
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fully bio-based cellulose nanofiber/epoxy composites with both sustainable production and selective matrix deconstruction towards infinite fiber recycling systems†
E.
Subbotina
*,
C.
Montanari
 ,
P.
Olsén
* and
Lars A.
Berglund
,
P.
Olsén
* and
Lars A.
Berglund
Department of Fibre and Polymer Technology, Wallenberg Wood Science Center, KTH Royal Institute of Technology, Teknikringen 56, 100 44 Stockholm, Sweden. E-mail: polsen@kth.se
First published on 13th December 2021
Abstract
Design of nanocellulose-based composite materials suitable for selective disintegration, recovery and recycling of individual components is of great scientific and technical interest. Cellulose nanofiber/epoxy (CNF/EP) composites are candidate bio-based substitutes for petroleum-based materials. However, chemical recovery of such intimately mixed nanocomposites has not been addressed, due to the limited chemical stability of nanocellulose and due to the covalently crosslinked epoxy network. In this work we develop CNF/EP composites designed for selective disintegration. Deconstruction is achieved by including two types of labile linkages to the polymer network; acetals and esters. Besides enabling recycling of the CNF reinforcement, the thermoset constituents were further depolymerized into valuable monomeric units in 63–95% yield. In addition, the preparation of both; epoxy monomers and final composite materials is performed using solely bio-derived materials and solvents.
Introduction
In order to meet global climate targets, a circular bioeconomy can contribute towards more efficient use of biomass.1 Increased focus on high-value materials is apparent in the literature, and cellulose/thermoset nanobiocomposites is an important example since they can serve as replacement for fossil-based plastics and composites and find use as engineering materials.2 There are, however, only limited efforts on end-of-life treatment and recycling of these nanostructured biocomposites. The reasons for that are the covalently cross-linked thermoset network, and sensitivity of cellulose to degradation during chemical treatment. Here, we design CNF/EP composites for selective disintegration and recovery of both components: the products of thermoset depolymerization and the nanocellulose reinforcement.The strongly increasing interest in cellulose nanofibers (CNFs)3–7 as “new” components in material systems is encouraging. Dense films from CNF can be prepared by casting from colloidal water suspensions8 or by filtration and drying.9–13 The films are flexible, and transparent with low thermal expansion, good gas barrier properties, and excellent mechanical properties. The Young's modulus can reach 20 GPa and the tensile strength 350 MPa for films with random-in-the-plane CNF orientation.12 CNF nanopaper limitations, however, include high hydrophilicity, limited possibilities for molding and their physical and chemical properties can only be modified within certain boundaries. These limitations provide arguments for cellulose-based polymer matrix composites, where the polymer phase extends possibilities for properties modulation.14–19 CNF/epoxy resin composites are of interest for applications in the automotive industry,20 as semi-structural composites,21 in electronics22 and as insulating materials.23 In general, cellulose biocomposites are often considered eco-friendly, simply due to the renewable resource origin of the cellulosic component. For CNF/epoxy, the epoxide component is commonly the diglycidyl ether of bisphenol A, DGEBA,22,24,25 which is petroleum-based. There are some exceptions in the form of cellulose/bio-based epoxy composites, although the materials are not degradable or recyclable.26,27 Progress in biobased epoxy thermosets is described in a recent review.28 Thermosets are often difficult to degrade29–31 in a controlled manner. Labile bonds can be introduced, to facilitate polymer degradation and partial recovery of chemical building blocks.32,33 It is difficult to find any study on degradable cellulose-epoxy biocomposites in the literature. One example, though, is on biodegradability of pineapple leaf/coir fiber reinforced epoxy composites in soil. Degradation of the composites occurred via partial depolymerization and bacterial degradation of cellulose and hemicellulose.34 In another example, biodegradation was investigated in CNF/EP composites for GaAs-based microwave electronics. The fungal biodegradation resulted in only 10% weight loss after 60 days for an epoxy-coated CNF film.22
Epoxy thermosets with triggered degradation were recently investigated based on the vanillin-derived diepoxide 1 (ref. 35) (Fig. 1). The curing of epoxide 1 with diamines resulted in a thermoset able to disintegrate via acetal-cleavage under mild acidic conditions. A drawback, however, of epoxy-amine systems is the inertness of C–N bonds towards acid hydrolysis. As a consequence, degradation products are a complex mixture which is difficult to reprocess. We investigate fully disintegrable CNF/epoxy composites via curing of diepoxide 1 with succinic anhydride, succinic acid, or citric acid, thus introducing labile acetals and ester linkages in the thermoset. Acetal moieties makes it possible to separate the partially depolymerized thermoset from the CNF reinforcement under relatively mild conditions, so that the CNF reinforcement structure is preserved. A complete depolymerization of the thermoset into monomeric products is performed with high selectivity at higher temperatures and longer reaction times. In addition, this processing concept is scalable and the starting material components (vanillin,36 succinic anhydride and acid,37 citric acid38) and solvents are from biomass and available on industrial or pilot-scale. Although we have shown that epoxies can be prepared directly from “technical lignin”,39 vanillin from lignin is preferable not only because of “designed degradability” investigated here, but also because of more homogeneous network structure of the final epoxy thermoset.
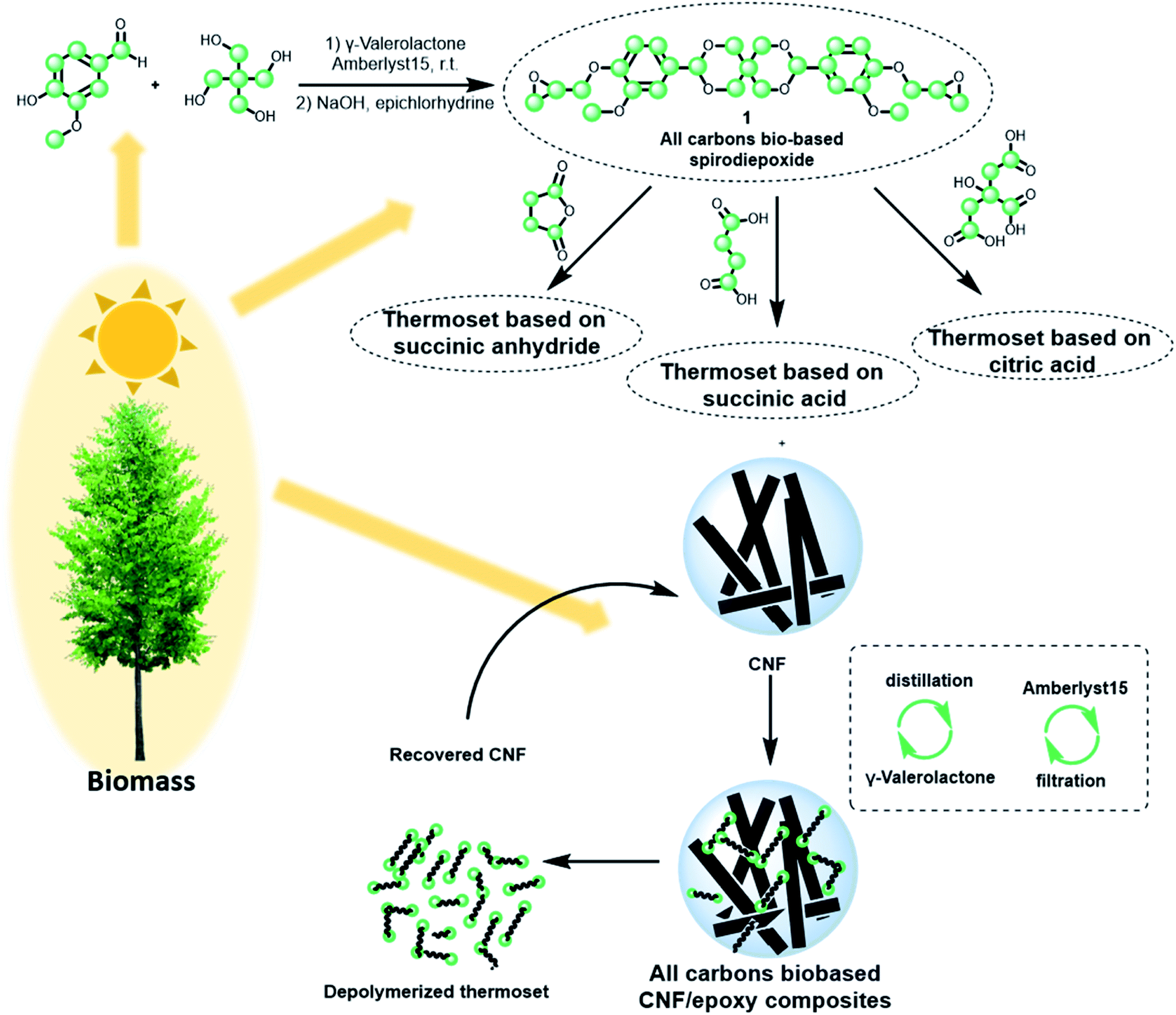 | ||
| Fig. 1 Schematic representation of life cycle of all biobased carbon CNF/epoxy composite materials from the synthesis of monomeric materials to the regeneration of the composite (this work). | ||
Results and discussion
There are three main steps in the investigation: (a) monomer and thermoset synthesis, (b) cellulose nanocomposite preparation by in situ polymerization in a porous cellulose nanofiber network and (c) recycling of cellulose nanofibers and epoxy thermoset components.Preparation of diepoxy monomer 1
The first step is modification of the synthetic pathway for diepoxy monomer 1 (Fig. 1). In previous studies, classical acetalization reactions are performed under Dean–Stark conditions using solvents such as benzene, toluene or DMF.35,40–42 The environmental and health concerns with these solvents,49 suggests the need for a greener acetalization procedure. We therefore performed acetalization of vanillin with pentaerythritol at room temperature using a biomass-derived solvent – γ-valerolactone (GVL)43 and using a heterogeneous acid catalyst – Amberlyst 15 (ESI, Table S1†). Vanillin and pentaerythritol are both commercially produced from biomass.36,44 Furthermore, a new electrocatalytic process for vanillin from lignin combines an eco-friendly process with significantly lowered cost.45 The product was isolated by precipitation from the reaction mixture by addition of water. GVL does not form azeotropic mixtures with water,46 which means that the solvent can be completely recovered by distillation. The catalyst was separated from the reaction mixture by filtration and can be reused in consecutive runs. Epoxidation to 1 was performed according to an existing procedure, using bio-based epichlorohydrin (ESI†).35 Note that 1 is an attractive alternative to diglycidyl ether of bisphenol A, the dominating petroleum-based epoxide in commercial use.Preparation of thermosets
A fully biobased and recyclable cellulose nanocomposite system is a significant challenge, and it is not straight-forward to select a suitable epoxide curing agent for the formation of a three-dimensional polymer network of sufficiently high glass transition temperature. We need a curing agent currently produced commercially from biomass, which forms degradable bonds during epoxide curing. Ester bonds were selected for the purpose of tailored degradation during recycling. An example of a degradable thermoset with ester bonds is described,47 although the degradation products were obtained as a complex mixture. Here, the curing behavior of spirodiepoxide 1 was investigated with the following curing agents: succinic anhydride, succinic and citric acids. Since our biocomposite material includes cellulose nanofibers as stiffening reinforcement, mild and selective polymerization (curing) mechanisms are desirable. In this regard, uncatalyzed curing is preferential to avoid cellulose nanofiber degradation and catalyst accumulation in the biocomposite.The thermal properties of the prepared thermosets as a function of curing conditions and bio-based hardeners are summarized in Table 1. The curing of spirodiepoxide 1 with succinic anhydride and citric acid resulted in thermosets with higher glass transition temperatures (Tg) than for succinic acid. Citric acid is known to also form intramolecular anhydride structures at elevated temperatures, which facilitates curing.48 The higher Tg's observed for succinic anhydride and citric acid is actually related to the difference in curing mechanism between anhydrides and acids, where acids must undergo condensation reactions to form a fully cross-linked network (ESI, Fig. S3†).
| Entry | Curinga agent | T g (cured at 170 °C)b | T g (cured at 200 °C)b |
|---|---|---|---|
| a The reactions were performed in melt. Molar ratio of epoxy groups to carboxyl groups (for acids): 1/2. Molar ratio of epoxy groups to anhydride (for succinic anhydride): 1/1. b Mid, point. | |||
| 1 | Succinic anhydride | 104 | 119 |
| 2 | Succinic acid | 74 | 77 |
| 3 | Citric acid | 95 | 109 |
Preparation and characterization of biocomposites
CNFs have high modulus and strength and may provide significant reinforcement to a thermoset matrix for increased mechanical properties. Nanofibers need to be well-dispersed, since agglomeration will reduce reinforcement efficiency. Chemical interactions between the CNFs and the polymer matrix are also important.49 The preparation procedure is vital in order to obtain well-dispersed CNFs in the polymer matrix. For CNF/epoxy biocomposites, high nanostructural control achieved by solvent exchange combined with impregnation of liquid epoxy precursors and in situ polymerization.50 A wet cake of porous CNFs, oriented random-in-plane, is obtained by vacuum filtration of a dilute colloidal CNF dispersion. It is exchanged with organic solvents to substitute water, followed by impregnation with a solution of epoxy precursors. After impregnation, the solvent evaporates and the nanostructured biocomposite is cured at elevated temperature. An organic solvent must fulfill several criteria. It should dissolve monomers at relatively high concentration, swell the CNF network to facilitate impregnation, and preferably have a low boiling point. Solvent screening revealed that the spirodiepoxide 1 has low solubility in the large majority of common organic solvents at ambient temperature. By increasing the temperature to 60–70 °C, it was possible obtain a 25 wt% solution of 1 in acetonitrile (MeCN). MeCN is classified as a first choice polar aprotic solvent according to the Sanofi green solvent guide,51 and as usable by Pfizer.52 Moreover, methods to produce bio-based MeCN are in active development.53 We used MeCN to prepare nanostructured cellulose biocomposites via the solvent exchange method from diepoxide 1 and succinic anhydride as a curing agent (ESI† and Fig. 2a). | ||
Fig. 2 (a) Preparation of biocomposites by solvent exchange to MeCN using succinic anhydride as a curing agent, and in water using citric acid and succinic acid as curing agents. (b) SEM images of CNF film and CNF/epoxy composites prepared via solvent exchange to MeCN or in all-water system. Number in parenthesis indicates wt% of CNF in the final material. (c) SEM/EDS images for modified composites prepared in all-water system containing sulfur in diepoxy monomer (left) and in the curing agent/curing agent (right). (d) tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ of neat epoxy cured with succinic anhydride (green), MeCN_succinic anhydride (33 wt% CNF) (purple), H2O_succinic acid 42 (wt% CNF) (black), H2O_citric acid (40 wt% CNF) (red). (e) Storage modulus (T)/storage modulus (T = 30 °C) of neat epoxy cured with succinic anhydride (green), MeCN_succinic anhydride (33 wt% CNF) (purple), H2O_succinic acid 42 (wt% CNF) (black), H2O_citric acid (40 wt% CNF) (red). δ of neat epoxy cured with succinic anhydride (green), MeCN_succinic anhydride (33 wt% CNF) (purple), H2O_succinic acid 42 (wt% CNF) (black), H2O_citric acid (40 wt% CNF) (red). (e) Storage modulus (T)/storage modulus (T = 30 °C) of neat epoxy cured with succinic anhydride (green), MeCN_succinic anhydride (33 wt% CNF) (purple), H2O_succinic acid 42 (wt% CNF) (black), H2O_citric acid (40 wt% CNF) (red). | ||
A significant improvement would be to omit organic solvents completely during biocomposites preparation. Water is an ideal medium in the context of sustainable development and can also swell the CNF network reinforcement. There are two problems: epoxy precursors are usually insoluble in water, and the precursors may lose their chemical functionality in water. The use of carboxylic acids rather than anhydrides allowed us to perform impregnation in water. Diepoxy monomer 1 is insoluble in water. It can, however, be sedimented in the water-phase together with the CNF dispersion. CNFs can stabilize colloids, which has been used for the preparation of, e.g., polystyrene/CNF composites in water from Pickering emulsions;54 however, there are no indications that this occurs in this system. Here we prepared a CNF/epoxy wet cake via vacuum filtration of an aqueous dispersion of CNF combined with diepoxide 1. The wet-cake was then immersed in an acid solution in water (succinic acid or citric acid). The acid diffused into the cake to serve as a curing agent (for determining the ratio of monomers infiltrated into the CNF wet cake, see ESI, Fig. S1 and S2†). Curing the pure-thermoset at different water concentrations does not influence the gel content (Table S2†), suggesting that the water evaporated before the reaction. The temperature was above the melting point of epoxy precursors (diepoxide) (170 °C) to promote homogeneous distribution of epoxide in the CNF network (ESI† and Fig. 2a). Possibly, the molten diepoxide was able to flow prior to gelation.
Fig. 2b shows SEM images of fracture surfaces of biocomposites prepared via solvent exchange to MeCN and also in water-based systems. Note that the CNF content is very high (Wf ≈ 33–42 wt%). For all biocomposites, the nanofibers are homogeneously distributed in the bio-epoxy thermoset matrix in images with a 500 nm scale bar. The layered structure characteristic for the neat CNF films (left hand image) is no longer present. Importantly, composites prepared in the water-based system exhibit even distribution of nanofibers similar to the composites prepared via classical solvent exchange. Two modified biocomposites containing sulfur in either the diepoxy monomer (thiirane) or the hardener (thiomalic acid) were prepared to confirm even matrix distribution for the all-water system. SEM/EDS imaging revealed uniform distribution of sulfur in both composites at the scale of 250 nm (Fig. 2c and ESI†).
Dynamic mechanical analysis can be used to obtain information concerning the epoxide thermoset structure in the biocomposite. Fig. 2d shows the temperature dependence of the mechanical loss factor tanδ of the neat bio-epoxy thermoset and CNF/bio-epoxy composites. The maximum on a tanδ–T curve is assumed to roughly correspond to the glass transition temperature (Tg) of the thermoset. The peak temperature of the epoxy matrix is strongly shifted to higher temperatures in the biocomposites. The only possible reason is that the epoxy thermoset is covalently linked to the cellulose nanofiber surface (Fig. S13†). As a consequence, the molecular mobility is decreased and Tg shifted to higher temperature, since the effective cross-link density of the epoxy is increased. The wide tanδ transitions for nanostructured biocomposites are caused by inhomogeneous crosslink density from epoxide-CNF reactions. In amine-cured epoxies, nanocellulose was found to have catalytic effects on epoxy curing.55Fig. 2e shows the normalized temperature dependence of storage modulus for all biocomposites. For a neat, sparsely cross-linked bio-epoxy thermoset, the storage modulus drops rapidly at temperatures above 100 °C. The decrease in storage modulus at high temperatures is much less pronounced for CNF reinforced thermoset matrices (for absolute storage modulus values, see ESI, Table S3†). The reason is not only strong thermoset reinforcement by the CNF network, but also an increased Tg for the epoxy matrix by increased cross-link density through reactions with the CNF nanofiber surfaces.
Recycling
To develop material streams suitable for a circular bioeconomy, we analyzed the recycling and degradation behavior of the MeCN_succinic_anhydride (33 wt% CNF) biocomposite in detail. The film was immersed into 0.5 M HCl acetone/water solution 1/1 (v/v) and kept overnight at 80 °C in order to detach the bio-epoxy matrix from CNF via its partial depolymerization into monomers and oligomers. The CNF film was then removed from the solution and washed with water to remove all depolymerized fragments of the thermoset. The residual solution (containing oligomeric and monomeric fragments of the thermoset) was kept at 90 °C for additional 12 hours for its complete depolymerization into solely monomeric compounds (Fig. 3a). To our delight, the depolymerization proceeded with high selectivity. After neutralization and EtOAc/water extraction, succinic acid (>95%) and pentaerythritol (77%) were quantified by NMR in the water phase (ESI, Fig. S6 and S7†). In contrast, the organic phase contained compound 2, a product of Aldol condensation between vanillin and acetone, in 63% yield (ESI, Fig. S3 and S4†). No other degradation products were detected in significant amounts. The lower yield observed for the vanillin-derived diepoxide is probably due covalent linkages between CNFs and diepoxy 1. Chemical bonds between diepoxides and C6–OH groups of CNFs during curing has been proposed.56 Notably, the recovered compound 2 is a versatile and highly functionalized building block. As a demonstration, we converted a crude mixture of 2 to triol 3 – a potential monomer for the preparation of thermosets and branched polyesters – via reduction with NaBH4 (Fig. 3b, ESI, Fig. S8 and S9†). In addition, we demonstrated a synthetic use of the activated double bond in product 2via a Michael addition of thiolmalic acid. This resulted in the formation of an another potential monomeric block for thermosets – the tetrafunctionalized product 4 (Fig. 3b, ESI, Fig. S10 and S11†). | ||
| Fig. 3 Recycling of biocomposites: (a) disintegration of a thermoset from CNF film. (b) Demonstration of synthetic utility of recovered product 2 (NaBH4 reduction of and Michael addition of thiomalic acid). | ||
The recovered CNF film structure was analyzed by FTIR (ESI, Fig. S12†) and SEM (Fig. 4a). SEM images of the recovered film reveal a layered structure characteristic of CNF nanofiber networks. A 2nd generation composite was prepared from the recovered CNF film. The CNF film was soaked in a solution of monomers (succinic anhydride and 1) and then cured (Fig. 4b and ESI†). SEM images of the 2nd generation CNF/epoxy composite showed equally good distribution of CNFs in the polymer matrix, comparted with the virgin biocomposite, without any apparent structural damage or coloration to the CNF network (Fig. 4b). A slight drop in tensile strength was observed for the 2nd generation biocomposites (from 127 to 105 MPa) compared to the 1st generation (Table S4†).
 | ||
| Fig. 4 (a) SEM images of CNF/epoxy biocomposite (MeCN_succinic_anhydride (33 wt% CNF)) (left), recovered CNF film (middle) and 2nd generation biocomposite (right). (b) Demonstration of recyclability and ability to reshape the recovered CNF film for preparation of 2nd generation composites. | ||
Conclusions
We have designed and studied recyclable CNF/bio-epoxy biocomposites, prepared exclusively from commercially available bio-based building blocks. For preparation of the biocomposites we developed an all-water system and compared it with a solvent-exchange procedure. An introduction of 2 types of labile linkages (acetal and ester) in an epoxy thermoset matrix allowed for a complete disintegration of CNF from the polymer with minimal structural damage to the CNF network. The selective depolymerization pathway of the bio-epoxy thermoset enables the recovery of monomeric building-blocks and the production of the 2nd generation CNF/bio-epoxy composite from the recovered CNF film. In addition, the chemical versatility of the depolymerization products was explored towards new types of multifunctional building-blocks.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
PO and LB acknowledge funding from the KAW Biocomposites program.Notes and references
- P. Stegmann, M. Londo and M. Junginger, Resour. Conserv. Recycl.: X, 2020, 6, 100029 Search PubMed.
- L. A. Berglund and T. Peijs, MRS Bull., 2010, 35, 201–207 CrossRef CAS.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- U. Ray, S. Zhu, Z. Pang and T. Li, Adv. Mater., 2021, 33, 2002504 CrossRef CAS PubMed.
- B. Thomas, M. C. Raj, A. K. B, R. M. H, J. Joy, A. Moores, G. L. Drisko and C. Sanchez, Chem. Rev., 2018, 118, 11575–11625 CrossRef CAS PubMed.
- T. Li, C. Chen, A. H. Brozena, J. Y. Zhu, L. Xu, C. Driemeier, J. Dai, O. J. Rojas, A. Isogai, L. Wågberg and L. Hu, Nature, 2021, 590, 47–56 CrossRef CAS PubMed.
- A. J. Benítez and A. Walther, J. Mater. Chem. A, 2017, 5, 16003–16024 RSC.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- M. Nogi, S. Iwamoto, A. N. Nakagaito and H. Yano, Adv. Mater., 2009, 21, 1595–1598 CrossRef CAS.
- M. Henriksson, L. A. Berglund, P. Isaksson, T. Lindström and T. Nishino, Biomacromolecules, 2008, 9, 1579–1585 CrossRef CAS PubMed.
- Y. Ogawa, Y. Nishiyama and K. Mazeau, Cellulose, 2020, 27, 9779–9786 CrossRef CAS.
- X. Yang, M. S. Reid, P. Olsén and L. A. Berglund, ACS Nano, 2020, 14, 724–735 CrossRef CAS PubMed.
- S. Galland, F. Berthold, K. Prakobna and L. A. Berglund, Biomacromolecules, 2015, 16, 2427–2435 CrossRef CAS PubMed.
- T. Kaldéus, A. Träger, L. A. Berglund, E. Malmström and G. Lo Re, ACS Nano, 2019, 13, 6409–6420 CrossRef PubMed.
- R. Wang, H. Yu, M. Dirican, L. Chen, D. Fang, Y. Tian, C. Yan, J. Xie, D. Jia, H. Liu, J. Wang, F. Tang, A. M. Asiri, X. Zhang and J. Tao, ACS Appl. Energy Mater., 2020, 3, 785–793 CrossRef CAS.
- M. Farooq, T. Zou, G. Riviere, M. H. Sipponen and M. Österberg, Biomacromolecules, 2019, 20, 693–704 CrossRef CAS PubMed.
- W. Hu, S. Chen, Z. Yang, L. Liu and H. Wang, J. Phys. Chem. B, 2011, 115, 8453–8457 CrossRef CAS PubMed.
- A. Mihranyan, M. Esmaeili, A. Razaq, D. Alexeichik and T. Lindström, J. Mater. Sci., 2012, 47, 4463–4472 CrossRef CAS.
- A. Carlmark and E. Malmström, J. Am. Chem. Soc., 2002, 124, 900–901 CrossRef CAS PubMed.
- S. Y. Park, S. Yook, S. Goo, W. Im and H. J. Youn, Nanomaterials, 2020, 10, 625 CrossRef CAS PubMed.
- F. Ansari and L. A. Berglund, Biomacromolecules, 2018, 19, 2341–2350 CrossRef CAS PubMed.
- Y. H. Jung, T.-H. Chang, H. Zhang, C. Yao, Q. Zheng, V. W. Yang, H. Mi, M. Kim, S. J. Cho, D.-W. Park, H. Jiang, J. Lee, Y. Qiu, W. Zhou, Z. Cai, S. Gong and Z. Ma, Nat. Commun., 2015, 6, 7170 CrossRef PubMed.
- H. Yang, Q. Chen, X. Wang, M. Chi, H. Liu and X. Ning, Polymers, 2019, 11, 1359 CrossRef CAS PubMed.
- F. Ansari, A. Sjöstedt, P. T. Larsson, L. A. Berglund and L. Wågberg, Composites, Part A, 2015, 74, 60–68 CrossRef CAS.
- H. Yano, J. Sugiyama, A. N. Nakagaito, M. Nogi, T. Matsuura, M. Hikita and K. Handa, Adv. Mater., 2005, 17, 153–155 CrossRef CAS.
- M. Shibata and K. Nakai, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 425–433 CrossRef CAS.
- S. S. Nair, C. Dartiailh, D. B. Levin and N. Yan, Polymers, 2019, 11, 612 CrossRef PubMed.
- R. Auvergne, S. Caillol, G. David, B. Boutevin and J.-P. Pascault, Chem. Rev., 2014, 114, 1082–1115 CrossRef CAS PubMed.
- S. Ma and D. C. Webster, Prog. Polym. Sci., 2018, 76, 65–110 CrossRef CAS.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- W. A. Ogden and Z. Guan, J. Am. Chem. Soc., 2018, 140, 6217–6220 CrossRef CAS PubMed.
- X. Chen, S. Chen, Z. Xu, J. Zhang, M. Miao and D. Zhang, Green Chem., 2020, 22, 4187–4198 RSC.
- Y. Liu, B. Wang, S. Ma, T. Yu, X. Xu, Q. Li, S. Wang, Y. Han, Z. Yu and J. Zhu, Composites, Part B, 2021, 211, 108654 CrossRef CAS.
- M. Mittal and R. Chaudhary, Mater. Res. Express, 2019, 6, 045301 CrossRef.
- S. Ma, J. Wei, Z. Jia, T. Yu, W. Yuan, Q. Li, S. Wang, S. You, R. Liu and J. Zhu, J. Mater. Chem. A, 2019, 7, 1233–1243 RSC.
- M. Fache, B. Boutevin and S. Caillol, ACS Sustainable Chem. Eng., 2016, 4, 35–46 CrossRef CAS.
- A. Carlson, B. Coggio, K. Lau, C. Mercogliano and J. Millis, Industrial Production of Succinic Acid, in Chemicals and Fuels from Bio-Based Building Blocks, 2016, pp. 173–190 Search PubMed.
- Z. Tong, X. Zheng, Y. Tong, Y.-C. Shi and J. Sun, Microb. Cell Fact., 2019, 18, 28 CrossRef PubMed.
- C. Gioia, G. Lo Re, M. Lawoko and L. Berglund, J. Am. Chem. Soc., 2018, 140, 4054–4061 CrossRef CAS PubMed.
- T.-S. Jin, T.-S. Li, Z.-H. Zhang and Y.-J. Yuan, Synth. Commun., 1999, 29, 1601–1606 CrossRef CAS.
- B. R. Jermy and A. Pandurangan, Appl. Catal., A, 2005, 295, 185–192 CrossRef CAS.
- T. Jin, M. Yang, X. Wang, G. Feng and T. Li, J. Chem. Res., 2004, 2004, 203–205 CrossRef.
- F. Kerkel, M. Markiewicz, S. Stolte, E. Müller and W. Kunz, Green Chem., 2021, 23, 2962–2976 RSC.
- N. Warlin, M. N. Garcia Gonzalez, S. Mankar, N. G. Valsange, M. Sayed, S.-H. Pyo, N. Rehnberg, S. Lundmark, R. Hatti-Kaul, P. Jannasch and B. Zhang, Green Chem., 2019, 21, 6667–6684 RSC.
- M. Zirbes, L. L. Quadri, M. Breiner, A. Stenglein, A. Bomm, W. Schade and S. R. Waldvogel, ACS Sustainable Chem. Eng., 2020, 8, 7300–7307 CrossRef CAS.
- D. Havasi, P. Mizsey and L. T. Mika, J. Chem. Eng. Data, 2016, 61, 1502–1508 CrossRef CAS.
- T. Liu, X. Guo, W. Liu, C. Hao, L. Wang, W. C. Hiscox, C. Liu, C. Jin, J. Xin and J. Zhang, Green Chem., 2017, 19, 4364–4372 RSC.
- B. A. J. Noordover, R. Duchateau, R. A. T. M. van Benthem, W. Ming and C. E. Koning, Biomacromolecules, 2007, 8, 3860–3870 CrossRef CAS PubMed.
- X. Yang, S. K. Biswas, J. Han, S. Tanpichai, M.-C. Li, C. Chen, S. Zhu, A. K. Das and H. Yano, Adv. Mater., 2020, 2002264 Search PubMed.
- F. Ansari, S. Galland, M. Johansson, C. J. G. Plummer and L. A. Berglund, Composites, Part A, 2014, 63, 35–44 CrossRef CAS.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- E. C. Corker, U. V. Mentzel, J. Mielby, A. Riisager and R. Fehrmann, Green Chem., 2013, 15, 928–933 RSC.
- S. Fujisawa, E. Togawa and K. Kuroda, Biomacromolecules, 2017, 18, 266–271 CrossRef CAS PubMed.
- A. Omrani, L. C. Simon and A. A. Rostami, Mater. Sci. Eng., A, 2008, 490, 131–137 CrossRef.
- F. Ansari, E. L. Lindh, I. Furo, M. K. G. Johansson and L. A. Berglund, Compos. Sci. Technol., 2016, 134, 175–183 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta07758a |
| This journal is © The Royal Society of Chemistry 2022 |