
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic solid oxide sorbents for CO2 capture: state-of-the art and future perspectives
Ribooga
Chang†
 a,
Xianyue
Wu†
bc,
Ocean
Cheung
*a and
Wen
Liu
*b
a,
Xianyue
Wu†
bc,
Ocean
Cheung
*a and
Wen
Liu
*b
aDepartment of Materials Science and Engineering, Nanotechnology and Functional Materials, Uppsala University, SE75121, Sweden. E-mail: ocean.cheung@angstrom.uu.se
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 62 Nanyang Drive, 637459, Singapore. E-mail: wenliu@ntu.edu.sg
cInterdisciplinary Graduate Programme, Nanyang Environmental & Water Research Institute, 1 Cleantech Loop, 637141, Singapore
First published on 20th December 2021
Abstract
Carbon capture is an important and effective approach to control the emission of CO2 from point sources such as fossil fuel power plants, industrial furnaces and cement plants into the atmosphere. For an efficient CO2 capture operation, many aspects of the CO2 capture steps need to be carefully considered. Currently the most mature CO2 capture technology is liquid amine scrubbing. Alternatively, solid sorbents can be used to effectively capture CO2 while alleviating the disadvantages associated with liquid amine sorbents. In this review, we critically assess solid metal oxide CO2 sorbents, especially oxides of group 1 (Li, Na and K) and group 2 (Mg, Ca, Sr and Ba) metals, for capturing CO2 at moderate to high temperatures. In particular, we focus on the recent advances in developing synthetic metal oxide sorbents, and the correlation between the design, synthetic approaches and their cyclic CO2 capture performance, which are characterised by CO2 uptake capacity, rate of carbonation and cyclic stability. The state-of-the-art, challenges, opportunities and future research directions for these metal oxide sorbents are discussed. By devoting more research effort to address the issues identified, there can be great potential to utilise Group 1 and 2 metal oxides as cost-effective, highly efficient sorbents for CO2 capture in a variety of carbon capture applications.
 Ribooga Chang | Ribooga Chang is a PhD student at the Department of Materials Science and Engineering at Uppsala University, Sweden. Ribooga holds a master's degree from Gwangju Institute of Science and Technology (GIST), Korea. Her PhD work has been focused on the development of high-temperature sorbents and porous materials for a number of applications e.g. gas separation, water treatment, and drug delivery. |
 Xianyue Wu | Xianyue Wu is a doctoral student in Chemical Engineering at the School of Chemical and Biomedical Engineering, Nanyang Technological University. Xianyue is also enrolled on an Interdisciplinary Graduate Programme in collaboration with Nanyang Environment & Water Research Institute (NEWRI), Residues, Resource and Reclamation Centre (R3C). She has a Bachelor of Engineering (Chemical and Biomolecular Engineering) with Honours (Highest Distinction) degree from Nanyang Technological University, Singapore. Her research interests include catalyst and process development in thermal catalysis of carbon monoxide and carbon dioxide. She is currently focusing on work regarding nickel-based dual function material for CO2 capture and conversion. |
 Ocean Cheung | Ocean Cheung is an assistant professor at the Division of Nanotechnology and Functional Materials at Uppsala University, Sweden. He holds a Master of Chemistry (MChem) degree from the University of Warwick, UK and a PhD in Materials Chemistry from Stockholm University, Sweden. His research is focused on the development of functional porous materials for environmental and bio-applications. Focused application areas within Dr Cheung's research include gas/vapour adsorption/separation, catalysis water treatment, and controlled drug delivery. |
 Wen Liu | Wen Liu completed his training as a chemical engineer at the University of Cambridge. He is currently an Assistant Professor at the School of Chemical and Biomedical Engineering, Nanyang Technological University. Dr Liu's research focuses on studying carbon capture and utilisation (CCU) processes, from understanding the atomic scale reaction mechanisms, to designing new CCU strategies using process intensification strategies. He has a special interest in using hypothesis-driven methods to develop high performance oxide materials for carbon capture, oxygen transport, thermochemical water splitting and heterogeneous catalysis. |
1 Introduction
1.1 Background
Global warming is one of the major challenges of the present century.1 The high concentration of CO2 in the atmosphere due to human activity is indisputably the main cause of global warming. In particular, the combustion of fossil fuels for electricity generation, transportation, oil refining, cement production and metallurgy results in the release of CO2 into the atmosphere at a rate that is far beyond what the ecosystem can take up.2 Therefore, decarbonisation technologies have been one of the focal points of research in science and engineering. In a sustainable and low-carbon future, renewable energies will eventually replace fossil fuels. In the meantime, the carbon capture, storage and utilisation scheme (CCSU) will play an important role as a transitional solution to provide low-emission hydrogen, electricity and CO2-derived products from fossil fuel feedstocks. After renewable energies have completely decarbonised the energy sector, CCSU will continue to be relevant to industrial processes such as steelmaking and cement production.3The first step in CCSU is the capture of CO2 from point sources of emissions or, in the case of direct air capture (DAC), from the atmosphere. When capturing from the combustion of fossil fuels, three main strategies exist: pre-combustion, post-combustion and oxy-fuel combustion. Pre-combustion capture involves extracting CO2 from syngas during hydrogen production (by gasification, reforming and downstream purification processes), followed by the combustion of the “blue hydrogen” produced. Post-combustion capture is an end-of-pipe strategy that extracts CO2 from the combustion flue gases. Oxy-fuel combustion is proceeds by burning carbonaceous fuels in the absence of nitrogen. The flue gas of oxy-fuel combustion, after the removal of water, NOx, SOx and any residue N2, can be considered captured. The interested reader is referred to Stanger et al.4 for detailed discussions of various carbon capture strategies.
Regardless of the strategy, CO2 capture requires gas separation, which can be achieved by various technologies including chemical scrubbing, physical adsorption (physisorption), cryogenic distillation, membrane separation, etc. Currently, chemical scrubbing using amine solutions is the most mature carbon capture technology, which can be deployed at commercially viable scales.5 However, CO2 capture by amine scrubbing is energy intensive (typically 4–6 MJ kg−1 CO2 captured by monoethanolamine).6 In addition, liquid amine solutions is expensive to produce, corrosive, and suffers from thermal and oxidative degradation during regeneration (although some priority amine formulations claim superior cyclic stability).7
Physical sorption processes utilise sorbents in either liquid form (e.g. selexol) or solid form (e.g. zeolites and metal–organic framework) and do not require high operating temperatures.8,9 Instead, the regeneration of physical sorbents requires strong vacuum and/or gentle heating, which are associated with unrecoverable energy penalty. Physisorbents such as zeolites, zeotypes, metal–organic frameworks (MOFs) tend to perform best at ambient temperatures with respect to CO2 uptake capacity: zeolites and zeotype typically have high CO2 uptake at low pressures in the range of approximate 4–25 wt% up to 1 bar at ambient temperatures.10 MOFs perform comparably better at high pressures, CO2 uptake at ambient temperatures of well over 100 wt% have been reported close to the saturation pressure of CO2 (e.g. NU-1000).11,12 It is important to note that these physisorbents do not have high CO2 uptake at high temperatures (i.e. the temperature of combustion flue gas) and therefore, the possible use of physisorbents in CO2 will require cooling of the flue gas. In addition, most physical sorption technologies are not specifically selective towards CO2 and must operate under dry conditions to avoid water competing for adsorption sites. This need to de-humidify the gas feed adds extra costs. Although hydrophobic sorbents have been developed to tackle the problem associated with selectivity towards water,13,14 there remains a need to reduce the production cost of these complex synthetic sorbents. As an alternative, cryogenic separation technologies exploit the difference in the boiling points of the gas molecules, such that CO2 can be extracted from the process gas mixtures via distillation or anti-sublimation.15 Therefore, cryogenic separation is capable of producing CO2 of very high purity that can be readily transported and utilised without further processing. However, the supply of cryogenic chill is often costly, both in terms of capital expenditure (CAPEX) and operational costs (OPEX), unless a low-cost cold energy source (e.g. the latent heat of the vaporisation of liquified nature gas) is available.16
Membrane technology offers process simplicity and scalability for extracting CO2 from gas mixtures.17 Unfortunately, the requirement for high pressure gradients across the membranes and the low durability of the membrane materials present notable technical challenges to the large-scale deployment of membrane-based for CO2 capture technologies.18
In order to address the practical issues (e.g. thermal degradation and corrosiveness) associated with liquid amine scrubbing, the use of solid sorbents, such as solid amines and alkali metal oxides, has been proposed. In the latter case, the solid oxide sorbents occupy a large compositional space with versatile chemical formulations that can be tailored for a variety of CO2 capture applications.19 For example, the temperatures at which the sorbents take up and release CO2 can be tuned by design. Different types of sorbents (e.g. physisorbents, liquid absorbents and high temperature solid sorbents) take up CO2via different mechanisms and have different operational conditions, it is therefore, not always possible to compare the different types of sorbents directly. Here, we review the recent developments of solid oxide sorbents, with a focus on synthetic strategies towards improved CO2 capture performance.
1.2 Reversible CO2 uptake by metal oxide sorbents
Metal oxides capable of chemically taking up CO2 typically contain alkali metals and, or alkaline earth metals. The reversible capture of CO2 by the metal oxide sorbents can be generalized by:| MxO + CO2 ⇌ MxCO3 | (1) |
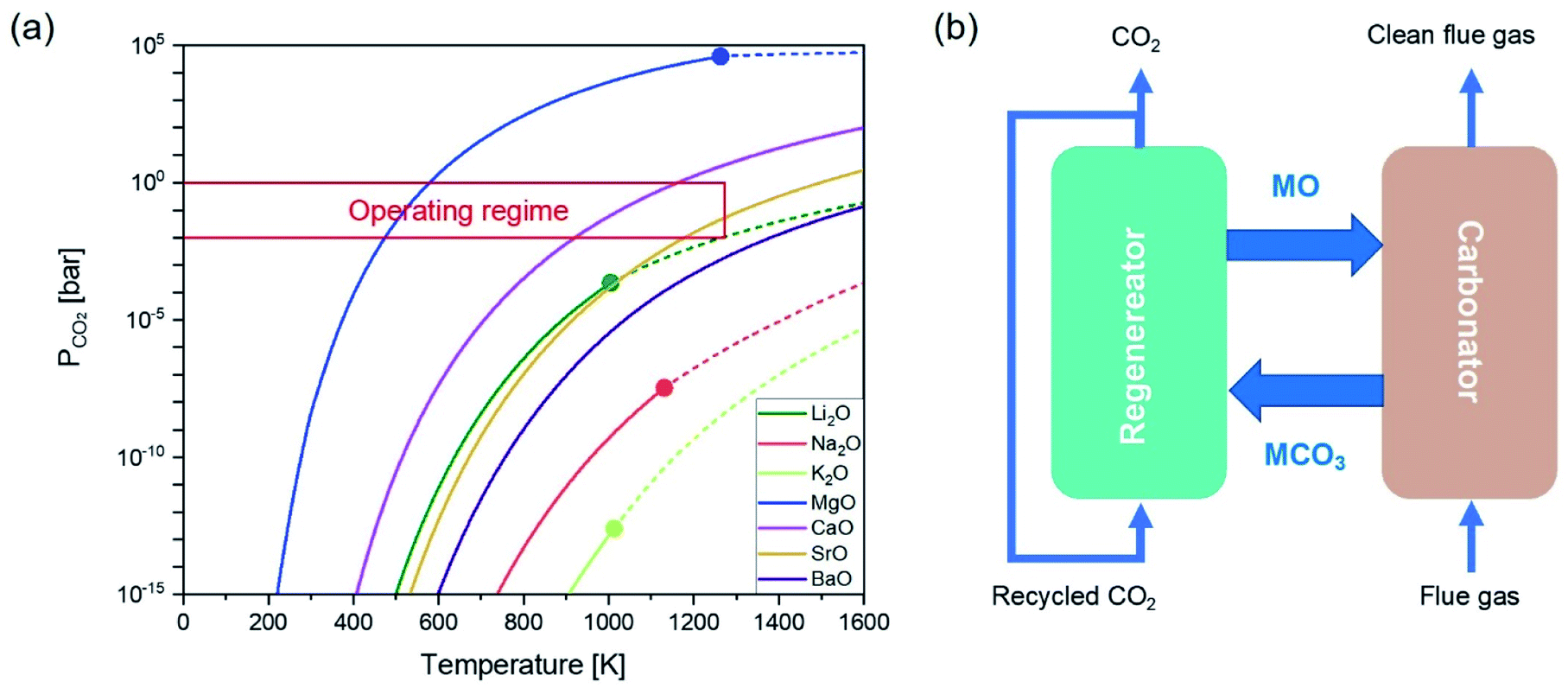 | ||
| Fig. 1 (a) Equilibrium curves of partial pressures of CO2 for the carbonation of the binary oxides of alkali metal/alkaline earth metal as functions of temperature. The red shaded area indicates CO2 partial pressures that are practically achievable in a continuous CO2 capture system. The discontinuities of the equilibrium curves changing from solid lines to dashed represents the melting point of the corresponding carbonates. (b) Schematic illustration of post-combustion CO2 capture by solid sorbents in interconnected fluidised beds. | ||
| Cation | Oxide | Basicity |
|---|---|---|
| Mg2+ | MgO | 0.78 |
| Ca2+ | CaO | 1.0 |
| Li+ | Li2O | 1.0 |
| Sr2+ | SrO | 1.1 |
| Ba2+ | BaO | 1.15 |
| Na+ | Na2O | 1.15 |
| K+ | K2O | 1.4 |
As there is a temperature gap between the carbonation and the calcination steps, continuous CO2 capture typically requires circulating the sorbents between two reactors, viz. a carbonator and a calciner. A common reactor configuration to achieve the swings between carbonation and regeneration is a set of interconnected fluidised beds, as depicted in Fig. 1b.20–22
Microscopically, the solid oxide sorbent particles undergo significant morphological changes as they cyclically take up and release CO2. Although the carbonation of different types of sorbents follows different reaction mechanisms, there are some commonalities. During CO2 capture, the sorbent typically develops a layer of carbonate, restricting the access of CO2 to the unreacted oxides, thus reducing the overall rates of CO2 uptake. In addition, the carbonates formed generally have low melting points, causing the sorbents to sinter, thereby hindering their performance in subsequent capture cycles. Because of these limitations, there are considerable gaps between the sorbents' stoichiometric CO2 uptake capacities and those practically accessible after multiple carbon capture cycles. To improve the overall efficiency and effectiveness of the oxide-based CO2 capture processes, a significant amount of research is required to understand the sorbent behaviour and subsequently to rationally design high performance sorbents, often by optimising the synthesis approach. In general, an ideal solid oxide sorbent should (i) be able to undergo reversible carbonation at temperatures that are closely matching the temperatures of the upstream/downstream processes, (ii) have fast uptake and release kinetics, (iii) exhibit stable performance over large number of CO2 capture cycles, (iv) have sufficient physical hardness to survive prolonged attrition in circulating fluidised beds and (v) can be produced in large quantities with relatively simple methods and low costs.
1.3 Objective and review outline
Over the past decades, there have been large number of studies that develop and investigate solid oxide CO2 sorbents, with focuses on both the process and materials aspects. The interested reader is referred to past reviews which provide broad overviews of CO2 capture technologies employing solid sorbents,25 and in particular metal oxide-based solid sorbents.26 It is worth noting that the studies addressing the materials challenge ubiquitously point towards the need to develop high-performance synthetic sorbents. Despite the abundant reports of synthetic sorbents showing improvements over natural sorbents such as limestone, the relationships between the sorbents' compositions, synthesis methods, structures and CO2 capture behaviour are still not fully understood. Therefore, given the rapid development in characterisation techniques and synthesis methods, we see the need for a critical assessment of the literature focusing the recent advances in synthetic strategies for preparing high performance metal oxide based CO2 sorbents with the following specific objectives:• Review the state-of-the-art understanding of the behaviour of the oxide sorbents over CO2 capture cycles.
• Review different synthetic approaches for preparing high performance oxide sorbents.
• Identify knowledge gaps in understanding the behaviour of synthetic sorbents under realistic CO2 capture conditions.
• Highlight emerging approaches for improving the performance of synthetic sorbents.
• Promote the need for standards for evaluating the performance of newly developed CO2 sorbents.
2 CaO based sorbents
CaO based sorbents are the most commonly discussed due to their high CO2 uptake capacity, fast carbonation and regeneration rates, high earth-abundance and low cost. In Table 2 we summarise the CO2 uptake properties of a number of recently reported CaO based CO2 sorbents. The reversible reaction between CaO and CO2 is also known as calcium looping or calcium carbonate looping:| CaO(s) + CO2(g) ⇌ CaCO3(s), ΔH298 = −178 kJ mol−1 | (2) |
| Sorbent | Method | Conditions | Capacity | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name of the sorbent | Base (B) | Support (S) | Fraction of S | Carbonation temperature [°C] | Calcination temperature [°C] | Carbonation time [min] | Calcination time [min] | CO2 [%] | BET [m2 g−1] | CO2 uptake | Cycle capacity | |||
| [wt%] | [mol%] | [loss%/χ cycle] | ||||||||||||
| a (a) Combustion, (b) impregnation, (c) precipitation, (d) sol–gel, (e) solid-state, (f) wet-mixing, (g) solution-combustion. | ||||||||||||||
| S20-4x | Ca(NO3)2·4H2O | Zr(NO3)2·6H2O | 0.1 | e | 675 | 850 | 20 | 10 | 20 | 12 | 41.8 | 53.3 | 0.8/20 | 53 |
| S20-6x | Ca(NO3)2·4H2O | Zr(NO3)2·6H2O | 0.1 | e | 675 | 850 | 20 | 10 | 20 | 20 | 49.7 | 63.4 | 7.3/20 | 53 |
| S20-8x | Ca(NO3)2·4H2O | Zr(NO3)2·6H2O | 0.1 | g | 675 | 850 | 20 | 10 | 20 | 23 | 48.9 | 62.2 | 5.7/50 | 53 |
| CL-CE-75 | Calcium L-lactate hydrate | CaO·Al2O3 cement | 0.25 | f | 650 | 900 | 30 | 10 | 15 | — | 36 | 45.9 | —/70 | 74 |
| CC-AN-80 | Ca3(C6H5O7)2·4H2O | Al(NO3)3 | 0.2 | b | 650 | 800 | 30 | 10 | 15 | 10 | 59 | 75.2 | 14/28 | 75 |
| CaO-SBA15 | Ca(CH3COO)2 | SBA | 0.5 | b | 700 | 910 | 60 | 30 | 100 | 155 | 43.1 | 55 | 20/40 | 76 |
| CaO-CC | CaCO3 | — | — | — | 650 | 120 | 950 | 30 | 15 | 1 | 71.2 | 90.7 | 23.75/11 | 77 |
| CaO-ES | CaCO3 | MgCO3 | — | — | 650 | 120 | 950 | 30 | 15 | 1 | 61.8 | 78.8 | 20.41/11 | 77 |
| CaO–P | CaCl2 | — | — | c | 650 | 120 | 950 | 30 | 15 | 5 | 48.6 | 61.9 | 2.97/11 | 77 |
| Ca–Al–CO3 | CaO | Al2O3 | 0.5 | c | 600 | 700 | 45 | 20 | 50 | — | 55.7 | 71 | —/10 | 66 |
| Ca–Al–CO3 | CaO | Al2O3 | 0.5 | c | 600 | 700 | 25 | 20 | 50 | — | 56.7 | 72.3 | —/30 | 66 |
| Ca1Ni0.1 | Ca(NO3)·4H2O | Ni(NO3)2·6H2O | 0.1 | d | 650 | 650 | 25 | 3 | 15 | 13 | 66.0 | 84.1 | —/20 | 55 |
| Ca1Ni0.1Ce0.033 | Ca(NO3)·4H2O | Ce(NO3)2·6H2O | 0.033 | d | 650 | 650 | 25 | 3 | 15 | 21 | 62.1 | 79.1 | —/20 | 55 |
| CNAN-SG | Ca(NO3) | Al(NO3)3·9H2O | 0.3 | d | 650 | 900 | 5 | 5 | 15 | 13 | 27.5 | 35 | 28/21 | 65 |
| CAAN-SG | Ca(CH3COO)2 | Al(NO3)3·9H2O | 0.3 | d | 650 | 900 | 5 | 5 | 15 | 12 | 13.6 | 17.3 | 40/21 | 65 |
| CaO-GS 2 mM | CaO | Gemini surfactant | 0.0008 | f | 600 | 850 | — | 30 | 15 | 16 | 29.0 | 37 | — | 72 |
| CaO-SDS 20 mM | CaO | Sodium dodecyl sulfate | 0.008 | f | 600 | 850 | — | 30 | 15 | 9 | 27.0 | 34.4 | — | 72 |
| CaO-(s-s) | CaO | ZrO2 | 0.03 | e | 650 | 780 | 120 | 60 | Pure | — | 57.7 | 73.5 | — | 58 |
| CaO-(c-sg) | CaO | ZrO2 | 0.03 | d | 650 | 780 | 120 | 60 | Pure | — | 77.5 | 98.7 | — | 58 |
| CaO/CaZrO3 | Ca(NO3)2·4H2O | ZrO(NO3)2·xH2O | 0.14 | d | 650 | 850 | 30 | 5 | 15 | — | ∼48.0 | 61.1 | 7/50 | 68 |
| CaO–ZrO2–SG | CaO | ZrO2 | 0.1 | d | 200 | 900 | — | — | — | 25 | 65.0 | 82.8 | —/20 | 59 |
| CaO–ZrO2–IM | CaO | ZrO2 | 0.1 | b | 200 | 900 | — | — | — | 19 | 64.0 | 81.6 | 6.25/14–20 | 59 |
| Ca-hyd_95:5_2 h | Ca(OH)2 | ZrO2 | 0.05 | d | 800 | 800 | 5 | 15 | 50 | 12 | 51.0 | 65 | 39.22/90 | 78 |
| Ca-hyd_95:10_2 h | Ca(OH)2 | ZrO2 | 0.1 | d | 750 | 750 | 20 | 10 | 40 | 11 | 52.0 | 66.3 | 5.77/10 | 78 |
| C74D26 | Carbide | Dolomite | 0.35 | a | 700 | 850 | 20 | 10 | 15 | 14 | 54.0 | 68.9 | —/10 | 79 |
| Mg/Ca-0.2 | Ca(CH3COO)2 | Mg(NO3)2 | 11 | d | 700 | 700 | 30 | 30 | Pure | — | 59.2 | 75.4 | 4.7/7 | 80 |
| Ca/Al-2.5–1% | CaO | Al2O3 | 0.4 | c | 600 | 700 | 45 | 20 | 100 | 6 | 59.1 | 75.3 | 5.9/220 | 48 |
| Ca-CNT | CaO nanotube | — | — | — | 650 | 850 | 30 | 10 | 15 | 12 | 58.0 | 73.9 | 34.48/15 | 81 |
The forward reaction typically takes place around 650 °C, whereas regeneration takes place in 1 bar CO2 at >900 °C.4 The CaO sorbents can be applied to both pre- and post-combustion CO2 capture at power stations, as well as CO2 capture from industrial processes such as cement production.27,28 In the case of pre-combustion capture,29 CaO facilitates the sorbent-enhanced water gas shift to produce highly concentrated H2:30
| CaO(s) + H2O(g) + CO(g) ⇌ CaCO3(s) + H2(g) | (3) |
In addition, CaO based sorbents can be used for capturing CO2 produced by cement plants.31 In fact, CaO based capture process can be efficiently integrated into the cement production process,32 requiring only minor modifications to the existing equipment and process, whilst reducing the fuel consumption and CO2 emissions by ∼75% and ∼85%, respectively.33 Because of the relative ease to prepare CaO based sorbents at large batches, the integration of CaO based carbon capture has been demonstrated at pilot scales (to up 1.9 MWth) by various research groups.34,35 Freshly prepared CaO sorbents, without any further modification, could easily take up CO2 close to the stoichiometric capacity of 78 wt% (based on the mass of the fully calcined CaO)36,37 however, the performance of unmodified CaO sorbents will rapidly deteriorate over uptake–regeneration cycles as a result of sintering,38,39 as the surface area and accessible pore volumes of the sorbents rapidly diminish. In continuous operation, the deactivated sorbents act as inert materials, which imposes significant energy penalty as they participate in the temperature swing cycles without capturing CO2. The deactivation mechanism during calcium looping cycles has been extensively studied.32,40–42 The biggest challenge of using CaO sorbents is to develop solutions that address their poor cyclic stability. The two general strategies to improve the performance of calcium looping systems are (i) optimising operating conditions and (ii) prepare high performance synthetic sorbents, as discussed in the following sections.
2.1 Optimising operating conditions
The performance of the CaO based sorbents depends on the operating conditions. Accordingly, researchers have exploited such dependence to delay or mitigate sorbent deactivation over calcium looping cycles. For example, the presence of steam in the calcium looping reactors is found to influence both reaction rates and mass transfer43,44 specifically, steam is known to enhance the rate of calcination.45,46 In addition, experimental observations suggest that operation in the presence of steam increases the porosity of the sorbents, and subsequently improve their CO2 capture performance in terms of both reaction kinetics and cyclic stability.47,48The issue of sorbent deactivation can also be circumvented by reactivating the spent sorbents. The recent study by Sun et al.49 provided insights into the effects of different reactivation methods on the cyclic performance of CaO/MgO (CaO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MgO mass ratio = 75:25) sorbents, obtained using calcium acetate monohydrate and magnesium acetate tetrahydrate as precursors. Sun et al.49 tested different sorbent reactivation methods: (i) hydration, (ii) hydration/impregnation and (iii) acidification. Good cyclic stability was observed when the sorbent was reactivated by hydration or hydration/impregnation, rendering CO2 uptake of 39 and 41.4 wt% (49.7 and 52.8 mol%) after 40 cycles, respectively. In both cases, the CO2 uptake increase with the number of cycles due to “self-activation”. Self-reactivation, sometimes referred to as “deep carbonation”, is a phenomenon that can be explained using the pore skeleton model by Manovic and Anthony,50 as depicted in Fig. 2. An inward hard skeleton is formed during calcination. However, the hard skeleton hampers the ion diffusion through the product layer of CaCO3, impeding the rate of carbonation during the initial cyclic operation. Over the carbonation–regeneration cycles, the soft skeleton is gradually activated and contributes to an increase rate of carbonation.50
:MgO mass ratio = 75:25) sorbents, obtained using calcium acetate monohydrate and magnesium acetate tetrahydrate as precursors. Sun et al.49 tested different sorbent reactivation methods: (i) hydration, (ii) hydration/impregnation and (iii) acidification. Good cyclic stability was observed when the sorbent was reactivated by hydration or hydration/impregnation, rendering CO2 uptake of 39 and 41.4 wt% (49.7 and 52.8 mol%) after 40 cycles, respectively. In both cases, the CO2 uptake increase with the number of cycles due to “self-activation”. Self-reactivation, sometimes referred to as “deep carbonation”, is a phenomenon that can be explained using the pore skeleton model by Manovic and Anthony,50 as depicted in Fig. 2. An inward hard skeleton is formed during calcination. However, the hard skeleton hampers the ion diffusion through the product layer of CaCO3, impeding the rate of carbonation during the initial cyclic operation. Over the carbonation–regeneration cycles, the soft skeleton is gradually activated and contributes to an increase rate of carbonation.50
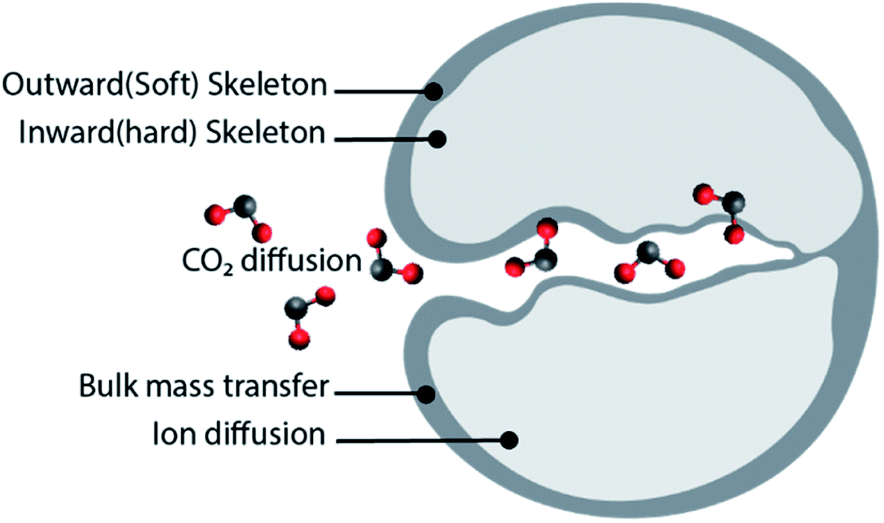 | ||
| Fig. 2 The pore skeleton model for CO2 and ion diffusion. Illustrated according to the finding presented in ref. 51. | ||
Sorbent reactivation by acidification is relevant for removing the irreversibly formed CaSO4 due to the presence of SO2 in the flue gas. For a CaO:MgO sorbent, reactivation by acidification resulted in a CO2 uptake of 23.3 wt% (29.7 mol%) after 40 cycles. Thus, acidification as a reactivation method needs further development to overcome the high cost and the potential release of toxic substances.49
2.2 Synthetic CaO based sorbents stabilised by inert supports
In addition to employing reactivation steps, the cyclic performance of CaO sorbents can be fundamentally improved by mixing CaO with inert refractory materials (e.g. Al2O3, ZrO2, MgO and their derivatives) to render additional sintering resistance and physical hardness. A number of studies have reported CaO based sorbents supported on varying amounts of refractory materials. The resulting CO2 uptake capacity also varied, partly due to the differences in the overall compositions, as shown in Fig. 4. The stabilising effects of adding different types and amounts of thermally stabilising cations, referencing notable recent works, are summarised in Table 2 and discussed below.Soleimanisalim et al.52 used CaCl2·2H2O and a small amount of ZrO(NO3)2·6H2O as precursors to prepare a stabilised CaO sorbent containing 10 wt% CaZrO3 perovskite by the solution combustion method. The resulting CaO sorbent showed a high CO2 uptake of ∼53.3 wt% (68.2 mol%) in the first cycle and ∼42.7 wt% (54.41 mol%) after 31 cycles. The stabilising effect of CaZrO3 was also reported by Hashemi et al.53 who found 20 wt% CaZrO3 to be optimal for maintaining cyclic stability (46.2 wt% (58.87 mol%) after 20 cycles). To maximise cyclic stability and CO2 capture performance, the type of stabiliser and the amount of stabiliser were optimized in the relevant studies.43,44 Specifically, Guo et al.39 showed that the Ca:Al ratio of Ca–Al mixed oxides (Ca2+/Al3+ = 2, 2.5, 3, 3.5, 4) synthesised by coprecipitation affected the CO2 uptake capacity. The Ca:Al ratio of 2.5 gave the highest CO2 uptake capacity of around 47 wt% (60 mol%) after 3 cycles. The high CO2 capacity and reasonable cyclic performance was attributed to the large pore size and the small particle size. Compared to the morphological properties, the amount of Ca12Al14O33 present was also found to be a crucial factor in determining the sorbent's CO2 capture capacity.48,52,54 Sun et al.55 revealed that CeO2 can be used as an effective physical barrier to prevent the sintering of CaO. Similarly, Vall et al.56 employed MgO and Al2O3 to stabilise a synthetic CaO sorbent, obtained by calcining highly porous amorphous calcium carbonate (HPACC–BET surface area of over 450 m2 g−1). Whilst MgO alone can effectively stabilise the CaO sorbent, the sorbent synergistically stabilised by both MgO and Al2O3 yielded even higher stability, showing only a 2.8% decrease in the CO2 uptake capacity after 23 cycles (1st cycle 54.2 wt%, 69.1 mol%). Scanning electron micrographs (SEM) (Fig. 3) of the CaO stabilised with both MgO and Al2O3 confirmed the effective hinderance of sintering after 23 cycles.
 | ||
| Fig. 3 Scanning electron micrograph of CaO stabilised with both MgO and Al2O3 (left) as-synthesised and (right) after 23 cycles of carbonation/calcination.56 | ||
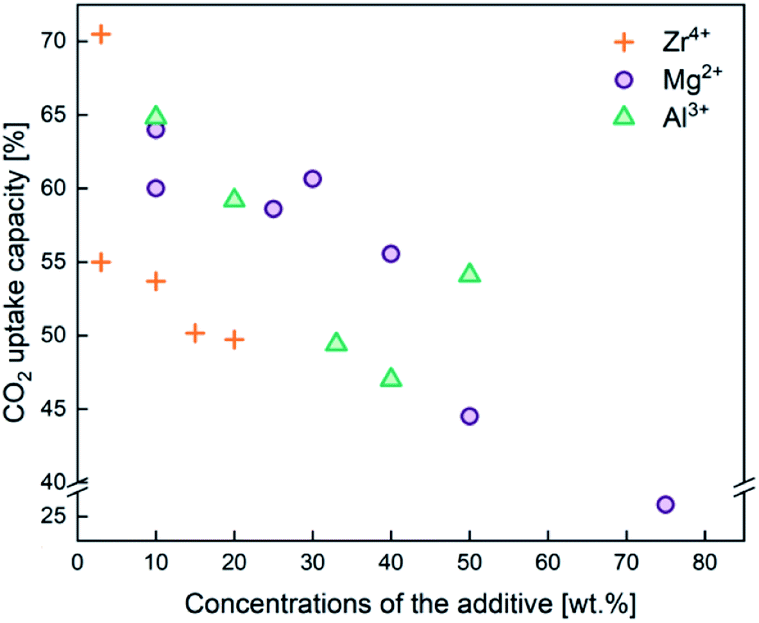 | ||
| Fig. 4 CO2 capture capacity of CaO based sorbents stabilised by the three different typical sintering-resistant, oxidic additives containing Zr4+, Al3+, and Mg2+. The effect of these additives on the cyclic stability of CaO sorbents is summarised in Table 2. | ||
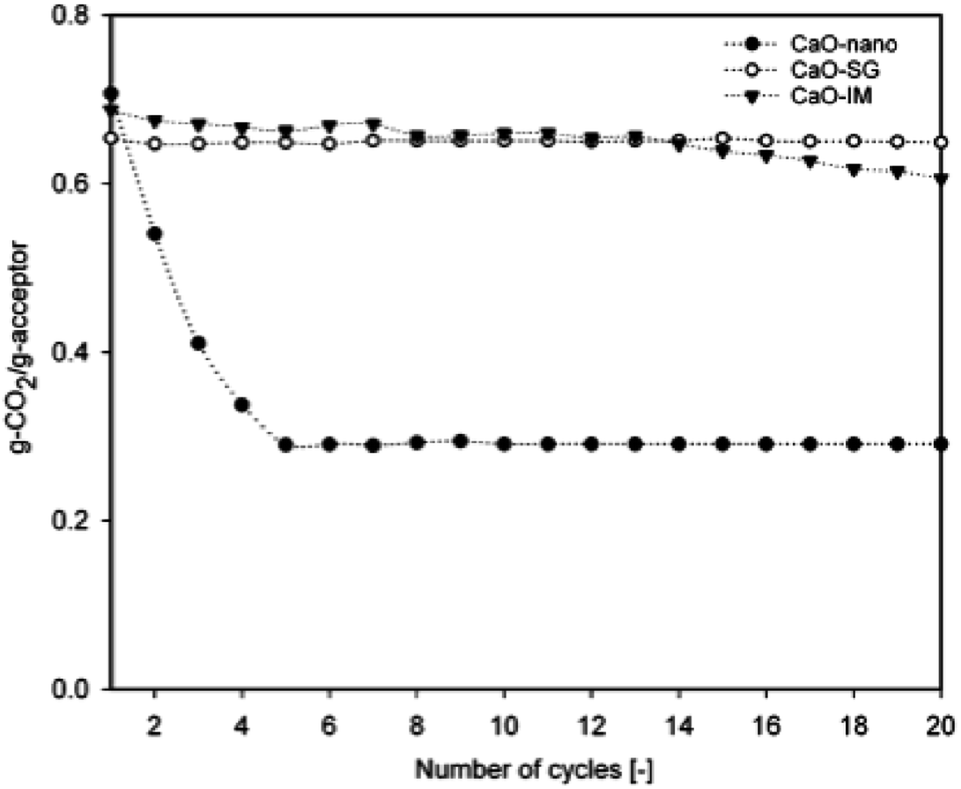 | ||
| Fig. 5 CO2 capture capacity of nano-CaO by sol–gel and wet impregnation methods. Reprinted with permission from ref. 59. Copyright 2021 American Chemical Society. | ||
Precursors. Although the exact roles and effects of the various chemical precursors may differ, they are nonetheless critical to the performance of the synthesised sorbents. Azimi et al.65 demonstrated that different combinations of Ca and Al precursors yield sorbents with noticeably different particle sizes, surface areas and morphologies. For instance, for the sol–gel synthesis of a CaO based sorbent, highly soluble nitrate precursors such as calcium nitrate (CN) and aluminium nitrate (AN) form inert phases such as Ca12Al14O33 and Ca9Al6O18 more easily than the nitrate-free precursors. The insoluble precursors such as nano-structured alumina (nA) formed a more compact structure than the ones prepared from aluminium nitrate (AN), which yielded a “fluffy structure” with a surface area of 13 m2 g−1 and a mean particle size ∼16.9 nm. Overall, the Al-supported CaO sorbent synthesised with soluble nitrate precursors showed the highest CO2 uptake (91.7 wt% (117 mol%), with 28 wt% loss after 21 cycles) than sorbents synthesised from other types of precursors.
Synthesis and calcination temperatures. Temperature is a factor that can be used to control the surface area and particle size of CaO based sorbents. Kou et al.66 observed that high synthesis temperature promoted crystal growth and particle aggregation in Ca–Al mixed oxides. The average particle size of the samples also increased from 29.5 nm for the synthesised at RT to 42.1 nm for the sorbent synthesised at 80 °C. In fact, the crystallinities of CaO, Ca–Al spinel and CaCO3 all increased with increasing synthesis temperature, as shown in Fig. 6. On the other hand, the increase in particle size is accompanied by a decrease in CO2 capture capacity.
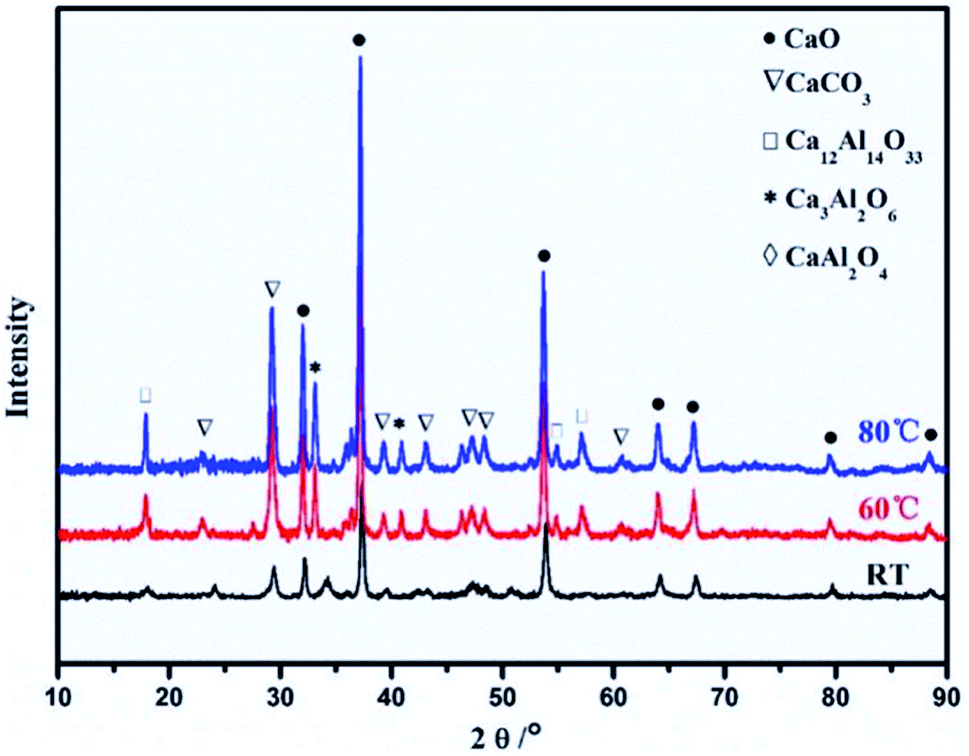 | ||
| Fig. 6 XRD patterns of the Ca–Al mixed oxides (Ca2+/Al3+ = 3) synthesised at room temperature (RT), 60 °C and 80 °C. Reprinted with permission66 copyright 2021 Elsevier. | ||
Apart from the temperature of the wet-synthesis, the calcination temperatures also affect the surface areas and pore volumes of the sorbents. Wang et al.67 observed during the synthesis of CaO/CaZrO3 hollow spheres that the heating rate of calcination significantly affected the cyclic stability of the sorbent – a slow heating rate of 5 °C min−1 resulted in a stable (capacity decreased by <5 wt% after 30 cycles) compared to a sorbent prepred with a calcination rate of 20 °C min−1 (capacity decreased by <20 wt%).67 Antzara et al.68 performed the calcination of ZrO2 promoted CaO at 750 and 800 °C and found that low calcination temperature (750 °C) reduced sintering during carbonation and resulted in CaO with high surface area and pore volume compared to CaO/ZrO2 sorbents calcined at 800 °C.68
Template. The templated synthesis of CaO based sorbent has been adopted for morphology control and to introduce macro porosity to the sorbent. Carbon based template is one of the most commonly used for the synthesis of structured CaO sorbents. Ping et al.69 found that the cage-like CaCO3 hollow spheres synthesised using Ca(NO3)2, urea and polysaccharide spheres template showed an excellent CO2 uptake of up to 78.6 wt% (100.2 mol%) in the first cycle, 45 wt% higher than the same composition synthesised without the template. Colloidal carbon spheres derived from glucose were used as the template for the synthesis of CaO/CaZrO3 and CaO sorbents.67,70 Wang et al.67 found that CaO/CaZrO3 hollow spheres synthesised with Ca(NO3)2 and Zr(NO3)4, templated with carbon sphere (from glucose) had improved cyclic stability over 100 cycles. In a similar study, Wang et al.70 prepared CaO based meshed hollow spheres by urea hydrolysis with carbon sphere (from glucose) template and demonstrated a high CO2 capacity of ∼75 wt% (96 mol%) in the first cycle, with a moderate capacity decrease of 12 wt% over 28 cycles. The authors suggested that the hollow sphere structures could produce porous CaO particles with improved CO2 uptake.70 In other similar studies, Naeem et al.71 incorporated a carbon template, prepared by poly-condensation of resorcinol with an aqueous formaldehyde solution, to the sol–gel synthesis of a Al2O3 and Y2O3 supported CaO sorbent. The resulting structure consisting of interconnected microspheres of supported CaO had an uptake of CO2 of 61 wt% (78 mol%), which remained unchanged after 10 cycles.71
Surfactants. Previous studies have shown the use of surfactant to be an effective way to control the particle size of CaO based sorbents. In a number of cases, such as Jamrunroj et al.72 and Chen et al.,73 there is an optimal amount of surfactant that is required to produce high performance sorbents. Jamrunroj et al.72 found that 2 mM of gemini (GS: 12-carbon hydrophobic chains and 3 carbon alkyl spacer, 12–3–12) yielded the best CO2 capture performance. The resulting CaO–GS-2 mM sorbent, having small particles of (∼37 nm) and high surface area (16 m2 g−1), showed the highest CO2 uptake (29 wt% (37 mol%)) amongst all the CaO–GS sorbents tested.72 Similarly, Chen et al.73 concluded that the best performing CuO/CaO sorbent was obtained with a low H2O
:cetyltrimethyl ammonium bromide (C19H42BrN, CTAB) molar ratio of 23:1. This low CTAB ratio is important for producing small particles (46.5 nm) with a CO2 uptake of 13 wt% (17 mol%), which decreased to 10 wt% (13 mol%) after 19 cycles.73 These studies exemplify that optimal levels of surfactant are crucial for producing sorbent materials with small particle sizes and high capture performance. However, in the case of CaO synthesised with sulphonic single chain (SDS), 40 mM of SDS yield the smallest particles with the lowest CO2 uptake of 18 wt% (23 mol%) compared to other SDS doping amount.72 Therefore, the surfactant effect cannot be easily generalised. In addition, factors such as the micellar structures and the interfacial adsorption ability of the surfactant should also be thoroughly understood and carefully considered during synthesis.
2.3 Summary and knowledge gap
CaO based CO2 sorbents are promising for capturing CO2 at high temperatures. These sorbents show high CO2 uptake capacities with rapid uptake kinetics. Significant advances have been made to improve the performance of the sorbents through carefully tuned synthetic procedures and formulations. However, major challenges remain for the efficient, robust, long term, cost-effective application of CaO based sorbents. Besides steam injection and sorbent reactivation, there are a number of synthetic approaches to enhance the uptake and cyclic stability of CaO sorbents, primarily involving supporting CaO with thermally stable support materials and optimising synthetic parameters. Challenges lie in gaining an in-depth understanding of the effectiveness of each approach, and how they can be synergistically combined to enhance the performance of the CaO sorbents. On that front, we find the use of surfactant in the synthesis of CaO sorbents a particularly interesting avenue for further research. Making good use of surfactants' surface chemistry could hold the key to producing stable, small and porous CaO particles.3 MgO based sorbents
Magnesium oxide (MgO) captures CO2 by:| MgO(s) + CO2(g) ⇌ MgCO3(s); ΔH298 = −118 kJ mol−1 | (4) |
The reversible CO2 capture can be carried out over the temperature range between 200 and 400 °C, making it suitable for heat integration with processes where abundant low-grade heat is available. Although the maximum theoretical CO2 uptake by MgO sorbent is high (110 wt%, 101 mol%), the practically accessible uptake capacity is no more than 10 wt%, owing to the formation of an impermeable layer of MgCO3, which effectively stops the encapsulated MgO from further carbonation. On the other hand, the low operating temperatures could mitigate sorbent deactivation by sintering. Therefore, the majority of the research efforts in developing synthetic MgO based sorbents focus on improving the CO2 uptake capacities through physical and chemical modifications, including (i) introducing dopants to promote the reaction with CO2 or (ii) preparing nanostructured MgO with very large specific surface areas to minimise the encapsulation of MgO by MgCO3. A recent review by Hu et al.82 focuses on the development of MgO sorbent for CO2 capture and provides a detailed insight in this topic. Here, we present a focused discussion on the synthesis and performance of these MgO sorbents. A list of recently developed MgO sorbents and their CO2 capture performance is shown in Table 3, with key papers discussed in the following sections.
| Sorbent | Method | Conditions | Capacity | Ref. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Name of the sorbent | Base (B) | Promotor (P) | Fraction of S | Carbonation temperature [°C] | Calcination temperature [°C] | Carbonation time [min] | Calcination time [min] | CO2 [%] | BET [m2 g−1] | CO2 uptake | Cycle stability | |||
| [wt%] | [mol%] | [loss%/cycle] | ||||||||||||
| a (a) Sol–gel, (b) wet mixing, (c) precipitation, (d) physical mixing, (HP) high purity, (UP) ultra purity. | ||||||||||||||
| MgO-AMS10-325 °C | MgO | LiNO3, KNO3, Na2CO3, K2CO3 | 0.44, 0.56, 0.5, 0.5 | c | 325 | 450 | 180 | 90 | HP | — | 83.9 | 76.8 | 17/30 | 88 |
| MgO-AMS10-350 °C | 350 | 450 | 240 | 60 | HP | — | 81.5 | 74.6 | — | 88 | ||||
| MgO–NaNO3 + NaNO2 | MgO | NaNO3, NaNO2 | 0.07, 0.15 | c | 350 | 400 | 30 | 20 | Pure | 23 | 55.0 | 50.4 | 55.2/15 | 84 |
| MgO–NaNO3 | MgO | NaNO3 | 0.07 | c | 325 | 400 | 30 | 20 | Pure | 29 | 83.2 | 76.2 | 54.5/15 | 84 |
| MgO–K2CO3 | Mg(NO3)2·6H2O | K2CO3 | 3 | b | 375 | 400 | 20 | 30 | 100 | 2 | 8.6 | 7.8 | 5.1/17 | 104 |
| MgO–KNO3 | MgO | KNO3 | 0.2 | c | 325 | 450 | 20 | 30 | 100 | 107 | 10.2 | 9.4 | 2412 | 105 |
| MgO–KNO3 | MgO | KNO3 | 0.2 | c | 375 | 450 | 20 | 30 | 100 | 107 | 8.3 | 7.6 | 10/12 | 105 |
| Na–Mg double salt | MgO | Na2CO3 | 0.1 | a | 375 | 500 | — | — | Pure | — | 15.4 | 14.1 | 0/7 | 106 |
| MgO–Na2CO3 | Mg(NO3)2·6H2O | Na2CO3 | 0.5 | b | 380 | 470 | 60 | 10 | 100 | — | 15.0 | 13.7 | —/9 | 107 |
| MgONaNa-0.1 | MgO | Na2CO3, NaNO3 | 0.1 | b | 325 | 450 | 60 | 10 | Pure | 39 | 45.2 | 41.4 | 34.96/14 | 91 |
| 1.00NaNO3/MgO | MgO | NaNO3 | 1 | b | 330 | 400 | — | — | 100 | — | 23.3 | 21.4 | —/15 | 108 |
| MgO–(Li/Na/K)NO3 | MgO | LiNO3, NaNO2, KNO2 | 0.1 | b | 300 | 450 | 240 | 60 | HP | 19 | 73.9 | 67.7 | — | 109 |
| MgO–(Li/Na/K)NO3 | MgO | 0.1 | b | 350 | 350 | 30 | 30 | HP | 19 | 14.1 | 12.9 | —/20 | 109 | |
| MgO–(Li/Na/K)NO3 | MgO | 0.1 | b | 340 | 450 | 240 | 60 | 100 | — | 69.1 | 63.3 | 14/20 | 86 | |
| Mg3Al1–CO3pH12 | Mg(NO3)2·6H2O | Al(NO3)3·9H2O | 0.3 | c | 200 | 400 | — | 60 | — | — | 3.7 | 3.3 | — | 110 |
| NaNO3-5Mg30-MgO | Mg(NO3)2·6H2O | NaNO3 | 0.2 | d | 300 | 450 | 180 | 60 | 100 | — | 11.5 | 10.5 | +19.02/8 | 111 |
| MgONaNO3 | Hydrotalcite | NaNO3 | 0.2 | d | 300 | 450 | 180 | 60 | 100 | — | 37.4 | 34.3 | 53.6/8 | 111 |
| MgO-10%CeO2 | MgO–CeO2 | LiNO3, KNO3, Na2CO3, K2CO3 | 0.17 | a | 325 | 425 | 60 | 15 | 100 | 252 | 35 | 32.1 | +1.22/30 | 112 |
| 5A5M | Mg(NO3)2·6H2O | Al(NO3)3·9H2O | 1 | a | 200 | 600 | 60 | 60 | 10 | 177 | 13.1 | 12 | 12/6 | 113 |
| MgO-AC | Mg(NO3)2·6H2O | NH4OH | 5 | c | 300 | 400 | 60 | — | UP | 362 | 5.2 | 4.7 | 42/27 | 96 |
3.1 Promoted MgO sorbents
Recently, the used of alkali metal nitrates has been in the spotlight as a means to improve CO2 capture performance of MgO based sorbents. The use of alkali metal nitrates exploits the fact that they melt during the CO2 capture process. The molten nitrate prevents the formation a solid MgCO3 layer on the surface of MgO. Fig. 7 illustrates the function of the molten nitrate to promote the carbonation of MgO. The most commonly used nitrate promoters are NaNO3 and KNO3. Because these nitrates do not directly react with CO2, the CO2 capture capacity is governed by the amount of active MgO available in the solid.83 In addition to alkali metal nitrates, alkali metal carbonates could also enhance the performance of MgO sorbents, rendering both faster kinetics and higher uptake capacity. Further enhancements can be achieved when carbonate and nitrate promoters are used concomitantly. These different approaches to promote MgO sorbents are discussed below.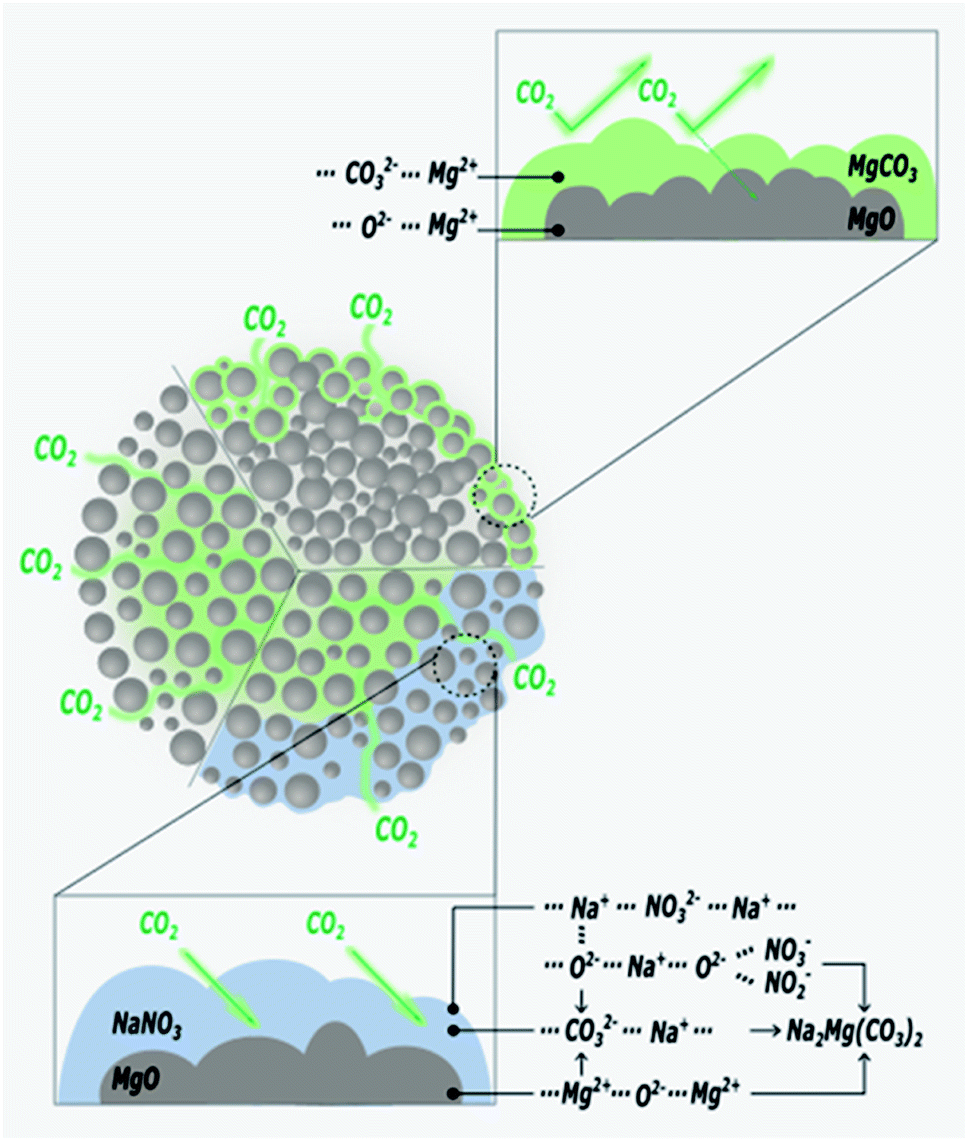 | ||
| Fig. 7 The role of the MgO sorbent with nitrate salts for improving CO2 capture capacity. | ||
:NaNO3 = 1:0.07) the CO2 uptake increased from 66.9 wt% (61.3 mol%) to over 87 wt% (80 mol%), whilst showing stable performance over 15 cycles.
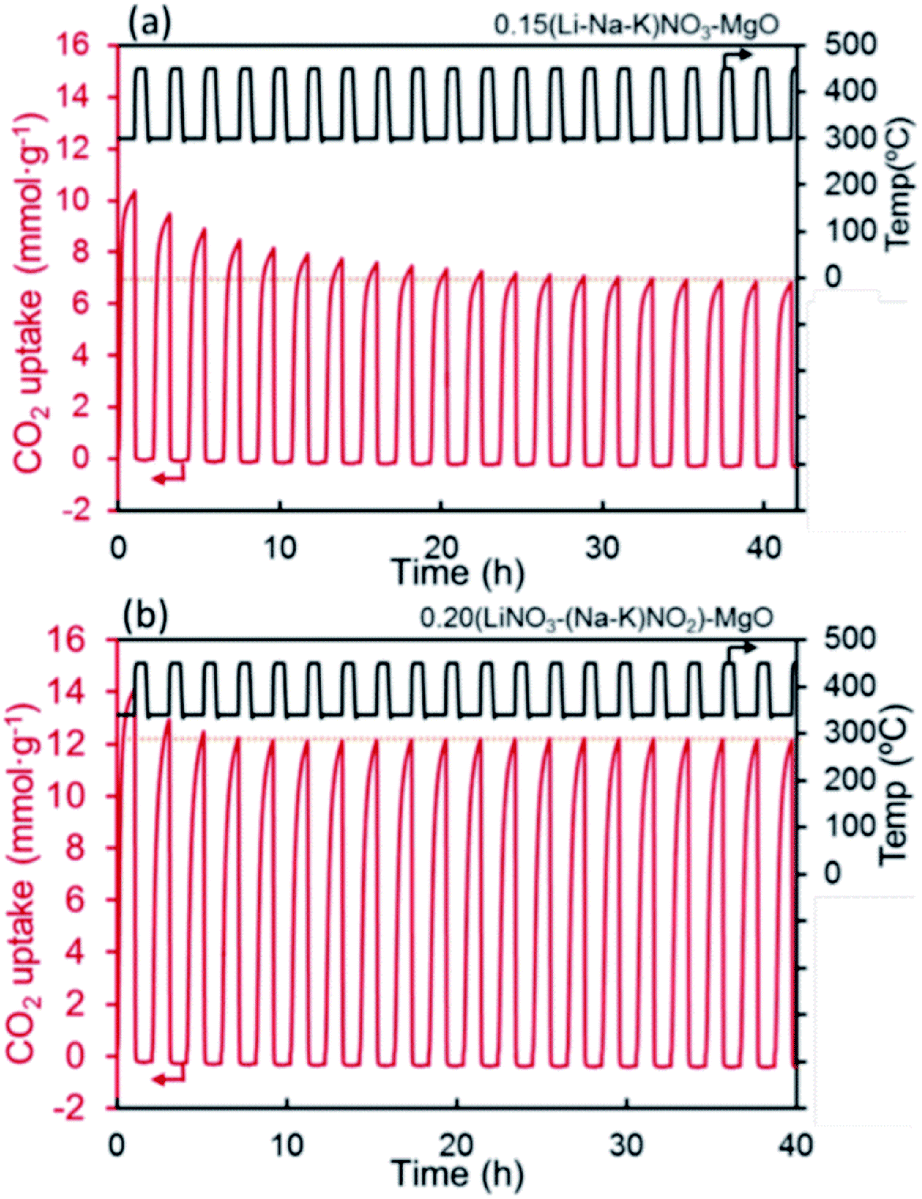 | ||
| Fig. 8 CO2 capture capacity of promoted MgO by alkali metal nitrates ((Li–Na–K)NO3) and nitrate/nitrite (LiNO3–(Na–K)NO2) salt. Reprinted with permission86 copyright 2021 American Chemical Society. | ||
:Na:K of 0.435:0.315:0.250). They classified the CO2 capture process into two steps: (1) “fast and large”, and (2) “slow and small”. The amount of TEC used affects the kinetics of these two steps differently – the rate of step 1 increases with increasing amount of TEC; but this was not observed for step 2. The optimal TEC amount was found to be 60 mol% (sample 60-TEC/MgO), which gave a CO2 uptake of 43.4 wt% (39.7 mol%) in step 1 and an additional 33.9 wt% (31.0 mol%) in step 2. It was also found that MgO with low TEC content (20–40 mol%) had better cyclic stability than those with high TEC content (60–100 mol%).
:nitrate ratio to be important. Co-doping 30 wt% K2CO3 with 10 wt% of NaNO3 or KNO3 both worked well, rendering 44 wt% (40 mol%) uptake at 300 °C in 20 atm CO2.83 Vu et al.91 promoted the performance of mesoporous MgO aerogels with double sodium salts (MgO·Na2CO3·NaNO3). During operation, Na2CO3 and NaNO3 facilitate a high concentration of O2− in the molten salts, which would enhance the CO2 capture capacity by accelerating the formation of MgCO3. Furthermore, the molten salts hinder the formation of the rate-limiting carbonate surface layers, whilst accelerating the production of CO32−. Therefore, it is important to dope MgO with an optimal amount of alkali salts to maximise the benefit of eutectic formation. Specifically, the amount of Na2CO3 should be equal to or less than the amount of NaNO3 for optimal CO2 capture performance. According to Vu et al., the highest CO2 uptake of 56 wt% (51 mol%) was achieved at 325 °C (1 atm, pure CO2) when the molar ratio of MgO:Na2CO3:NaNO3 was 1:0.05:0.2.91 Wang et al.92 also showed the benefit of having both nitrate and carbonate doped on MgO nano-sheets through a series of experiments and found that the optimal sorbent composition to be [(LiNO3, KNO3)2–(Na2CO3, K2CO3)]0.15/MgO, which showed a high CO2 uptake of 73 wt% (67 mol%) (350 °C, 1 atm, 100% CO2). They proposed a reaction mechanism between CO2 and the sorbent, involving the following steps:| NO−3 + K2CO3 + CO2 → NO−2 + K2C2O6 | (5) |
| K2C2O6 + MgO → K2Mg(CO3)2 + 0.5O2 | (6) |
| Na2CO3 + MgO + CO2 → Na2Mg(CO3)2 | (7) |
The finding of Wang et al.92 was in agreement with Ding et al.,88 who revealed that the reaction between CO2 and MgO promoted with 10 mol% [(Li0.44K0.56)NO3]2[(Na0.5K0.5)CO3] takes place in three stages: (1) the formation of carbonate in the first 7 minutes – at this stage, CO2 is dissolved in the molten salt and then reacts with MgO and formed a layer of carbonate (CO32−), producing K2C2O6 and K2Mg(CO3)2. (2) The restriction of the rigid carbonate layer and the formation of the high concentration of oxygen ion within 10 minutes – oxygen ions (O2−) are produced by the dissolution of MgO and the decomposition of the molten alkali nitrates and nitrites. (3) The nucleation of magnesium carbonate occurs between 10 and 60 minutes after contacting the CO2.
3.2 Structured MgO sorbents
Morphological features, such as surface area, pore volume, size and thickness of the particle are all critical to the performance of MgO sorbents.93 Therefore, researchers have explored various synthetic approaches to improve the morphological properties of MgO sorbents to enhance their CO2 uptake.94–96 In general, the CO2 uptake correlates well to the sorbent's surface area. Guo et al.94 showed that solid state chemical reaction (SR) method produced MgO with, smaller particle sizes, higher BET surface area (100 m2 g−1), smaller pore sizes and higher pore volume (0.67 cm3 g−1) than those produced by other synthesis methods, such as direct calcination (DC), direct precipitation (DP) and sol–gel (SG) – when MgCl2 was used as the Mg precursor in all the synthesis methods. As a result, MgO synthesised by SR showed higher CO2 uptake (10.5 wt%, 9.6 mol%) than those by the DC, DP and SG methods, which yielded CO2 uptakes of 2.95–6.8 wt% (2.7–6.2 mol%). In addition, the CO2 capture capacities are also correlated to the sorbents' basicity, which are classified as weak, medium and strong. Accordingly, the SR method yielded the highest number of medium and strong basic sites (responsible for 2.54 and 1.24 mmol CO2 per g of uptake, respectively) as well as the highest total amount of basic sites (with a total uptake of 5.49 mmol CO2 per g). The abundant basic sites enhanced the CO2 uptake of the SR sorbent. Similarly, Ding et al.95 showed that MgO synthesised from magnesium acetate tetrahydrate (Mg(CH3COO)2) and oxalic acid (C2H2O4) dihydrate solution (MgO–MO) had a high BET surface area of 252 m2 g−1, high pore volume of 0.763 cm3 g−1 and CO2 uptake capacity of 7.59 wt% (7.0 mol%) at 50 °C. The CO2 uptake capacity of MgO–MO was superior compared to MgO synthesised by calcining magnesium carbonate (MgO–BMC), the rehydration of MgO–BMC (MgO–RF) and commercial MgO (MgO–CA).95 Hanif et al.96 found that MgO obtained by ammonia precipitation (AC) showed higher surface area (362 m2 g−1, 0.701 cm3 g−1) and CO2 uptake 7.53 wt% (6.9 mol%) than those by urea hydrolysis (UC) and thermal degradation (TC).Researchers have also developed porous MgO with structural regularities as a means to control the morphologies of the synthetic sorbents. The performance of the various MgO sorbents with well-defined morphologies is summarised in Fig. 9. Notable morphologies are discussed in details below (Fig. 10).
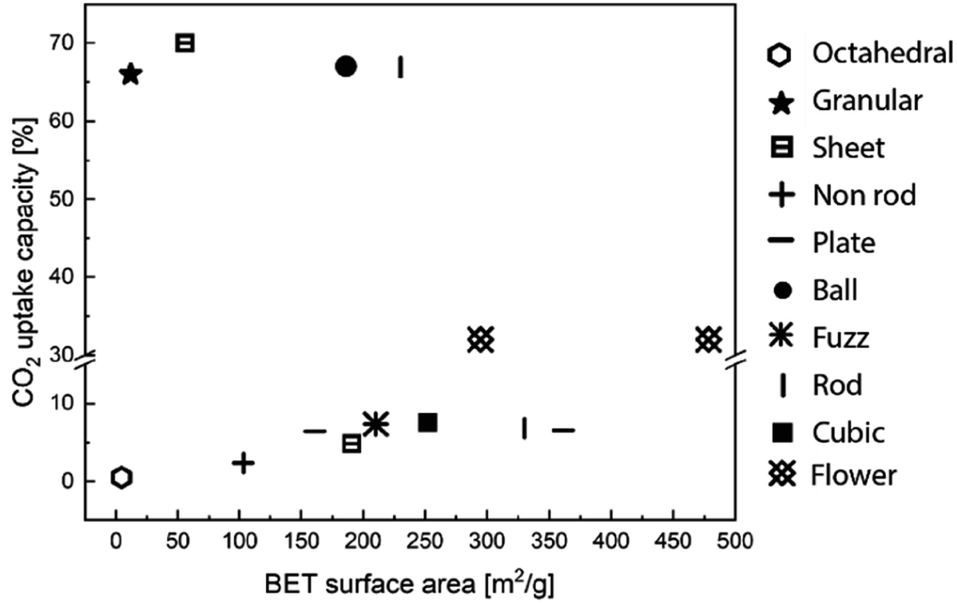 | ||
| Fig. 9 CO2 uptake capacity and BET surface area according to the various structure of MgO sorbents. The data points entries are obtained from ref. 84, 92, 95, 96, 98 and 99. | ||
 | ||
| Fig. 10 SEM images of the structure of MgO sorbents: plate,96 rod,97 and fuzz-like structure of MgO sorbents95 and TEM image of the flower-like plate,98 Reprinted with permission copyright 2021 American Chemical Society.96 Reprinted with permission copyright 2021 Royal Society of Chemistry,95 Reprinted with permission copyright 2021 John Wiley and Son, Inc.97,98 | ||
3.3 Summary and knowledge gap
MgO based materials are promising CO2 sorbents for applications at moderately high temperatures. When low-grade heat (<400 °C) is abundantly available on-site, using MgO to capture CO2 may be more advantageous than calcium looping.101 Additionally, MgO has the potential to become a highly effective CO2 sorbent if its stoichiometric capture capacity can be fully exploited. In practice, the performance of MgO based sorbents is significantly hindered by its unfavourable carbonation mechanism and sluggish rates. Many different approaches have been studied to promote the uptake of CO2 by MgO. Whilst high surface area, morphologically well-defined MgO sorbents could significantly improve CO2 uptake, the experimentally accessible capacities are still substantially lower than the CaO based sorbents. Also, carbonation–calcination cycles could induce drastic morphological changes to the sorbents, making the morphologies of the freshly-prepared materials less relevant to the long-term performance.102 Alternatively, the doping of alkaline metal salts to form low-melting eutectics is a much more effective to tackle the issue of the slow CO2 uptake kinetics. However, the underlying science governing the promotional effects, including the interaction between the different salts in the eutectics, the interplay between different phases (e.g. molten salt, MgO and MgCO3) and the mechanisms of ionic diffusion in the molten salt, must be further studied and better understood.103 Lastly, many MgO sorbents suffer from poor cyclic stability. Therefore, further development is required to improve the effectiveness of the MgO based sorbents.4 Li, Na, K, Sr and Ba based sorbents
As briefly discussed in Section 1, the carbonation of Li2O, Na2O, K2O, SrO and BaO is practically irreversible. However, irreversible CO2 sorbents are economically unattractive as they cannot be recycled and reused in continuous processes. Therefore, Li, Na, K, Sr and Ba based sorbents are often formulated as ternary oxides consisting of (i) at least one alkali metal or alkaline earth metal and (ii) at least one transition metal or semi-metal. The presence of transition metals and semi-metals introduces acidity to the ternary oxides and modulates the overall basicity of the sorbents, making them thermally regenerable at practically achievable temperatures. This section discusses the recent development in the formulation, synthesis and the CO2 capture performance of synthetic Li, Na, K, Sr and Ba based sorbents. Fig. 11 summarises the stoichiometric and experimental CO2 uptake capacities of the Li, Na, K, Sr and Ba based sorbents reported in the recent literature. A more elaborated summary is provided in Table 4.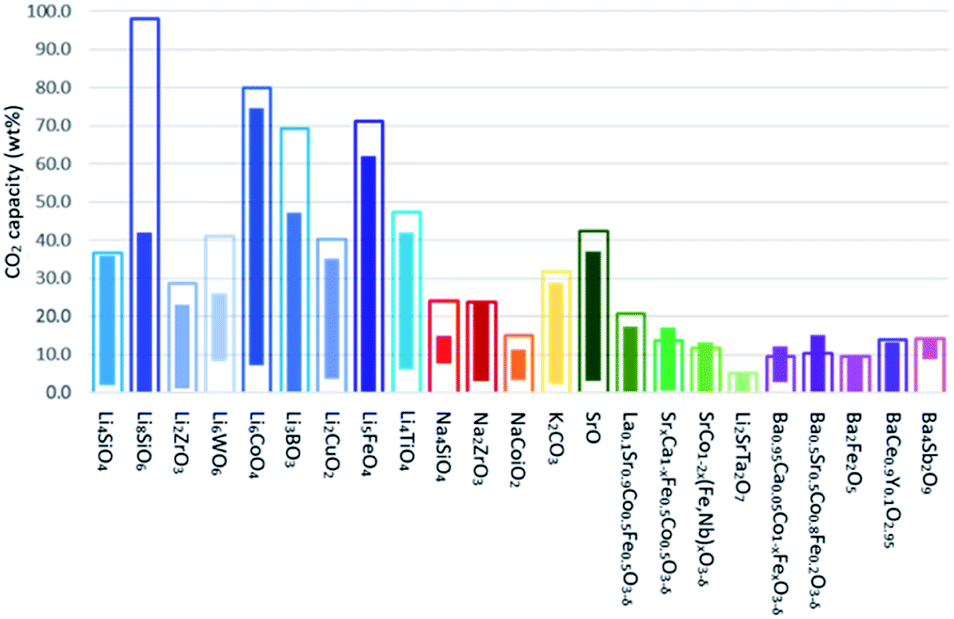 | ||
| Fig. 11 Theoretical and experimental CO2 uptake capacities of various Li-, Na-, K-, Sr- and Ba- based CO2 sorbents. The hollow bars represent the stoichiometric capture capacities, whereas the solid bars represent the range of experimentally measured CO2 uptakes. The sorbents are classified according to the chemical composition of the active components, i.e. excluding the formulation of the support. | ||
| Sorbent | Synthesis method | BET [m2 g−1] | Stoichiometric CO2 capacity [wt%] | Experimental CO2 uptake | Carbonation temperature [°C] | Calcination temperature [°C] | Carbonation CO2 partial pressure [bar] | Calcination CO2 partial pressure [bar] | Max no. of cycles | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| [wt%] | [mol%] | ||||||||||
| a Synthesis methods: (a) solid state; (b) impregnation suspension; (c) doping; (d) wet chemistry; (e) calcination; (f) citrate sol–gel method. b Indicates temperature programmed CO2 capture experiments in a TGA. | |||||||||||
| Li4SiO4 | a, b, c | 0.06–10 | 36.7 | 1.8–36.0 | 4.9–98.1 | RT-700b | 550–900 | 0.04–1.0 | 0 | 250 | 114 and 116–121 |
| Li8SiO6 | a | — | 98.0 | 0–42.0 | 0–42.9 | 25–776b | — | 1.5 | — | — | 115 |
| Li2ZrO3 | a, b, c | 2–3 | 28.7 | 0.01–23.0 | 0–80.1 | 500–575 | 600–700 | 0.10–2.0 | 0–0.585 | 30 | 122–124 |
| Li6WO6 | a | 8 | 41.1 | 3.6–25.9 | 8.8–63.0 | 30–710b | 730–760 | 0.60 | 0 | 4 | 130 |
| Li6CoO4 | a | 1 | 80.0 | 5.0–76.4 | 6.3–95.5 | 300–700 | 700–750 | 0.20–1.0 | 0 | 10 | 127 |
| Li3BO3 | c, d | 1–3 | 69.1 | 0–47.2 | 0–68.3 | 500–650 | 650 | 0.20–1.0 | 0 | 10 | 131 |
| Li2CuO2 | a, c | 1–6 | 40.2 | 3.6–37.4 | 9.0–93.0 | 30–750b | 750–850 | 1.0–5.0 | 0 | — | 128 and 129 |
| Li5FeO4 | a | — | 71.2 | 0–62.0 | 0–87.1 | 30–700b | — | 1.0 | — | — | 126 |
| Li4TiO4 | a | — | 47.3 | 0–42.0 | 0–88.8 | 300–856 | — | 1.0 | — | — | 125 |
| Na4SiO4 | d | — | 23.9 | 7.7–19.2 | 32.2–80.3 | 50–840b | — | 0.80 | — | — | 133 |
| Na2ZrO3 | a, d | 4–5 | 23.8 | 4.5–23.8 | 18.9–100.0 | 150–800 | 680–900 | 0.025–1.0 | 0 | 70 | 134–137 |
| NaCoO2 | a | — | 15.0 | 3.3–11.1 | 22.0–73.3 | 100–800 | — | 0.050–1.0 | — | — | 132 |
| K2CO3 | b, c, e | 0.4–459 | 31.8 | 4.3–28.7 | 13.5–90.3 | 50–100 | 60–300 | 0.010–0.18 | 0–1.0 | 10 | 138–143 |
| SrO | e | 1–3 | 42.5 | 3.0–37.1 | 7.1–87.3 | 1100–1200 | 1100–1200 | 0.30–0.50 | 0 | 10 | 144 |
| La0.1Sr0.9Co0.5Fe0.5O3−δ | f | — | 20.7 | 0–17.3 | 0–83.6 | 600–800 | — | 0.10–1.0 | — | — | 145 |
| SrxCa1−xFe0.5Co0.5O3−δ | f | — | 27.3 | 0.4–17.0 | 1.5–62.3 | RT-950b | — | 1.0 | — | — | 146–148 |
| SrCo1−2x(Fe, Nb)xO3−δ | a | — | 23.6 | 0–13.1 | 0–55.5 | 925 | — | 0.75 | — | — | 149 |
| Li2SrTa2O7 | — | — | 5.1 | 0–4.9 | 0–96.1 | 140 | 700 | — | — | 6 | 151 |
| Ba0.95Ca0.05Co1−xFexO3−δ | f | — | 19.2 | 2.8–12.0 | 14.6–62.5 | 850 | — | 0.10 | — | — | 148 |
| Ba0.5Sr0.5Co0.8Fe0.2O3−δ | a | — | 20.9 | 0–14.9 | 0–71.4 | RT-950b | — | 1.0 | — | — | 149 |
| Ba2Fe2O5 | a | — | 18.8 | 0–9.4 | 0–50.0 | 1000 | 1000 | 1.0 | 0 | — | 153 and 154 |
| BaCe0.9Y0.1O2.95 | a | — | 13.8 | 0–13.1 | 0–94.9 | 600–1000 | — | 1.0 | — | — | 152 |
| Ba4Sb2O9 | a | — | 14.1 | 9.5–14.0 | 67.4–99.3 | 650–750 | 950 | 0.42 | 1 | 100 | 155 |
4.1 Li based sorbents
| Li4SiO4(s) + CO2(g) ⇌ Li2CO3(s) + Li2SiO3(s) | (8) |
The two solid products arrange themselves following a double-shell model, in which Li2CO3 and Li2SiO3 form an outer shell and an inner shell, respectively, as depicted in Fig. 12a.114 The inner Li2SiO3 shell encapsulates the unreacted Li4SiO4 core. Once the shells have fully developed, further carbonation requires the migration of CO2 inward through the Li2CO3 layer, and the migration of Li+ and O2− ions outward through the Li2SiO3 layer. Therefore, the interface between Li2CO3 and Li2SiO3 is regarded as the reaction front, where new Li2CO3 forms. As both shells grow thicker, the diffusion resistance increases and slows down the rate. It is believed that the diffusion of CO2 through the Li2CO3 layer is the rate limiting step, because the diffusion of CO2 through the carbonate is considered slower than the diffusion of Li+ and O2− through the silicate. Owing to the product layer diffusion resistances, the experimentally accessible uptake capacities are always lower than the stoichiometric values. For the chemically unmodified lithium silicates, the surface area of the sorbent becomes the effective rate limiting factor, which subsequently determines the practically achievable uptake capacity.
Using a lithium-richer compound such as Li8SiO6 could potentially improve the CO2 uptake to 98 wt%. The carbonation of Li8SiO6 consists of 3 steps, as illustrated in Fig. 12b and confirmed by in situ synchrotron powder X-ray diffraction (SPXRD):115
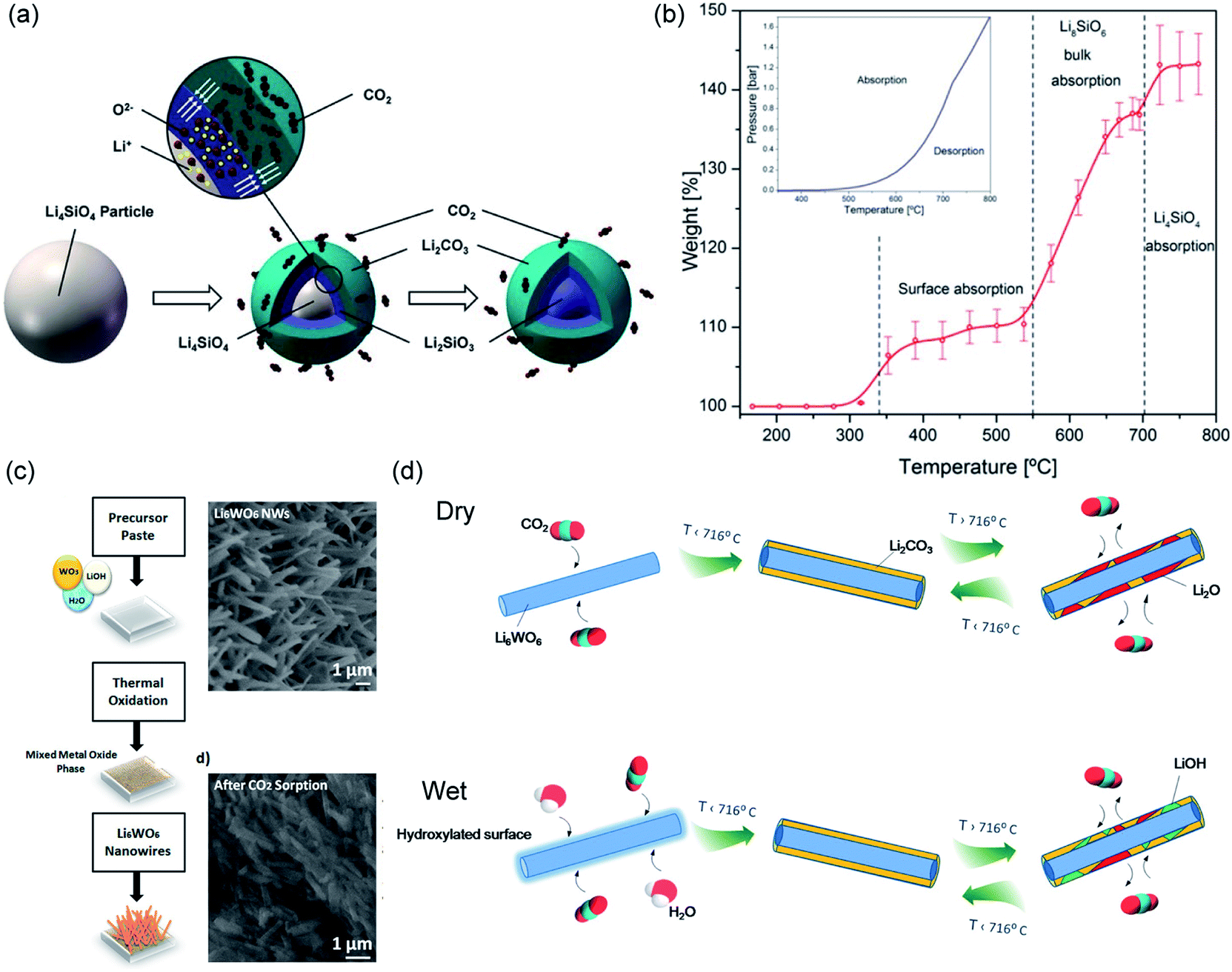 | ||
| Fig. 12 (a) Schematic illustration of the “double shell” model describing Li4SiO4 carbonation. Reprinted with permission114 copyright 2021 John Wiley and Son, Inc. (b) Temperature-programmed carbonation of Li8SiO6 in a TGA. Reprinted with permission115 copyright 2021 American Chemical Society. (c) Illustration of synthesis of Li6WO6 NWs and its morphological change 30 min of carbonation at 700 °C. Reprinted with permission91 copyright 2021 American Chemical Society. (d) Schematic illustration of CO2 capture and release on Li6WO6 NWs under dry and wet conditions. Reprinted with permission1 copyright 2021 American Chemical Society. | ||
Steps 1 and 2 involve the uptake of CO2 by Li8SiO6 on the surface and in the bulk:
| Li8SiO6(s) + 2CO2(g) ⇌ 2Li2CO3(s) + Li4SiO4(s) | (9) |
Step 3 involves the uptake of CO2 by the formed Li4SiO4:
| Li4SiO4(s) + CO2(g) ⇌ Li2CO3(s) + Li2SiO3(s) | (10) |
Lithium silicates can be easily synthesised by solid-state reactions. A typical synthesis involves mechanically mixing lithium and silicon precursors with desired stoichiometric ratios, followed by drying and calcination (at 800–900 °C). Typical lithium precursors include Li2CO3,116,117 Li2O (ref. 118) and LiNO3,119 whereas the silicon precursor is almost always SiO2. Regardless of the precursor used, solid-state synthesised lithium silicates exhibit low specific surface areas (<1 m2 g−1) and low experimental CO2 uptake capacities: 28–35 wt% (76–95 mol%) for Li4SiO4 (ref. 116 and 117) and 42 wt% (43 mol%) for Li8SiO6.115 In addition, the poor sintering resistance of the solid product, Li2SiO3, results in rapid performance decay over CO2 capture cycles.117
Given the dependence on product layer diffusion during carbonation, it is intuitive to prepare Li4SiO4 sorbents with high surface areas using wet chemistry methods such as impregnation suspension,114,120 which involves mixing an aqueous solution of lithium precursors (e.g. lithium acetate and lithium lactate) with a SiO2 sol suspension, which is subsequent dried and calcined.114 Indeed, the resulting Li4SiO4 samples exhibit higher surface areas (1.6–2 m2 g−1), higher CO2 uptake (up to 36 wt%, i.e. 98 mol%) and better cyclic durability (over 40 cycles) than those synthesised by solid-state reactions.114
Alternatively, the performance of Li4SiO4 can be improved by doping. Inert dopants (e.g. CaCO3) can increase the sorbents' specific surface area and sintering resistance.117 Low melting-point dopants, e.g. Na2CO3, K2CO3, NaNO3 and KNO3, can accelerate the transport of CO2 through the Li2CO3 product layer, analogous to the case of alkali metal salt doped MgO sorbents.119–121 Lastly, redox-active dopants, such as Fe3+, can improve the transport of O2− ions through the Li2SiO3 product layer.118 Synthetically, doping can be achieved by including dopant precursors during the solid state synthesis of the Li4SiO4 sorbent,116,117 impregnating the solid-state synthesised Li4SiO4 with soluble dopants (e.g. Na2CO3, K2CO3, NaNO3 and KNO3), or performing a secondary solid state reaction between the as-synthesised Li4SiO4 and the dopant precursor. Two-step solid state synthesis was used for the synthesis of Li4+xSi1−xFexO4 sorbents from Li4SiO4 and Li5FeO4.118 As an economic and sustainable alternative, Wang et al. used acid-leached blast furnace slag, which contains TiO2, Fe2O3, Al2O3, CaO and K2O, as the silicon precursor for the solid-state synthesis of Li4SiO4.116 The inherent presence of dopants in the silica precursor offers a cost-effective means to prepare high performance Li4SiO4 sorbents. In all cases, the dopant-promoted Li4SiO4 showed increased specific surface area (from 0.06 to 0.3 m2 g−1), improved CO2 uptake (up to 35 wt%, i.e. 95 mol%),117 improved cyclic stability (up to 250 cycles)121 and faster rate of carbonation at low reaction temperatures in low CO2 concentrations (in 20% CO2 at 200 °C).118
| Li2ZrO3(s) + CO2(g) ⇌ Li2CO3(s) + ZrO2(s) | (11) |
The experimentally reported carbonation temperatures are in the range of 450–650 °C. Similar to lithium silicates, the carbonation of Li2ZrO3 also proceeds via a double shell model, in which the inner and outer product layers are ZrO2 and Li2CO3, respectively. The CO2 diffusion through the Li2CO3 inner layer is considered to be the rate limiting step. Li2ZrO3 can be simply synthesised by solid-state reaction from Li2CO3 and ZrO2. However, solid-state synthesised Li2ZrO3 show slow carbonation and low uptake capacities (5.9–10 wt%, i.e. 20.6–34.8 mol%).122,123 Therefore, synthetic methods used to improve the performance of Li4SiO4 have also been adopted to promote the Li2ZrO3 sorbents, such as (i) doping low-melting point salts to improve the CO2 mobility through the carbonate product layer,123 (ii) doping redox-active Fe3+ to improve the O2−–CO32− bi-ionic diffusivity through the Li2CO3 product layer,122 and (iii) preparing Li2ZrO3–Na2ZrO3 solid–solution sorbents by impregnation suspension.124 Methods (i) and (iii) appear to be more effective in enhancing CO2 uptake (20–22 wt%, i.e. 70–77 mol%), whereas the improvement by Fe-doping is marginal (7.0–9.2 wt%, i.e. 24.4–32.1 mol%).
Ternary oxide phases containing Li and transition metals have also been prepared, primarily by solid-state synthesis, as CO2 sorbents.125–130 A summary of the carbonation chemistries of these Li-transition metal oxides is presented in Table 5. Amongst these ternary oxides, Li6CoO4 has both the highest stoichiometric CO2 uptake and the highest experimental CO2 uptake of 80.0 and 74.5 wt%, respectively,127 whereas Li6WO6 nanowires showed the ability to rapidly capture CO2 at ambient temperatures in the presence of moisture (relative humidity = 58 ± 3%). The high performance of Li6WO6 nanowires at ambient temperatures is attributed to the hydration and activation of the surface of the nanowires for accelerated carbonation, as illustrated in Fig. 12d.127 For sorbents containing redox-active metals, e.g. Co and Fe, the redox environment during carbonation will also affect the CO2 capture performance. For instance, the presence of O2 promotes CO2 uptake of Li5FeO4 by enhancing the ionic diffusion through the product layer.118 In contrast, the performance of Li6CoO4 deteriorates quickly over cycles (76.4 wt%, i.e. 95.5 mol%, in the first cycle to 30.5 wt%, i.e. 38.1 mol% after 10 cycles), owing to the irreversible oxidation of Co2+ to Co3+ by the O2 present under the testing conditions.127
| Active sorbent | Carbonation reactions | Stoichiometric CO2 uptake capacity (wt%) |
|---|---|---|
| Li4TiO4 | Li4TiO4 + 4CO2 = 4Li2CO3 + TiO2 | 41.5–47.3 |
| Li5FeO4 | Li5FeO4 + 2CO2 = LiFeO2 + 2Li2CO3 | 71.2 |
| LiFeO2 + CO2 = Li2CO3 + Fe2O3 | ||
| Li6CoO4 | Li6CoO4 + 3CO2 = 3Li2CO3 + CoO | 80 |
| Li2CuO2 | Li2CuO2 + CO2 = Li2CO3 + CuO | 40.2 |
| Li6WO6 | Li6WO6 + CO2 = Li4WO5 + Li2CO3 | 41.1 |
| Li4WO5 + CO2 = Li2WO4 + Li2CO3 | ||
| Li2WO4 + CO2 = WO3 + Li2CO3 |
The cyclic stability of Li-transition metal oxides is either poor or unreported. Doping a second transition metal (e.g. Mn, Fe or Ni)128 and alkali metal nitrates (MNO3, M = Li, Na, K)129 has been proven effective for improving the capture capacity (from 23 wt% to 35 wt%, i.e. 57 to 87.1 mol%) and cyclic stability of Li2CuO2. Again, the improvement is by virtue of improving the ionic conductivity of the product layers.
Lastly, trilithium borate (Li3BO3) has been synthesised by precipitation using LiOH and H3BO3 as the precursors.131Li3BO3 captures CO2 with a stoichiometric uptake capacity of 41.4 wt%:
| 4Li3BO3 + 3CO2 ⇌ 3Li2CO3 + Li6B4O9 | (12) |
With a reported experimental CO2 uptake of 35.4 wt% (51.2 mol%), measured after 4 h in pure CO2 at 580 °C. The poor cyclic stability of Li3BO3 could be improved by NaNO2 or KNO2 doping, which significantly accelerates product layer diffusion as the dopant salts form a molten layer. Interestingly, the 10 mol% (Na–K)NO2 doped Li3BO3 showed an experimental uptake of 47.2 wt% (68.3 mol%), exceeding the stoichiometric maximum capacity predicted by eqn (16). This was because the alkali metal nitrites promoted the deep carbonation of lithium borate:
| 4Li3BO3 + 5CO2 ⇌ 5Li2CO3 + Li2B4O7 | (13) |
| Li3BO3 + CO2 ⇌ Li2CO3 + LiBO2 | (14) |
Resulting in improved stoichiometric uptake capacities of 69.1 and 55.4 wt%, respectively.
4.2 Na based sorbents
Na based sorbents have similar chemical formulae as the Li based ones (see Section 4.1). Because sodium is earth-abundant, Na based sorbents are potentially more economical. On the other hand, sodium is heavier and therefore, the stoichiometric CO2 uptake capacity of Na based sorbents are generally lower than that of the Li equivalents in terms of wt%. Experimentally, sodium orthosilicate, sodium zirconate and other sodium metal oxides have shown promising CO2 capture performance over a wide temperature range (from near-ambient temperatures to 850 °C):| Na4SiO4 + CO2 ⇌ Na2CO3 + Na2SiO3 | (15) |
| Na2ZrO3 + CO2 ⇌ Na2CO3 + ZrO2 | (16) |
These compounds generally exhibit good cyclic stability, with the exception of undoped Na2CoO2:
| Na2CoO2 + CO2 ⇌ Na2CO3 + CoO, | (17) |
Similar to Li based sorbents, Na based sorbents can be prepared by solid-state reactions between metal precursors, e.g. Na2SiO3 + NaOH,133 Na2CO3 + ZrO2,134 and Na2CO3 + CoCO3,132 as well as by wet-chemistry methods such as sol–gel synthesis.135,136
Na4SiO4 was found to perform well above 750 °C, showing a maximum uptake of 19.2 wt% (80.3 mol%, with a stoichiometric uptake capacity of 23.9 wt%) at 840 °C.133 One approach to improve the low-temperatures performance of Na4SiO4 is doping alkali carbonates (M2CO3, M = Li, Na, K), which promotes the diffusion of O2− through the molten carbonate layer. The promotional effects follow the order K2CO3 > Na2CO3 > Li2CO3, probably because of the increased basicity from Li to K.133 Although the dopants decrease the mass-based CO2 uptake capacity of the Na4SiO4 sorbent, doping significantly improved carbonation rates even below 700 °C. In particular, K2CO3-doped Na4SiO4 showed 4.7 wt% (19.7 mol%) CO2 uptake at 300 °C, doubling that of undoped Na4SiO4.
During the carbonation of Na2ZrO3, the solid products could form a mesoporous layer with high gas permeability, as depicted in Fig. 13a and b. As a result, the Na2ZrO3 sorbents do not experience significant product layer diffusion resistance, as long as the product layer does not sinter.137 Nevertheless, increasing the initial porosity of Na2ZrO3, e.g. by synthesising the sorbent using sol–gel method, could still render improvements in CO2 uptake and cyclic stability during high temperature operations.135 The best performance was achieved by the Na2ZrO3 prepared from sodium oxalate and Zr(NO3)4, showing an uptake of 22.6 wt% (95.0 mol%, with a stoichiometric uptake of 23.8 wt%), which was largely maintained over 5 successive cycles. Ji et al. attributed the high performance to the partly sintered product layer, which facilitates fast sodium diffusion as a form of mass transfer enhancement, as depicted in Fig. 13c.
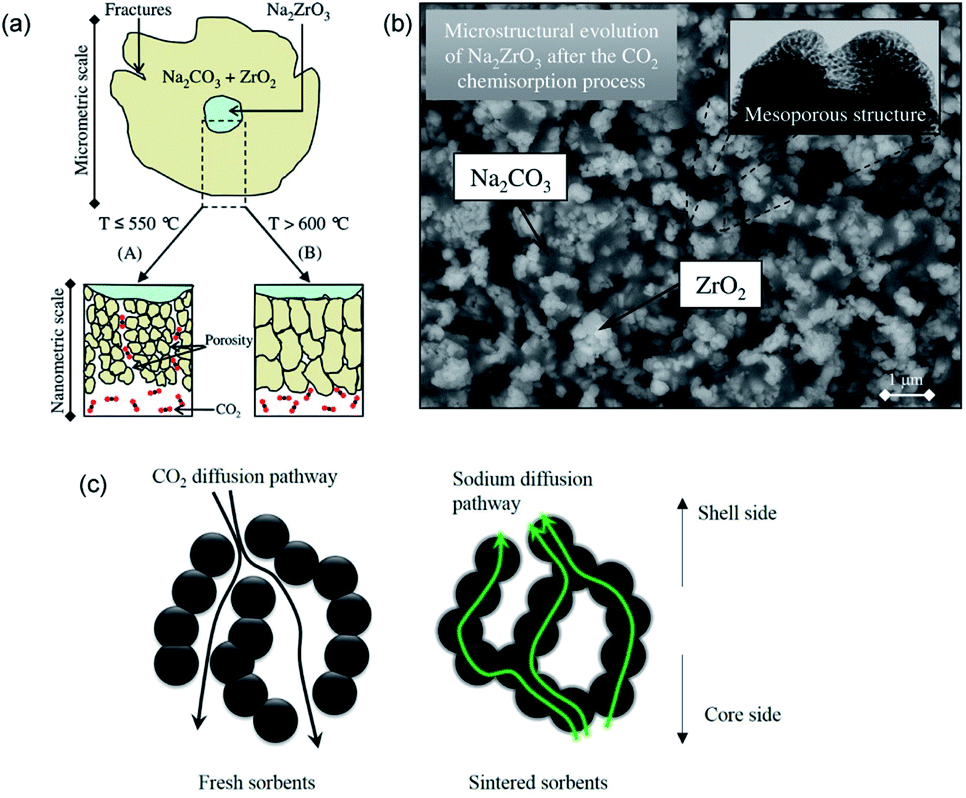 | ||
| Fig. 13 (a) Scheme of the CO2 uptake on Na2ZrO3 at different temperatures. Reprinted with permission134 copyright 2021 American Chemical Society (b) SEM image of growth of ZrO2 and Na2CO3 after the carbonation of Na2ZrO3. Reprinted with permission134 copyright 2021 American Chemical Society (c) Scheme of the CO2 and sodium diffusion in the sorbent under fresh and sintered state. Reprinted with permission135 copyright 2021 Elsevier. | ||
For NaCoO2, substitutionally doping the Co sites with Fe could substantially improve the redox activity, ionic conductivity and therefore CO2 capture performance of the ternary oxide. For instance, NaCo0.7Fe0.3O2 showed improved capacity of 10.6 wt% (70.7 mol%) in 20% CO2 at 700 °C (cf. 3.3 wt%, 22.0 mol% without doping).132 The CO2 uptake could be further improved to 11.1 wt% (74.0 mol%) in the presence of 5% O2 at 800 °C, because the presence of O2 would stabilise the NaCoO2 phase.
4.3 K based sorbents
Unlike lithium and sodium based sorbents, whose functional forms are mixed oxides, potassium based sorbents primarily function in the form of K2CO3 and take up CO2 by forming bicarbonate in the presence of moisture with a stoichiometric CO2 uptake capacity of 31.8 wt%:| K2CO3 + CO2 + H2O ⇌ 2KHCO3 | (18) |
Experimentally, the carbonation and regeneration of K2CO3 occur at 50–60 °C and around 200 °C, respectively. This makes K2CO3 an excellent candidate for CO2 capture at near-ambient temperatures. The practically achievable CO2 uptake by K2CO3 is as high as 28.7 wt% (90.3 mol%).138 However, the cyclic stability of K2CO3 is limited.
To improve the cyclic stability, researchers have impregnated K2CO3 on various types of high melting point supports. The most readily available support material is alumina.139 However, the interaction between Al2O3 and K2CO3 would form the thermally stable KAl(CO3)(OH)2:140
| K2CO3 + Al2O3 + CO2 + 2H2O ⇌ KAl(CO3)(OH)2, | (19) |
Alternatively, support materials that do not react with K2CO3 can be used, such as silicates (e.g. Al6Si2O13, CaSiO3, ZrSiO4, which inert towards K2CO3),141 ZrO2 (ref. 139) and porous carbon.143 In all cases, stable performance over 10 cycles can be achieved at the expense of reduced CO2 uptake (6.5–11.8 wt%, i.e. 20.4–37.1 mol% cf. the stoichiometric maximum of 31.8 wt%). Most interestingly, Yang et al.143 impregnated K2CO3 on carbon aerogels, which possess extremely specific high surface areas (350 to 450 m2 g−1) and provide additional physisorption sites for CO2. As a result, they reported the highest CO2 uptake of 9.2–11.8 wt% (28.9–37.1 mol%), amongst the highest of all supported K2CO3 sorbents. Nevertheless, the application of the carbon based supports for CO2 capture must be further validated under realistic operation conditions, considering the lack of CO2 selectivity by the physisorption process and the lack of stability of carbon in the presence of oxygen, a common component in combustion flue gases.
4.4 Sr based sorbents
SrO can function by itself as a high temperature (>1000 °C) CO2 sorbent with relatively high initial CO2 uptake. Indeed, Miccio et al.144 prepared SrO sorbents with mean particle size of 0.5 mm and 5.0 mm by calcining SrCO3 and wet-granulation (Fig. 14a). The SrO sorbents with mean particle size of 0.5 mm and 5.0 mm had specific surface areas of 1.3 and 2.2 m2 g−1, respectively.144 Both the fine, low surface area and coarse, high surface area SrO particles showed high initial CO2 uptake capacities of 37.1 and 30.5 wt% (87.3 and 48.2 mol%), respectively, when tested in 50% CO2 at 1100 °C. However, the uptake capacity rapidly dropped over cycles due to sintering. The attempt to support SrO2 on Al2O3 by mechanical mixing resulted in the formation of Sr3Al2O6 and SrAl2O4, both appeared beneficial to the sorbent's cyclic stability, at the expense of reducing the capture capacity to 8.7–9.3 wt% (20.5–21.9 mol%).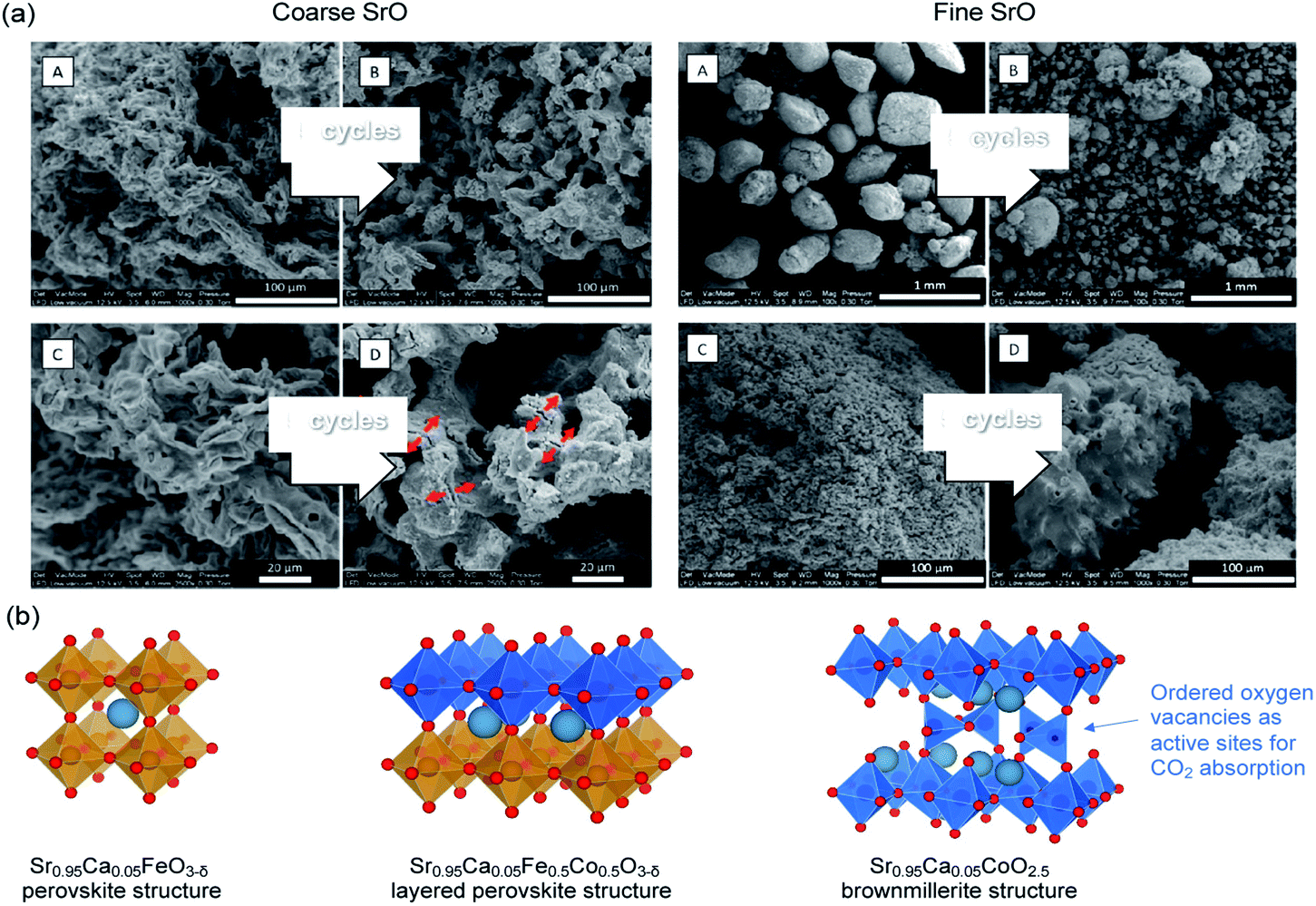 | ||
| Fig. 14 (a) SEM images of coarse and fine SrO particles before (A & C) and after five cycles (B & D) of carbonation−calcination at 1100 °C (b) Sr0.95Ca0.05Fe0.5Co0.5O3−δ perovskite with a layered structure and its phase segregation to a Fe-rich and a Co-rich phase. The undercoordinated Co sites are considered to be the preferential sites for CO2 absorption. Reprinted with permission144 copyright 2021 American Chemical Society. | ||
Alternatively, Sr-containing perovskites, in which Sr occupies the A sites in the ABO3 lattice, can be used directly as CO2 sorbents. These perovskite structured sorbents are typically prepared via sol–gel synthesis.145 The A site and B site occupancies of the perovskites can be flexibly adjusted by varying the compositions of the metal precursors during the sol–gel synthesis. The site occupancies in turn influence the CO2 capture performance. For example, La0.1Sr0.9Co0.5Fe0.5O3−δ was found to capture more CO2 (17.3 wt%, i.e. 83.6 mol%) than Sr0.95Ca0.05Fe0.5Co0.5O3−δ (4.48 wt%, i.e. 18.3–33.2 mol%).146,147 However, further increasing the Ca occupancy in the A site (replacing more Sr) to Sr0.5Ca0.5Fe0.5Co0.5O3−δ could improve the uptake to 17 wt% (62.3 mol%).147 Such improvement was attributed to the fact that Sr0.5Ca0.5Fe0.5Co0.5O3−δ with δ ≈ 0.53 corresponds to a brownmillerite structure (see Fig. 14b) with ordered oxygen vacancies, which are regarded by many to be beneficial to CO2 uptake:146,148
| 2(Sr, Ca)(Fe, Co)O2.5 + CO2 ⇌ (Sr, Ca)CO3 + (Sr, Ca)(Fe, Co)2O4 | (20) |
Adjusting the B-site occupancies could also influence the capture performance. Lu et al.149 synthesised SrCo1−2x(Fe, Nb)xO3−δ with equimolar amount of Fe and Nb by solid-state reactions between SrCO3, Co3O4 (or Co2O3), Fe2O3, and Nb2O5. For x = 0.05 and 0.10, the freshly synthesised sorbents showed CO2 uptake of 13.1 wt% (55.5 mol%) and 11.1 wt% (47.0 mol%), respectively, when evaluated in 75% CO2 at 925 °C. Here, the increased B site occupancy by Nb enhanced the acidity and reduced the activity of the perovskite towards carbonation.150
Beyond the cubic-perovskite structure, a Ruddlesden–Popper (RP) structured (A′2[An−1BnO3n+1]) Li2SrTa2O7 sorbent was prepared by Galven et al. by solid state synthesis.151 This RP phase achieved 4.90 wt% (96.1 mol%) uptake (with a stoichiometric uptake of 5.11 wt%) after 15 h in humid CO2 at 140 °C, 30 bar pressure:
| Li2SrTa2O7 + 0.5CO2 + 0.5H2O ⇌ LiHSrTa2O7 + 0.5Li2CO3 | (21) |
In addition, the Li2SrTa2O7 sorbent exhibited excellent cyclability with negligible capacity loss over 6 cycles of isothermal carbonation at 140 °C and regeneration up to 700 °C. Such performance merits further development and investigation.
4.5 Ba based sorbents
Like Sr based sorbents, most studies on Ba based sorbents focus on the use of perovskite-type oxides with Ba occupying the A sites. These Ba based perovskites can be prepared by either sol–gel synthesis148 or solid state reactions.152 Likewise, the performance of the Ba based sorbents also depends on the site occupancies. For example, Nomura et al.148 found that changing the Fe occupancy in (Ba0.95Ca0.05)(Co1−xFex)O3−δ from 0.2 to 0.9 resulted in a reduction in CO2 uptake from 10.0 wt% (52.5 mol%) to 8.7 wt% (45.3 mol%). Given that BaFeO3−δ (0 < δ < 0.5) type materials are capable of reversibly changing phase between perovskite (ABO3) and brownmillerite (A2B2O5), facilitated by oxygen uptake and release, their CO2 capture performance would also dependent on the oxygen partial pressure and the temperature. In general, unstable BaFeO3−δ structures correspond to improved CO2 capture performance. For example, carbonating Ba0.95Ca0.05Co0.8Fe0.2O3−δ in the absence of oxygen renders an improved capture capacity of 12 wt% (62.5 mol%), owing to the thermal decomposition of perovskite to brownmillerite, which appeared as a better sorbent; this finding is in agreement with Lu et al.149 and Fujishiro et al.153 Yi et al. doped the B-sites of BaFeO3−δ with Nb,154 and reported low CO2 uptake (1.3 wt%, 6.9 mol%) of the doped perovskite. We suspect that the Nb dopant donates electrons to the perovskite structure, which becomes chemically stabilised and unreactive towards CO2. The most outstanding Ba based sorbents is perhaps the 6H-perovskite Ba4Sb2O9 reported by Dunstan et al.155 The authors prepared the sorbents by solid-state reaction between BaCO3 and Sb2O3. Ba4Sb2O9 reacts with CO2 with a stoichiometric CO2 uptake capacity of 14.1 wt%:| Ba4Sb2O9 + 3CO2 ⇌ BaSb2O6 + 3BaCO3 | (22) |
Experimentally, near-stoichiometric CO2 uptake of 13.75 wt% (99.3 mol%) was observed upon carbonation at 750 °C. During long-term cyclic operation, Ba4Sb2O9 showed >10 wt% (70.9 mol%) CO2 uptake over 100 uptake–regeneration cycles (Fig. 15a). The excellent cyclic stability of Ba4Sb2O9 originated from the sorbent's ability to fully regenerate its complete pore structure in each cycle, despite the fact that the fully carbonated sorbent showed virtually no porosity (Fig. 15b). The same authors also found Ba4Nb2−xTaxO9 materials to be poor CO2 sorbents, showing uptake <0.07 wt% in 100% CO2 at up to 1000 °C.156
 | ||
| Fig. 15 (a) CO2 uptake by 6H perovskite Ba4Sb2O9 over 100 carbonation–calcination cycles in at TGA. (b) Surface morphology Ba4Sb2O9 as it evolves over carbonation–calcination cycles. Reprinted with permission155 copyright 2021 American Chemical Society. | ||
4.6 Summary and knowledge gap
Compared to the CaO and MgO based sorbents, Li, Na, K, Sr and Ba based sorbents are less commonly studied. Owing to the high alkalinity of the metal sites, Li, Na, K, Sr and Ba based oxides require chemical modifications to function as reversible CO2 sorbents at reasonable temperatures. These chemical modifications refer to a large pool of candidate sorbent compositions. Whilst many of these sorbents behave similarly during CO2 uptake (e.g., the double shell model for lithium silicates, lithium zirconates, sodium silicates and sodium zirconates), significantly more research is needed to fully understand and rationalise the behaviour of these mixed oxide sorbents in relation to their chemical composition, structure and synthesis methods. Following the discussion above, we summarise the following knowledge gaps for Li, Na, K, Sr and Ba based sorbents:• The effect of strain development on the product layers during carbonation (e.g. following the double shell model depicted in Fig. 12a) is not well understood.
• Most of the sorbents discussed in this section have not been tested under practical CO2 capture conditions, i.e. carbonation in 5–15% CO2 (in the presence of moisture) and calcination in pure CO2.
• The test conditions employed by different studies are inconsistent, making it difficult to make fair comparisons.
• Most CO2 uptake tests were done in TGA, whilst some sorbents have only been studied for 1 carbonation cycle or by temperature programme carbonation.
• The synthesis parameters have not be sufficiently explored to identify optimal synthesis procedures.
• The cost-benefit of synthesising sorbents with complex structures and compositions must be justified.
5 Challenges and opportunities
This review discusses the recent advances in developing synthetic oxide sorbents for CO2 capture. The CO2 uptake ability of these sorbents originates from the alkalinity of the group 1 and group 2 metals. Because of reasons associated with cost, natural abundance and the performance of the natural mineral-derived sorbents, different classes of solid oxide sorbents have received varying extents of research attention and have reached different levels of technological maturity. Specifically, CaO based sorbents can be produced at large quantities from low-cost Ca sources (e.g. limestone), and have been extensively tested over a wide range of scales (from TGA to pilot plants) in realistic flue gas compositions. In contrast, all non-CaO based sorbents require specific chemical modifications to achieve improved CO2 capture kinetics and capacity. Furthermore, the lack of testing standards for the non-CaO sorbents makes it difficult to have meaningful comparisons between different studies, even if they report similar chemical formulations. Addressing the lack of testing standards with priority will allow the field to develop faster and more efficiently.There is also much commonality between different classes of sorbents. For instance, the carbonation of almost all solid oxide sorbents depends on the porosity and the mass transfer through the carbonated product layer. However, maintaining the porous structure of a solid material while it undergoes many cycles of phase changes (i.e. between oxides and carbonates) is technically challenging. Indeed, much research has been devoted to improving the cyclic stability of the solid oxide sorbents through synthetic approaches. One of the key methods involves mixing the active sorbent phases with thermal stabilisers such as Al2O3 and ZrO2. Here, the chemical interaction between the active component (solid bases) and the thermal stabilizer phases (typically acidic oxides) must be understood and taken into consideration, as these interactions could potentially form inert phases (e.g. Ca12Al14O33 and KAl(CO3)(OH)2) to dilute the CO2 uptake capacity of the composite sorbents.
The synthetic approaches to improve the CO2 uptake kinetics are also equally applicable across different classes of sorbents. For example, doping the sorbent surface with low-melting point salts could result in the formation of a molten salt layer during high temperature operation. Consequently, the mass transfer of CO2 through the carbonate product layer can be drastically improved. Doping redox active metals such as Fe3+ could further enhance the mass transfer, which is governed by ionic diffusion through the molten layer, thereby achieving fast CO2 capture and high CO2 uptake capacity. For sorbents that do not rely on the functioning of the molten salt layer, it is generally beneficial to prepare sorbents with high specific surface area and high porosity. To this end, the knowledge in preparing CaO based sorbents, e.g. the effect of synthesis parameters on the morphological properties of the final products, can be adapted for improving the performance of other types of oxide sorbents for a wide range of process applications.
For applications where the capturing and regeneration should take place at moderate to low temperatures, there is also an opportunity to hybridise metal oxides with membranes, polymers, and carbon-based materials to yield synergistic effects and enhanced sorbent performance.157–159 For instance, it has been reported that doping membranes with metal oxides resulted in improved CO2 uptake. Doped membranes with polar –OH group on the surfaces can increase the membrane sorbent's reactivity with CO2 molecules – as CO2 molecules and metal oxide act as electron acceptors and donors, respectively, the selectivity and permeability of the oxide-doped membrane towards CO2 molecules can be effectively increased.160–162 On the other hand, more research effort is required to improve the cyclic regenerability and durability of these composite sorbent materials.
Beyond experimental approaches, the emerging of computational chemistry has offered opportunity to accelerate sorbent formulation with unprecedented efficiencies. In fact, computational approaches have been widely adopted by chemists developing solid sorbents based on MOFs, zeolites and carbon materials,163–166 including high-throughput screening studies that explored candidate sorbent materials in computational databases.167–170 For solid oxide sorbents, ab initio computational studies would in the first instance rely on density functional theory (DFT) calculations to predict the properties of the oxides and the active sites for CO2 uptake. Dunstan et al. has conducted a pioneering high throughput screening of oxidic CO2 sorbent materials using the Materials Project database and identified Na3SbO4 and Li5FeO4 to be promising CO2 sorbents.19 Similarly, the computational screening by Gaultois et al. found Li5SbO5 as a potential sorbent, which showed satisfactory CO2 uptake performance (∼72% of its stoichiometric uptake capacity) over 25 cycles.130 At present, there remains a gap between the thermochemical material properties that are computationally predicted and those experimentally measured. Such gap could potentially hinder the effective of the computational approaches to discover and develop new sorbents. However, with the rapid advancement in computational methods to simulate detailed chemical and physical phenomena, we anticipate computer-aided materials design methods to play major roles in the future development of solid oxide CO2 sorbents.
Author contributions
Ribooga Chang: data curation, formal analysis, writing – original draft, writing – review & editing; Xianyue Wu: data curation, formal analysis, writing – original draft, writing – review & editing; Ocean Cheung: conceptualization, funding acquisition, supervision, writing – review & editing; Wen Liu: conceptualization, funding acquisition, supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. C. and O. C. thank the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) (Grant no. IB2019-8184), the Swedish Research Council (Grant no. 2020-04029) and Swedish Research Council for Sustainable Development (FORMAS, Grant no. 2018-00651) for their financial support. W. L. acknowledges the financial support by National Research Foundation of Singapore under its Campus of Research Excellence and Technological Enterprise (CRETE) programme.References
- M. Z. Akram, V. Atla, A. Nambo, B. P. Ajayi, J. B. Jasinski, J. He, J. R. Gong and M. Sunkara, Nano Lett., 2018, 18, 4891–4899 CrossRef CAS PubMed.
- G. D. Farquhar, M. L. Goulden, M. J. R. Fasham, M. Heimann, V. J. Jaramillo, H. S. Kheshgi, C. Le Quéré, R. J. Scholes and D. W. R. Wallace, IPCC, 2001 Search PubMed.
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- R. Stanger, T. Wall, R. Spörl, M. Paneru, S. Grathwohl, M. Weidmann, G. Scheffknecht, D. McDonald, K. Myöhänen, J. Ritvanen, S. Rahiala, T. Hyppänen, J. Mletzko, A. Kather and S. Santos, Int. J. Greenhouse Gas Control, 2015, 40, 55–125 CrossRef CAS.
- M. Finkenrath, Chem. Eng. Technol., 2012, 35, 482–488 CrossRef CAS.
- A. Brunetti, F. Scura, G. Barbieri and E. Drioli, J. Membr. Sci., 2010, 359, 115–125 CrossRef CAS.
- F. Vega, A. Sanna, B. Navarrete, M. M. Maroto-Valer and V. J. Cortés, Greenhouse Gases: Sci. Technol., 2014, 4, 707–733 CrossRef CAS.
- S. Kumar, R. Srivastava and J. Koh, J. CO2 Util., 2020, 41, 101251 CrossRef CAS.
- Z. Hu, Y. Wang, B. B. Shah and D. Zhao, Adv. Sustainable Syst., 2019, 3, 1800080 CrossRef.
- O. Cheung and N. Hedin, RSC Adv., 2014, 4, 14480–14494 RSC.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- L. H. Xie, M. M. Xu, X. M. Liu, M. J. Zhao and J. R. Li, Adv. Sci., 2020, 7, 1901758 CrossRef CAS PubMed.
- S. Mukherjee, S. Sharma and S. K. Ghosh, APL Mater., 2019, 7, 050701 CrossRef.
- N. Ghasem, in Advances in Carbon Capture, 2020, pp. 479–501 Search PubMed.
- C. Song, Q. Liu, S. Deng, H. Li and Y. Kitamura, Renewable Sustainable Energy Rev., 2019, 101, 265–278 CrossRef CAS.
- G. Ji and M. Zhao, Recent Advances in Carbon Capture and Storage, 2017, ch. 3, DOI: DOI:10.5772/65723.
- U. W. R. Siagian, A. Raksajati, N. F. Himma, K. Khoiruddin and I. G. Wenten, J. Nat. Gas Sci. Eng., 2019, 67, 172–195 CrossRef CAS.
- M. T. Dunstan, A. Jain, W. Liu, S. P. Ong, T. Liu, J. Lee, K. A. Persson, S. A. Scott, J. S. Dennis and C. P. Grey, Energy Environ. Sci., 2016, 9, 1346–1360 RSC.
- J. Blamey, E. J. Anthony, J. Wang and P. S. Fennell, Prog. Energy Combust. Sci., 2010, 36, 260–279 CrossRef CAS.
- C. C. Dean, J. Blamey, N. H. Florin, M. J. Al-Jeboori and P. S. Fennell, Chem. Eng. Res. Des., 2011, 89, 836–855 CrossRef CAS.
- M. Reitz, M. Junk, J. Ströhle and B. Epple, Int. J. Greenhouse Gas Control, 2016, 54, 272–281 CrossRef CAS.
- J. A. Duffy, J. Am. Ceram. Soc., 1997, 80, 1416–1420 CrossRef CAS.
- J. A. Duffy, Geochim. Cosmochim. Acta, 1993, 57, 3961–3970 CrossRef CAS.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 RSC.
- M. T. Dunstan, F. Donat, A. H. Bork, C. P. Grey and C. R. Müller, Chem. Rev., 2021, 121, 12681–12745 CrossRef CAS PubMed.
- A. Perejón, L. M. Romeo, Y. Lara, P. Lisbona, A. Martínez and J. M. Valverde, Appl. Energy, 2016, 162, 787–807 CrossRef.
- M. C. Romano, I. Martínez, R. Murillo, B. Arstad, R. Blom, D. C. Ozcan, H. Ahn and S. Brandani, Energy Procedia, 2013, 37, 142–150 CrossRef CAS.
- A. M. Kierzkowska, R. Pacciani and C. R. Muller, J. Mater. Chem. A, 2013, 6, 1130–1148 CAS.
- T. Xu, X. Wang, B. Xiao and W. Liu, Chem. Eng. J., 2021, 425, 131522 CrossRef CAS.
- IEA, The challenge of reaching zero emissions in heavy industry, IEA, Paris, 2020, https://www.iea.org/articles/the-challenge-of-reaching-zero-emissions-in-heavy-industry Search PubMed.
- S. Gardarsdottir, E. De Lena, M. Romano, S. Roussanaly, M. Voldsund, J.-F. Pérez-Calvo, D. Berstad, C. Fu, R. Anantharaman, D. Sutter, M. Gazzani, M. Mazzotti and G. Cinti, Energies, 2019, 12, 542 CrossRef CAS.
- M. C. Romano, M. Spinelli, S. Campanari, S. Consonni, G. Cinti, M. Marchi and E. Borgarello, Energy Procedia, 2013, 37, 7091–7099 CrossRef CAS.
- B. Arias, M. E. Diego, J. C. Abanades, M. Lorenzo, L. Diaz, D. Martínez, J. Alvarez and A. Sánchez-Biezma, Int. J. Greenhouse Gas Control, 2013, 18, 237–245 CrossRef CAS.
- M.-H. Chang, W.-C. Chen, C.-M. Huang, W.-H. Liu, Y.-C. Chou, W.-C. Chang, W. Chen, J.-Y. Cheng, K.-E. Huang and H.-W. Hsu, Energy Procedia, 2014, 63, 2100–2108 CrossRef CAS.
- B. Arias, M. Alonso and C. Abanades, Ind. Eng. Chem. Res., 2017, 56, 2634–2640 CrossRef CAS.
- M. Erans, M. Jeremias, V. Manovic and E. J. Anthony, J. Visualized Exp., 2017, 128, e56122 Search PubMed.
- A. de la Calle Martos, J. M. Valverde, P. E. Sanchez-Jimenez, A. Perejon, C. Garcia-Garrido and L. A. Perez-Maqueda, Phys. Chem. Chem. Phys., 2016, 18, 16325–16336 RSC.
- C. Zhongxiang, H. S. Song, M. Portillo, C. J. Lim, J. R. Grace and E. J. Anthony, Energy Fuels, 2009, 23, 1437–1444 CrossRef.
- H. Sun, C. Wu, B. Shen, X. Zhang, Y. Zhang and J. Huang, Mater. Today Sustain., 2018, 1–2, 1–27 Search PubMed.
- Y. Hu, H. Lu, W. Liu, Y. Yang and H. Li, Chem. Eng. J., 2020, 396, 125253 CrossRef CAS.
- J. M. Valverde, J. Nanopart. Res., 2018, 20, 39 CrossRef.
- V. Manovic and E. J. Anthony, Ind. Eng. Chem. Res., 2010, 49, 9105–9110 CrossRef CAS.
- S. Yang and Y. Xiao, Ind. Eng. Chem. Res., 2008, 47, 4043–4048 CrossRef CAS.
- J. Yin, C. Zhang, C. Qin, W. Liu, H. An, G. Chen and B. Feng, Chem. Eng. J., 2012, 198–199, 38–44 CrossRef CAS.
- B. Arias, G. S. Grasa and J. C. Abanades, Chem. Eng. J., 2010, 163, 324–330 CrossRef CAS.
- A. N. Antzara, A. Arregi, E. Heracleous and A. A. Lemonidou, Chem. Eng. J., 2018, 333, 697 CrossRef CAS.
- H. Guo, S. Yan, Y. Zhao, X. Ma and S. Wang, Chem. Eng. J., 2019, 359, 542–551 CrossRef CAS.
- J. Sun, W. Wang, Y. Yang, S. Cheng, Y. Guo, C. Zhao, W. Liu and P. Lu, Fuel, 2020, 266, 117056 CrossRef CAS.
- V. Manovic and E. J. Anthony, Environ. Sci. Technol., 2008, 42, 4170–4174 CrossRef CAS PubMed.
- K. Labus, Materials, 2021, 14, 548 CrossRef CAS PubMed.
- A. H. Soleimanisalim, M. H. Sedghkerdar, D. Karami and N. Mahinpey, J. Nat. Gas Sci. Eng., 2016, 36, 1056–1061 CrossRef CAS.
- S. M. Hashemi, D. Karami and N. Mahinpey, Fuel, 2020, 269, 117432 CrossRef CAS.
- A. Kurlov, A. Armutlulu, F. Donat, A. R. Studart and C. R. Müller, Ind. Eng. Chem. Res., 2019, 59, 7182–7188 CrossRef.
- H. Sun, J. Wang, J. Zhao, B. Shen, J. Shi, J. Huang and C. Wu, Appl. Catal., B, 2019, 244, 63–75 CrossRef CAS.
- M. Vall, J. Hultberg, M. Strømme and O. Cheung, RSC Adv., 2019, 9, 20273–20280 RSC.
- C. Luo, Y. Zheng, N. Ding, Q. L. Wu and C. G. Zheng, Chin. Chem. Lett., 2011, 22, 615–618 CrossRef CAS.
- H. J. Yoon and K. B. Lee, Chem. Eng. J., 2019, 355, 850–857 CrossRef CAS.
- K. S. Sultana, D. T. Tran, J. C. Walmsley, M. Rønning and D. Chen, Ind. Eng. Chem. Res., 2015, 54, 8929–8939 CrossRef CAS.
- N. Gao, K. Chen and C. Quan, Fuel, 2020, 260, 116411 CrossRef CAS.
- R. Han, J. Gao, S. Wei, Y. Su and Y. Qin, J. Mater. Chem. A, 2018, 6, 3462–3470 RSC.
- A. Armutlulu, M. A. Naeem, H. J. Liu, S. M. Kim, A. Kierzkowska, A. Fedorov and C. R. Muller, Adv. Mater., 2017, 29, 1702896 CrossRef PubMed.
- W. Peng, Z. Xu, C. Luo and H. Zhao, Environ. Sci. Technol., 2015, 49, 8237–8245 CrossRef CAS PubMed.
- W. Liu, H. An, C. Qin, J. Yin, G. Wang, B. Feng and M. Xu, Energy Fuels, 2012, 26, 2751–2767 CrossRef CAS.
- B. Azimi, M. Tahmasebpoor, P. E. Sanchez-Jimenez, A. Perejon and J. M. Valverde, Chem. Eng. J., 2019, 358, 679–690 CrossRef CAS.
- X. Kou, C. Li, Y. Zhao, S. Wang and X. Ma, Fuel Process. Technol., 2018, 177, 210–218 CrossRef CAS.
- Y. Wang, W. Zhang, R. Li, W. Duan and B. Liu, Energy Fuels, 2016, 1248–1255 CAS.
- A. N. Antzara, A. Arregi, E. Heracleous and A. A. Lemonidou, Chem. Eng. J., 2018, 333, 697–711 CrossRef CAS.
- H. Ping and S. Wu, RSC Adv., 2015, 5, 65052–65057 RSC.
- S. Wang, H. Shen, S. Fan, Y. Zhao, X. Ma and J. Gong, Chem. Eng. Sci., 2015, 135, 532–539 CrossRef CAS.
- M. A. Naeem, A. Armutlulu, Q. Imtiaz and C. R. Müller, ChemPhysChem, 2017, 18, 3280 CrossRef CAS PubMed.
- P. Jamrunroj, S. Wongsakulphasatch, A. Maneedaeng, C. K. Cheng and S. Assabumrungrat, Powder Technol., 2019, 344, 208–221 CrossRef CAS.
- J. Chen, T. Shi, L. Duan, Z. Sun and E. J. Anthony, Chem. Eng. J., 2020, 393, 124716 CrossRef CAS.
- C. Qin, W. Liu, H. An, J. Yin and B. Feng, Environ. Sci. Technol., 2012, 46, 1932–1939 CrossRef CAS PubMed.
- Z. Zhou, Y. Qi, M. Xie, Z. Cheng and W. Yuan, Chem. Eng. Sci., 2012, 74, 172–180 CrossRef CAS.
- C.-H. Huang, K.-P. Chang, C.-T. Yu, P.-C. Chiang and C.-F. Wang, Chem. Eng. J., 2010, 161, 129–135 CrossRef CAS.
- M. Olivares-Marín, E. M. Cuerda-Correa, A. Nieto-Sánchez, S. García, C. Pevida and S. Román, Chem. Eng. J., 2013, 217, 71–81 CrossRef.
- M. Broda and C. R. Müller, Fuel, 2014, 127, 94–100 CrossRef CAS.
- X. Yan, Y. Li, X. Ma, J. Zhao, Z. Wang and H. Liu, New J. Chem., 2019, 43, 5116–5125 RSC.
- L. Huang, Q. Zheng, B. Louis and Q. Wang, Energy Technol., 2018, 6, 2469–2478 CrossRef CAS.
- S. Wei, R. Han, Y. Su, J. Gao, G. Zhao and Y. Qin, Energy Fuels, 2019, 33, 5398–5407 CrossRef CAS.
- Y. Hu, Y. Guo, J. Sun, H. Li and W. Liu, J. Mater. Chem. A, 2019, 7, 20103–20120 RSC.
- B. W. Hwang, J. H. Lim, H. J. Chae, H.-J. Ryu, D. Lee, J. B. Lee, H. Kim, S. C. Lee and J. C. Kim, Process Saf. Environ. Prot., 2018, 116, 219–227 CrossRef CAS.
- X. Zhao, G. Ji, W. Liu, X. He, E. J. Anthony and M. Zhao, Chem. Eng. J., 2018, 332, 216–226 CrossRef CAS.
- A. Dal Pozzo, A. Armutlulu, M. Rekhtina, P. M. Abdala and C. R. Müller, ACS Appl. Energy Mater., 2019, 2, 1295–1307 CrossRef.
- T. Harada and T. A. Hatton, Chem. Mater., 2015, 27, 8153–8161 CrossRef CAS.
- J. S. Kwak, K. R. Oh, K. Y. Kim, J. M. Lee and Y. U. Kwon, Phys. Chem. Chem. Phys., 2019, 21, 20805–20813 RSC.
- J. Ding, C. Yu, J. Lu, X. Wei, W. Wang and G. Pan, Appl. Energy, 2020, 263, 114681 CrossRef CAS.
- S. Jin, K. Ho and C.-H. Lee, Chem. Eng. J., 2018, 334, 1605–1613 CrossRef CAS.
- H. Joo, S. J. Cho and K. Na, J. CO2 Util., 2017, 19, 194–201 CrossRef CAS.
- A.-T. Vu, K. Ho, S. Jin and C.-H. Lee, Chem. Eng. J., 2016, 291, 161–173 CrossRef CAS.
- L. Wang, Z. Zhou, Y. Hu, Z. Cheng and X. Fang, Ind. Eng. Chem. Res., 2017, 56, 5802–5812 CrossRef CAS.
- Y. Yoo, D. Kang, E. Choi, J. Park and I.-S. Huh, Chem. Eng. J., 2019, 370, 237–250 CrossRef CAS.
- Y. Guo, C. Tan, P. Wang, J. Sun, W. Li, C. Zhao and P. Lu, Chem. Eng. J., 2020, 379, 122277 CrossRef CAS.
- Y.-D. Ding, G. Song, X. Zhu, R. Chen and Q. Liao, RSC Adv., 2015, 5, 30929–30935 RSC.
- A. Hanif, S. Dasgupta and A. Nanoti, Ind. Eng. Chem. Res., 2016, 55, 8070–8078 CrossRef CAS.
- V. A. Tuan and C. H. Lee, Vietnam J. Chem., 2018, 56, 197–202 CrossRef CAS.
- P. Li, W. Liu, J. S. Dennis and H. C. Zeng, ACS Appl. Mater. Interfaces, 2017, 9, 9592–9602 CrossRef CAS PubMed.
- P. Li, Y. Lin, R. Chen and W. Li, Dalton Trans., 2020, 49, 5183–5191 RSC.
- P. Li and H. C. Zeng, Environ. Sci. Technol., 2017, 51, 12998–13007 CrossRef CAS PubMed.
- H.-H. Lee, J.-C. Lee, Y.-J. Joo, M. Oh and C.-H. Lee, Appl. Energy, 2014, 131, 425–440 CrossRef CAS.
- H. Cui, Q. Zhang, Y. Hu, C. Peng, X. Fang, Z. Cheng, V. V. Galvita and Z. Zhou, ACS Appl. Mater. Interfaces, 2018, 10, 20611–20620 CrossRef CAS PubMed.
- A. H. Bork, M. Rekhtina, E. Willinger, P. Castro-Fernández, J. Drnec, P. M. Abdala and C. R. Müller, Proc. Natl. Acad. Sci., 2021, 118, e2103971118 CrossRef CAS PubMed.
- G. Xiao, R. Singh, A. Chaffee and P. Webley, Int. J. Greenhouse Gas Control, 2011, 5, 634–639 CrossRef CAS.
- A.-T. Vu, Y. Park, P. R. Jeon and C.-H. Lee, Chem. Eng. J., 2014, 258, 254–264 CrossRef CAS.
- C. H. Lee, S. Mun and K. B. Lee, Chem. Eng. J., 2014, 258, 367–373 CrossRef CAS.
- K. Zhang, X. S. Li, Y. Duan, D. L. King, P. Singh and L. Li, Int. J. Greenhouse Gas Control, 2013, 12, 351–358 CrossRef CAS.
- S. I. Jo, Y. I. An, K. Y. Kim, S. Y. Choi, J. S. Kwak, K. R. Oh and Y. U. Kwon, Phys. Chem. Chem. Phys., 2017, 19, 6224–6232 RSC.
- Y. Qiao, J. Wang, Y. Zhang, W. Gao, T. Harada, L. Huang, T. A. Hatton and Q. Wang, Ind. Eng. Chem. Res., 2017, 56, 1509–1517 CrossRef CAS.
- Q. Wang, H. H. Tay, Z. Guo, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Appl. Clay Sci., 2012, 55, 18–26 CrossRef CAS.
- M. L. T. Trivino, V. Hiremath and J. G. Seo, Environ. Sci. Technol., 2018, 52, 11952–11959 CAS.
- S. Jin, K.-J. Ko and C.-H. Lee, Chem. Eng. J., 2019, 371, 64–77 CrossRef CAS.
- K. K. Han, Y. Zhou, Y. Chun and J. H. Zhu, J. Hazard. Mater., 2012, 203–204, 341–347 CrossRef CAS PubMed.
- X. Yang, W. Liu, J. Sun, Y. Hu, W. Wang, H. Chen, Y. Zhang, X. Li and M. Xu, ChemSusChem, 2016, 9, 1607–1613 CrossRef CAS PubMed.
- F. Cova, G. Amica, K. Kohopää and M. V. Blanco, Inorg. Chem., 2019, 58, 1040–1047 CrossRef CAS PubMed.
- H. Wang, J. Zhang, G. Wang, Q. Wang and T. Song, J. Therm. Anal. Calorim., 2018, 133, 981–989 CrossRef CAS.
- X. Chen, Z. Xiong, Y. Qin, B. Gong, C. Tian, Y. Zhao, J. Zhang and C. Zheng, Int. J. Hydrogen Energy, 2016, 41, 13077–13085 CrossRef CAS.
- H. A. Lara-García, O. Ovalle-Encinia, J. Ortiz-Landeros, E. Lima and H. Pfeiffer, J. Mater. Chem. A, 2019, 7, 4153–4164 RSC.
- K. Wang, J. Hong, Z. Zhou, Z. Lin and P. Zhao, Energy Technol., 2019, 7, 325–332 CrossRef CAS.
- X. Yang, W. Liu, J. Sun, Y. Hu, W. Wang, H. Chen, Y. Zhang, X. Li and M. Xu, ChemSusChem, 2016, 9, 2480–2487 CrossRef CAS PubMed.
- M. Seggiani, E. Stefanelli, M. Puccini and S. Vitolo, Chem. Eng. J., 2018, 339, 51–60 CrossRef CAS.
- N. Gómez-Garduño and H. Pfeiffer, Thermochim. Acta, 2019, 673, 129–137 CrossRef.
- D. Peltzer, J. Mùnera, L. Cornaglia and M. Strumendo, Chem. Eng. J., 2018, 336, 1–11 CrossRef CAS.
- D. Peltzer, J. Múnera and L. Cornaglia, J. Environ. Chem. Eng., 2019, 7, 102927 CrossRef CAS.
- N. Togashi, T. Okumura and K. Oh-Ishi, J. Ceram. Soc. Jpn., 2007, 115, 324–328 CrossRef CAS.
- M. V. Blanco, K. Kohopää, I. Snigireva and F. Cova, Chem. Eng. J., 2018, 354, 370–377 CrossRef CAS.
- E. Bernabé-Pablo, F. Plascencia-Hernández, A. Yañez-Aulestia and H. Pfeiffer, Chem. Eng. J., 2020, 384, 123291 CrossRef.
- M. A. Martínez-Cruz, A. Yañez-Aulestia, G. Ramos-Sánchez, M. Oliver-Tolentino, M. Vera, H. Pfeiffer, D. Ramírez-Rosales and I. González, Dalton Trans., 2020, 49, 4549–4558 RSC.
- A. Yañez-Aulestia, Q. Wang and H. Pfeiffer, Phys. Chem. Chem. Phys., 2020, 22, 2803–2813 RSC.
- M. W. Gaultois, M. T. Dunstan, A. W. Bateson, M. S. C. Chan and C. P. Grey, Chem. Mater., 2018, 30, 2535–2543 CrossRef CAS.
- T. Harada and T. A. Hatton, J. Mater. Chem. A, 2017, 5, 22224–22233 RSC.
- E. Vera, S. García, M. M. Maroto-Valer and H. Pfeiffer, Adsorption, 2020, 26, 781–792 CrossRef CAS.
- J. Liu, Z. Wang, Z. Wang, J. Song, G. Li, Q. Xu, J. You, H. Cheng and X. Lu, Phys. Chem. Chem. Phys., 2019, 21, 13135–13143 RSC.
- L. Martínez-dlCruz and H. Pfeiffer, J. Phys. Chem. C, 2012, 116, 9675–9680 CrossRef.
- G. Ji, M. Z. Memon, H. Zhuo and M. Zhao, Chem. Eng. J., 2017, 313, 646–654 CrossRef CAS.
- T. Zhao, E. Ochoa-Fernández, M. Rønning and D. Chen, Chem. Mater., 2007, 19, 3294–3301 CrossRef CAS.
- S. Munro, M. Åhlén, O. Cheung and A. Sanna, Chem. Eng. J., 2020, 388, 124284 CrossRef CAS.
- C. Zhao, X. Chen and C. Zhao, Int. J. Greenhouse Gas Control, 2010, 4, 655–658 CrossRef CAS.
- S. C. Lee, H. J. Chae, S. J. Lee, Y. H. Park, C. K. Ryu, C. K. Yi and J. C. Kim, J. Mol. Catal. B: Enzym., 2009, 56, 179–184 CrossRef CAS.
- S. C. Lee and J. C. Kim, Catal. Surv. Asia, 2007, 11, 171–185 CrossRef CAS.
- M. S. Cho, S. C. Lee, H. J. Chae, Y. M. Kwon, J. B. Lee and J. C. Kim, Process Saf. Environ. Prot., 2018, 117, 296–306 CrossRef CAS.
- S. Boonprasop, B. Chalermsinsuwan and P. Piumsomboon, J. Taiwan Inst. Chem. Eng., 2018, 88, 215–225 CrossRef CAS.
- G. Yang, H. Luo, T. Ohba and H. Kanoh, Int. J. Chem. Eng., 2016, 2016, 4012967 Search PubMed.
- F. Miccio, A. N. Murri and E. Landi, Ind. Eng. Chem. Res., 2016, 55, 6696–6707 CrossRef CAS.
- Q. Yang and Y. S. Lin, Ind. Eng. Chem. Res., 2006, 45, 6302–6310 CrossRef CAS.
- Z. Homonnay, K. Nomura, G. Juhász, M. Gál, K. Sólymos, S. Hamakawa, T. Hayakawa and A. Vértes, Chem. Mater., 2002, 14, 1127–1135 CrossRef CAS.
- G. Juhasz, Z. o. Homonnay, K. Nomura, T. Hayakawa, S. Hamakawa and A. Vértes, Solid State Ionics, 2001, 139, 219–231 CrossRef CAS.
- K. Nomura, Y. Ujihira, T. Hayakawa and K. Takehira, Appl. Catal., A, 1996, 137, 25–36 CrossRef CAS.
- H. Lu, J. P. Kim, S. H. Son and J. H. Park, Mater. Lett., 2011, 65, 2858–2860 CrossRef CAS.
- J. Yi, M. Schroeder, T. Weirich and J. Mayer, Chem. Mater., 2010, 22, 6246–6253 CrossRef CAS.
- C. Galven, J.-L. Fourquet, E. Suard, M.-P. Crosnier-Lopez and F. Le Berre, Dalton Trans., 2010, 39, 4191–4197 RSC.
- B. R. Sneha and V. Thangadurai, J. Solid State Chem., 2007, 180, 2661–2669 CrossRef CAS.
- F. Fujishiro, K. Fukasawa and T. Hashimoto, J. Am. Ceram. Soc., 2011, 94, 3675–3678 CrossRef CAS.
- J. Yi, T. E. Weirich and M. Schroeder, J. Membr. Sci., 2013, 437, 49–56 CrossRef CAS.
- M. T. Dunstan, W. Liu, A. F. Pavan, J. A. Kimpton, C. D. Ling, S. A. Scott, J. S. Dennis and C. P. Grey, Chem. Mater., 2013, 25, 4881–4891 CrossRef CAS.
- M. T. Dunstan, P. D. Southon, C. J. Kepert, J. Hester, J. A. Kimpton and C. D. Ling, J. Solid State Chem., 2011, 184, 2648–2654 CrossRef CAS.
- M. Li, K. Huang, J. A. Schott, Z. Wu and S. Dai, Microporous Mesoporous Mater., 2017, 249, 34–41 CrossRef CAS.
- Y. Zhou, K. Yan, Z. Yang, R. A. Bauer, N. Hong and H. Verweij, ACS Appl. Nano. Mater., 2020, 3, 6654–6663 CrossRef CAS.
- H. R. Amedi and M. Aghajani, J. Nat. Gas Sci. Eng., 2016, 35, 695–702 CrossRef CAS.
- N. Azizi, T. Mohammadi and R. M. Behbahani, J. Energy Chem., 2017, 26, 454–465 CrossRef.
- W. Zhu, F. Liu, M. Gou, R. Guo and X. Li, Green Chem. Eng., 2021, 2, 132–143 CrossRef.
- W. M. Aframehr, B. Molki, R. Bagheri, P. Heidarian and S. M. Davodi, Chem. Eng. Res. Des., 2020, 153, 789–805 CrossRef CAS.
- C. Cazorla, S. A. Shevlin and Z. X. Guo, J. Phys. Chem. C, 2011, 115, 10990–10995 CrossRef CAS.
- J. Yu, L.-H. Xie, J.-R. Li, Y. Ma, J. M. Seminario and P. B. Balbuena, Chem. Rev., 2017, 117, 9674–9754 CrossRef CAS PubMed.
- B. Vujic and A. P. Lyubartsev, Chem. Eng. Sci., 2017, 174, 174–188 CrossRef CAS.
- X. Huang, J. Lu, W. Wang, X. Wei and J. Ding, Appl. Surf. Sci., 2016, 371, 307–313 CrossRef CAS.
- G. Avci, I. Erucar and S. Keskin, ACS Appl. Mater. Interfaces, 2020, 12, 41567–41579 CrossRef CAS PubMed.
- G. Avci, S. Velioglu and S. Keskin, ACS Appl. Mater. Interfaces, 2018, 10, 33693–33706 CrossRef CAS PubMed.
- H. Fang, A. Kulkarni, P. Kamakoti, R. Awati, P. I. Ravikovitch and D. S. Sholl, Chem. Mater., 2016, 28, 3887–3896 CrossRef CAS.
- C. Shi, L. Li and Y. Li, J. CO2 Util., 2020, 42, 101346 CrossRef CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |