CAS.
- S. Cheng, M. Søgaard, L. Han, W. Zhang, M. Chen, A. Kaiser and P. V. Hendriksen, Chem. Commun., 2015, 51, 7140–7143 RSC.
- W. Fang, F. Liang, Z. Cao, F. Steinbach and A. Feldhoff, Angew. Chem., Int. Ed., 2015, 54, 4847–4850 CrossRef CAS PubMed.
- W. Fang, F. Steinbach, C. Chen and A. Feldhoff, Chem. Mater., 2015, 27, 7820–7826 CrossRef CAS.
- X. Bi, X. Meng, P. Liu, N. Yang, Z. Zhu, R. Ran and S. Liu, J. Memb. Sci., 2017, 522, 91–99 CrossRef CAS.
- K. Kobayashi, M. Nishioka, K. Sato, T. Inoue, S. Hamakawa and T. Tsunoda, J. Solid State Electrochem., 2006, 10, 629–634 CrossRef CAS.
- K. Partovi, M. Bittner and J. Caro, J. Mater. Chem. A, 2015, 3, 24008–24015 RSC.
- Y. He, L. Shi, F. Wu, W. Xie, S. Wang, D. Yan, P. Liu, M. R. Li, J. Caro and H. Luo, J. Mater. Chem. A, 2017, 6, 84–92 RSC.
- H. Luo, T. Klande, Z. Cao, F. Liang, H. Wang and J. Caro, J. Mater. Chem. A, 2014, 2, 7780–7787 RSC.
- X. Wang, Y. Huang, D. Li, L. Zeng, Y. He, M. Boubeche and H. Luo, J. Memb. Sci., 2021, 633, 119403 CrossRef CAS.
- G. Chen, B. Tang, M. Widenmeyer, L. Wang, A. Feldhoff and A. Weidenkaff, J. Memb. Sci., 2020, 595, 117530 CrossRef CAS.
- H. Luo, H. Jiang, T. Klande, F. Liang, Z. Cao, H. Wang and J. Caro, J. Memb. Sci., 2012, 423–424, 450–458 CrossRef CAS.
- G. Chen, Z. Zhao, M. Widenmeyer, R. Yan, L. Wang, A. Feldhoff and A. Weidenkaff, Membranes, 2020, 10, 183 CrossRef CAS PubMed.
- S. Wang, L. Shi, Z. Xie, Y. He, D. Yan, M. R. Li, J. Caro and H. Luo, J. Eur. Ceram. Soc., 2019, 39, 4882–4890 CrossRef CAS.
- Y. Huang, C. Zhang, X. Wang, D. Li, L. Zeng, Y. He, P. Yu and H. Luo, Ceram. Int., 2022, 48(1), 415–426 CrossRef CAS.
- L. Shi, S. Wang, T. Lu, Y. He, D. Yan, Q. Lan, Z. Xie, H. Wang, M. R. Li, J. Caro and H. Luo, J. Alloys Compd., 2019, 806, 500–509 CrossRef CAS.
- L. Shi, S. Wang, T. Lu, Y. He, D. Yan, Q. Lan, Z. Xie, H. Wang, M. Boubeche and H. Luo, Ceram. Int., 2019, 45, 20033–20039 CrossRef CAS.
- C. Zhang, Y. Huang, L. Zeng, Y. He, P. Yu and H. Luo, Processes, 2021, 9, 1–15 Search PubMed.
- F. Liang, H. Luo, K. Partovi, O. Ravkina, Z. Cao, Y. Liu and J. Caro, Chem. Commun., 2014, 50, 2451–2454 RSC.
- P. Wang, H. Cheng, Y. Wang and X. Lu, MATEC Web Conf., 2016, 67, 06001–06007 CrossRef.
- T. Chen, H. Zhao, N. Xu, Y. Li, X. Lu, W. Ding and F. Li, J. Memb. Sci., 2011, 370, 158–165 CrossRef CAS.
- S. Fang, C. Chen and L. Winnubst, Solid State Ionics, 2011, 190, 46–52 CrossRef CAS.
- J. Yi, Y. Zuo, W. Liu, L. Winnubst and C. Chen, J. Memb. Sci., 2006, 280, 849–855 CrossRef CAS.
- B. Wang, J. Yi, L. Winnubst and C. Chen, J. Memb. Sci., 2006, 286, 22–25 CrossRef CAS.
- Z. Wang, W. Sun, Z. Zhu, T. Liu and W. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 11038–11043 CrossRef CAS PubMed.
- T. Liu, W. He, H. Huang, S. Wang, H. J. M. Bouwmeester and C. Chen, Ind. Eng. Chem. Res., 2014, 53, 6131–6136 CrossRef CAS.
- W. Li, T. F. Tian, F. Y. Shi, Y. S. Wang and C. S. Chen, Ind. Eng. Chem. Res., 2009, 48, 5789–5793 CrossRef CAS.
- T. Chen, H. Zhao, Z. Xie, L. Feng, X. Lu, W. Ding and F. Li, Int. J. Hydrogen Energy, 2012, 37, 5277–5285 CrossRef CAS.
- H. Li, Y. Liu, X. Zhu, Y. Cong, S. Xu, W. Xu and W. Yang, Sep. Purif. Technol., 2013, 114, 31–37 CrossRef CAS.
- H. Li, X. Zhu, Y. Liu, W. Wang and W. Yang, J. Memb. Sci., 2014, 462, 170–177 CrossRef CAS.
- X. Zhu, H. Liu, Y. Cong and W. Yang, Chem. Commun., 2012, 48, 251 RSC.
- W. Yang, F. Li and Q. Li, Chem. Eng. Sci., 2019, 199, 210–219 CrossRef CAS.
- S. Huang, W. Li, Z. Cao, H. Li, H. Ma and X. Zhu, J. Memb. Sci., 2019, 579, 342–350 CrossRef CAS.
- K. Partovi, C. H. Rüscher, F. Steinbach and J. Caro, J. Memb. Sci., 2016, 503, 158–165 CrossRef CAS.
- Z. Zhang, W. Zhou, Y. Chen, D. Chen, J. Chen, S. Liu, W. Jin and Z. Shao, ACS Appl. Mater. Interfaces, 2015, 7, 22918–22926 CrossRef CAS PubMed.
- S. Guo, Z. Liu, J. Zhu, X. Jiang, Z. Song and W. Jin, Fuel Process. Technol., 2016, 154, 19–26 CrossRef CAS.
- H. Zhou, W. Liang, F. Liang and H. Jiang, Catal. Today, 2019, 331, 2–6 CrossRef CAS.
- K. J. Yoon and O. A. Marina, J. Memb. Sci., 2016, 499, 301–306 CrossRef CAS.
- X. Zhu, Q. Li, Y. He, Y. Cong and W. Yang, J. Memb. Sci., 2010, 360, 454–460 CrossRef CAS.
- Q. Li, X. Zhu and W. Yang, J. Memb. Sci., 2008, 325, 11–15 CrossRef CAS.
- Z. Cao, X. Zhu, W. Li, B. Xu, L. Yang and W. Yang, Mater. Lett., 2015, 147, 88–91 CrossRef CAS.
- H. Takamura, T. Kobayashi, T. Kasahara, A. Kamegawa and M. Okada, J. Alloys Compd., 2006, 408–412, 1084–1089 CrossRef CAS.
- C. Gaudillere, J. Garcia-Fayos, M. Balaguer and J. M. Serra, ChemSusChem, 2014, 7, 2554–2561 CrossRef CAS PubMed.
- H. Wang, W. S. Yang, Y. Cong, X. Zhu and Y. S. Lin, J. Memb. Sci., 2003, 224, 107–115 CrossRef CAS.
- A. L. Shaula, V. V. Kharton, F. M. B. Marques, A. V. Kovalevsky, A. P. Viskup and E. N. Naumovich, J. Solid State Electrochem., 2006, 10, 28–40 CrossRef CAS.
- A. S. Yu, T.-S. Oh, R. Zhu, A. Gallegos, R. J. Gorte and J. M. Vohs, Faraday Discuss., 2015, 182, 213–225 RSC.
- K. S. Yun, C. Y. Yoo, S. G. Yoon, J. H. Yu and J. H. Joo, J. Memb. Sci., 2015, 486, 222–228 CrossRef CAS.
- Y. Il Kwon, J. H. Park, S. M. Kang, G. D. Nam, J. W. Lee, J. H. Kim, D. Kim, S. M. Jeong, J. H. Yu and J. H. Joo, Energy Environ. Sci., 2019, 12, 1358–1368 RSC.
- S. Lia, W. Jin, N. Xu and J. Shi, J. Memb. Sci., 2001, 186, 195–204 CrossRef CAS.
- B. Wang, M. Zhan, D. Zhu, W. Liu and C. Chen, J. Solid State Electrochem., 2006, 10, 625–628 CrossRef CAS.
- W. Fang, Y. Zhang, J. Gao and C. Chen, Ceram. Int., 2014, 40, 799–803 CrossRef CAS.
- T. Liu, Y. Wang, R. Yuan, J. Gao, C. Chen and H. J. M. Bouwmeester, ACS Appl. Mater. Interfaces, 2013, 5, 9454–9460 CrossRef CAS PubMed.
- Y. Zhang, R. H. Yuan, Z. Y. He, J. F. Gao and C. S. Chen, Solid State Ionics, 2016, 288, 342–346 CrossRef CAS.
- T. Liu, W. Zhao and Y. Wang, ACS Appl. Nano Mater., 2018, 1, 3774–3778 CrossRef CAS.
- C. Li, X. Ban, C. Chen and Z. Zhan, Solid State Ionics, 2020, 345, 115176 CrossRef CAS.
- W. He, H. Huang, J. fen Gao, L. Winnubst and C. sheng Chen, J. Memb. Sci., 2014, 452, 294–299 CrossRef CAS.
- W. Li, J. J. Liu and C. S. Chen, J. Memb. Sci., 2009, 340, 266–271 CrossRef CAS.
- Z. Dehaney-Steven, D. Papargyriou and J. T. S. Irvine, Solid State Ionics, 2016, 288, 338–341 CrossRef CAS.
- J. H. Park, S. M. Kang, Y. il Kwon, G. D. Nam, K. S. Yun, S. J. Song, J. H. Yu and J. H. Joo, J. Memb. Sci., 2019, 117620 Search PubMed.
- S. Gupta, J. J. Adams, J. R. Wilson, E. G. Eddings, M. K. Mahapatra and P. Singh, Appl. Energy, 2016, 165, 72–80 CrossRef CAS.
- S. Pirou, J. Gurauskis, V. Gil, M. Søgaard, P. V. Hendriksen, A. Kaiser, S. Ovtar and R. Kiebach, Fuel Process. Technol., 2016, 152, 192–199 CrossRef CAS.
- S. Pirou, J. M. Bermudez, P. V. Hendriksen, A. Kaiser, T. R. Reina, M. Millan and R. Kiebach, J. Memb. Sci., 2017, 543, 18–27 CrossRef CAS.
- F. Liang, H. Luo, K. Partovi, O. Ravkina, Z. Cao, Y. Liu and J. Caro, Chem. Commun., 2014, 50, 2451–2454 RSC.
- X. Zhu, Q. Li, Y. Cong and W. Yang, Catal. Commun., 2008, 10, 309–312 CrossRef CAS.
- J. Li, in DOE/NETL Gasification Systems and Coal & Coal-Biomass to Liquid Project Review Meeting, Pittsburgh, PA, 2018 Search PubMed.
- M. Ramasamy, E. S. Persoon, S. Baumann, M. Schroeder, F. Schulze-Küppers, D. Görtz, R. Bhave, M. Bram and W. A. Meulenberg, J. Memb. Sci., 2017, 544, 278–286 CrossRef CAS.
- J. Xue, L. Chen, Y. Wei and H. Wang, Chem. Eng. J., 2017, 327, 202–209 CrossRef CAS.
- K. S. Yun, J. H. Park, Y. Il Kwon, D. Y. Kim, C. Y. Yoo, J. H. Yu and J. H. Joo, J. Mater. Chem. A, 2016, 4, 13549–13554 RSC.
- J. A. Escribano, J. García-Fayos and J. M. Serra, J. Eur. Ceram. Soc., 2017, 37, 5223–5231 CrossRef CAS.
- Z. Zhu, J. Xiao, W. He, T. Wang, Z. Wei and Y. Dong, J. Eur. Ceram. Soc., 2015, 35, 3187–3194 CrossRef CAS.
- A. S. Sihar, N. H. Othman, N. H. Alias, M. Z. Shahruddin, S. S. Asghrar Syed-Hassan, M. A. Rahman, A. F. Ismail and Z. Wu, Ceram. Int., 2019, 45, 13086–13093 CrossRef CAS.
- Y. Liu and L. Hong, J. Memb. Sci., 2003, 224, 137–150 CrossRef CAS.
- H. Middleton, S. Diethelm, R. Ihringer, D. Larrain, J. Sfeir and J. Van Herle, J. Eur. Ceram. Soc., 2004, 24, 1083–1086 CrossRef CAS.
- M. Lipińska-Chwałek, L. Kiesel and J. Malzbender, J. Eur. Ceram. Soc., 2014, 34, 2519–2524 CrossRef.
- D. K. Ramachandran, M. Søgaard, F. Clemens, J. Gurauskis and A. Kaiser, Sep. Purif. Technol., 2015, 147, 422–430 CrossRef CAS.
- Y. Xing, S. Baumann, D. Sebold, M. Rüttinger, A. Venskutonis, W. A. Meulenberg and D. Stöver, J. Am. Ceram. Soc., 2011, 94, 861–866 CrossRef CAS.
- X. Chang, C. Zhang, W. Jin and N. Xu, J. Memb. Sci., 2006, 285, 232–238 CrossRef CAS.
- A. V. Kovalevsky, V. V. Kharton, F. M. M. Snijkers, J. F. C. Cooymans, J. J. Luyten and F. M. B. Marques, J. Memb. Sci., 2007, 301, 238–244 CrossRef CAS.
- A. A. Yaremchenko, V. V. Kharton, A. A. Valente, F. M. M. Snijkers, J. F. C. Cooymans, J. J. Luyten and F. M. B. Marques, J. Memb. Sci., 2008, 319, 141–148 CrossRef CAS.
- A. B. Haugen, L. M. Aguilera, K. Kwok, T. Molla, K. B. Andersen, A. Kaiser, P. V. Hendriksen and R. Kiebach, Ceramics, 2018, 229–245 CrossRef CAS.
- S. Zhang, C. Li, X. Meng, X. Tan, Z. Zhu, J. Sunarso and S. Liu, MATEC Web Conf., 2020, 15, 1–11 Search PubMed.
- T. F. Tian, W. Li, T. Liu and C. S. Chen, Solid State Ionics, 2012, 225, 690–694 CrossRef CAS.
- T. Chen, Z. Wang, J. Hu, M. H. Wai, S. Kawi and Y. S. Lin, J. Memb. Sci., 2020, 597, 117770 CrossRef CAS.
- P. Cousin and R. A. Ross, Mater. Sci. Eng. A, 1990, 130(1), 119–125 CrossRef.
- U. Nigge, H.-D. Wiemhöfer, E. W. J. Römer, H. J. M. Bouwmeester and T. R. Schulte, Solid State Mater. Sci., 2002, 146, 163–174 CAS.
- D. Segal, J. Mater. Chem., 1997, 7, 1297–1305 RSC.
- E. Lesley. Smart and Elaine A. Moore, Solid State Chemistry, CRC PRess, Boca Raton, 2005 Search PubMed.
- T. Ishihara, T. Yamada, H. Arikawa, H. Nishiguchi and Y. Takita, Solid State Ionics, 2000, 135, 631–636 CrossRef CAS.
- X. Zhu, Y. Liu, Y. Cong and W. Yang, Solid State Ionics, 2013, 253, 57–63 CrossRef CAS.
- S. Tao and J. T. S. Irvine, J. Electrochem. Soc., 2004, 151, A252 CrossRef CAS.
- P. Babilo, T. Uda and S. M. Haile, J. Mater. Res., 2007, 22, 1322–1330 CrossRef CAS.
- Y. Yamazaki, R. Hernandez-Sanchez and S. M. Haile, J. Mater. Chem., 2010, 20, 8158 RSC.
- M. Ramasamy, Dual Phase Oxygen Transport Membrane for Efficient Oxyfuel Combustion, Jülich, 2016, vol. 351 Search PubMed.
- X. Zhu, H. Wang and W. Yang, J. Memb. Sci., 2008, 309, 120–127 CrossRef CAS.
- H. Cheng, N. Zhang, X. Xiong, X. Lu, H. Zhao, S. Li and Z. Zhou, ACS Sustain. Chem. Eng., 2015, 3, 1982–1992 CrossRef CAS.
- L. Zhang, N. Xu, X. Li, S. Wang, K. Huang, W. H. Harris and W. K. S. Chiu, Energy Environ. Sci., 2012, 5, 8310 RSC.
- X. Xi, A. Kondo and M. Naito, J. Alloys Compd., 2016, 688, 1047–1052 CrossRef CAS.
- P. Jana, V. A. De La Peña O'Shea, J. M. Coronado and D. P. Serrano, Int. J. Hydrogen Energy, 2010, 35, 10285–10294 CrossRef CAS.
- X. Yao, P. Li, B. Yu, F. Yang, J. Li, Y. Zhao and Y. Li, Int. J. Hydrogen Energy, 2017, 42, 22192–22200 CrossRef CAS.
- B. Kim, K. Cho, J. Choi and D. Shin, J. Nanosci. Nanotechnol., 2015, 15, 536–539 CrossRef CAS PubMed.
- G. Ding, T. Gan, J. Yu, P. Li, X. Yao, N. Hou, L. Fan, Y. Zhao and Y. Li, Catal. Today, 2017, 298, 250–257 CrossRef CAS.
- D. S. Jung, S. K. Hong, J. S. Cho and Y. C. Kang, J. Eur. Ceram. Soc., 2008, 28(1), 109–115 CrossRef CAS.
- A. Purwanto, W. N. Wang, I. W. Lenggoro and K. Okuyama, J. Eur. Ceram. Soc., 2007, 27(16), 4489–4497 CrossRef CAS.
- G. L. Chiarello, I. Rossetti, P. Lopinto, G. Migliavacca and L. Forni, Catal. Today, 2006, 117, 549–553 CrossRef CAS.
- G. Messing, S.-C. Zhang and V. Jayanthi, J. Am. Ceram. Soc., 1993, 76, 2707–2726 CrossRef CAS.
- J. Leng, Z. Wang, J. Wang, H. H. Wu, G. Yan, X. Li, H. Guo, Y. Liu, Q. Zhang and Z. Guo, Chem. Soc. Rev., 2019, 48, 3015–3072 RSC.
- M. Rafique, H. Nawaz, M. Shahid Rafique, M. Bilal Tahir, G. Nabi and N. R. Khalid, Int. J. Energy Res., 2019, 43, 2423–2446 CrossRef CAS.
- S. Ovtar, M. Søgaard, K. Norrman and P. V. Hendriksen, J. Electrochem. Soc., 2018, 165, F220–F231 CrossRef CAS.
- N. H. Menzler, J. Malzbender, P. Schoderböck, R. Kauert and H. P. Buchkremer, Fuel Cells, 2014, 14, 96–106 CrossRef CAS.
- F. Le Goupil, A. Baker, F. Tonus, A. Berenov, C. A. Randall and N. M. N. Alford, J. Eur. Ceram. Soc., 2019, 39, 3315–3319 CrossRef CAS.
- J. Schnell, F. Tietz, C. Singer, A. Hofer, N. Billot and G. Reinhart, Energy Environ. Sci., 2019, 12, 1818–1833 RSC.
- R. E. Mistler and E. R. Twiname, Tape Casting: Theory and Practice, The American Ceramic Society, Westerville, Ohio, 2000 Search PubMed.
- X. Chen, M.-A. Einarsrud and T. Grande, J. Eur. Ceram. Soc., 2015, 35, 309–315 CrossRef CAS.
- R. Fernández-González, T. Molina, S. Savvin, R. Moreno, A. Makradi and P. Núñez, J. Eur. Ceram. Soc., 2014, 34, 953–959 CrossRef.
- F. Schulze-Küppers, S. Baumann, W. A. Meulenberg, D. Stöver and H. P. Buchkremer, J. Memb. Sci., 2013, 433, 121–125 CrossRef.
- A. Julian, E. Juste, P. M. Geffroy, V. Coudert, S. Degot, P. Del Gallo, N. Richet and T. Chartier, J. Memb. Sci., 2009, 333, 132–140 CrossRef CAS.
- S. Pirou, Development of Dual-Phase Oxygen Transport Membranes for Carbon Capture Processes, Technical University of Denmark, 2017 Search PubMed.
- A. Roosen, J. Eur. Ceram. Soc., 2001, 21, 1993–1996 CrossRef CAS.
- F. Schulze-Küppers, U. V. Unije, H. Blank, M. Balaguer, S. Baumann, R. Mücke and W. A. Meulenberg, J. Memb. Sci., 2018, 564, 218–226 CrossRef.
- A. B. Haugen, A. Geffroy, A. Kaiser and V. Gil, J. Eur. Ceram. Soc., 2018, 38, 3279–3285 CrossRef CAS.
- J. W. Phair, M. Lundberg and A. Kaiser, Rheol. Acta, 2009, 48, 121–133 CrossRef CAS.
- J. Garcia-Fayos, M. P. Lobera, M. Balaguer and J. M. Serra, Front. Mater., 2018, 5, 1–11 CrossRef.
- M. P. Lobera, J. M. Serra, S. P. Foghmoes, M. Søgaard and A. Kaiser, J. Memb. Sci., 2011, 385–386, 154–161 CrossRef CAS.
- M. Zhu, X. Dong, Y. Chen, F. Xue, J. Lian, L. Xiao, G. Ding and G. Wang, Ceram. Int., 2016, 42, 13925–13931 CrossRef CAS.
- Y. Xi, K. Gao, X. Pang, H. Yang, X. Xiong, H. Li and A. A. Volinsky, Ceram. Int., 2017, 43, 11992–11997 CrossRef CAS.
- B. J. H. Stadler, in Materials Processing: a Unified Approach of Metals, Ceramics and Polymers, Elsevier Inc., 2016, pp. 513–588 Search PubMed.
- V. S. K. Chakravadhanula, C. Kübel, T. Hrkac, V. Zaporojtchenko, T. Strunskus, F. Faupel and L. Kienle, Nanotechnology, 2012, 23(49), 495701–495708 CrossRef.
- J. Xiong, M. Z. Ghori, B. Henkel, T. Strunskus, U. Schürmann, L. Kienle and F. Faupel, Acta Mater., 2014, 74, 1–8 CrossRef CAS.
- C. Solís, M. Balaguer, J. Garcia-Fayos, E. Palafox and J. M. Serra, Sustain. Energy Fuels, 2020, 4, 3747–3752 RSC.
- C. Solís, F. Toldra-Reig, M. Balaguer, S. Somacescu, J. Garcia-Fayos, E. Palafox and J. M. Serra, ChemSusChem, 2018, 11, 2818–2827 CrossRef PubMed.
- M. Knarr and R. Bayer, Carbohydr. Polym., 2014, 111, 80–88 CrossRef CAS PubMed.
- R. Hoffmann, U. Pippardt and R. Kriegel, J. Memb. Sci., 2019, 570–571, 61–68 CrossRef CAS.
- G. Pećanac, J. Malzbender, F. Pauly, M. L. Fontaine, P. Niehoff, S. Baumann, T. Beck and L. Singheiser, Ceram. Int., 2015, 41, 2411–2417 CrossRef.
- R. Hoffmann, U. Pippardt and R. Kriegel, J. Memb. Sci., 2019, 581, 270–282 CrossRef CAS.
- U. Pippardt, J. Böer, L. Kiesel, R. Kircheisen, R. Kriegel and I. Voigt, AIChE J., 2014, 60, 15–21 CrossRef CAS.
- P. I. Dahl, M. L. Fontaine, F. Ahouanto, C. Denonville, O. Paulsen, Y. Larring, T. Peters, P. P. Henriksen and R. Bredesen, in Energy Procedia, Elsevier Ltd, 2012, vol. 23, pp. 187–196 Search PubMed.
- B. Tjaden, Understanding Mass Transport Mechanisms in Oxygen Transport Membrane Porous Support Layers: Correlating 3D Image-Based Modelling with Diffusion Measurements, University College London, 2016 Search PubMed.
- R. Prasad, J. M. Schwartz, E. T. Robinson and C. F. Gottzmann, Syngas production method utilizing an oxygen transport membrane, US Pat., US6695983, 2004 Search PubMed.
- X. Yin, L. Hong and Z. L. Liu, Appl. Catal. A Gen., 2006, 300, 75–84 CrossRef CAS.
- X. Yin, L. Hong and Z.-L. Liu, J. Phys. Chem. C, 2007, 11(26), 9194–9202 CrossRef.
- D. V. Rosato, D. V. Rosato and M. V. Rosato, Plastic Product Material and Process Selection Handbook, Elsevier Ltd, 2004 Search PubMed.
- M. Jacobs, M.-L. Fontaine, R. Bredesen, B. Michielsen, V. Middelkoop, Y. Larring and F. Snijkers, J. Memb. Sci., 2015, 477, 58–64 CrossRef CAS.
- D. K. Ramachandran, K. Kwok, M. Søgaard, F. Clemens, A. J. Glasscock and A. Kaiser, J. Eur. Ceram. Soc., 2015, 35, 1527–1537 CrossRef CAS.
- D. K. Ramachandran, F. Clemens, A. J. Glasscock, M. Søgaard and A. Kaiser, Ceram. Int., 2014, 40, 10465–10473 CrossRef CAS.
- S. Ovtar, J. Gurauskis, A. Bjørnetun Haugen, C. Chatzichristodoulou, A. Kaiser and P. V. Hendriksen, J. Memb. Sci., 2017, 523, 576–587 CrossRef CAS.
- Y. Du, N. Hedayat, D. Panthi, H. Ilkhani, B. J. Emley and T. Woodson, Materialia, 2018, 1, 198–210 CrossRef.
- J. Seuba, J. Leloup, S. Richaud, S. Deville, C. Guizard and A. J. Stevenson, J. Eur. Ceram. Soc., 2017, 37, 2423–2429 CrossRef CAS.
- J. Seuba Torreblanca, Improving OTM mechanical properties by controlling the pore architecture, Material chemistry, Université Montpellier, 2015, English. ffNNT : 2015MONTS081ff. fftel-01629613f Search PubMed.
- A. B. Haugen, J. Gurauskis, A. Kaiser and M. Søgaard, J. Eur. Ceram. Soc., 2016, 37, 1039–1047 CrossRef.
- J. Puetz and M. A. Aegerter, in Sol-Gel Technologies for Glass Producers and Users, Springer US, 2004, pp. 37–48 Search PubMed.
- S. Cheng, Oxygen transport membranes for biomass gasification and cement industry, Technical University of Denmark, 2015 Search PubMed.
- C. J. Brinker, in Chemical Solution Deposition of Functional Oxide Thin Films, Springer, Vienna, 2013 Search PubMed.
- Webpage, Dip Coating Theory: Film Thickness, https://www.ossila.com/blogs/news/new-guide-dip-coating-film-thickness, last visited December 2021.
- J. Gurauskis, Ø. F. Lohne, H. L. Lein and K. Wiik, J. Eur. Ceram. Soc., 2012, 32, 649–655 CrossRef CAS.
- J. T. Ritchie, J. T. Richardson and D. Luss, AIChE J., 2001, 47, 2092–2101 CrossRef CAS.
- Z. Liu, G. Zhang, X. Dong, W. Jiang, W. Jin and N. Xu, J. Memb. Sci., 2012, 415–416, 313–319 CrossRef CAS.
- X. Shao, D. Dong, G. Parkinson and C. Z. Li, J. Mater. Chem. A, 2013, 1, 9641–9644 RSC.
- B. F. K. Kingsbury and K. Li, J. Memb. Sci., 2009, 328, 134–140 CrossRef CAS.
- X. Tan, Y. Liu and K. Li, Ind. Eng. Chem. Res., 2005, 44, 61–66 CrossRef CAS.
- N. H. Othman, Z. Wu and K. Li, J. Memb. Sci., 2014, 468, 31–41 CrossRef CAS.
- X. Shao, D. Dong, G. Parkinson and C. Z. Li, J. Memb. Sci., 2014, 454, 444–450 CrossRef CAS.
- M. Salehi, E. M. Pfaff, A. Kaletsch, T. Graule, F. Clemens and B. Grobéty, Int. J. Appl. Ceram. Technol., 2015, 12, 13–18 CrossRef.
- M. Salehi, Fabricating of Thin Planar and Tubular BSCF Oxygen Separation Membranes by Thermoplastic Processing, University of Fribourg, 2013 Search PubMed.
- R. V. B. Oliveira, V. Soldi, M. C. Fredel and A. T. N. Pires, J. Mater. Process. Technol., 2005, 160, 213–220 CrossRef CAS.
- S. K. Robinson and M. R. Paul, Met. Powder Rep., 2001, 56, 24–26 CrossRef.
- M. N. Rahaman, Sintering of Ceramics, CRC Press, 2003, vol. 56 Search PubMed.
- M. Nygren and Z. Shen, Solid State Sci., 2003, 5, 125–131 CrossRef CAS.
- J. D. Nicholas and L. C. De Jonghe, Solid State Ionics, 2007, 178, 1187–1194 CrossRef CAS.
- D. P. Fagg, A. L. Shaula, V. V. Kharton and J. R. Frade, J. Memb. Sci., 2007, 299, 1–7 CrossRef CAS.
- M. Balaguer, C. Solís and J. M. Serra, Chem. Mater., 2011, 23, 2333–2343 CrossRef CAS.
- D. Ghosh, H. Han, J. C. Nino, G. Subhash and J. L. Jones, J. Am. Ceram. Soc., 2012, 95, 2504–2509 CrossRef CAS.
- X. He, F. Ye, H. Zhang and L. Liu, Mater. Sci. Eng. A, 2010, 527, 5268–5272 CrossRef.
- T. T. Molla, D. K. Ramachandran, D. W. Ni, V. Esposito, F. Teocoli, E. Olevsky, R. Bjørk, N. Pryds, A. Kaiser and H. L. Frandsen, J. Eur. Ceram. Soc., 2014, 35, 941–950 CrossRef.
- A. Kaiser, S. Foghmoes, C. Chatzichristodoulou, M. Søgaard, J. A. Glasscock, H. L. Frandsen and P. V. Hendriksen, J. Memb. Sci., 2011, 378, 51–60 CrossRef CAS.
- T. T. Molla, R. Bjørk, E. Olevsky, N. Pryds and H. L. Frandsen, Comput. Mater. Sci., 2014, 88, 28–36 CrossRef.
- D. W. Ni, V. Esposito, C. G. Schmidt, T. T. Molla, K. B. Andersen, A. Kaiser, S. Ramousse and N. Pryds, J. Am. Ceram. Soc., 2013, 96, 972–978 CrossRef CAS.
- C. Faber, Y. Allahverdiyeva-Rinne, V. Artero, L. Baraton, A. Barbieri, H. Bercegol, M. Fleischer, H. Huynhthi, J. Kargul, H. Lepaumier, L. Lopez, A. Magnuson and A. Roth, SUNRISE: Solar Energy for a Circular Economy: Technological Roadmap, 2020 Search PubMed.
- A. A. Plazaola, A. C. Labella, Y. Liu, N. B. Porras, D. A. P. Tanaka, M. V. S. Annaland and F. Gallucci, Processes, 2019, 7(3), 128 CrossRef CAS.
- W. Deibert, M. E. Ivanova, S. Baumann, O. Guillon and W. A. Meulenberg, J. Memb. Sci., 2017, 543, 79–97 CrossRef CAS.
- R. Yuan, Z. He, Y. Zhang, W. Wang and C. Chen, AIChE J., 2016, 62, 2170–2176 CrossRef CAS.
- G. E. Keller and M. M. Bhasin, J. Catal., 1982, 73, 9–19 CrossRef CAS.
- M. P. Lobera, S. Escolástico and J. M. Serra, ChemCatChem, 2011, 3, 1503–1508 CrossRef CAS.
- M. P. Lobera, M. Balaguer, J. Garcia-Fayos and J. M. Serra, ChemCatChem, 2012, 4, 2102–2111 CrossRef CAS.
- M. P. Lobera, S. Escolástico, J. Garcia-Fayos and J. M. Serra, ChemSusChem, 2012, 5, 1587–1596 CrossRef CAS PubMed.
- Z. Cao, H. Jiang, H. Luo, S. Baumann, W. A. Meulenberg, H. Voss and J. Caro, Catal. Today, 2012, 193, 2–7 CrossRef CAS.
- H. Naito and H. Arashi, Solid State Ionics, 1995, 79, 366–370 CrossRef CAS.
- G. He, W. Liang, C. L. Tsai, X. Xia, S. Baumann, H. Jiang and W. A. Meulenberg, iScience, 2019, 19, 955–964 CrossRef CAS.
- S.-J. Song, J.-H. Moon, H.-W. Ryu, T. H. Lee, S. E. Dorris and U. Balachandran, J. Ceram. Process. Res., 2008, 9, 123–125 Search PubMed.
- W. Liang, H. Zhou, J. Caro and H. Jiang, Int. J. Hydrogen Energy, 2018, 43, 14478–14485 CrossRef CAS.
- W. Liang, Z. Cao, G. He, J. Caro and H. Jiang, ACS Sustain. Chem. Eng., 2017, 5, 8657–8662 CrossRef CAS.
- G. Chen, F. Buck, I. Kistner, M. Widenmeyer, T. Schiestel, A. Schulz, M. Walker and A. Weidenkaff, Chem. Eng. J., 2020, 392, 123699 CrossRef CAS.
- A. Bogaerts and E. C. Neyts, ACS Energy Lett, 2018, 3, 1013–1027 CrossRef CAS.
- T. L. Ochs, D. B. Oryshchyn and C. A. Summers, Proceedings of Electric Utilities, Tucson, 2005 Search PubMed.
- P. Kauranen, “Nanotechnology”, Presentation at Materials and New Production – NMP, Helsinki, 2008 Search PubMed.
- A. Seltzer, Z. Fan and H. Hack, in 7th Annual COAL-GEN Conference, Milwaukee, 2007 Search PubMed.
- J. Davison, Energy, 2007, 32, 1163–1176 CrossRef CAS.
- J. Hong, G. Chaudhry, J. G. Brisson, R. Field, M. Gazzino and A. F. Ghoniem, Energy, 2009, 34, 1332–1340 CrossRef CAS.
- R. Kneer, D. Toporov, M. Förster, D. Christ, C. Broeckmann, E. Pfaff, M. Zwick, S. Engels and M. Modigell, Energy Environ. Sci., 2010, 3, 198–207 RSC.
- E. S. Rubin, A. B. Rao and M. B. Berkenpas, Technical Documentation : Oxygen-Based Combustion Systems (Oxyfuels) with Carbon Capture and Storage, CCS, 2007 Search PubMed.
- H. Stadler, F. Beggel, M. Habermehl, B. Persigehl, R. Kneer, M. Modigell and P. Jeschke, Int. J. Greenh. Gas Control, 2011, 5, 7–15 CrossRef CAS.
- S. C. Bhatia, Advanced Renewable Energy Systems, 2014, 437–489 Search PubMed.
- J. Li, Advanced Gasification and Novel Transformational Coal Conversion Technologies Development, 2018 Search PubMed.
- S. Chakravarti, B. R. Kromer, K. A. Craigie, S. M. Kelly and J. Li, US Pat., US9365422, 2016 Search PubMed.
- S. M. Kelly, S. M. Swami and J. D. Peck, US Pat., US2017/0166445A1, 2017 Search PubMed.
- J. Lin and S. M. Kelly, US Pat., US9938146B2, 2015 Search PubMed.
- J. Lin, P. Plonczak, S. M. Kelly, U. R. Doraswami and J. A. Lane, US Pat., US10441922B2, 2019 Search PubMed.
- J. A. Kilner and M. Burriel, Annu. Rev. Mater. Res., 2014, 44, 365–393 CrossRef CAS.
- R. Bredesen and J. Sogge, A Technical and Economic Assessment of Membrane Reactors for Hydrogen and Syngas Production, 1996 Search PubMed.
- M. J. den Exter, W. G. Haije and J. F. Vente, in Inorganic Membranes for Energy and Environmental Applications, 2009, p. 31 Search PubMed.
- V. V. Kharton, F. M. Figueiredo, L. Navarro, E. N. Naumovich, A. V. Kovalevsky, A. A. Yaremchenko, A. P. Viskup, A. Carneiro, F. M. B. Marques and J. R. Frade, J. Mater. Sci., 2001, 36, 1105–1117 CrossRef CAS.
- F. Schulze-Küppers, S. Baumann, F. Tietz, H. J. M. Bouwmeester and W. A. Meulenberg, J. Eur. Ceram. Soc., 2014, 34, 3741–3748 CrossRef.
- J. D. Kim, G. D. Kim, J. W. Moon, Y. il Park, W. H. Lee, K. Kobayashi, M. Nagai and C. E. Kim, Solid State Ionics, 2001, 143, 379–389 CrossRef CAS.
- R. Barfod, A. Hagen, S. Ramousse, P. V. Hendriksen and M. Mogensen, Fuel Cells, 2006, 6, 141–145 CrossRef CAS.
- J. T. S. Irvine, J. W. L. Dobson, T. Politova, S. G. Martín and A. Shenouda, Faraday Discuss., 2007, 134, 41–49 RSC ; discussion 103–118, 415–419..
| This journal is © The Royal Society of Chemistry 2022 |