
Coupling isolated Ni single atoms with sub-10 nm Pd nanocrystals embedded in porous carbon frameworks to boost oxygen electrocatalysis for Zn–air batteries†
Shangzhi
Wang‡
,
Zinan
Lin‡
,
Mengmeng
Li
,
Zehan
Yu
,
Minjun
Zhang
,
Mingxing
Gong
,
Yawen
Tang
 * and
Xiaoyu
Qiu
*
* and
Xiaoyu
Qiu
*
Jiangsu Key Laboratory of New Power Batteries, Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing, 210023, China. E-mail: tangyawen@njnu.edu.cn; 07255@njnu.edu.cn
First published on 18th November 2021
Abstract
Developing effective bifunctional catalysts for the oxygen reduction and evolution reaction (ORR/OER) is essential for accelerating the cathode efficiency of Zn–air batteries. Herein, atomically dispersed Ni single atoms are supported by sub-10 nm Pd nanocrystals embedded in N-doped carbon frameworks (Ni SAs-Pd@NC), in an effort to achieve superior bifunctional activity in both the ORR and OER. The key synthetic point depends on the protection mechanism of 1-naphthylamine, which could provide a carbon source for Ni SAs and restrict the Pd size under sub-10 nm during 600 °C pyrolysis, simultaneously. The synergistic effect of sub-10 nm Pd with superior ORR activity and Ni–N4 SAs with favourable OER activity leads to bifunctional catalytic performance, meanwhile the rod-like carbon frameworks with ultrathin, porous and N-doped features contribute to accelerated electron transfer and structural robustness. As a proof-of-concept application, Ni SAs-Pd@NC demonstrates ultrahigh ORR activity with a positive half-wave potential of 0.84 V and a low OER overpotential of 380 mV at 10 mA cm−2, in an alkaline medium. For a rechargeable Zn–air battery, the Ni SAs-Pd@NC cathode delivers a low charge–discharge voltage gap of 0.87 V, a high energy density of 884.6 W h kgZn−1, a high power density of 134.2 mW cm−2 and remarkable long-term cyclability for operation over 700 cycles, outperforming commercial Pt/C + RuO2 benchmarks. This work successfully integrates single atom sites with small-sized noble metals to break out their incompatibility in synthesis and to improve their thermostability, which offers a versatile approach to develop single atom-based bifunctional catalysts for energy devices.
1. Introduction
Zn–air batteries have been identified as promising energy storage devices by virtue of their low cost, high security, and theoretically large energy density (1084 W h kg−1).1,2 Considering the sluggish kinetics of reversible oxygen reactions at the air–cathode electrode, efficient and stable electrocatalysts are essential to improve the performance of charge and discharge cycles.3–5 Currently, physical mixing of ORR active species and OER active species together has been regarded as a conventional approach to acquire bifunctional air–cathode catalysts, such as the state-of-the-art Pt/C + RuO2/IrO2.6 However, this strategy often leads to poor compatibility and possible inhomogeneous distribution of the two components, which inevitably cause the dissolution and agglomeration of active components during long-term operation.7,8 Accordingly, the exploitation of a bifunctional electrocatalyst to simultaneously boost the ORR and OER is crucial to improve the performance of rechargeable Zn–air batteries.9 Besides, great efforts have been devoted to searching for candidates to replace state-of-the-art electrocatalysts, by considering the opposite reaction pathways of ORR and OER catalysis. For the ORR, it is revealed that the ORR activity of Pd/C is close to that of Pt/C due to the similar crystal structure and electronic configuration of Pd and Pt, which provides higher volcano–plot activity and better stability than transition metal-based candidates.10 For the OER, besides Ru and Ir, the intrinsic activity follows the trend of Ni > Co > Mo > W > Fe, making Ni-based nanomaterials the most promising candidates for the OER in alkaline media.11,12Downsizing Ni-based nanomaterials to the single-atom level has been regarded as an effective way to obtain superior selectivity and excellent activity towards the OER, as well as unique size quantum effects and higher atom utilization efficiency.13,14 The rational coupling of Ni single atoms (SAs) with different substrates has been regarded as an effective way to develop polyfunctionality, prevent atom aggregation and enhance the stability of Ni SAs.15,16 Until now, different substrates for Ni SAs have been studied, such as graphene, metal oxides (e.g., TiO2, CeO2 and MgO), metallic sulphides (e.g., MoS2), and metal–organic frameworks.17–19 However, the coexistence of Ni SAs with small-sized noble metal nanoparticles (NPs) has been rarely reported due to the widely divergent on synthetic methods and thermostability of Ni SAs and NPs. Normally, the synthesis of Ni SAs requires a high-temperature (300–900 °C) pyrolysis of Ni, N, and C.20,21 However, small-sized noble metals can easily undergo agglomeration, reconstruction and merging under a high temperature environment (>300 °C), generating stable sintering products with a larger particle size.22 Therefore, it remains difficult to realize the simultaneous combination of Ni SAs and small-sized noble metals in one synthetic system.
Herein, we report a novel incorporation of atomically dispersed Ni SAs with sub-10 nm Pd nanocrystals embedded in N-doped porous carbon frameworks (Ni SAs-Pd@NC). The 1-naphthylamine protected pyrolysis mechanism is responsible for the formation, which involves the steps of (i) coordination to form PdII–(C10H7–NH2); (ii) π–π stacking with NiPc; (iii) in situ pyrolysis of the NiPc@PdII–(C10H7–NH2) complex. Electrochemical tests and characterization verify that the in situ embedded sub-10 nm Pd nanocrystals are favourable for the ORR and the Ni SAs are highly active for the ORR. Meanwhile, the ultrathin N-doped C layers enable accelerated electron transfer, which could facilitate both the ORR and OER processes. Due to these attributes, Ni SAs-Pd@NC demonstrates encouraging activity and stability for the ORR and OER in an alkaline medium, showing a half-wave potential of 0.84 V for the ORR and an overpotential of 380 mV at 10 mA cm−2 for the OER. Moreover, the Ni SAs-Pd@NC cathode displays superior performance in a rechargeable Zn–air battery prototype with a high specific output power, a low charge–discharge voltage gap of 0.87 V and an outstanding long-term cyclability for over 700 cycles.
2. Experimental
2.1 Materials and chemicals
1-Naphthylamine (C10H9N) was purchased from Shanghai Macklin Biological Technology CO., Ltd. (Shanghai, China). Nickel(II) phthalocyanine (NiPc) was supplied by Alfa Aesar chemical Co., Ltd. (Shanghai, China). Palladium chloride (PdCl2) was derived from Shanghai Dibo Chemical Technology Co., Ltd. (Shanghai, China). Tetrahydrofuran (THF) and HNO3 were bought from the Sinopharm chemical reagent Co., Ltd. (Shanghai, China). The commercial Pd/C, Pt/C and RuO2 were obtained from Johnson Matthey Chemicals Ltd. (Shanghai, China). All reagents were of analytical reagent grade and directly used without further purification.2.2 Synthesis of Ni SAs-Pd@NC
Typically, 0.5 mL of PdCl2 solution (50 mM) and 360 mg of 1-naphthylamine were added into a mixture solvent of 5.0 mL of DI water and 5.0 mL of ethanol, and then kept under continuous sonication for 5 min. The obtained complex was separated by centrifugation at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 6 min and washed with ethanol three times to remove the excess 1-naphthylamine. Then the products were dried at 40 °C for 10 h to obtain the PdII–(C10H7–NH2) nanosheets. Afterwards, 100 mg of PdII–(C10H7–NH2) nanosheet powder was added into 30 mL of DI water. Meanwhile, 50 mg of NiPc was dispersed into 30 mL of THF. Then the above two suspensions were mixed together and stirred overnight to obtain NiPc@PdII–(C10H7–NH2). The resulting complex was separated by centrifugation at 15000 rpm for 6 min, vacuum dried, and ground into fine powder. Finally, the powder was put into a tube furnace and calcined at 650 °C for 3 h under a flow of N2 (ramp rate: 3 °C min−1) to acquire Ni SAs-Pd@NC, with a yield of ca. 8.16 mg. In order to evaluate the influence of Pd and Ni contents on the catalytic performance of the catalyst, the feeding ratio of PdII–(C10H7–NH2) and NiPc was changed to 1:1 (100 mg of PdII–(C10H7–NH2) vs. 100 mg of NiPc) and 4:1 (100 mg of PdII–(C10H7–NH2) vs. 25 mg of NiPc) to make a comparison, keeping the other steps unchanged.
000 rpm for 6 min and washed with ethanol three times to remove the excess 1-naphthylamine. Then the products were dried at 40 °C for 10 h to obtain the PdII–(C10H7–NH2) nanosheets. Afterwards, 100 mg of PdII–(C10H7–NH2) nanosheet powder was added into 30 mL of DI water. Meanwhile, 50 mg of NiPc was dispersed into 30 mL of THF. Then the above two suspensions were mixed together and stirred overnight to obtain NiPc@PdII–(C10H7–NH2). The resulting complex was separated by centrifugation at 15000 rpm for 6 min, vacuum dried, and ground into fine powder. Finally, the powder was put into a tube furnace and calcined at 650 °C for 3 h under a flow of N2 (ramp rate: 3 °C min−1) to acquire Ni SAs-Pd@NC, with a yield of ca. 8.16 mg. In order to evaluate the influence of Pd and Ni contents on the catalytic performance of the catalyst, the feeding ratio of PdII–(C10H7–NH2) and NiPc was changed to 1:1 (100 mg of PdII–(C10H7–NH2) vs. 100 mg of NiPc) and 4:1 (100 mg of PdII–(C10H7–NH2) vs. 25 mg of NiPc) to make a comparison, keeping the other steps unchanged.
2.3 Synthesis of sub-10 nm Pd@NC
The synthesis of sub-10 nm Pd@NC was similar to that of Ni SAs-Pd@NC, except for cancelling the addition of NiPc. Typically, 0.5 mL of PdCl2 solution (50 mM) and 360 mg of 1-naphthylamine were added into a mixture solvent of 5.0 mL of DI water and 5.0 mL of ethanol, and then kept under continuous sonication for 5 min. The obtained complex was separated by centrifugation and washed with ethanol to remove excess 1-naphthylamine. Then the resultant products were dried under 40 °C for 10 h, followed by heat treatment in a tube furnace at 650 °C under a flow of N2 for 3 h (ramp rate: 3 °C min−1) to acquire sub-10 nm Pd@NC.2.4 Synthesis of Ni SAs@NC
20 mg of fresh-made Ni SAs-Pd@NC was soaked into 40 mL of concentrated nitric acid (16 M) for 12 h to remove the sub-10 nm Pd nanocrystals. The products were separated and washed by centrifugation at 15000 rpm for 6 min to obtain Ni SAs@NC.
2.5 Characterization
Scanning electron microscopy (SEM) images were acquired using a Hitachi S5500 SEM/STEM. High-resolution TEM (HRTEM) and energy dispersive X-ray (EDX) elemental analyses were carried out on a JEOL JEM-2100F transmission electron microscope operating at an accelerating voltage of 200 kV. The crystallinity of the samples was determined by recording X-ray diffraction (XRD) patterns on a Model D/max-rC X-ray diffractometer using a Cu Kα radiation source (λ = 1.5406 Å) and operating at 40 kV and 100 mA. X-ray photoelectron spectroscopy (XPS) measurements were performed with a Thermo VG Scientific ESCALAB 250 spectrometer equipped with a monochromatic Al Kα X-ray source. The binding energy was calibrated by means of the C 1s peak energy of 284.6 eV. The Raman spectrum was recorded on a Raman spectrometer (LabRAM HR800, λ = 514.4 nm). Synchrotron-based X-ray absorption spectroscopy (XAS) was performed at the beamline 1W1B station of the National Synchrotron Radiation Research Centre (NSRRC, Taiwan, China). The metal contents were determined by Inductively Coupled Plasma Mass Spectrometry (ICP-MS) measurements.2.6 Electrochemical measurements
All electrochemical measurements were performed on a CHI 760D electrochemical work station (Shanghai Chenhua Co). A standard three-electrode system was used, including a rotating ring-disk electrode (RRDE) covered with the catalysts as the working electrode (0.196 cm2), a saturated calomel electrode (SCE) protected by the Luggin capillary, and a platinum wire serving as the auxiliary electrode. The preparation of the working electrode involves the following steps: 600 μL of DI water, 300 μL of ethanol, 5 mg of catalyst, and 100 μL of Nafion solution (5 wt%) were mixed together and ultrasonicated to form the catalyst ink. Subsequently, 10 μL of the above ink was pipetted, spread on the electrode, and dried at 40 °C to prepare the working electrode. For the ORR measurement, the RRDE was used to acquire linear sweep voltammograms (LSVs), rotating in 0.1 M KOH electrolyte at a predetermined speed of 1600 rpm and a scan rate of 5 mV s−1. The electron transfer number (n) and the yield of H2O2 were calculated using the following formula: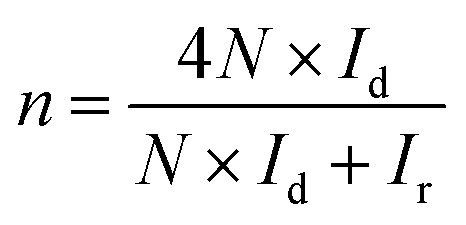 | (1) |
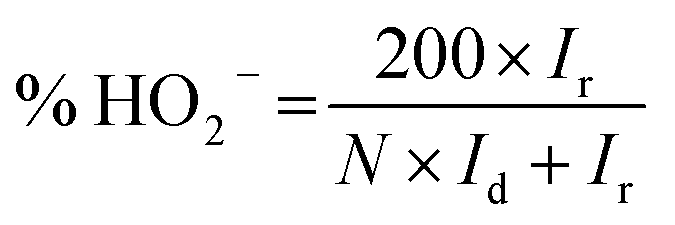 | (2) |
2.7 Zn–air battery tests
The Zn–air battery performance was tested using a homemade Zn–air cell. The air cathode includes the hydrophobic carbon paper with a catalyst layer on the electrolyte-facing side and a gas-diffusion layer on the air-facing side. The catalyst ink was dropped and painted onto the carbon paper (1.0 cm2) to fabricate the catalyst layer with a loading of 10 mg cm−2. The gas diffusion layer enables the O2 to effectively permeate from the ambient air to the catalyst sites. A well-polished Zn plate with a thickness of 0.3 mm was used as the anode. The 0.2 M ZnCl2 + 6 M KOH mixed solution was used as the electrolyte. The generated hydroxyl ions migrated in the electrolyte from the air cathode to the metallic anode and completed the battery reaction. The reaction pathways were reversed upon recharge with the reduction of zincate ions back to zinc and the release of oxygen, which could be summarized as follows:Discharge:
Anode:
| Zn + 4OH− → [Zn(OH)4]2− | (3) |
| [Zn(OH)4]2− → ZnO + H2O + 2OH− | (4) |
Cathode:
| O2 + 2H2O + 4e− → 4OH− | (5) |
Overall reaction:
| 2Zn + O2 → 2ZnO | (6) |
Parasitic reaction:
| Zn + 2H2O → Zn(OH)2 + H2 | (7) |
Recharge:
Anode:
| ZnO + H2O + 2OH− → [Zn(OH)4]2− | (8) |
| [Zn(OH)4]2− + 2e− → Zn + 4OH− | (9) |
Cathode:
| 4OH− → O2 + 2H2O + 4e− | (10) |
Overall reaction:
| 2ZnO → 2Zn + O2 | (11) |
The cycling stability test was carried out using a Land CT2001A system with 5 min rest time between each discharge and charge at a current density of 10 mA cm−2. Each discharge and charge period was set to be 10 min.
3. Results and discussion
The formation pathway of Ni SAs-Pd@NC is displayed in Fig. 1. Firstly, the PdII was coordinated with 1-naphthylamine (a p–π compound with –NH2 groups) to form the planar PdII–(C10H7–NH2) complex with a flake-like shape, as shown in the TEM images in Fig. S1a and b.† Then the newly formed PdII–(C10H7–NH2) complex was coupled with rigid NiPc macrocyclic molecules through the π–π stacking interaction, forming the NiPc@PdII–(C10H7–NH2) complex. The interaction between the PdII–(C10H7–NH2) complex and NiPc is confirmed by the UV-vis spectrum. As shown in Fig. S2,† after the addition of NiPc, the characteristic peak of PdII–(C10H7–NH2) at 310 nm shows a negative shift to 302 nm, along with the increased intensity, indicating their π–π stacking interaction. Meanwhile, the flake-like shape of PdII–(C10H7–NH2) was fractured and assembled into a rod-like structure after continuous stirring, as shown in the TEM images in Fig. S1c and d.† At last, pyrolysis at 600 °C was used to transform the NiPc@PdII–(C10H7–NH2) complex into Ni SAs-Pd@NC. Three component transformations occurred simultaneously: (i) the NiII anchored by macrocyclic molecules was carbothermally reduced into atomically dispersed Ni sites; (ii) the PdII sites were reduced to Pd nanocrystals with a small size of sub-10 nm because the C10H7–NH2 surrounded the PdII sites to prevent their aggregation through the space confinement effect; (iii) C10H7–NH2 was transformed into N-doped carbon frameworks to encapsulate the sub-10 nm Pd and support the Ni SAs. Meanwhile, the C10H7–NH2 could release gaseous byproducts (e.g., CO2 and H2O) at 600 °C, resulting in a rich porous structure with volume shrinkage.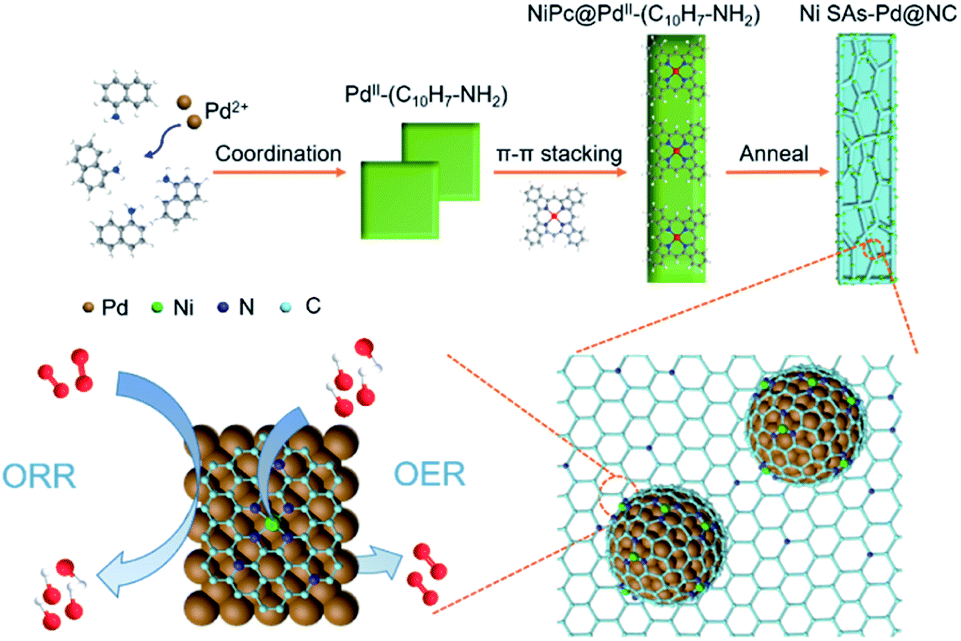 | ||
| Fig. 1 Schematic illustration showing the formation pathway of Ni SAs-Pd@NC. | ||
Fig. 2 displays the structural features of Ni SAs-Pd@NC. The large-scale SEM image shows that the product displays a rod-like structure with a diameter of 2.8 μm (Fig. 2a). The magnified SEM image validates the homogeneous distribution of numerous Pd nanoparticles embedded in the virgulate carbon skeleton (Fig. 2b). The HAADF-STEM image shows the three-dimensional spatial configuration of the carbon framework, with randomly dispersed sub-10 nm Pd nanocrystals (Fig. 2c). However, it is hard to distinguish the Ni SAs of Ni SAs-Pd@NC due to the Z-contrast between the heavier Pd atom and lighter Ni atom. The HRTEM image of Ni SAs-Pd@NC demonstrates porous features with interconnected carbon frameworks (Fig. 2d), which could promote the penetration of electrolyte and enhance the surface accessibility of active sites.25 Measured from a random area, the average diameter of Pd nanoparticles was determined to be 7.2 nm, with a narrow size distribution (Fig. S3†). Fig. 2e confirms the existence of ultrathin carbon layers with a thickness of 1.25 nm around the sub-10 nm Pd nanocrystals. They could serve as a protective cover to prevent the segregation, dissolution, and Ostwald ripening of sub-10 nm Pd nanocrystals during the electrocatalytic process.26Fig. 2f shows the monocrystalline fringes of an individual sub-10 nm Pd nanocrystal, and the interplanar spacing was measured to be 0.224 nm, corresponding to the (111) facets of fcc-Pd. The elemental composition of Ni SAs-Pd@NC was investigated by line scanning and EDX mapping images shown in Fig. 2g. It verifies the homogeneous distribution of Ni, Pd, N, and C elements across the entire surface of the porous carbon nanorod, verifying the successful coupling of Ni sites with sub-10 nm Pd nanocrystals.
 | ||
| Fig. 2 Characteristics of Ni SAs-Pd@NC. (a and b) SEM images. (c) HADDF-STEM image. (d–f) HRTEM images recorded at different resolutions. (g) EDX line scan and corresponding mapping images. | ||
The XRD pattern was obtained to investigate the crystal structure of Ni SAs-Pd@NC (Fig. 3a). It displays five diffraction peaks located at 40.14°, 46.69°, 68.17°, 82.17° and 86.69°, respectively, corresponding to the (111), (200), (220), (311) and (222) planes of fcc-Pd (JCPDS No. 65-6174). Importantly, no obvious diffraction peak ascribed to Ni-based species can be observed, confirming the atomically dispersed state of Ni sites in Ni SAs-Pd@NC. In order to show the compositional evolution of Ni SAs-Pd@NC under different atomic ratios of Pd and Ni, products with different feeding ratios of PdII–(C10H7–NH2):NiPc = 4:1 and 1:1 were prepared by keeping the dosage of PdII–(C10H7–NH2) unchanged. From TEM images (Fig. S4†) and XRD patterns (Fig. S5†), it is found that the product with a feed ratio of 4:1 shows a similar rod-like structure to Ni SAs-Pd@NC and the corresponding XRD characteristic peaks are located at the same position, indicating that the decrease of Ni content could not cause the morphology and composition change. However, TEM images of the products with a feed ratio of 1:1 display rod-like carbon frameworks with an inconspicuous sinnet of sub-10 nm Pd nanocrystals. The corresponding XRD pattern shows the characteristic peaks corresponding to NiCx species (JCPDS No. 45-0979). This indicates that the excess NiPc formed the Ni-based species through pyrolysis, thus decreasing the purity of atomically distributed Ni sites. The Raman spectrum of Ni SAs-Pd@NC shows the characteristic peaks of the D band carbon located at 1356.2 cm−1 and the G band located at 1578.6 cm−1, respectively (Fig. 3b). The value of ID/IG was determined to be 1.16, indicative of the high graphitization degree of Ni SAs-Pd@NC. Fig. 3c shows the EDX spectrum of Ni SAs-Pd@NC, displaying a Ni content of 1.69% and Pd content of 20.41%, respectively. This result is basically consistent with the elemental contents from ICP-AES, which shows a value of 1.86 at% for Ni and 20.12 at% for Pd, respectively. The pore size distribution of Ni SAs-Pd@NC was analyzed by N2 adsorption–desorption isotherms as shown in Fig. S6†. It displays a characteristic type II isotherm with a pronounced hysteresis loop, suggesting the presence of meso-pores. The pore-size distribution curve (inset) shows a pore-size distribution of around 8.2 nm, indicating the existence of micropores.
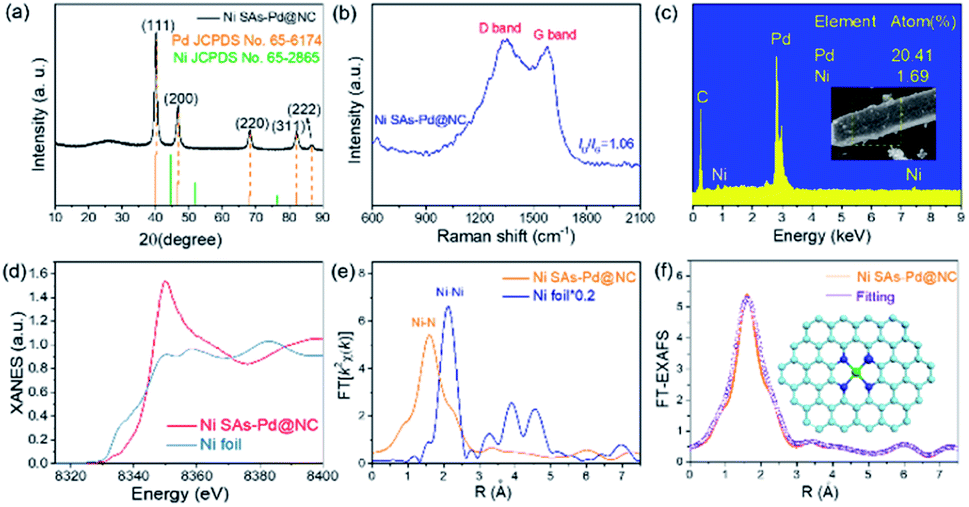 | ||
| Fig. 3 Structural characteristics of Ni SAs-Pd@NC. (a) XRD pattern. (b) Raman spectrum. (c) EDX analysis. (d) XANES spectra of the Ni K-edge. (e) FT-EXAFS of the Ni K-edge. (f) EXAFS fitting curves at the R space (inset: atomic structure model of Ni sites). | ||
The XANES and the EXAFS spectra of Ni SAs-Pd@NC were recorded to confirm the coordination bonding configurations and SA state of Ni sites. The Ni K-edge XANES spectra show a stronger main peak at 8349.8 eV and a more positive absorption at the pre-edge position as shown in Fig. 3d. It reveals the distinct electronic configuration of Ni in Ni SAs-Pd@NC compared to that of the Ni foil, indicating the presence of the +2 valence state for the formation of Ni–Nx coordination. The Ni K-edge EXAFS spectra of Ni SAs-Pd@NC (Fig. 3e) display the sole peak located at 1.60 Å, while the Ni foil shows Ni–Ni metallic bonds at 2.12 Å. The formation of Ni–N bonds with high purity is identified, implying the atomically dispersed Ni–Nx SAs in Ni SAs-Pd@NC. Moreover, the Ni K-edge EXAFS spectra of Ni SAs-Pd@NC could be well distinguished from that of the dual-atom and small-cluster, further confirming the existence of Ni–N bonds in Ni SAs-Pd@NC.27,28 The least-square EXAFS fitting was performed to depict quantitative parameters of Ni in Ni SAs-Pd@NC (Fig. 3f, S7 and Table S1†). The coordination number of Ni was calculated to be 4.1 and the bond length was determined to be 2.07 Å, indicating that all the atomic Ni sites are four-coordinated by nitrogen species, as illustrated in the atomic models in Fig. 3f. The electronic structure in Ni SAs-Pd@NC was investigated by XPS analysis (Fig. S8†). The full survey indicates the coexistence of Pd, Ni, N, O and C elements in Ni SAs-Pd@NC (Fig. S8a†). The Pd 3d spectrum of Ni SAs-Pd@NC displays two characteristic peaks located at 335.3 and 340.6 eV, respectively, corresponding to the metallic state of dominating Pd0 (Fig. S8b†). Specifically, the binding energy of Pd 3d shows an obvious positive-shift of 0.4 eV compared to that of the standard Pd (3d5/2 = 334.9 eV, 3d3/2 = 340.1 eV), which could be derived from the incorporation of Ni SAs and carbon layers that change the electronic structure of Pd. Such regulation of the electron density of Pd could downshift the d-band centre, which is beneficial for changing the binding strength of intermediates during the electro-catalytic process.29,30 The Ni 2p spectra of Ni SAs-Pd@NC show two main peaks at 855.6 and 872.9 eV, accompanied by satellites at 861.6 and 880.3 eV, respectively (Fig. S8c†). They can be assigned to the typical Ni–Nx species, which is consistent with the EXAFS analysis. The N 1s spectrum of Ni SAs-Pd@NC shows three deconvoluted peaks at 400.3, 399.4 and 398.7 eV, which could be assigned to graphitic N, pyrrolic N, and pyridinic N, respectively (Fig. S8d†). The C 1s spectrum of Ni SAs-Pd@NC shows three deconvoluted peaks at 288.2, 285.4 and 284.5 eV, corresponding to the sp2 hybrid carbon (30.4%), nitrogen-bonded carbon (47.9%), and carbon-oxygen species (21.7%), respectively. The high percentage of nitrogen-bonded carbon confirms the incorporation of N atoms into the carbon framework, which is favourable for high electrical conductivity to facilitate both the ORR and OER kinetics.31
In order to identify the origin of ORR and OER activity in Ni SAs-Pd@NC, the pure Pd sample (denoted as Pd@NC) and pure Ni SAs sample (denoted as Ni SAs@NC) were synthesized to make a comparison. The synthetic procedure of Pd@NC was similar to that of Ni SAs-Pd@NC, except for without the addition of NiPc. As shown in Fig. S9,† Pd@NC displays a porous lamellar structure with uniformly distributed sub-10 nm Pd nanocrystals, along with the XRD characteristic peaks related to fcc-Pd. The synthesis of Ni SAs@NC was based on the acid etching of as-obtained Ni SAs-Pd@NC to remove the sub-10 nm Pd nanocrystals, leaving the Ni SAs and carbon frameworks. As shown in Fig. S10,† no obvious character of sub-10 nm Pd nanocrystals could be observed in the TEM image of Ni SAs@NC. Meanwhile, only the C (004) peak could be observed in the XRD pattern, confirming the removal of sub-10 nm Pd nanocrystals. The preservation of Ni single atoms after acid etching was confirmed by XPS analysis. As shown in Fig. S11,† the full survey indicates the coexistence of Ni, N, O, and C elements in Ni SAs-Pd@NC. Meanwhile, no obvious signal referring to Pd could be observed, indicating the good removal of Pd nanocrystals. The Ni 2p spectra of Ni SAs@NC show two main peaks at 855.7 and 873.4 eV, accompanied by satellites at 861.2 and 879.7 eV, respectively. They can be assigned to the typical Ni–Nx species, which is similar to the result of Ni-SAs-Pd@NC. The above results verify the preservation of Ni SAs after acid etching.
In order to shed light on the influence of Ni content on the ORR activity, the electro-catalytic performances of Ni SAs-Pd@NC (4:1), Ni SAs-Pd@NC (2:1), and NiCx-Pd@NC (1:1) with the same dosage of PdII–(C10H7–NH2) but incremental dosage of NiPc were taken for comparison. Similar to the cyclic voltammograms (CV) in N2 and O2-saturated 0.1 M KOH solution, well-defined cathodic peaks are clearly observed for all samples in O2-saturated solution, while featureless CV curves are observed in N2-saturated electrolyte (Fig. 4a). Specifically, the reduction peak position of Ni SAs-Pd@NC (2:1) is similar to that of Ni SAs-Pd@NC (4:1) and more positive than that of NiCx-Pd@NC (1:1), indicative of the preferential reduction capacity of Ni SAs-Pd@NC at 2:1 and 4:1. Fig. 4b shows the LSV polarization curve of Ni SAs-Pd@NC for the ORR, which is comparable with those of the samples with different feed ratios, the pure samples (Pd@NC and Ni SAs@NC), and commercial Pd/C under the same conditions. Obviously, Ni SAs-Pd@NC (2:1) demonstrates the highest onset potential (Eonset = 0.94 V) and half-wave potentials (E1/2 = 0.84 V). This point is further confirmed by the histogram in Fig. 4c, which shows that both the Eonset and E1/2 of Ni SAs-Pd@NC (2:1) are more positive than those of Ni SAs-Pd@NC (4:1), NiCx-Pd@NC (1:1), Pd@NC, and Pd/C, respectively. A similar trend is observed in the corresponding Tafel slope, which is a critical factor to evaluate the reaction kinetics. As shown in Fig. 4d, the Tafel slope of Ni SAs-Pd@NC (2:1) was calculated to be 55 mV dec−1, which is lower than that of Ni SAs-Pd@NC (+4:1) (71 mV dec−1), NiCx-Pd@NC (1:1) (88 mV dec−1), Pd@NC (73 mV dec−1), and Pd/C (64 mV dec−1), respectively, indicating a fast kinetics conducive to the positively shifted ORR process. Meanwhile, the average number of electrons transferred (n) and hydroxyl coverage ratio on Ni SAs-Pd@NC (2:1) were calculated to be ca. 3.90 and 3.4%, respectively, while the compressive samples exhibit n values of 3.82, 3.27, 3.70 and 3.69, together with a hydroxyl coverage ratio of 5.5%, 28.2%, 10.5%, and 10.8%, respectively (Fig. 4e). This suggests that the oxygen was reduced through a straightforward four-electron pathway on Ni SAs-Pd@NC in an alkaline medium. Overall, focusing on the Pd:Ni ratio, the ORR performance follows the order of Ni SAs-Pd@NC (2:1) ≈ Ni SAs-Pd@NC (4:1) > NiCx-Pd@NC (1:1). Considering that Ni SAs-Pd@NC with the feed ratios of 2:1 and 4:1 shows better ORR performance than NiCx-Pd@NC (1:1), we can conclude that the exorbitant Ni content in Ni SAs-Pd@NC is adverse to the ORR activity. Meanwhile, the ORR performance of Ni SAs-Pd@NC is slightly superior to that of Pd@NC, illustrating that the incorporation of Ni SAs with Pd@NC could enable a synergistic effect to enhance the ORR activity. However, the pure Ni sample (Ni SAs@NC) shows feeble activity towards the ORR, which is significantly inferior to that of Ni SAs-Pd@NC. Such poor ORR activity of the pure Ni sample reveals that the main ORR active sites in Ni SAs-Pd@NC could be the sub-5 nm Pd nanocrystals. The ORR stability of Ni SAs-Pd@NC (2:1) was investigated using the chronoamperometric curve (Fig. 4f). After operating for 20000s, the current density of Ni SAs-Pd@NC (2:1) remains at 85.6% of the initial value, while the commercial Pd/C shows more pronounced attenuation to 58.9%. Meanwhile, the accelerated durability tests (ADTs) further confirm the remarkable ORR durability and structural robustness of Ni SAs-Pd@NC (2:1) (Fig. S12†). After 1000 cycles, Ni SAs-Pd@NC (2:1) exhibits a negligible decrease in current density, together with an inappreciable morphology change (Fig. S13†).
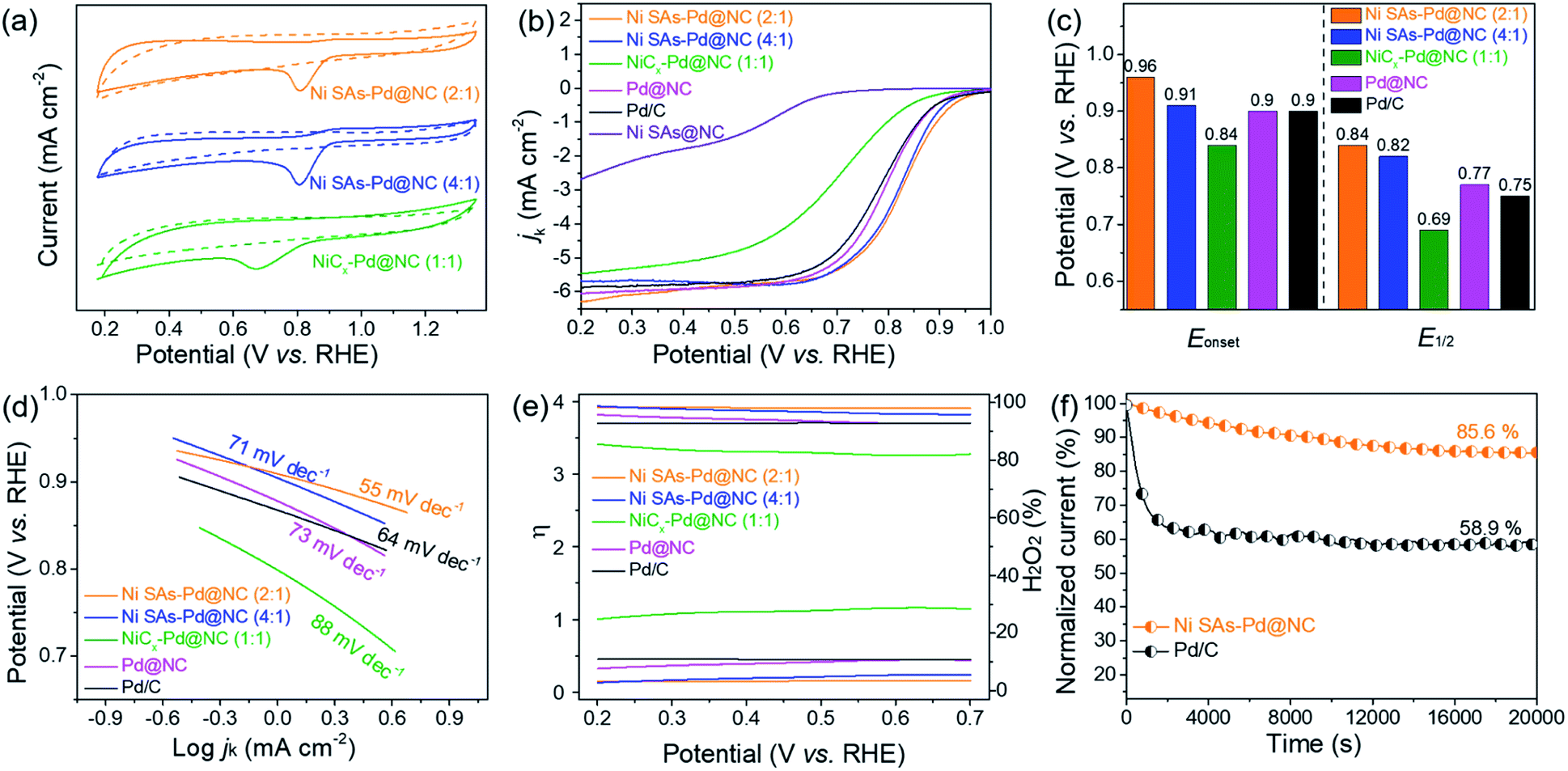 | ||
| Fig. 4 (a) CV curves recorded in N2 or O2 saturated 0.1 M KOH solution. (b) ORR polarization curves recorded in O2-saturated 0.1 M KOH. (c) Histogram of Eonset and E1/2. (d) Corresponding Tafel plots. (e) Curves of the average electron transfer number and percentage of peroxide. (f) Chronoamperometric curve recorded at 0.7 V. | ||
The OER performance of Ni SAs-Pd@NC (2:1) was measured in O2-saturated 0.1 M KOH solution, and compared with those of Ni SAs-Pd@NC (4:1), NiCx-Pd@NC (1:1), Ni SAs@NC, Pd@NC and commercial RuO2 (Fig. 5a). The polarization curve of Ni SAs-Pd@NC (2:1) shows a small overpotential of 380 mV at the current density of 10 mA cm−2, close to that of RuO2 and Ni SAs@NC, and much lower than that of Ni SAs-Pd@NC (4:1), NiCx-Pd@NC (1:1), and Pd@NC (as shown in the histogram in Fig. 5b). Moreover, the Tafel slope of Ni SAs-Pd@NC shows a value of 79 mV dec−1 (Fig. 5c), which is much lower than that of Ni SAs-Pd@NC (4:1) (83 mV dec−1), NiCx-Pd@NC (1:1) (89 mV dec−1), Ni SAs@NC (87 mV dec−1), and commercial RuO2 (97 mV dec−1). The lowest Tafel slope of Ni SAs-Pd@NC (2:1) indicates that the first discharge step controls the reaction kinetics of the OER and a larger output of current density could be acquired on Ni SAs-Pd@NC (2:1) in a wide range of operating voltages. Overall, the OER performance of Ni SAs-Pd@NC (2:1) is similar to that of Ni SAs@NC but much better than that of Pd@NC. This trend indicates that the atomically dispersed Ni–N4 species are the main active sites for the OER because the removal of sub-10 nm Pd nanocrystals from Ni SAs-Pd@NC could not cause any obvious degeneration of the OER performance. Meanwhile, focusing on the Ni content, the OER performance follows the order of Ni SAs-Pd@NC (2:1) > NiCx-Pd@NC (1:1) > Ni SAs-Pd@NC (4:1). This indicates that a higher content of Ni SAs refers to better OER performance. However, the superabundant addition of NiPc induces the formation of NiCx species, thus leading to a decreased activity for the OER. The long-term OER stability of Ni SAs-Pd@NC (2:1) was revealed by chronoamperometry and accelerated durability tests. As shown in Fig. 5d, the current density of Ni SAs-Pd@NC (2:1) shows a weak decay with retaining 72.3% of the initial current density at a constant potential for 20000 s, while only 44.5% of the initial current density is retained on commercial RuO2. Meanwhile, the LSV curve of Ni SAs-Pd@NC (2:1) shows a slight decay of 11 mV after an ADT for 1000 cycles, further confirming its excellent electrocatalytic stability for the OER (Fig. S14†). The E1/2 (for ORR) and the Ej10 (for OER) of Ni SAs-Pd@NC (2:1) are compared to those of the representative bifunctional catalysts reported to date (Fig. 5e). It is shown that both the E1/2 and Ej10 of Ni SAs-Pd@NC (2:1) are highly active to surpass most of the reference samples, showing its potential to serve as a bifunctional cathode catalyst for the Zn–air battery. The potential gap (ΔE = Ej10 − E1/2) was calculated to investigate the overall bifunctional oxygen electrocatalysis property of Ni SAs-Pd@NC (2:1) (Fig. 5f). The ΔE of Ni SAs-Pd@NC (2:1) shows a value of 0.77 V, which is much lower than that of Ni SAs-Pd@NC (4:1) (0.88 V) and NiCx-Pd@NC (1:1) (0.99 V), and also outperforms the ΔE values of previously reported state-of-the-art bifunctional electrocatalysts (Table S2†). Such a small ΔE value of Ni SAs-Pd@NC (2:1) indicates its superiority in both the bifunctionality and electrode reversibility, enabling it to exhibit promising discharging and charging capability for Zn–air batteries.32
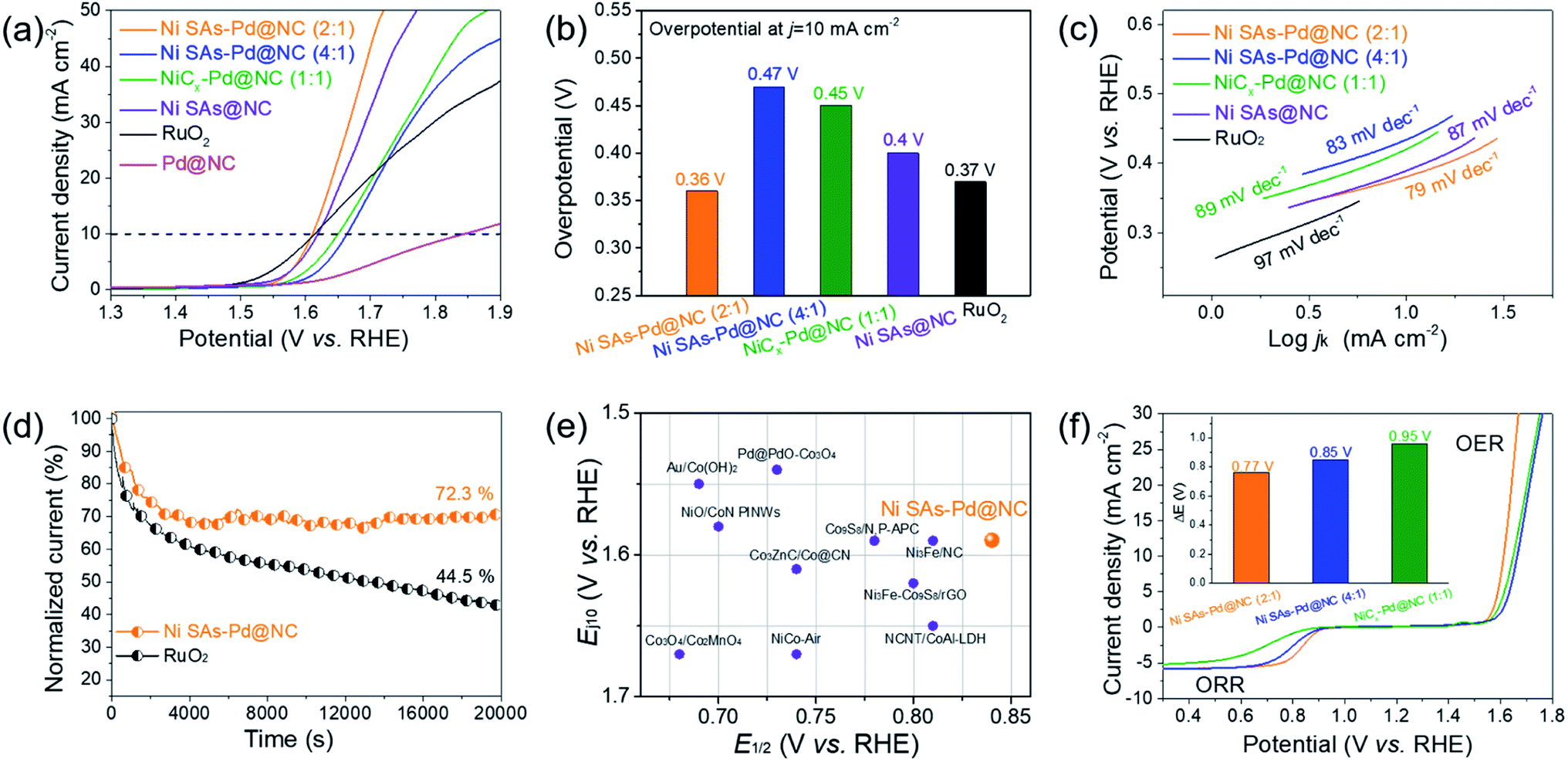 | ||
| Fig. 5 (a) OER polarization curves recorded in O2-saturated 0.1 M KOH. (b) Bar plots of overpotentials obtained at 10 mA cm−2. (c) Corresponding Tafel plots. (d) Chronoamperometric curve recorded at 1.6 V. (e) Plot of comparison of catalysts from references. (f) Overall polarization curves. | ||
The above results verify Ni SAs-Pd@NC with the optimal feed ratio of 2:1 as a promising bifunctional ORR and OER catalyst, which is qualified to serve as an air–cathode material in rechargeable Zn–air batteries (ZABs). Accordingly, a rechargeable Zn–air battery was assembled by using Ni SAs-Pd@NC as the air cathode material, a Zn plate as the anode, and a mixed solution of 0.2 M ZnCl2 + 6 M KOH as the electrolyte. As shown in Fig. 6a and b, an LED bulb powered by three Ni SAs-Pd@NC-based Zn–air batteries could work continuously for more than 5 h, suggesting the standardized specific capacity and energy density of the Ni SAs-Pd@NC cathode. The open circuit voltage (OCV) curve of Zn–air batteries assembled with Ni SAs-Pd@NC shows a value of 1.44 V, which is close to the theoretical threshold of 1.65 V for the Zn–air battery (Fig. S15†). Moreover, the Ni SAs-Pd@NC ZAB exhibits a maximum power density of 134.2 mW cm−2, which is much better than that of the Pt/C + RuO2-constructed ZAB (109.7 mW cm−2, Fig. S16†). Fig. 6c displays the galvanostatic discharge curves of Ni SAs-Pd@NC and Pt/C + RuO2 at a constant current density of 5 mA cm−2. Through normalizing with the consumed Zn mass, the specific capacity of the Ni SAs-Pd@NC cathode was determined to be 719.2 mA h gZn−1, surpassing that of the Pt/C + RuO2 cathode (622.9 mA h gZn−1). Meanwhile, the Ni SAs-Pd@NC cathode delivers a higher energy density of ca. 884.6 W h kgZn−1, which is much larger than that of the Pt/C + RuO2 cathode (741.3 W h kgZn−1). The cycling stability of the Ni SAs-Pd@NC cathode was evaluated by galvanostatic charge and discharge tests (Fig. 6d). The Ni SAs-Pd@NC cathode possesses excellent stability with no obvious decay in charge (≈1.96 V) and discharge (≈1.09 V) voltages over 700 cycles (230 h), while the Pt/C + RuO2 cathode failed in less than 60 cycles. This indicates the excellent long-range stability of the Ni SAs-Pd@NC cathode for continuous recharge in Zn–air batteries. After running for 700 cycles, the round-trip overpotential of the Ni SAs-Pd@NC cathode is about 0.88 V, with a high voltaic efficiency of 55.6% (Fig. S17†). In contrast, the Pt/C + RuO2 cathode shows a low round-trip efficiency of 44.9% after running for 50 cycles, indicating a poor cycle life compared to the Ni SAs-Pd@NC cathode. Moreover, as shown in Fig. 6e, the OCV value of the Ni SAs-Pd@NC cathode could be well recovered without any detectable decrease while the current density is again decreased back. This illustrates the high rate capability and fast-dynamic response of the Ni SAs-Pd@NC cathode.
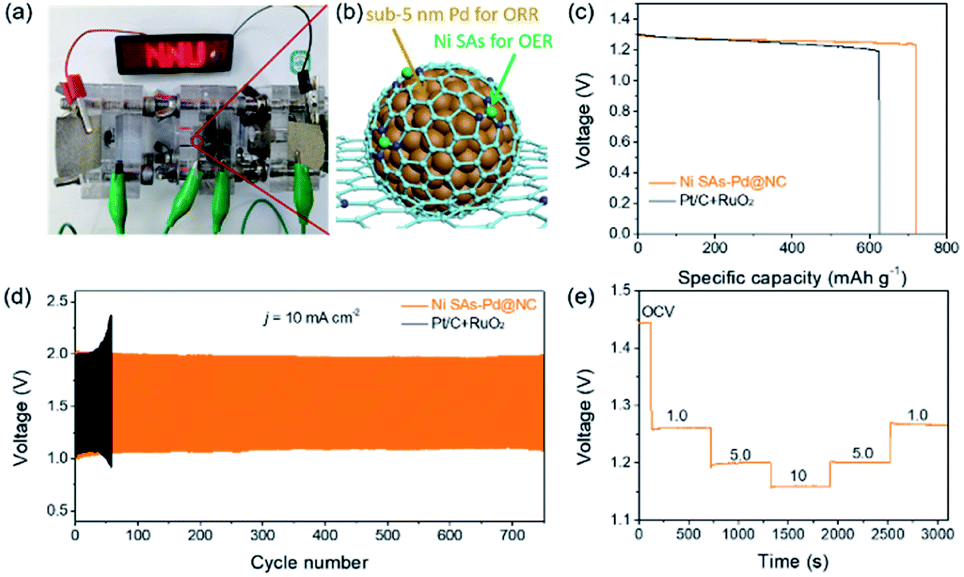 | ||
| Fig. 6 (a) An LED bulb (∼5.0 V) powered by three Zn–air batteries. (b) Schematic configuration of a Zn–air battery assembled with Ni SAs-Pd@NC. (c) Discharge curves at a constant current density of 5 mA cm−2. (d) The discharge and charge voltage profiles of Zn–air batteries with Ni SAs-Pd@NC at 10 mA cm−2. (e) Discharge profiles at different current densities (open-circuit voltage (OCV), 1.00, 5.00, and 10.00 mA cm−2, and recovery to OCV). | ||
The outstanding activity and stability of Ni SAs-Pd@NC for the ORR, OER and rechargeable Zn–air batteries could be attributed to the following features: (i) it has been widely recognized that the ORR and OER are two reversible electrochemical reactions, and thus the ORR or OER activity is generally compromised owing to the competition between the ORR and OER on the same catalyst surface.33 In Ni SAs-Pd@NC, the sub-10 nm Pd nanocrystals dominantly contribute to the ORR activity, while the Ni–N4 sites with low catalytic barriers and high atom utilization efficiency are responsible for the OER, thus resulting in a higher selectivity towards the respective ORR and OER.34,35 The superior ORR selectivity of Pd and OER selectivity of Ni are confirmed by electro-catalytic data. As revealed, the pure Pd sample shows superior ORR activity but poor OER activity, however the pure Ni sample shows superior OER activity but poor ORR activity. (ii) As confirmed by the XPS survey and electrocatalytic data, the incorporation of Ni–N4 SAs with sub-10 nm Pd nanocrystals could generate a synergistic effect. On one hand, Ni–N4 SAs could modify the electronic structure of sub-10 nm Pd nanocrystals, thus tuning the adsorption of the intermediate for the ORR.36 On the other hand, the sub-10 nm Pd nanocrystals are more resistant to corrosion than the transition metal carrier, which could improve the electro-chemical stability of Ni–N4 SAs.37,38 (iii) Ni SAs-Pd@NC with hierarchical, interconnected, and porous architecture could increase the contact area for the electrolyte and micromolecules, thus delivering fast mass transport with low ion diffusion barriers.39 (iv) The ultrathin N-doped carbon frameworks not only ensure high conductivity for fast electron transfer, but also provide strong metal–support binding with both the Ni SAs and sub-10 nm Pd nanocrystals to achieve outstanding structural stability in the device.40
4. Conclusion
In summary, we report the integration of isolated Ni SAs with sub-10 nm Pd nanocrystals embedded in N-doped carbon frameworks, in an effort to achieve a single-atom-based bifunctional catalyst for Zn–air batteries. The synthetic strategy depends on the in situ pyrolysis of the NiPc@PdII–(C10H7–NH2) complex, which could simultaneously generate atomically dispersed Ni sites, form the porous carbon frameworks, and protect the sub-10 nm Pd sites from growing. Such combination features Ni–N4 SAs to offer active sites for the OER and the sub-10 nm Pd sites to favour the ORR activity. Meanwhile, the N-doped porous carbon framework could accelerate diffusion/transfer kinetics and offer strong metal–support binding with both Ni SAs and sub-10 nm Pd. Accordingly, Ni SAs-Pd@NC with an optimal proportion of 2:1 exhibits excellent electro-catalytic activity towards the ORR and OER in an alkaline medium, and could be qualified as an air cathode to provide the rechargeable Zn–air battery with outstanding energy efficiency and cycling stability. This work paves a new avenue for coupling single-atom sites with small-sized noble metals, which offers a practical strategy for developing high-performance and selective electro-catalysts.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
X. Q. acknowledges the financial support from the National Natural Science Foundation of China (21902078), and Natural Science Foundation of Jiangsu Higher Education Institutions of China (19KJB150033). Y. T. acknowledges the financial support from the Natural Science Foundation of Jiangsu Province (BK20171473) and National Natural Science Foundation of China (21875112). We also acknowledge the support from the National and Local Joint Engineering Research Centre of Biomedical Functional Materials and a project sponsored by the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- M. Luo, Z. Zhao, Y. Zhang, Y. Sun, Y. Xing, F. Lv, Y. Yang, X. Zhang, S. Hwang, Y. Qin, J. Y. Ma, F. Lin, D. Su, G. Lu and S. Guo, Nature, 2019, 574, 81–85 CrossRef CAS PubMed.
- J. Diao, Y. Qiu, S. Liu, W. Wang, K. Chen, H. Li, W. Yuan, Y. Qu and X. Guo, Adv. Mater., 2020, 32, 1905679 CrossRef CAS PubMed.
- M. Ge, S. Wi, X. Liu, J. Bai, S. Ehrlich, D. Lu, W. K. Lee, Z. Chen and F. Wang, Angew. Chem., Int. Ed., 2021, 60, 17350–17355 CrossRef CAS PubMed.
- S. Huang, O. Ting, B. Zheng, M. Dan and Z. Liu, Angew. Chem., Int. Ed., 2021, 60, 9546–9552 CrossRef CAS.
- O. Ting, X. Wang, X. Mai, A. Chen, Z. Tang and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 11948–11957 CrossRef.
- X. He, X. Yi, F. Yin, B. Chen, G. Li and H. Yin, J. Mater. Chem. A, 2019, 7, 6753–6765 RSC.
- J. Lee, A. Kumar, T. Yang, X. Liu, A. R. Jadhav, G. Park, Y. Hwang, J. Yu, C. T. K. Nguyen, Y. Liu, S. Ajmal, M. G. Kim and H. Lee, Energy Environ. Sci., 2020, 13, 5152–5164 RSC.
- X. Wang, O. Ting, L. Wang, J. Zhong and Z. Liu, Angew. Chem., Int. Ed., 2020, 59, 6492–6499 CrossRef CAS PubMed.
- J. Park, C. Lee, J. Ju, J. Lee, J. Seol, S. Lee and J. Kim, Adv. Funct. Mater., 2021, 31, 2101727 CrossRef CAS.
- L. Xie, X. P. Zhang, B. Zhao, P. Li, J. Qi, X. Guo, B. Wang, H. Lei, W. Zhang, U. P. Apfel and R. Cao, Angew. Chem., Int. Ed., 2021, 60, 7576–7581 CrossRef CAS PubMed.
- S. Ren, X. Duan, S. Liang, M. Zhang and H. Zheng, J. Mater. Chem. A, 2020, 8, 6144–6182 RSC.
- Z. Li, H. He, H. Cao, S. Sun, W. Diao, D. Gao, P. Lu, S. Zhang, Z. Guo, M. Li, R. Liu, D. Ren, C. Liu, Y. Zhang, Z. Yang, J. Jiang and G. Zhang, Appl. Catal., B, 2019, 240, 112–121 CrossRef CAS.
- H. Zhang, Y. Liu, T. Chen, J. Zhang, J. Zhang and X. Lou, Adv. Mater., 2019, 31, 1904548 CrossRef CAS.
- Y. Li, Z. Wu, P. Lu, X. Wang, W. Liu, Z. Liu, J. Ma, W. Ren, Z. Jiang and X. Bao, Adv. Sci., 2020, 7, 1903089 CrossRef CAS PubMed.
- S. Tang, X. Zhou, T. Liu, S. Zhang, T. Yang, Y. Luo, E. Sharman and J. Jiang, J. Mater. Chem. A, 2019, 7, 26261–26265 RSC.
- Q. Wang, X. Huang, Z. Zhao, M. Wang, B. Xiang, J. Li, Z. Feng, H. Xu and M. Gu, J. Am. Chem. Soc., 2020, 142, 7425–7433 CrossRef CAS.
- W. Lai, L. Zhang, W. Hua, S. Indris, Z. Yan, Z. Hu, B. Zhang, Y. Liu, L. Wang, M. Liu, R. Liu, Y. Wang, J. Wang, Z. Hu, H. Liu, S. Chou and S. Dou, Angew. Chem., Int. Ed., 2019, 58, 11868–11873 CrossRef CAS.
- M. Millet, G. Algara-Siller, S. Wrabetz, A. Mazheika, F. Girgsdies, D. Teschner, F. Seitz, A. Tarasov, S. Levchenko, R. Schlogl and E. Frei, J. Am. Chem. Soc., 2019, 141, 2451–2461 CrossRef CAS PubMed.
- L. Zhang, D. Liu, Z. Muhammad, F. Wan, W. Xie, Y. Wang, L. Song, Z. Niu and J. Chen, Adv. Mater., 2019, 31, 1903955 CrossRef CAS PubMed.
- E. Meza, R. E. Diaz and C. W. Li, ACS Nano, 2020, 14, 2238–2247 CrossRef CAS.
- C. Cheng, S. Li, Y. Xia, L. Ma, C. Nie, C. Roth, A. Thomas and R. Haag, Adv. Mater., 2018, 30, 1802669 CrossRef PubMed.
- J. Yang, W. Li, D. Wang and Y. Li, Adv. Mater., 2020, 32, 2003300 CrossRef CAS.
- W. Wan, Y. Zhao, S. Wei, C. A. Triana, J. Li, A. Arcifa, C. S. Allen, R. Cao and G. R. Patzke, Nat. Commun., 2021, 12, 5589 CrossRef CAS.
- W. Feng, J. Yuan, F. Gao, B. Weng, W. Hu, Y. Lei, X. Huang, L. Yang, J. Shen, D. Xu, X. Zhang, P. Liu and S. Zhang, Nano Energy, 2020, 75, 104990 CrossRef CAS.
- Q. Ji, L. Bi, J. Zhang, H. Cao and X. S. Zhao, Energy Environ. Sci., 2020, 13, 1408–1428 RSC.
- M. Balogun, H. Yang, Y. Luo, W. Qiu, Y. Huang, Z. Liu and Y. Tong, Energy Environ. Sci., 2018, 11, 1859–1869 RSC.
- Xi. Han, X. Ling, D. Yu, D. Xie, L. Li, S. Peng, * C. Zhong, N. Zhao, Y. Deng and W. Hu, Adv. Mater., 2019, 31, 1905622 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS.
- E. Wang, M. Chen, X. Guo, S. L. Chou, B. Zhong and S. X. Dou, Small Methods, 2020, 4, 1900163 CrossRef CAS.
- X. Chen, D. Ma, B. Chen, K. Zhang, R. Zou, X. Wu and Q. Zhu, Appl. Catal., B, 2020, 267, 118720 CrossRef CAS.
- C. Han, F. Liu, J. Liu, Q. Li, J. Meng, B. Shao, Q. He, X. Wang, Z. Liu and L. Mai, J. Mater. Chem. A, 2018, 6, 6220–6224 RSC.
- O. Ting, Y. Ye, C. Wu, K. Xiao and Z. Liu, Angew. Chem., Int. Ed., 2019, 58, 4923–4928 CrossRef.
- T. Ling, T. Zhang, B. Ge, L. Han, L. Zheng, F. Lin, Z. Xu, W. B. Hu, X. W. Du, K. Davey and S. Z. Qiao, Adv. Mater., 2019, 31, 1807771 CrossRef.
- A. M. Harzandi, S. Shadman, A. S. Nissimagoudar, D. Y. Kim, H. D. Lim, J. H. Lee, M. G. Kim, H. Y. Jeong, Y. Kim and K. S. Kim, Adv. Energy Mater., 2021, 11, 2003448 CrossRef CAS.
- Z. Liu, X. Luo, L. Qin, G. Fang and S. Liang, Adv. Powder Mater., 2021 DOI:10.1016/j.apmate.2021.10.002.
- A. Han, B. Wang, A. Kumar, Y. Qin, J. Jin, X. Wang, C. Yang, B. Dong, Y. Jia, J. Liu and X. Sun, Small Methods, 2019, 3, 1800471 CrossRef.
- D. M. Morales, M. A. Kazakova, S. Dieckhöfer, A. G. Selyutin, G. V. Golubtsov, W. Schuhmann and J. Masa, Adv. Funct. Mater., 2020, 30, 1905992 CrossRef CAS.
- J. Zhang, B. Song, C. Cai, L. Zhang and Y. Shi, Adv. Powder Mater. DOI:10.1016/j.apmate.2021.10.001.
- H. Xiong, G. Sun, Z. Liu, L. Zhang, L. Li, W. Zhang, F. Du and Z. Qiao, Angew. Chem., Int. Ed., 2021, 60, 10334–10341 CrossRef CAS PubMed.
- Y. Zhu, W. Sun, J. Luo, W. Chen, T. Cao, L. Zheng, J. Dong, J. Zhang, M. Zhang, Y. Han, C. Chen, Q. Peng, D. Wang and Y. Li, Nat. Commun., 2018, 9, 3861 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta06897k |
| ‡ S. Wang and Z. Lin contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |