
Direct evidence of cobalt oxyhydroxide formation on a La0.2Sr0.8CoO3 perovskite water splitting catalyst†
Anthony
Boucly
 a,
Luca
Artiglia
*a,
Emiliana
Fabbri
*a,
Dennis
Palagin
a,
Dino
Aegerter
a,
Daniele
Pergolesi
a,
Zbynek
Novotny
ab,
Nicolò
Comini
b,
J. Trey
Diulus
b,
Thomas
Huthwelker
a,
Markus
Ammann
a and
Thomas J.
Schmidt
ac
a,
Luca
Artiglia
*a,
Emiliana
Fabbri
*a,
Dennis
Palagin
a,
Dino
Aegerter
a,
Daniele
Pergolesi
a,
Zbynek
Novotny
ab,
Nicolò
Comini
b,
J. Trey
Diulus
b,
Thomas
Huthwelker
a,
Markus
Ammann
a and
Thomas J.
Schmidt
ac
aPaul Scherrer Institut, Forschungsstrasse 111, CH-5232 Villigen PSI, Switzerland. E-mail: luca.artiglia@psi.ch; emiliana.fabbri@psi.ch
bPhysik-Institut, Universität Zürich, CH-8057 Zürich, Switzerland
cLaboratory of Physical Chemistry, ETH Zürich, CH-8093 Zürich, Switzerland
First published on 13th October 2021
Abstract
Understanding the mechanism of the oxygen evolution reaction (OER) on perovskite materials is of great interest for the development of more active catalysts. Despite a lot of literature reports, the complexity of catalytic systems and scarce in situ and operando surface sensitive spectroscopic tools render the detection of active sites and the understanding of reaction mechanisms challenging. Here, we carried out and compared in situ and ex situ ambient pressure X-ray photoelectron spectroscopy experiments on a La0.2Sr0.8CoO3−δ perovskite OER catalyst. The experimental results show that segregated surface strontium, which is present in the as prepared sample, is leached into the electrolyte after immersion, leading to surface cobalt active site enrichment. Such a cobalt-enriched oxide surface evolves into a new phase, whose spectral feature is detected in situ and after the OER. With the help of theoretical simulations, such a species is assigned to cobalt oxyhydroxide, providing direct evidence of its formation and surface segregation during the oxygen evolution reaction.
Introduction
Hydrogen-based systems have the capability to be a viable solution to compensate the intermittent nature of renewable energy. In this scenario, when a surplus of renewable energy is available, water electrolyzers can convert it into hydrogen, which can be reused to produce electricity or fuels when needed. For this reason, great efforts have been recently dedicated to the understanding of water electrolysis and the improvement of catalytic materials towards the development of efficient and cost-effective water electrolyzers.1 Particularly, the anodic reaction, i.e., the oxygen evolution reaction (OER), is affected by slow kinetics and high overpotential, reducing the efficiency of the whole device.2 Thus, a widespread commercialization of water electrolyzers urges the development of highly active, stable and cost-effective electrocatalysts.Perovskite-type oxides are promising materials for the OER in an alkaline environment. Such materials allow a broad variety of elemental compositions and, thus, fine-tuning of their properties.3–6 However, despite the large scientific interest, there is still no unequivocal consensus on the reaction mechanism occurring on the surface of perovskite oxides. The “conventional” mechanism is based on four concerted proton–electron transfer steps.1,2,7,8 Recently, a new oxygen evolution mechanism, called the lattice oxygen evolution reaction (LOER), has been proposed. It involves the oxidation of perovskite lattice oxygen for the production of molecular oxygen.2,5,6,9,10 In addition, both proposed reaction mechanisms seem to be often accompanied by catalyst surface reconstruction, leading to the loss of crystallinity and the development of a superficial oxyhydroxide phase.5,11–14 Such a phase, dynamically assembled during the electrocatalytic reaction, is believed to be extremely active toward the OER.5,6,12–15Operando X-ray absorption spectroscopy (XAS) studies on perovskite oxide catalysts have already indicated that an oxyhydroxide phase can form during the OER.5 However, with XAS being a bulk technique, the analysis had to be limited to catalysts with a high surface to bulk ratio undergoing a significant surface modification. Furthermore, XAS does not allow quantification of the thickness of the oxyhydroxide layer, i.e. if such a phase forms at the solid–liquid interface or extends toward the bulk. A quantitative detection/understanding of how such a phase forms, and what the main properties leading to high OER activity are, remain open questions of paramount importance. This calls for advances in surface sensitive operando and/or in situ characterization studies.
Recent progress in ambient pressure X-ray photoelectron spectroscopy (APXPS) allows the in situ surface investigation of OER catalysts. Particularly, by means of the “dip and pull” method, which makes use of a standard electrochemical three electrode setup, it is possible to simultaneously perform electrochemical and XPS characterization experiments.16–19 In the “dip and pull” method, a thin liquid electrolyte layer (10–30 nm thickness) is stabilized at the sample surface. This provides potential control of the working electrode while performing XPS measurements. However, in order to probe the buried interface of the electrode, it is necessary to generate photoelectrons with high kinetic energies, which can travel through the thin electrolyte layer and a vapor ambient pressure of ∼30 mbar. For this reason, tender X-rays (3000–10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 eV) are typically used.17,20 Under OER conditions, OH− ions in the thin electrolyte layer are quickly consumed and diffusion limitations from the bulk solution can lead to their depletion. This creates a high ohmic resistance that makes real operando measurements challenging. A second possibility for investigating catalyst surface changes induced by the OER is by performing XPS measurements ex situ directly after electrochemical measurements. In this case, XPS is not performed while a potential is applied to the working electrode. However, the possibility to carry out electrochemical measurements in an XPS chamber while carefully controlling the reaction environment during the analysis can still allow for preserving and observing irreversible catalyst surface modifications induced by an applied potential. This ex situ approach overcomes drawbacks associated with the thin electrolyte meniscus, required for “dip and pull” measurements. Soft X-rays (hν < 2000 eV) can be utilized and their energy can be tuned to be sensitive to the topmost layers of the electrode, where active species form.
000 eV) are typically used.17,20 Under OER conditions, OH− ions in the thin electrolyte layer are quickly consumed and diffusion limitations from the bulk solution can lead to their depletion. This creates a high ohmic resistance that makes real operando measurements challenging. A second possibility for investigating catalyst surface changes induced by the OER is by performing XPS measurements ex situ directly after electrochemical measurements. In this case, XPS is not performed while a potential is applied to the working electrode. However, the possibility to carry out electrochemical measurements in an XPS chamber while carefully controlling the reaction environment during the analysis can still allow for preserving and observing irreversible catalyst surface modifications induced by an applied potential. This ex situ approach overcomes drawbacks associated with the thin electrolyte meniscus, required for “dip and pull” measurements. Soft X-rays (hν < 2000 eV) can be utilized and their energy can be tuned to be sensitive to the topmost layers of the electrode, where active species form.
In the present study, we have compared the results obtained by in situ and ex situ APXPS measurements on a La0.2Sr0.8CoO3−δ (LSCO) thin film perovskite. LSCO is known to be an interesting, highly OER-active material.5,15 Among the La1−xSrxCoO3 series (with x < 1), the La0.2Sr0.8CoO3−δ composition displays the highest OER activity.21,22 As shown by Mefford et al.,21 the higher the covalent character of the Co–O bond in La1−xSrxCoO3−δ catalysts, the higher the vacancy concentration, and also the OER activity. The La1−xSrxCoO3−δ series was also investigated by Lopes et al.13 who demonstrated that the more oxygen vacancies in LSCO, the faster the A-site (Sr and La) dissolution and, consequently, the perovskite surface conversion into a highly active CoOOH layer.
In our previous study23 we have shown that depending on the preparation temperature, LSCO thin films present different degrees of strontium segregation. Strontium containing species segregated on the surface (hydroxides and/or carbonates) are soluble in water, and are almost quantitatively removed when immersing the electrodes in water.23 In addition, we found that the line shape of the lanthanum photoemission peak (La 4d) evolves as well, indicating a loss of the surface species albeit in a much lower amount compared to strontium. Finally, we observed that the removal of the strontium segregated species leads to more cobalt active sites being exposed/available for the OER. As a result, we found that the most active OER catalyst is the one with the highest initial amount of strontium surface segregation.23 In this work, such a LSCO thin film23 has been investigated by in situ and ex situ APXPS, showing not only the removal of superficial, segregated strontium upon contact with the liquid electrolyte, but also the formation of a completely new oxyhydroxide phase, as demonstrated by combining the experimental results with theoretical simulations of the core electron binding energies. To our knowledge, this is the first direct spectroscopic observation of such surface-active species involved in the OER, which was postulated in several research contributions.5,13,14 Our results also highlight the possibility to get important information about the nature of active sites making use of ex situ investigations carried out under well controlled conditions.
Results
Fig. 1 shows a sketch that summarizes the main measurements performed in the present study using ex situ and in situ APXPS and an electrochemical three electrode cell (WE: working electrode, CE: counter electrode, and RE: reference electrode). The electrochemical protocol, carried out before the XPS measurements with the WE fully immersed in the electrolyte solution, consisted of 50 cyclic voltammetry (CV) cycles performed between 1 and 1.65 V vs. RHE at 100 mV s−1 (see Fig. 1 and S1†). Ex situ measurements were carried out by pulling the WE completely out of the solution after the 50 CV cycles and using soft X-rays after rinsing in ultra-pure water. In this case, the electrolyte must be removed because low kinetic energy photoelectrons cannot be measured at a background pressure as high as 20 mbar. In situ measurements were carried out using tender X-rays and the so-called “dip and pull” method after obtaining the 50 CVs, under a nominal applied potential of 1.65 V vs. RHE.17–20 In the latter case, APXPS measurements could be performed under an applied potential with the CE and RE fully immersed in the electrolyte and the WE partially immersed in the electrolyte with the upper part (measured spot) only covered by a thin layer of electrolyte able to maintain electrical contact. High kinetic energy photoelectrons generated by tender X-rays can be measured at background pressures higher than 20 mbar and contain information about the interface of the WE buried under the thin layer of electrolyte. The detailed experimental approach is described in the Methods. As shown in Fig. 1 and S1,† an increase in the capacitive current (in the potential region between 1 and ∼1.6 V vs. RHE) over the 50 cycles and a slight decrease in the OER current (above ∼1.6 V vs. RHE) were observed for LSCO. This suggests that the sample surface undergoes some modifications. Furthermore two small features, which can be attributed to the Co3+/Co4+ redox couple, appear as the CV cycling continues. This is better highlighted in Fig. S1,† displaying the reduction feature appearing at 1.55 V vs. RHE and the oxidation one at 1.45 V. To unveil such (electro)chemically driven surface modifications, the electrode has been investigated in situ and ex situ by means of APXPS.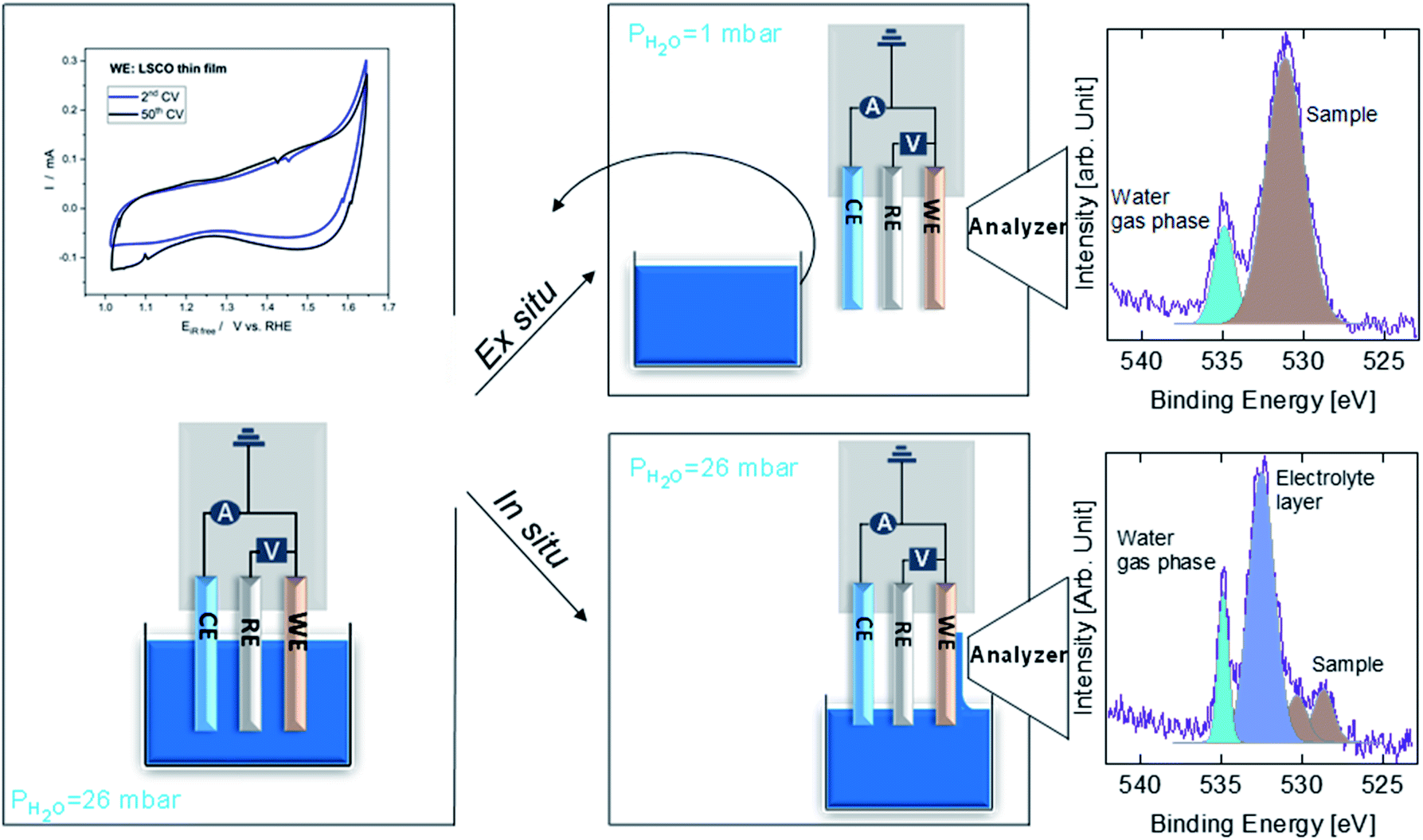 | ||
| Fig. 1 Schematics of the experiment. During the initial step, the sample is fully dipped in the electrolyte and the electrochemical measurements are done. Once these measurements are done, in the case of the in situ experiment (tender X-rays) we pull the sample directly in the measurement position while holding the potential and proceed with XPS measurements probing through the thin electrolyte. In the case of the ex situ experiment (soft X-rays), we pull out the sample, put it in storage under a partial pressure of 26 mbar and remove the beaker containing electrolyte before reintroducing the sample and bringing it to the XPS measurement position under a partial pressure of 1 mbar. | ||
As shown in Fig. 2a, initially the Sr 3d core level spectrum acquired in situ shows two features that can be deconvolved into two doublets (spin orbit splitting of 1.8 eV and branching ratio of 1.5).23 The first one, at a binding energy of 133.3 eV (Sr 3d5/2, orange color), corresponds to surface-segregated strontium species. The second one, centered at 131.5 eV (Sr 3d5/2, green color), is ascribed to bulk-like (lattice) strontium.16,24,25 Using a higher photon excitation energy, a second core-level peak of strontium, Sr 2p, could be acquired and is shown in Fig. S2a.† In this case, two components, corresponding to surface and bulk strontium, can be detected. The KEs of Sr 2p and Sr 3d shown in Fig. S2a† are approximately 3050 and 2160 eV (corresponding to 8.8 nm and 6.6 nm probing depths), respectively, and thus the former contains information from a larger depth. The relative intensity of the surface component in the case of Sr 2p (Srsurf/SrBulk=0.27) is lower than that calculated from Sr 3d (0.43), supporting the fact that such species segregate at the surface. The Sr 3d photoemission signal, shown in Fig. S2b,† was acquired ex situ with a KE of approximately 760 eV. This corresponds to approx. 3 nm probing depth, and allows us to focus the analysis on segregated strontium, whose relative intensity is the maximum (Srsurf/SrBulk = 2.45) among all the spectra shown in Fig. S2.† Upon immersion of the sample in the electrolyte solution and the application of a potential of 1.65 V vs. RHE, the signal of Sr 2p acquired in situ (Fig. S2a†) displays a clear evolution. A single peak (light green color), centered at 1938.8 eV, is present. The same line shape evolution is detected ex situ with soft X-rays (Fig. S2b†): a single doublet (Sr 3d5/2 at 133.0 eV, light green) is used to fit the spectrum collected after electrochemical measurements. It is important to highlight that the binding energy of the new doublet matches neither that of surface nor that of bulk strontium. This suggests a modification of the electronic state during CV cycles. The presence of a single component both under bulk (Sr 2p, Fig. S2a†) and surface (Sr 3d, Fig. S2b†) sensitive conditions suggests the formation of a new strontium-containing phase, promoted by the interaction with the electrolyte and OER cycles. Most likely, the local coordination environment of strontium changes after the reaction, and its binding energy is slightly affected. In addition, Fig. 2a and b show that, after electrochemistry, the intensity of Sr 3d decreases in a relevant way, whereas that of cobalt (Co 2p core-level peak) increases. This indicates that immersion of the electrode in the electrolyte solution followed by CV measurements leads to leaching of the segregated strontium layer, modification of its local structure, and cobalt enrichment at the surface. Indeed, after 50 CV cycles, the redox couple close to the OER region (possibly Co3+/Co4+)3 becomes more pronounced, most likely due to the surface Co enrichment.
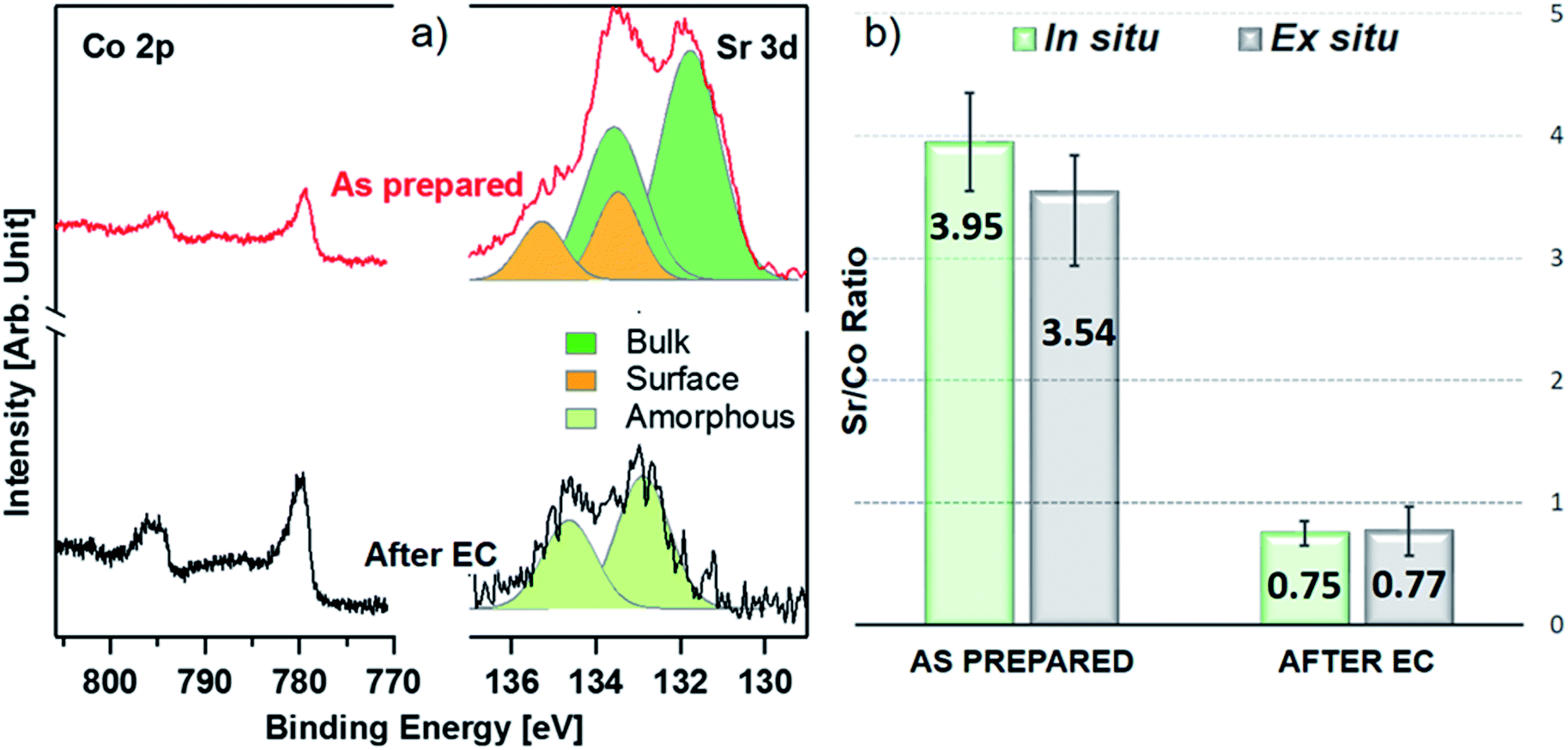 | ||
| Fig. 2 Photoemission spectra (a) acquired at hν = 2300 eV during the in situ experiment showing the collapse of the Sr 3d signal peak (right) compared to the Co 2p signal (left) with red and black curves corresponding to the “as prepared” and “after electrochemistry” state, respectively. The dark green component of the strontium deconvolution corresponds to the bulk signal, orange corresponds to the surface signal, and the light green single component seen after electrochemistry corresponds to the new single species displaying a shift in the binding energy. The Sr/Co ratios calculated from these spectra are shown as histograms (b) with the in situ experiment plotted in light green and the ex situ one plotted in gray. | ||
The evolution of the Sr 3d/Co 2p ratio (corrected by photon flux and photo-ionization cross sections), evaluated both during in situ and ex situ measurements, is shown in Fig. 2b and Table S3.† After introduction in the analysis chamber (“as prepared”), the electrode shows a ratio of 3.95 and 3.54, measured in situ and ex situ, respectively. After EC, such values decrease to 0.75 and 0.77. In both experiments the system ends up with more cobalt exposed at the surface, in fair agreement with previous observations.23 The fact that the Sr/Co ratios acquired in situ and ex situ show similar values (within the error) suggests that strontium leaching takes place within the probing depth of in situ measurements. Co 2p photoemission peaks are shown in Fig. 2a, and display the line shape of Co(III) in the lattice of perovskites (main peak, Co 2p3/2, centered at approximately 780 eV).26–29 When looking for in situ chemical changes of cobalt during the OER while applying 1.65 V, the main peak (Co 2p3/2) at 780 eV remains unchanged (see Fig. S3a†) compared to the “as prepared” conditions, while a slight modification of the shake-up satellite structure line shape is observed. Indeed, the satellite feature is centered at about 788 eV under the “as prepared” conditions, while during EC (“under 1.65 V”) it shifts towards a higher binding energy (around 790 eV). This change might indicate an increase of the oxidation state of Co,30 coherent with the increase of potential towards the OER. Indeed, cobalt oxidation does not lead to binding energy shifts of the main peak (2p3/2) and its spin–orbit satellite (2p1/2), whereas clear changes are observed in the structure of shake-up satellites.30,31 A shift of the latter toward a higher binding energy reflects cobalt oxidation. However due to the low signal/noise ratio, it is difficult to make a clear statement about this change. A further modification is also observed after the OER, with a more pronounced bump in the satellite structure at a binding energy of approx. 786 eV, lower BE than that observed under the “as prepared” conditions. This possibly indicates a slight evolution of the oxidation state towards reduction, compatible with our protocol where the potential decreases from 1.65 V to OCV before stopping the EC experiment and, then, measuring under the “after EC” conditions. However, like the line shape modification seen under 1.65 V potential, this change is too small to make a clear and definitive/quantitative statement on its nature. Ex situ experiments (Fig. S3b and c†), have been performed to cross check the in situ results. Both the Co 2p and Co L2,3 edges show negligible line shape modifications, suggesting that negligible changes of the electronic state of cobalt take place within the probed depth, which is 2.2 nm and 2.8 nm for Co 2p and Co L2,3, respectively. This is not surprising, because ex situ experiments were carried out outside the electrolyte solution and with no voltage applied. In addition, oxy-hydroxylation of the cobalt topmost layer does not change its oxidation state. Other reports show small line shape modifications of the shake-up satellite peak features in bulk cobalt hydroxide samples,32,33 which are challenging to detect if hydroxylation is limited to the sample surface.
Fig. 3 displays the comparison between O 1s measurements performed in situ (Fig. 3a) and ex situ (Fig. 3b). The signal of oxygen is of particular importance because it contains information about lattice components (oxygen in the lattice of LSCO surface termination) and surface species that are present or formed during the reaction. O 1s spectra of the “as prepared” sample were separated into two components. The first one, at a binding energy of 528.9 eV, corresponds to oxygen in the perovskite lattice, as commonly reported in the literature.24,34,35 The second component, at a binding energy of 531.5 eV, is assigned to oxygen in the termination layer.34 While Stoerzinger et al.16 separated the specific signal of carbonate from that of other oxygen surface species (like undercoordinated oxygen and peroxo-groups) in the O 1s spectrum, in this work, due to the small chemical shift, we combine all surface oxygen species in a single component. This result reflects the Sr 3d and 2p core level peak deconvolution (Fig. 2a and S2†), where a surface component due to strontium soluble species (such as carbonates) is present. A spectral component assigned to carbonates is also detected in the C 1s spectra (Fig. S4†). Based on the C 1s carbonate component intensity, we estimate that about 40% of the O 1s surface signal is generated from strontium carbonate in the “as prepared” sample. Indeed, considering the C/O ratio in the CO32− ion and upon normalization to the photon flux and cross section, the area of the C 1s carbonate component provides the carbonate fraction in the O 1s termination layer (531.5 eV). Such a value decreases to 27% after electrochemistry (“after EC” state). Such a difference in the proportion and the loss of the strontium surface component clearly indicate that the nature of the species comprising the O 1s surface signal is different between the “as prepared” and the “after EC” state. In situ measurements (Fig. 3a) were performed with two excitation energies (2300 and 5000 eV), in order to vary the information depth within the same measurement. The relative intensity of the 531.5 eV component shows an increasing trend passing from 5000 to 2300 eV, and becomes the main component of O 1s acquired ex situ (Fig. 3b). This proves that oxygen in the termination layer has a thickness distribution comparable/limited to the probing depth of ex situ measurements (approx. 2.2 nm). Due to the experimental geometry setup adopted in this work, 99% of the O 1s photoelectrons are generated within 2.2 nm depth. However, already more than 86% of the signal comes from the first 1.1 nm depth, proving the surface sensitivity of ex situ measurements. Such values are obtained from the inelastic mean free path (IMFP) of photoelectrons having specific kinetic energies in LSCO,36 corrected by the measurement geometry to give the attenuation length (AL). In this specific case 1 × AL corresponds to 1.1 nm (2.2 nm corresponds to 2 × AL). In situ measurements performed during EC show a complex line shape due to the presence of a thin liquid layer on top of the WE. As shown in Fig. 3a, the peak at 532.6 eV corresponds to condensed water and that at 535.0 eV to water vapor in equilibrium with the liquid (background in the analysis chamber). Liquid water is not visible during ex situ measurements (Fig. 3b), because spectra were acquired by dosing a 1 mbar water vapor background, without a stabilized electrolyte layer on top of the electrode and with no potential applied. According to the literature,37 1 mbar water vapor pressure should lead to adsorption of approx. 3 monolayers of water. The photoemission signal is probably not detected during ex situ experiments, either because of superimposition (to that of the O 1s surface component) or because at such a low relative humidity water is physisorbed. Three more components, centered at 529.0 eV, 530.6 eV and 531.6 eV, were used to deconvolve O 1s acquired in situ during EC. The first component (green color) and the third one (orange color) are still assigned to oxygen in the lattice and in the termination layer of LSCO, respectively. The middle one (red color), displaying an intermediate binding energy value, reflects the presence of a new oxygen-containing species. Interestingly, the O 1s spectrum acquired ex situ shows primarily this new component, at a binding energy of approx. 531.0 eV. Once again, we want to stress that O 1s acquired ex situ has a KE of approximately 480 eV, whereas the KE in situ is 4460 eV. This corresponds to a relevant change of the probing depth (from 2.2 nm to 6 nm), and demonstrates that newly formed species are located at the surface. We tentatively attribute them to hydroxyls, which form at the surface of the sample during electrochemistry.
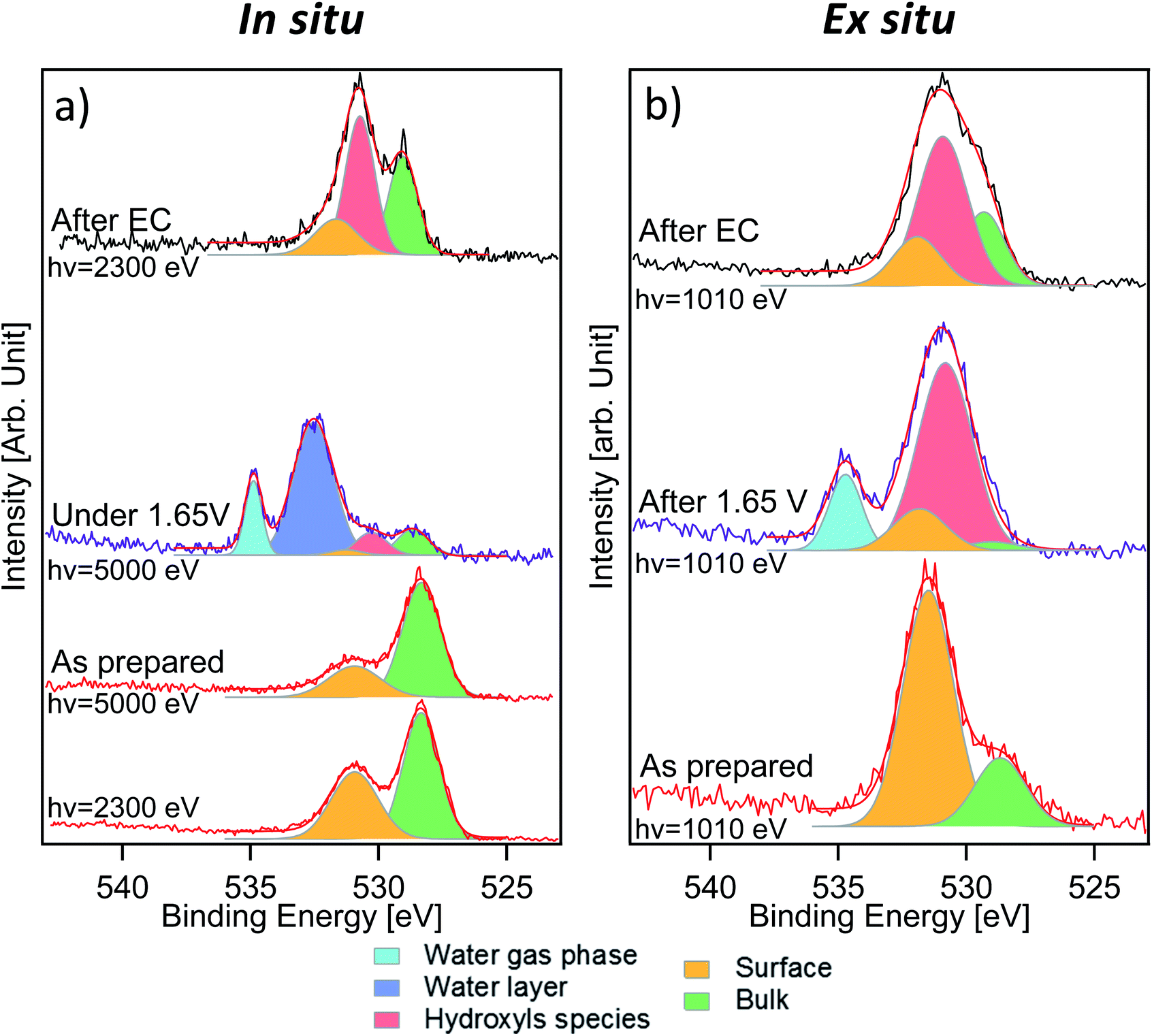 | ||
| Fig. 3 O 1s XPS spectra from the in situ experiment (a) and the ex situ experiment (b) with red, purple and black curves corresponding to the “as prepared”, “under 1.65 V vs. RHE/after 1.65 V vs. RHE” and “after electrochemistry” states, respectively. The green, orange, red, light blue and dark blue components correspond to the perovskite bulk, surface, hydroxyls, water gas phase and electrolyte signal, respectively. | ||
Measurements performed after EC give other interesting information about the evolution of the sample surface. Photoemission peaks can be separated into three components, assigned to oxygen in the lattice of LSCO (529.0 eV, green color), surface hydroxyl groups (530.6 eV, red color) and oxygen in the termination layer of LSCO (531.6 eV, orange color), which are detected in both measurement conditions. The increase of oxygen in the termination layer of LSCO suggests that hydroxyls may originate from such species during the OER, while some of them convert back under vacuum conditions. It is generally assumed that for LSCO perovskites, cobalt is the active site for the OER.2,38 Experimental data described above suggest the formation of hydroxyls during the reaction, while the surface is enriched in cobalt. Therefore, the formation of a cobalt oxyhydroxide phase during the reaction can be speculated. Such species are well visible in the O 1s spectrum, but not in the Co 2p spectrum. As mentioned above, line shape modifications of Co 2p upon formation of hydroxide are extremely small, and thus difficult to detect in a surface layer.32,33
To further support our hypothesis, theoretical simulations of the core electron binding energies of different oxygen species on LSCO have been performed and compared with the experimental data. Due to the complexity of the system, and its evolution upon an electrochemical process, a precise simulation of the investigated sample is challenging. Consequently, we do not expect a perfect agreement between the experimental and simulated peak positions. However, theoretical calculations of electron binding energies have already been used to help assigning new photoemission peaks,39–41 comparing their relative positions with respect to other “reference” components.
Fig. 4 shows the spectrum of O 1s acquired ex situ after EC measurements, deconvolved using the same peak components described above. The core electron binding energy value of lattice oxygen calculated for a (001) orientated bulk LSCO structure (Fig. 4, right) oscillates between 528.8 and 529.0 eV (O atoms computationally split in two groups, depending on their location with respect to La and Sr, see Fig. S5 in the ESI†), and is in good agreement with the position of the green component detected experimentally (528.9–529.0 eV). The structure of strontium carbonate was used to simulate oxygen in the termination layer of LSCO (strontium segregations soluble in water, as shown in Fig. 4, left), which should correspond to the highest BE peak (orange color in the O 1s). Simulations estimate a binding energy value between 531.6 and 531.9, in fair agreement with the experimental value of 531.5 eV. A series of possible defects that could form on cobalt oxide-terminated LSCO during EC have been simulated adsorbing hydroxyl groups on the CoO2 topmost layer. Three possible structures are presented in Fig. 4 (top) and correspond to different configurations of the adsorbed hydroxyl. With this, we aimed at simulating the electron binding energies corresponding to the O 1s orbitals of the CoOOH species. However, the exact configuration of these species, and whether they form a separate phase or are formed on the CoO2-terminated surface, remains an open question. Here, for the sake of simplicity, we either adsorb hydroxyl groups on the top layer of the CoO2-terminated surface, or place a hydroxyl group in the place of the oxygen vacancy, in both cases effectively forming the CoOOH species. These structures represent some of the possible geometrical structures of the surface hydroxyl groups, and correspond to the following situations: (a) formation of the surface OH− group in the place of a surface oxygen defect; (b) adsorption of the OH− group on the CoO2-terminated surface; and (c) formation of multiple OH− groups in the place of multiple surface oxygen defects. Although these three structures by no means represent a comprehensive set of possible surface structures (some additional structures are presented in Fig. S6 and S7 in the ESI†), they provide sufficient variability to check whether they may in principle correspond to the observed experimental data. The simulated binding energy of OH− in the three structures varies in the 530.2–531.0 eV range. The centroid of the new O 1s peak component detected experimentally is at 530.6 eV (red color), at the center of the simulated BE range. Most likely, a collection of different hydroxyl species form on top of the electrode during EC, and thus the experimental peak is a convolution of different species displaying similar BE values. In addition, simulations of hydroxylated (110) and (111) orientations (Fig. S8 and S9,† respectively) of LSCO were performed and gave BE values similar to those found on the (001) orientation, with a BE value between 530.5 and 530.9. In summary, an excellent agreement between the experiment and theory is found, and theory supports the hypothesis of cobalt oxyhydroxide active phase formation during the reaction.
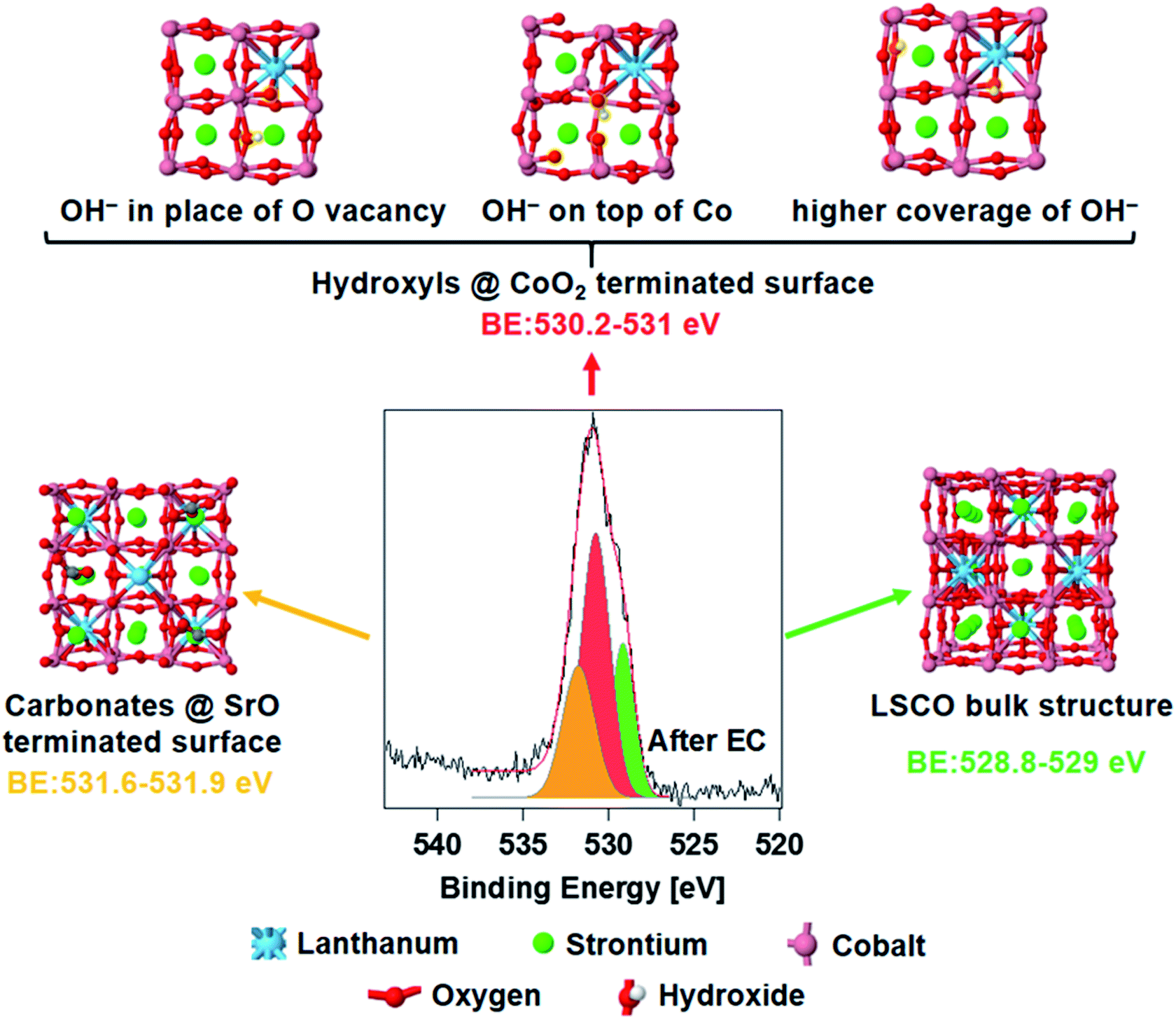 | ||
| Fig. 4 Oxygen binding energy simulation results compared to the experimental results. Colored binding energy values correspond to the calculated data, with green being the bulk perovskite oxygen (O2−), orange being the surface signal corresponding to the adsorbed carbonate species on the strontium segregated layer, and red being the hydroxyl groups formed during the electrochemical process. | ||
Discussion and conclusion
A perovskite LSCO catalyst for the OER has been fully characterized by means of APXPS in situ, probing the buried solid–liquid interface with tender X-rays, and ex situ, carrying out surface-sensitive measurements with soft X-rays in a controlled water vapor background after the reaction. Different experimental approaches provide complementary information that allow a complete characterization of the surface during and after the OER. Immersion of LSCO in the electrolyte solution leads to the leaching of surface segregated strontium species, leaving cobalt active sites exposed. Thanks to in situ and ex situ photoemission of O 1s performed during and after the OER, respectively, a new phase has been detected and assigned to surface cobalt oxyhydroxide. Such results have been confirmed by means of theoretical calculations of core electron binding energies. The literature about LSCO surface modifications during the OER is contrasting. Stoerzinger et al.16 reported no evidence of surface changes after the OER on La1−xSrxCoO3−δ catalysts with x = 0, 0.2, 0.4, 0.5, and 0.6 (i.e., not the LSCO composition studied in the present work). May et al.42 were able to observe amorphization on Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) perovskite by means of high-resolution transmission electron microscopy (HRTEM), whereas no changes were observed on La0.4Sr0.6CoO3−δ after the OER (note that also in this case the LSCO composition was different from ours). Conversely, a recent study by Lopes et al.13 shows surface amorphization upon the OER for the La1−xSrxCoO3−δ series (x = 0–0.3) perovskites. This demonstrates how difficult it is to properly address such challenges on La1−xSrxCoO3−δ materials, and how relevant are in situ measurements. In our case, clear modifications ascribable to surface amorphization have been detected in situ and consistently proven/reproduced by ex situ measurements.The direct observation of perovskite surface reconstruction during the OER, and the clear detection of a spectroscopic signature, undoubtedly indicate that the real active site for the perovskite OER activity is a Co-based oxyhydroxide layer. However, the perovskite structure is still visible underneath Co oxyhydroxide, suggesting an equilibrium between the two under the present experimental conditions. This study represents a clear example of in situ and ex situ surface sensitive measurements combined together to detect active species formed during a reaction. In particular, the ex situ experimental protocol can be easily reproduced in several facilities to detect non-transient modifications and avoids well known drawbacks connected to the use of the “dip and pull” method. On the other hand, in situ measurements are needed to identify transient modifications of the sample surface during an electrochemical process (e.g. cobalt reversible oxidation). Theoretical calculations of the core electron binding energy are a powerful tool to facilitate the interpretation of complex spectroscopic data. The first direct spectroscopic proof of cobalt oxyhydroxide formation represents an advancement in the understanding of the reaction mechanism of the OER on perovskite materials and will have relevant implications for future studies concerning this topic.
Methods
Materials preparation
La0.2Sr0.8CoO3−δ (LSCO) thin film samples were prepared by Pulsed Laser Deposition (PLD) using a KrF excimer laser (Lambda Physik LPX 300, 25 ns pulses, λ = 248 nm) with a laser fluence at the target surface set at ∼1.25 J cm−2 and a frequency of 10 Hz. The films were grown using a background pressure of 0.1 mbar of O2 for 30 min. The distance between the target and the substrate was fixed at 4 cm. A platinum paste spot was painted next to the substrate to monitor the temperature with a pyrometer. The samples were prepared on Al2O3(0001) substrates (Crystek) at a temperature of 550 °C. After deposition, the samples were annealed in an oxygen-saturated atmosphere for 30 min at 350 °C. Fig. S10† shows a representative XRD plot of one of our films in comparison with that of the LSCO powders. The diffraction peak of the Al2O3 substrate is marked with an asterisk. It can be seen that the film is polycrystalline but textured along the (110) and (111) out-of-plane orientation. Previous scanning electron microscopy characterization experiments of LSCO samples prepared with the same approach demonstrated excellent stability upon immersion in water and after CV cycles.23To ensure good electrical conductivity, a gold frame (150 nm) with a titanium sticking layer (8 nm) was deposited by sputtering along the perimeter of the substrate on top of the films. The sample was fixed at the bottom of a 5 × 1 cm2 glass lamella using double face copper tape. The electrical connection with the manipulator was made through a gold wire. A sketch of the electrode design is shown in Fig. S11.† Tests of such a setup performed in a previous work23 showed that both the gold layer and copper tape had negligible activity compared to the exposed perovskite.
XPS characterization
A sketch of the experimental protocols employed to investigate LSCO in situ and ex situ by means of APXPS is shown in Fig. 1. Details about the experimental chamber and the three electrode setup used have been described in ref. 19. Tender X-ray APXPS experiments (hereafter called in situ) were carried out at the PHOENIX I beamline of the Swiss Light Source Synchrotron (SLS), making use of the solid liquid interface endstation.19 The sample was first analyzed under ultra-high vacuum (UHV) conditions at different excitation energies: 2300 eV and 5000 eV (hereafter referred to as “as prepared”). The estimated inelastic mean free paths (IMFPs) by using the QUASES-IMFP-TPP2M program36 at hν = 2300 eV and hν = 5000 eV are 2.8 nm (KE = 1760 eV) and 6 nm (KE = 4460 eV) for O 1s, 2.5 nm (KE = 1510 eV) and 5.7 nm (KE = 4210 eV) for Co 2p, 3.3 nm (KE = 2160 eV) and 6.4 nm (KE = 4860 eV) for Sr 3d and 4.4 nm (KE = 3060 eV, only at hν = 5000 eV) for Sr 2p. The attenuation length (AL) can be obtained by correcting the IMFP by using the measurement geometry of the photoemission setup. In this work, around 99% of the XPS signal comes within 2 × AL giving a probing depth for energies of 5.6 and 12 nm for O 1s, 5 nm and 11.4 nm for Co 2p, 6.6 nm and 12.8 nm for Sr 3d and 8.8 nm for Sr 2p (only at hν = 5000 eV). The sample was then dipped in a beaker containing 0.1 M KOH and the potential was increased by cyclic voltammetry (CV) at 100 mV s−1 to reach 1.65 V vs. RHE. 50 CV cycles were then performed at 100 mV s−1, ending the protocol at 1.65 V vs. RHE. While the potential was held at 1.65 V vs. RHE, the LSCO thin film was slowly pulled out from the electrolyte until a thin electrolyte meniscus was stably formed allowing electrical connection between LSCO, the counter electrode (CE), and reference electrode (RE) via the electrolyte solution. In this configuration, the sample surface is investigated (called “under 1.65 V”) by tender X-ray APXPS at hν = 5000 eV, through the electrolyte meniscus still covering the sample surface and ensuring the potential control (in situ characterization). Afterwards, the sample was fully dipped into the beaker containing KOH and the potential was brought back to open circuit voltage (OCV). The sample was then cleaned in ultra-pure water before being characterized under UHV at hν = 2300 eV to attain the final state (called “after EC”) under the most surface sensitive conditions available.Soft X-ray APXPS experiments (hereafter called ex situ) were carried out at the in situ spectroscopy beamline (X07DB) of the SLS, making use of the same endstation mentioned above.19 Like in tender X-ray experiments, the sample was first analyzed under UHV conditions at two different photoelectron kinetic energies (KEs): 480 eV and 780 eV. For those kinetic energies, the estimated inelastic mean free paths are 1.1 nm and 1.5 nm giving a probing depth of 2.2 and 3 nm for 480 eV and 780 eV, respectively. At each probed depth, Sr 3d, La 4d, O 1s and either Co 2p (at KE ≈ 480 eV) or Co 3p (at KE ≈ 780 eV) core level spectra were acquired. Two different core level peaks of cobalt were used due to the limitation in the maximum excitation energy available at the beamline. Once the initial UHV characterization was performed (hereafter named “as prepared”), the sample was dipped in a beaker containing 0.1 M KOH. After 50 CV cycles (ending at 1.65 V vs. RHE), the potential was held at 1.65 V vs. RHE while the sample was completely removed from the electrolyte solution (no longer under potential control). Prior to APXPS measurements, the sample was dipped into a beaker containing ultra-pure water to rinse the KOH present at the sample surface. The sample was then brought into storage, both the beakers containing KOH and ultra-pure water were taken out and the sample was reintroduced into the analysis chamber. Finally, the LSCO surface was characterized by APXPS under a water partial pressure of 1 mbar (hereafter named “after 1.65 V”). After characterization, the sample was brought back once again into storage and both the beakers containing KOH and ultra-pure water were reintroduced. Then the sample was also reintroduced and dipped into KOH to end the electrochemical protocol (bringing the potential back to OCV). Using the same method described above, we removed both beakers and characterized the sample under UHV to have its final state (hereafter named “after EC”). The difference between the first and last CV cycle, showing the appearance of the Co redox couple is shown in Fig. 1.
For both tender and soft X-ray APXPS experiments, photoemission spectra were normalized to the photon flux and to the total ionization cross-section. The aliphatic centroid component of the C 1s peak (Fig. S4†) was used as a reference, with its binding energy fixed at 284.8 eV. All peaks were fitted with Gaussian functions after subtraction of a Shirley background. Deconvolution fitting parameters are shown in Tables S1 and S2† for soft and tender X-ray experiments, respectively.
Simulation of the core-electron binding energy
All theoretical calculations in this work have been performed with the all-electron full-potential DFT code FHI-aims43,44 within the periodic boundary condition model. Geometry optimizations of periodic models were carried out using the standard generalized gradient approximation with the PBE functional45 and the “tier2” atom-centered basis set using “tight” settings for numerical integrations. For the subsequent binding energy calculations, electronic exchange and correlation was treated on the hybrid functional level with the PBE0 functional.46For all considered models, the La/Sr ratio is assumed to be 1/3 to minimize the required unit cell size, with the resulting perovskite composition corresponding to La0.25Sr0.75CoO3 for the bulk systems and La0.25Sr0.75CoO2.5 for the surface-terminated systems. All initial geometries correspond to a cubic unit cell. During the geometry optimization the symmetry was allowed to relax freely. The (001) orientation of the LSCO slab was chosen for the simulations shown in Fig. 4, because it gives the simplest structure with either Co or La/Sr at the surface. Hydroxylated (110) and (111) orientations were also simulated, to compare the BE obtained with the experimental value.
In order to follow the changes in the nature of the surface species, the electron binding energy of O 1s electrons uses standard definitions of ionization energies from the respective core levels.47 These calculations are carried out at a hybrid functional level for the fully periodic structural models. We have shown recently that this methodology yields reliable results that can be compared to those of experimental X-ray photoelectron spectroscopy.40,48
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
The authors declare no competing interest.Acknowledgements
The post-doc position of AB was funded by the Energy and Environment division of the Paul Scherrer Institute. ZN, NC and JTD acknowledge Swiss National Science Foundation for project grant 200020_172641. JTD additionally acknowledges the support from European Union's Horizon 2020 program (FP-RESOMUS – MSCA 801459). This work was performed at the PHOENIX I (X07MB) beamline and In situ Spectroscopy (X07DB) beamline of the Swiss Light Source, Paul Scherrer Institut, Villigen PSI, Switzerland, using the SLIC chamber (19) D. P. gratefully acknowledges the use of computational resources provided by the Swiss National Supercomputing Centre (CSCS).Notes and references
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Developments and perspectives of oxide-based catalysts for the oxygen evolution reaction, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- E. Fabbri and T. J. Schmidt, Oxygen Evolution Reaction-The Enigma in Water Electrolysis, ACS Catal., 2018, 8(10), 9765–9774 CrossRef CAS.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y.-L. Lee and L. Giordano, et al., Activating lattice oxygen redox reactions in metal oxides to catalyse oxygen evolution, Nat. Chem., 2017, 9, 457 CrossRef CAS PubMed.
- Y. Pan, X. Xu, Y. Zhong, L. Ge, Y. Chen and J.-P. M. Veder, et al., Direct evidence of boosted oxygen evolution over perovskite by enhanced lattice oxygen participation, Nat. Commun., 2020, 11(1), 2002 CrossRef CAS PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim and J. Durst, et al., Dynamic surface self-reconstruction is the key of highly active perovskite nano-electrocatalysts for water splitting, Nat. Mater., 2017, 16(9), 925–931 CrossRef CAS PubMed.
- C. E. Beall, E. Fabbri and T. J. Schmidt, Perovskite Oxide Based Electrodes for the Oxygen Reduction and Evolution Reactions: The Underlying Mechanism, ACS Catal., 2021, 11, 3094–3114 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Electrolysis of water on (oxidized) metal surfaces, Chem. Phys., 2005, 319(1), 178–184 CrossRef CAS.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez and N. G. Inoglu, et al., Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3(7), 1159–1165 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, Role of Lattice Oxygen Participation in Understanding Trends in the Oxygen Evolution Reaction on Perovskites, ACS Catal., 2018, 8(5), 4628–4636 CrossRef CAS.
- J. S. Yoo, Y. Liu, X. Rong and A. M. Kolpak, Electronic Origin and Kinetic Feasibility of the Lattice Oxygen Participation During the Oxygen Evolution Reaction on Perovskites, J. Phys. Chem. Lett., 2018, 9(7), 1473–1479 CrossRef CAS PubMed.
- T. Binninger, R. Mohamed, K. Waltar, E. Fabbri, P. Levecque and R. Kötz, et al., Thermodynamic explanation of the universal correlation between oxygen evolution activity and corrosion of oxide catalysts, Sci. Rep., 2015, 5, 12167 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. Farinazzo Bergamo Dias Martins, H. He, T. Kawaguchi and P. Zapol, et al., Dynamic stability of active sites in hydr(oxy)oxides for the oxygen evolution reaction, Nat. Energy, 2020, 5(3), 222–230 CrossRef.
- P. P. Lopes, D. Y. Chung, X. Rui, H. Zheng, H. He and P. Farinazzo Bergamo Dias Martins, et al., Dynamically Stable Active Sites from Surface Evolution of Perovskite Materials during the Oxygen Evolution Reaction, J. Am. Chem. Soc., 2021, 7, 2741–2750 CrossRef.
- C. Baeumer, J. Li, Q. Lu, A. Y.-L. Liang, L. Jin and H. P. Martins, et al., Tuning electrochemically driven surface transformation in atomically flat LaNiO3 thin films for enhanced water electrolysis, Nat. Mater., 2021, 20, 674–682 CrossRef CAS PubMed.
- B. J. Kim, E. Fabbri, D. F. Abbott, X. Cheng, A. H. Clark and M. Nachtegaal, et al., Functional Role of Fe-Doping in Co-Based Perovskite Oxide Catalysts for Oxygen Evolution Reaction, J. Am. Chem. Soc., 2019, 141(13), 5231–5240 CrossRef CAS PubMed.
- K. A. Stoerzinger, X. Renshaw Wang, J. Hwang, R. R. Rao, W. T. Hong and C. M. Rouleau, et al., Speciation and Electronic Structure of La1−xSrxCoO3−δ During Oxygen Electrolysis, Top. Catal., 2018, 61(20), 2161–2174 CrossRef CAS.
- S. Axnanda, E. J. Crumlin, B. Mao, S. Rani, R. Chang and P. G. Karlsson, et al., Using “Tender” X-ray Ambient Pressure X-Ray Photoelectron Spectroscopy as A Direct Probe of Solid-Liquid Interface, Sci. Rep., 2015, 5, 9788 CrossRef CAS.
- H. Ali-Löytty, M. W. Louie, M. R. Singh, L. Li, H. G. Sanchez Casalongue and H. Ogasawara, et al., Ambient-Pressure XPS Study of a Ni–Fe Electrocatalyst for the Oxygen Evolution Reaction, J. Phys. Chem. C, 2016, 120(4), 2247–2253 CrossRef.
- Z. Novotny, D. Aegerter, N. Comini, B. Tobler, L. Artiglia and U. Maier, et al., Probing the solid-liquid interface with tender X rays: A new ambient-pressure X-ray photoelectron spectroscopy endstation at the Swiss Light Source, Rev. Sci. Instrum., 2020, 91(2), 023103 CrossRef CAS PubMed.
- M. Favaro, F. Abdi, E. Crumlin, Z. Liu, R. van de Krol and D. Starr, Interface Science Using Ambient Pressure Hard X-ray Photoelectron Spectroscopy, Surfaces, 2019, 2(1), 78–99 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai and A. M. Kolpak, et al., Water electrolysis on La(1−x)Sr(x)CoO(3-delta) perovskite electrocatalysts, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- X. Cheng, E. Fabbri, M. Nachtegaal, I. E. Castelli, M. El Kazzi and R. Haumont, et al., Oxygen Evolution Reaction on La1−xSrxCoO3 Perovskites: A Combined Experimental and Theoretical Study of Their Structural, Electronic, and Electrochemical Properties, Chem. Mater., 2015, 27(22), 7662–7672 CrossRef CAS.
- A. Boucly, E. Fabbri, L. Artiglia, X. Cheng, D. Pergolesi and M. Ammann, et al., Surface Segregation Acts as Surface Engineering for the Oxygen Evolution Reaction on Perovskite Oxides in Alkaline Media, Chem. Mater., 2020, 32(12), 5256–5263 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, W. T. Hong, M. D. Biegalski, H. M. Christen and Z. Liu, et al., In situ Ambient Pressure X-ray Photoelectron Spectroscopy of Cobalt Perovskite Surfaces under Cathodic Polarization at High Temperatures, J. Phys. Chem. C, 2013, 117(31), 16087–16094 CrossRef CAS.
- E. J. Crumlin, E. Mutoro, Z. Liu, M. E. Grass, M. D. Biegalski and Y.-L. Lee, et al., Surface strontium enrichment on highly active perovskites for oxygen electrocatalysis in solid oxide fuel cells, Energy Environ. Sci., 2012, 5(3), 6081 RSC.
- J. Yang, S. Hu, Y. Fang, S. Hoang, L. Li and W. Yang, et al., Oxygen Vacancy Promoted O2 Activation over Perovskite Oxide for Low-Temperature CO Oxidation, ACS Catal., 2019, 9(11), 9751–9763 CrossRef CAS.
- H. Seim, M. Nieminen, L. Niinistö, H. Fjellvåg and L.-S. Johansson, Growth of LaCoO3 thin films from β-diketonate precursors, Appl. Surf. Sci., 1997, 112, 243–250 CrossRef CAS.
- L. Armelao, M. Bettinelli, G. Bottaro, D. Barreca and E. Tondello, LaCoO3 Nanopowders by XPS, Surf. Sci. Spectra, 2001, 8(1), 24–31 CrossRef CAS.
- I. Álvarez-Serrano, G. J. Cuello, M. L. López, A. Jiménez-López, C. Pico and E. Rodríguez-Castellón, et al., Magnetic behaviour governed by Co spin transitions in LaCo1−xTixO3 (0 ⩽x⩽ 0.5) perovskite oxides, J. Phys. D: Appl. Phys., 2008, 41(19), 195001 CrossRef.
- Y. Lykhach, S. Piccinin, T. Skala, M. Bertram, N. Tsud and O. Brummel, et al., Quantitative Analysis of the Oxidation State of Cobalt Oxides by Resonant Photoemission Spectroscopy, J. Phys. Chem. Lett., 2019, 10(20), 6129–6136 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe, Co and Ni, Appl. Surf. Sci., 2011, 257(7), 2717–2730 CrossRef CAS.
- A. Kudielka, M. Schmid, B. P. Klein, C. Pietzonka, J. M. Gottfried and B. Harbrecht, Nanocrystalline cobalt hydroxide oxide: Synthesis and characterization with SQUID, XPS, and NEXAFS, J. Alloys Compd., 2020, 824, 153925 CrossRef CAS.
- J. Yang, H. Liu, W. N. Martens and R. L. Frost, Synthesis and Characterization of Cobalt Hydroxide, Cobalt Oxyhydroxide, and Cobalt Oxide Nanodiscs, J. Phys. Chem. C, 2010, 114(1), 111–119 CrossRef CAS.
- K. A. Stoerzinger, W. T. Hong, G. Azimi, L. Giordano, Y.-L. Lee and E. J. Crumlin, et al., Reactivity of Perovskites with Water: Role of Hydroxylation in Wetting and Implications for Oxygen Electrocatalysis, J. Phys. Chem. C, 2015, 119(32), 18504–18512 CrossRef CAS.
- P. A. W. van der Heide, Systematic X-ray photoelectron spectroscopic study of La1−xSrx-based perovskite-type oxides, Surf. Interface Anal., 2002, 33(5), 414–425 CrossRef CAS.
- S. Tougaard, QUASES-IMFP-TPP2M Code for the Calculation of the Inelastic Electron Mean Free Path, Version 3.0, available from: http://www.quases.com/ Search PubMed.
- G. Lole, V. Roddatis, U. Ross, M. Risch, T. Meyer and L. Rump, et al., Dynamic observation of manganese adatom mobility at perovskite oxide catalyst interfaces with water, Communications Materials, 2020,(1), 68 CrossRef.
- A. Badruzzaman, A. Yuda, A. Ashok and A. Kumar, Recent advances in cobalt based heterogeneous catalysts for oxygen evolution reaction, Inorg. Chim. Acta, 2020, 511, 119854 CrossRef CAS.
- L. Artiglia, J. Edebeli, F. Orlando, S. Chen, M.-T. Lee and P. Corral Arroyo, et al., A surface-stabilized ozonide triggers bromide oxidation at the aqueous solution–vapour interface, Nat. Commun., 2017, 8(1), 700 CrossRef PubMed.
- L. Artiglia, V. L. Sushkevich, D. Palagin, A. J. Knorpp, K. Roy and J. A. van Bokhoven, In Situ X-ray Photoelectron Spectroscopy Detects Multiple Active Sites Involved in the Selective Anaerobic Oxidation of Methane in Copper-Exchanged Zeolites, ACS Catal., 2019, 9(8), 6728–6737 CrossRef CAS.
- I. Gladich, S. Chen, M. Vazdar, A. Boucly, H. Yang and M. Ammann, et al., Surface Propensity of Aqueous Atmospheric Bromine at the Liquid–Gas Interface, J. Phys. Chem. Lett., 2020, 11(9), 3422–3429 CrossRef CAS PubMed.
- K. J. May, C. E. Carlton, K. A. Stoerzinger, M. Risch, J. Suntivich and Y.-L. Lee, et al., Influence of Oxygen Evolution during Water Oxidation on the Surface of Perovskite Oxide Catalysts, J. Phys. Chem. Lett., 2012, 3(22), 3264–3270 CrossRef CAS.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu and X. Ren, et al., Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun., 2009, 180(11), 2175–2196 CrossRef CAS.
- X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko and A. Sanfilippo, et al., Resolution-of-identity approach to Hartree–Fock, hybrid density functionals, RPA, MP2 andGWwith numeric atom-centered orbital basis functions, New J. Phys., 2012, 14, 053020 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868 CrossRef CAS PubMed.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: The PBE0 model, J. Chem. Phys., 1999, 110(13), 6158–6170 CrossRef CAS.
- S. Tardio and P. J. Cumpson, Practical estimation of XPS binding energies using widely available quantum chemistry software, Surf. Interface Anal., 2018, 50(1), 5–12 CrossRef CAS.
- D. Palagin, V. L. Sushkevich and J. A. van Bokhoven, Water Molecules Facilitate Hydrogen Release in Anaerobic Oxidation of Methane to Methanol over Cu/Mordenite, ACS Catal., 2019, 9(11), 10365–10374 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ta04957g |
| This journal is © The Royal Society of Chemistry 2022 |