
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure formation of PNIPAM microgels in foams and foam films†
Matthias Kühnhammer a,
Kevin Gräffa,
Edwin Lorana,
Olaf Soltwedela,
Oliver Löhmann‡
a,
Henrich Frielinghausb and
Regine von Klitzing*a
a,
Kevin Gräffa,
Edwin Lorana,
Olaf Soltwedela,
Oliver Löhmann‡
a,
Henrich Frielinghausb and
Regine von Klitzing*a
aInstitute for Condensed Matter Physics, Technische Universität Darmstadt, Hochschulstraße 8, 64289 Darmstadt, Germany. E-mail: klitzing@smi.tu-darmstadt.de
bJülich Center for Neutron Science at the Heinz Maier Leibnitz Zentrum, Forschungszentrum Jülich GmbH, Lichtenbergstrasse 1, 85747 Garching, Germany
First published on 19th October 2022
Abstract
Responsive aqueous foams are very interesting from a fundamental point of view and for various applications like foam flooding or foam flotation. In this study thermoresponsive microgels (MGs) made from poly(N-isopropyl-acrylamide) (PNIPAM) with varying cross-linker content, are used as foam stabilisers. The foams obtained are thermoresponsive and can be destabilised by increasing the temperature. The structuring of MGs inside the foam films is investigated with small-angle neutron scattering and in a thin film pressure balance. The foam films are inhomogeneous and form a network-like structure, in which thin and MG depleted zones with a thickness of ca. 30 nm are interspersed in a continuous network of thick MG containing areas with a thickness of several 100 nm. The thickness of this continuous network is related to the elastic modulus of the individual MGs, which was determined by atomic force microscopy indentation experiments. Both, the elastic moduli and foam film thicknesses, indicate a correlation to the network elasticity of the MGs predicted by the affine network model.
1 Introduction
Foams are abundant in every day life in the form of food, personal care products or household cleaning agents and are also important for industrial processes like flotation or textile manufacturing.1–4 Aqueous foams are dispersions of gas in water and the structure of their continuous liquid phase is often described as hierarchical with differently sized building blocks at different length scales. These building blocks are: foam films, which are liquid films separating two bubbles, Plateau borders that are made up of three foam films and nodes, which are the connection of Plateau borders.5,6 Foams are most commonly stabilised by surfactants but can also be stabilised by solid particles,7,8 proteins9–11 or polymer/surfactant mixtures.12,13 Because of their large interfacial area and resulting high surface energy, foams are thermodynamically unstable.14 The main mechanisms governing foam destabilisation are coarsening (diffusion of gas between bubbles),15 gravitational drainage16 and coalescence (merging of bubbles by rupturing of films).17 Both, from a fundamental point of view and also for some applications, like extraction processes, the control of foam stability by external stimuli is very interesting. By carefully choosing the composition, foams responsive to temperature, pH, magnetic fields or light irradiation can be obtained.18–21 Microgels (MGs) are cross-linked polymer particles, which can swell in an appropriate solvent and combine characteristics of polymers and particles.22 By choosing a specific polymer, MGs can be tailored to be stimuli-responsive. Using the thermoresponsive polymer poly(N-isopropylacrylamide) (PNIPAM), which has a lower critical solution temperature of approximately 32 °C,23–25 temperature responsive MGs are obtained, which collapse when the temperature is increased above the volume phase transition temperature (VPTT).26PNIPAM MGs synthesised using the well-established precipitation polymerisation method,27 have a heterogeneous structure with a densely cross-linked core and a loosely cross-linked shell with dangling polymer strands.28,29 These MGs are surface active and readily adsorb to air/water or oil/water interfaces.30–32 This inspired the fabrication of a range of stimuli-responsive emulsions which can be broken by changing the pH or the temperature.33–37 Originally the stimuli-response of these emulsions was hypothesised to be related to a lateral collapse of the MGs at the interface, which would reduce the surface coverage and facilitate the coalescence of emulsion droplets.32,37 Recent studies by Harrer et al. and Bochenek et al. investigating the temperature response of MG monolayers adsorbed at the air (oil)/water interface rebutted this interpretation as they found that adsorbed MGs only show a size reduction in the direction normal to the interface when heated above the VPTT.38,39 More precisely, the size of the flat corona adsorbed at the interface is not affected by temperature, whereas the core shrinks when the system is heated above the VPTT.39,40 The structure of PNIPAM MGs adsorbed at interfaces was also studied thoroughly. Given their soft nature, MGs deform upon adsorption at an interface.41–45 Picard et al. and Rey et al. studied the lateral ordering of MGs at the air/water interface by analysing monolayers transferred to a solid substrate along the compression isotherm in a Langmuir trough.43,44 In both studies very long ranged hexagonal ordering of the MGs even at very low surface pressures was observed. Upon compression, the inter-particle distance decreases gradually and eventually a phase transition characterised by an abrupt change in inter-particle distance is observed. In both studies this phase transition is explained by a change from shell-shell to core-core contact of the MGs. The phase transition seems to be facilitated by a higher amount of cross-linker used during the MG synthesis. Furthermore, Rey et al. observed the phase transition at lower surface pressures for higher cross-linker concentrations.44 Considering the large amount of publications dealing with PNIPAM MG stabilised emulsions, there are only very few publications investigating foams stabilised by PNIPAM MGs. Horiguchi et al. demonstrated that MG stabilised foams can be obtained at temperatures below the VPTT, whereas no foam formation is observed at temperatures above the VPTT.46 In addition the foams are temperature responsive as they rapidly collapse when the temperature is increased above the VPTT. Maestro et al. reported similar results and were able to detect a transition from a solid-like (T < VPTT) to a liquid-like (T > VPTT) behaviour of the interfacial MG films by passive interface rheology.47 According to the authors the liquid-like film should have a higher permeability, which could explain the decrease in foam stability at temperatures above the VPTT.
This article aims to deepen the understanding of aqueous foams stabilised by PNIPAM MGs. A series of MGs with varying cross-linker content is used to study macroscopic foam properties like foam stability and foamability and the influence of temperature on them. Furthermore, the structure of MG stabilised foams is investigated by analysing single horizontal foam films in a thin film pressure balance (TFPB) and macroscopic foams by small-angle neutron scattering (SANS). In addition, the deformation of MGs inside foam films is correlated with their elastic properties measured by atomic force microscopy (AFM).
2 Experimental section
Materials
N-Isopropylacrylamide (NIPAM, ≥99%) and N,N′-methylenebisacrylamide (BIS, ≥99.5%) were purchased from Sigma-Aldrich (USA). 2,2′-Azobis(2-methyl-propanimidamide) dihydrochloride (AAPH, ≥98%) was purchased from Cayman Chemical Company (USA). D2O (99.9% D) was purchased from Eurisotop (France). All chemicals were used as received. Deionised water from a MilliQ water purification system (Merck KGaA, Germany) with a specific resistance of 18.2 MΩ cm was used.Microgel synthesis
Microgels were synthesized by surfactant-free precipitation polymerization.27 In a custom-built, double walled reaction vessel, equipped with a reflux condenser and stirrer, NIPAM and BIS (total amount 20 mmol with varying NIPAM/BIS ratios, see Table 1) were dissolved in 120 mL water. The solution was heated to 80 °C and sparged with nitrogen gas for 1 h. Under constant stirring (1000 rpm) the reaction was initiated by injecting AAPH (33.9 mg, 0.125 mmol) dissolved in 1 mL water into the reaction mixture. After 1.5 h the turbid suspension was cooled down to room temperature and dialysed for 10 days against water with daily water exchange. The cleaned microgel suspension was lyophilised.| Name | c(BIS) (mol%) | n(NIPAM) (mmol) | n(BIS) (mmol) | R h (20 °C) (nm) | R h (50 °C) (nm) | h(AFM,dry) (nm) | ζ (20 °C) (mV) | ζ (50 °C) (mV) |
|---|---|---|---|---|---|---|---|---|
| MG2.0 | 2.0 | 19.60 | 0.40 | 393 ± 8 | 133 ± 4 | 66 ± 11 | 13 ± 4 | 44 ± 6 |
| MG3.5 | 3.5 | 19.36 | 0.70 | 319 ± 5 | 128 ± 2 | 78 ± 19 | 7 ± 4 | 34 ± 5 |
| MG5.0 | 5.0 | 19.01 | 1.00 | 320 ± 11 | 142 ± 2 | 113 ± 11 | 6 ± 4 | 35 ± 5 |
| MG7.5 | 7.5 | 18.56 | 1.50 | 348 ± 7 | 189 ± 8 | 184 ± 19 | 10 ± 4 | 31 ± 6 |
| MG10.0 | 10.0 | 18.03 | 2.00 | 419 ± 17 | 246 ± 7 | 244 ± 18 | 11 ± 4 | 36 ± 5 |
Microgel characterization
The hydrodynamic radii Rh of all microgels (MGs) at 20 °C and 50 °C were determined using a multi-angle dynamic light scattering (DLS) setup (LS Instruments, Switzerland). The ζ potentials of all MGs at 20 °C and 50 °C in water were measured with a Zetasizer Nano (Malvern Panalytical, UK) under the Smoluchowski approximation. MG samples for atomic force microscopy (AFM) measurements were prepared by spin coating 0.5 mL of 0.05 wt% MG dispersion at 1000 rpm for 90 s and 2000 rpm for 30 s onto 10 × 10 mm2 silicon wafers (Soitec, France) at room temperature. Immediately before the spin coating procedure the silicon wafers were cleaned by exposure to oxygen plasma (Diener Femto, Germany). AFM scans in air of spin coated MG samples were performed with a MFP-3D AFM (Oxford Instrument, UK) situated in an acoustic enclosure using an AC160TS cantilever (Olympus, Japan) with a typical spring constant of k = 26 N m−1 and a resonance frequency of fres = 300 kHz in tapping mode. The height of the adsorbed MGs in the dried state was determined by analyzing at least 50 individual MGs per sample with the analysis tool included in the AFM control software.AFM indentation measurements were performed with a Cypher AFM (Oxford Instruments, UK) equipped with a BL-AC40TS cantilever (Oxford Instruments, UK, k = 0.09 N m−1, fres = 110 kHz). Before each measurement the exact spring constants of the cantilevers were determined to be 0.076–0.112 N m−1 by the method described by Sader et al.48 The MG sample, spin coated as described above, and the cantilever were submerged in water and the temperature was set to 20.0 °C. After equilibration for at least 30 min, force curves were recorded by indenting the tip of the cantilever into the center of the adsorbed MGs. At least 8 force curves from different MG particles were recorded for each sample. Elastic properties of the MGs were extracted by fitting the force curves with the Hertz model:
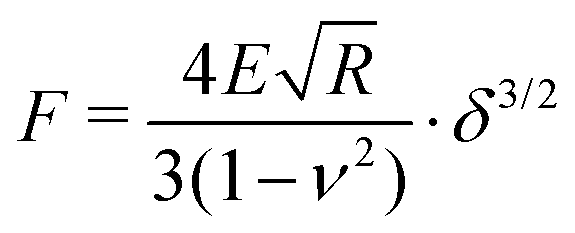 | (1) |
Foaming experiments
Foams were generated using the Bartsch method49 in which a defined amount of liquid is shaken for a defined amount of time in a container of defined geometry. In the experiments presented in this article foams were generated by vigorously shaking 8 mL of microgel (MG) dispersion in a 40 mL glass vial (inner diameter: 27.5 mm, height: 95 mm) by hand for 30 s. The foam decay was monitored by taking photos in regular time intervals and subsequent image analysis. Image analysis was performed using the Fiji distribution of the open-source image analysis software ImageJ.50 Pixel sizes were converted into real-space lengths by using the height of the foaming vial as a reference. The foam height was measured as the distance between the upper edge of the foaming solution and the upper edge of the foam. The foamability is the initial foam height immediately after the foaming procedure. The foam stability is judged by the half life time t1/2, which is the time it took for the initial foam height to be cut in half. Dispersions in water of all MGs mentioned in Table 1 were studied at three different concentrations (w = 0.3, 0.5 and 1.0 wt%). In order to investigate the influence of temperature on the foamability, the MG dispersions were preheated for 30 min in a 50 °C water bath and shaken according to the procedure described above. The temperature dependent foam stability was studied by placing MG dispersions, which were foamed at 22 °C into a 50 °C water bath and subsequent photo monitoring. Every foaming experiment was repeated at least three times. Prior to use, the glass vials were soaked in diluted alkaline Q9 cleaning solution (Bernd Kraft, Germany) overnight and thoroughly rinsed with water.Small angle neutron scattering
Small-angle neutron scattering (SANS) experiments were carried out at the KWS-1 instrument at the Heinz Maier-Leibnitz-Zentrum (MLZ, Garching, Germany).51,52 All measurements were performed with a 10 × 10 mm2 neutron beam with a wavelength of 0.49 nm and a wavelength resolution (FWHM) of 10%. Scattering patterns were recorded with a 2D 6Li-scintillation detector with photomultiplier tubes and a spatial resolution of 5.3 × 5.3 mm2 at sample–detector distances of 7.615 m (collimation length 8 m, data acquisition time 5 min) and 19.615 m (collimation length 20 m, data acquisition time 15 min), covering a q-range of ≈0.04–1.1 nm−1. Radial averaging of the 2D detector images, processing and merging of the data was done with the QtiSAS software.53 A custom-built sample cell was used specifically designed for studying macroscopic foams with SANS. Technical details are given in a previous publication.54 Foams were produced from microgel dispersions in D2O (w = 0.3, 0.5 and 1.0 wt%) by bubbling nitrogen gas at a rate of 10 mL min−1 through a porous glass plate (pore size 10–16 μm, porosity P16 (ISO 4793)) at the bottom of the cell. Once the foam level reached the top of the column the flow rate was reduced to a value to obtain a constant foam height over time and to prevent overflowing of the foam column. The flow rates were in a range of 1–3 mL min−1 with the exact value depending on the sample studied. Once this steady-state was reached, measurements at three different foam heights (h = 2 cm, 9.5 cm and 16 cm above the foaming solution) were performed.The SANS data were analysed using a self-written program and the following model:
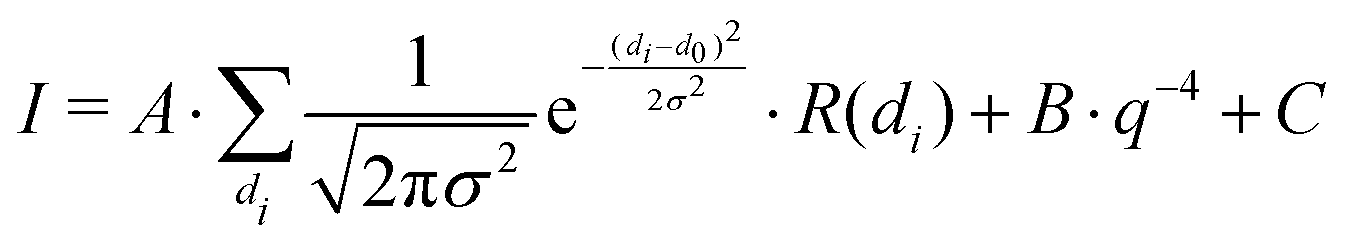 | (2) |
The model uses an incoherent superposition of reflectivity curves of a set of normal distributed film thicknesses and a Porod contribution. A and B are scaling factors for the reflectivity and Porod contribution, respectively. d0 and σ are the mean and standard deviation of the normally distributed film thicknesses. R(di) is an individual reflectivity curve for a layer in air with thickness di and C is the constant, incoherent background. This model is an adaptation of a model described in detail in another publication.55
Thin film pressure balance
Individual, horizontal foam films of MG dispersions (w = 0.3 wt%) were studied with a thin film pressure balance (TFPB) using the porous plate technique at 22 °C.56,57 In our custom-built setup, the film is formed in a 1 mm hole drilled into a porous glass disc (pore size 10–16 μm, porosity P16 (ISO 4793)). The film holder is placed in a sealed stainless steel chamber, which also contains a reservoir of the studied MG dispersion to ensure a saturated atmosphere and thereby preventing film drying. Before each measurement, the film holder was immersed in the MG dispersion for at least 2 h to equilibrate the porous glass disc with the solution. Right before the film formation, the film holder is raised from the sample solution and equilibrated for 30 min in the saturated atmosphere inside the pressure chamber. Upon increasing the pressure inside the sample cell, inhomogeneous foam films were formed. Disjoining pressure isotherms Π(d) were recorded by interferometrically measuring the equilibrium film thickness d as a function of pressure inside the sample cell. The equilibrium film thickness was assumed to be reached 5 min after the desired pressure was reached. Since the foam films studied are inhomogeneous, the whole film area is illuminated by a cold filtered halogen lamp and imaged by a colour CMOS camera (JAI Go-2400-USB, pixel size: 5.86 μm × 5.86 μm, Stemmer Imaging, Germany). In combination with the optics (reflected light microscope, focal reducer, extension tube), the resolution of the camera system is 1.07 pixel μm−1. Since the films are colourful (thickness > 100 nm), a purely intensity based thickness calculation approach cannot be used. Instead, a colour comparing algorithm was used to determine the film thickness. The algorithm is based on a modeled spectrum assuming a free standing water slab surrounded by air.58 The spectrum is represented in the HSV colour space (hue, saturation, value) and is stored in a lookup table with the regarding thickness values in 1 nm steps. For each film thickness in the range from 100 nm to 800 nm there is a corresponding set of unambiguously HSV values. Single spots with distinguishable features were selected for every measurement and spatially tracked over the whole pressure ramp. The film thickness was determined by an automated comparison between the experimental and theoretical hue colour values from the lookup table. Being grey, the thickness of the interspersed film areas can be assumed to be below 100 nm and therefore within the first interference order. The disjoining pressure isotherms presented in this article are an average from at least three individual measurements. Since the TFPB technique requires rather large sample volumes (around 30 mL), only the lowest MG concentration was investigated.3 Results
3.1 Microgel characterization
The hydrodynamic radius, ζ potential and height in the dried state of all microgels (MGs) are summarised in Table 1. A temperature-induced collapse of all MGs is observed. This volume phase transition is well-known for NIPAM MGs26 and is linked to the lower critical solution temperature (LCST) of linear PNIPAM, which is approximately 32 °C.23,24 All MGs have a small, positive ζ potential in the swollen state at 20 °C, which is due to the positively charged initiator fragments incorporated during the MG synthesis. In the collapsed state at 50 °C the ζ potential increases notably, which is attributed to the reduced surface area of the collapsed particles, while the total charge of the individual MGs stays constant. The height in the dried state h(AFM,dry) decreases with decreasing cross-linker content, which can be explained by the higher deformability of MGs with smaller cross-linker content.44 Representative height profiles of all MGs are shown in Fig. S2 in the ESI.†Fig. 1 shows the swelling ratio of the MGs studied as a function of the cross-linker concentration c(BIS). The swelling ratio is calculated as the hydrodynamic volume increase upon swelling (R20/R50)3. The swelling ratio decreases with increasing cross-linker concentration, which is an indirect indication for decreasing deformability with increasing cross-linker concentration.
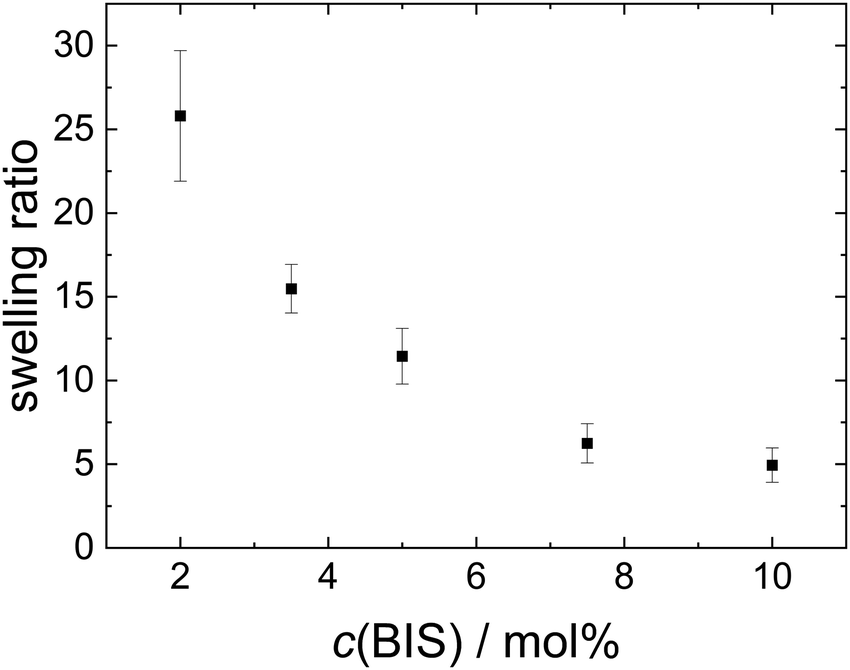 | ||
| Fig. 1 Temperature induced swelling ratio of the MGs as a function of cross-linker concentration. | ||
Fig. S1 in the ESI,† shows AFM scans of spin-coated MG samples under ambient conditions in air. All MGs have a spherical shape and a low polydispersity.
3.2 Foaming experiments
The foamability, which is the initial foam height immediately after foam formation, of the different MG dispersions at 22 °C is summarised in Fig. 2. The foamability of all MG dispersion increases approximately linear with the MG concentration w (see Fig. 2(a)). This observation indicates the absence of any aggregation processes in the studied concentration range and can be rationalised by the consideration that a larger number of MGs are capable of stabilising a larger interfacial area. Assuming a constant bubble size for the different MG concentrations leads to a linear relation between w(MG) and the foamability. Despite the seemingly imprecise foaming procedure, all foamability values are well reproducible. This suggests that during the foaming procedure some type of steady-state regarding the foamability is reached, which is not influenced by further shaking cycles. The existence of this steady-state in foamability was also reported in literature and is explained by an equilibrium between bubble formation and coalescence during the foaming procedure.59,60 When the foamability is plotted against the cross-linker concentration c(BIS) (see Fig. 2(b)), a non-monotonous trend is observable at all three MG concentrations studied. The foamability is maximal for c(BIS) = 2.0 mol%, decreases to its minimum around 5.0 mol% and increases slightly again for higher cross-linker concentrations.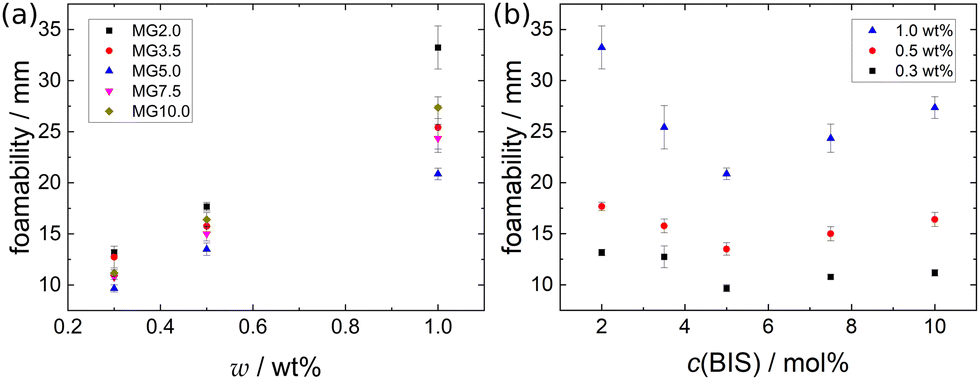 | ||
| Fig. 2 Foamability of the MG dispersions at T = 22 °C (a) as a function of MG concentration w and (b) as a function of cross-linker concentration c(BIS). | ||
 | ||
| Fig. 3 Foamability of a 1.0 wt% dispersion of MG2.0 (a) at T = 22 °C and (b) at T = 50 °C. Red dotted lines highlight the foam height, i.e. foamability. | ||
Besides the foamability, also the foam half life time t1/2 was measured. Fig. S3 in the ESI† shows a selection of photographs used to determine the half life time t1/2 at T = 22 °C of MG dispersions with w = 1.0 wt%. Fig. 5 (closed symbols) shows t1/2 at T = 22 °C of all MG dispersions studied. There is no clear trend observable for t1/2, neither as a function of MG concentration, nor as a function of cross-linker concentration. Most of the studied foams have half life times in the range of 10 ± 5 h with standard deviations of several h.
Fig. 4 shows a series of photographs used to determine t1/2 at T = 50 °C of a 1.0 wt% dispersion of MG2.0. Fig. 4(a), (b) and (c) show photographs of the foamed dispersions directly after foam formation (t = 0), after t = 5 min and t = 12 min, respectively. Fig. 5 summarises the half life times at T = 22 °C (closed symbols) and at T = 50 °C (open symbols) of all MG dispersions studied. t1/2 at T = 50 °C is independent of MG and cross-linker concentration and is approximately 15 ± 5 min for every foam studied, meaning that t1/2 at 50 °C is around two orders of magnitude lower than t1/2 at 22 °C.
 | ||
| Fig. 4 Exemplary foam decay T = 50 °C of a foam formed from a w = 1.0 wt% dispersion of MG2.0 subjected to a T = 50 °C water bath at t = 0. (a) Immediately after foam formation, (b) after 5 min and (c) after 12 min. Red dotted lines highlight the foam height. | ||
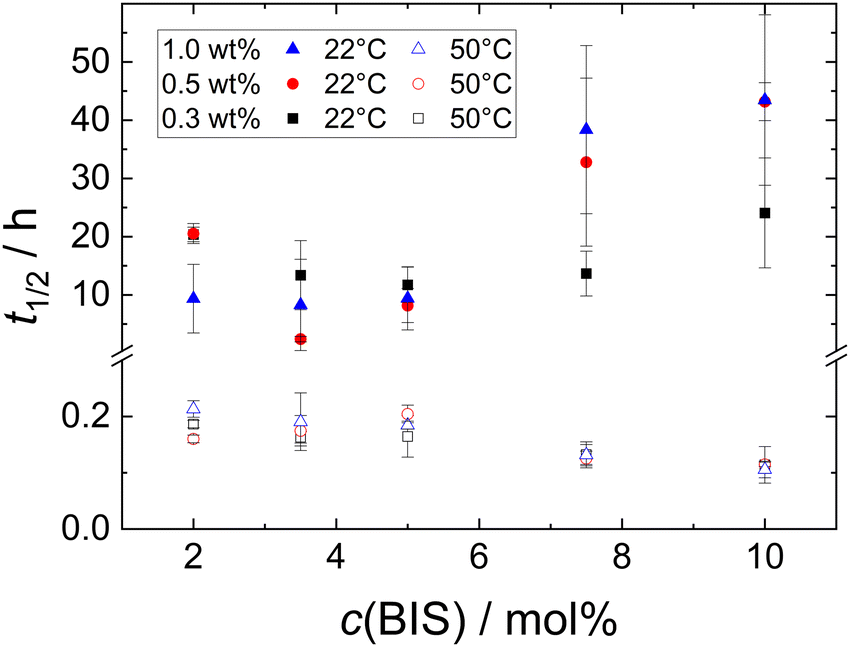 | ||
| Fig. 5 Half life time t1/2 at T = 22 °C (closed symbols) and T = 50 °C (open symbols) of foams produced from MG dispersions at three different concentrations as a function of cross-linker concentration c(BIS). | ||
3.3 SANS measurements
Fig. 6 shows SANS data of a foam prepared from a 0.3 wt% dispersion of MG2.0 recorded at different foam heights ((a) h = 2 cm, (b) h = 9.5 cm, (c) h = 16 cm) together with the model fits obtained using eqn (2) with a scattering length density (SLD) of ρfilm = 4.5 × 10−6 Å−2 for the foam films in the reflectivity contribution. For all foam heights the overall scattered intensity is dominated by the Porod contribution (I ∝ q−4) at low q. At q ≈ 0.1–0.2 nm−1 the scattered intensity is set off from the strict Porod behaviour (dashed green line) to higher intensities by the reflectivity contribution (dotted blue line). The magnitude of this intensity offset is increased for the two higher measurement positions (h = 9.5 cm and h = 16 cm). The fit parameters used for the model fits in Fig. 6 are summarised in Table 2. The fit parameters are: reflectivity scale factor A, mean foam film thickness d0, standard deviation of the foam film thickness σ, Porod scale factor B and incoherent background C.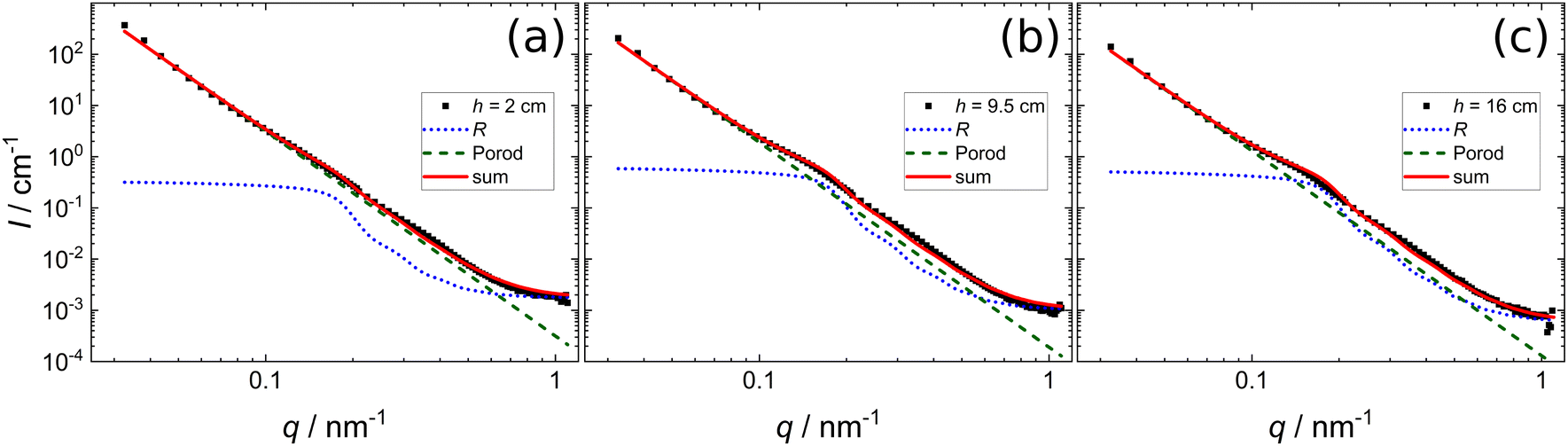 | ||
| Fig. 6 SANS data of a foam prepared from a 0.3 wt% dispersion of MG2.0, measured at different foam heights. (a) h = 2 cm, (b) h = 9.5 cm, (c) h = 16 cm. Black squares are experimental data. Lines are model fits according to eqn (2). For clarity the full curve (solid red line) is shown together with the Porod (dashed green line) and the reflectivity (dotted blue line) contribution. | ||
| h (cm) | A (cm−1) | d 0 (nm) | σ (nm) | B × 10−8 (nm4 cm−1) | C × 10−3 (cm−1) |
|---|---|---|---|---|---|
| 2 | 5.89 | 24.7 | 291 | 3.15 | 1.77 |
| 9.5 | 8.48 | 24.5 | 225 | 1.90 | 1.03 |
| 16 | 6.53 | 23.2 | 199 | 1.30 | 0.60 |
With increasing foam height, both the Porod scale factor B and the incoherent background C decrease. The reflectivity scale factor A first increases from 5.89 cm−1 to 8.48 cm−1 when the foam height is increased from 2.0 cm to 9.5 cm and then decreases again to 6.53 cm−1 for the highest foam height measured (h = 16.0 cm). The mean foam film thickness d0 is around 23 nm to 25 nm for all foam heights. The standard deviation decreases from 291 nm to 199 nm with increasing foam height. This large standard deviation is further discussed in Section 4.2.
Since all scattering curves are dominated by the Porod contribution and the features arising from the reflectivity contribution are barely visible in Fig. 6, the SANS data of foams prepared from 0.3 wt% dispersions of four different MGs ((a) MG2.0, (b) MG3.5, (c) MG5.0, (d) MG7.5) are presented in Porod plots (I·q4vs. q) in Fig. 7. For comparison a theoretical reflectivity curve of a free-standing 30 nm thick film in air with a SLD contrast of ρfilm = 4.5 × 10−6 Å−2 is shown (black line). Here, the q−4 decay corresponds to the Fresnel reflectivity of a bare interface. The Porod plots of foams prepared from 0.5 wt% and 1.0 wt% MG dispersions, are shown in the ESI† (Fig. S4 and S5).
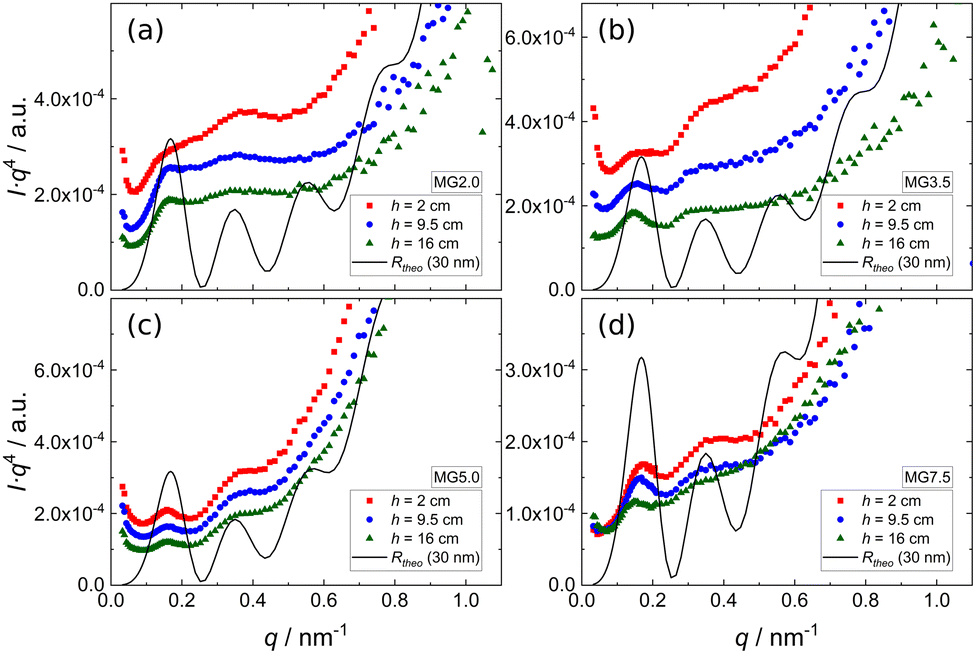 | ||
| Fig. 7 Porod plots of SANS data of foams prepared from 0.3 wt% dispersions of (a) MG2.0, (b) MG3.5, (c) MG5.0, (d) MG7.5, measured at different foam heights (red squares (h = 2 cm), blue circles (h = 9.5 cm) and green triangles (h = 16 cm)). The curves are shifted in intensity for clarity. For comparison a theoretical reflectivity curve of a 30 nm film in air with a SLD contrast of ρfilm = 4.5 × 10−6 Å−2 is shown (black line). | ||
All scattering curves display a first maximum at q ≈ 0.15 nm−1 and a very weak, in some cases barely visible, second maximum at q ≈ 0.33 nm−1. Albeit much less pronounced, both maxima and the intermediate minimum in the experimental SANS curves are at the same positions as the corresponding extrema of the theoretical reflectivity curve. It is important to note that for sufficiently thick films (d > 20 nm) the position of the first maximum of the theoretical reflectivity curve in the Porod plot corresponds to its critical edge and its q-position is therefore only governed by the SLD contrast between the film (ρfilm) and the surrounding medium (air, ρair) via the following relation:
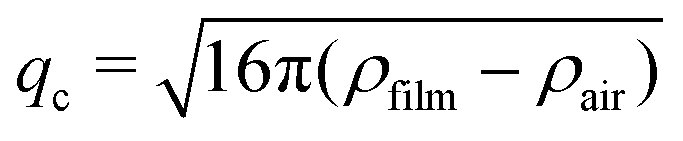 | (3) |
Assuming ρair = 0, the position of qc (i.e. the first maximum in the SANS curves) allows to calculate ρfilm. The first maximum in the SANS curves of the MG foams appears at qc ≈ 0.15 nm−1, which means that the foam films consisting of MGs and D2O have a SLD ρfilm of approximately 4.5 × 10−6 Å−2. An estimation of the foam films' expected SLD is not possible, because their composition (MG concentration inside the foam film) and the composition of the individual D2O swollen MGs is unknown. There are however reference values in literature which were determined using neutron reflectometry. Widmann et al. reported a SLD of a thin film of PNIPAM MGs on a solid substrate in a saturated D2O atmosphere of around 3 × 10−6 Å−2.61 Assuming that the MGs swell more in pure D2O than in a D2O vapour and taking into account the increased D2O concentration (ρD2O = 6.34 × 10−6 Å−2) in the foam films, a SLD of 4.5 × 10−6 Å−2 for the MG stabilised foam films is realistic.
3.4 TFPB measurements
Fig. 8 shows photographs of individual foam films formed in the TFPB using (a) MG2.0, (b) MG3.5, (c) MG5.0, (d) MG7.5, (e) MG10.0 at a concentration of w = 0.3 wt% at intermediate pressures of the isotherms. In all cases, the films consist of a coloured area which is interspersed with small, grey spots with varying brightness. The colours of the films do not originate from the usage of a filter or image post processing, they originate from interference of white light. Consequently, the thickness of the colourful areas is in the range of several 100 nm and can be analysed using the colour comparing algorithm described in Section 2. The exact thickness of the grey areas is more difficult to determine because this would require an intensity calibration, which is very challenging in the case of inhomogeneous films. However, as they appear to be colourless their thickness should be less then 100 nm. | ||
| Fig. 8 Photographs of foam films inside the TFPB, stabilised by different MG dispersions at a concentration of w = 0.3 wt% (a) MG2.0, (b) MG3.5, (c) MG5.0, (d) MG7.5, (e) MG10.0. Pictures are taken at intermediate pressures of the isotherms shown in Fig. 9, which are ≈500 Pa for MG2.0, MG3.5, MG5.0 and MG7.5 and ≈150 Pa for MG10.0. Scale bars are 200 μm. | ||
The disjoining pressure isotherms of the colourful areas of the foam films, stabilised by the different MGs, are shown in Fig. 9. In the case of MG2.0, the foam film thickness d decreases notably with increasing disjoining pressure Π. The foam film thicknesses in the case of the other MGs do not decrease significantly with increasing Π. The foam films with MG10.0 were only stable until Π ≈ 200 Pa, whereas all other foam films were stable up to Π > 1000 Pa. The initial foam film thickness d increases with the cross-linker concentration c(BIS) from d ≈ 300 nm for MG2.0 to d ≈ 540 nm for MG10.0.
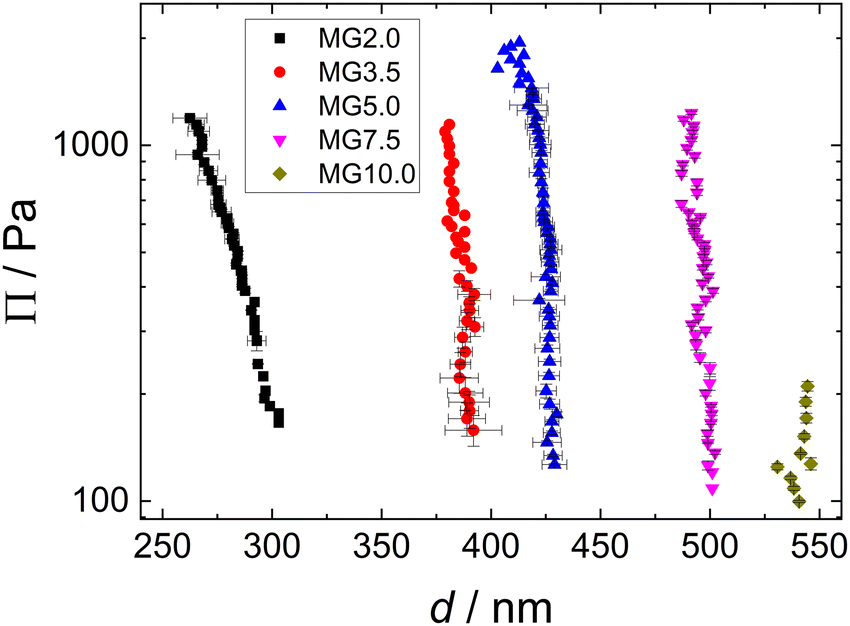 | ||
| Fig. 9 Disjoining pressure isotherms of coloured area of foam films stabilised by different MG dispersions (w = 0.3 wt%). | ||
3.5 AFM indentation measurements
In Fig. 10(a) exemplary force curves (open symbols) are shown together with the corresponding fits using the Hertz model (solid lines, eqn (1)). The slope of the force increase upon indentation increases with the cross-linker concentration. This shows the influence of the cross-linker concentration on the mechanical properties of the MGs. The negative forces measured close to the point of contact between AFM tip and MG (indentation = 0) are due to attractive interactions between tip and MG.62,63 The elastic moduli E of the different MGs (given in Fig. 10(b)) increase from 100 kPa for MG2.0 to 400 kPa for MG10.0. All force curves used for the determination of E for every MG are summarised in Fig. S7 in the ESI.† Every force curve was recorded at the apex, i.e. height maximum, of a individual MG, because of the radial inhomogeneity of the elastic properties of MGs synthesised with the batch method used here.64–68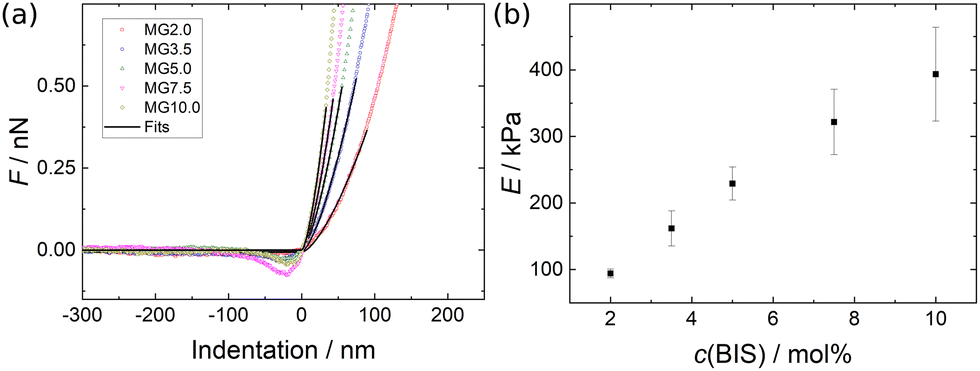 | ||
| Fig. 10 (a) Exemplary force curves for all MGs studied (open symbols) with corresponding fits using the Hertz model (solid lines, eqn (1)). (b) Elastic moduli as a function of cross-linker concentration extracted from Hertz model fits to the force curves shown in Fig. S7 (ESI†). | ||
Similar experiments were reported in literature, which yielded elastic moduli in the range from 5 kPa to 550 kPa for PNIPAM MGs prepared with varying amounts of cross-linker and synthesis conditions.64,69–73
4 Discussion
4.1 Macroscopic foam properties
The foamability increases with the MG concentration, which indicates that a fast coverage of the surface by MGs supports the foam formation. A possible explanation for the non-monotonous trend in foamability with increasing cross-linker concentration described in Section 3 is the difference in size of the MGs. This possible correlation is investigated in Fig. 11. Here, the foamability at different concentrations (black squares: w = 0.3 wt%, red circles: w = 0.5 wt% and blue triangles: w = 1.0 wt%, left ordinate) is plotted together with the hydrodynamic radius Rh(20 °C) (connected green diamonds, right ordinate) as a function of the cross-linker concentration c(BIS).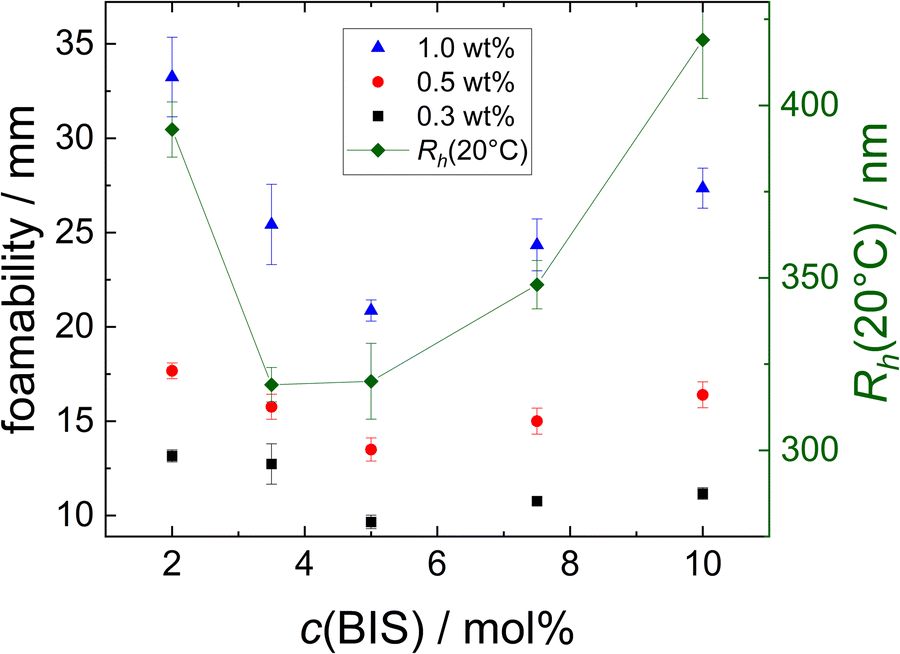 | ||
| Fig. 11 Foamability of the MG dispersions at different concentrations (black squares: w = 0.3 wt%, red circles: w = 0.5 wt% and blue triangles: w = 1.0 wt%, left ordinate) and hydrodynamic radius Rh(20 °C) (connected green diamonds, right ordinate) as a function of cross-linker concentration c(BIS). | ||
R h(20 °C) also shows a non-monotonous trend with respect to c(BIS), displaying a minimum between c(BIS) = 3.5 mol% and 5.0 mol%. Although the trend is similar for Rh and foamability, a direct correlation is not obvious. In this context it is important to keep in mind that the concentrations of the MG dispersions are given in wt%. Assuming that all MGs have a similar density, the number of MG particles present in a dispersion with a certain mass concentration w should be inversely proportional to the size of the MGs. This relation would suggest that the foamability of the resulting dispersions also should be inversely proportional to the size of the MGs, as the MGs' total surface area is higher at a given mass concentration. Even if there are differences in the density of the MGs with varying cross-linker concentration, this effect should not suffice to invert the expected trend described above. Furthermore, MGs deform upon adsorption to an air/water interface. The extent of this deformation is, among other things, influenced by the cross-linker concentration.43,44 Consequently, the area occupied by a single MG adsorbed to the air/water interface cannot be predicted only considering its size in bulk. Furthermore, a part of the MGs will not be adsorbed to the air/water interface during the foaming procedure, but remain in the bulk liquid. The distribution of MGs between foam and bulk liquid might again depend on parameters like the cross-linker concentration and MG size.
The exact origin of the observed trend in foamability remains unclear, mainly because of the complexity of foam formation itself and the deformability of MGs. However, it seems that the MG size is not the dominating factor. There are several parameters governing foamability discussed in literature like viscosity of the bulk solution,74,75 dynamic surface tension76–79 or dilatational surface viscosity.80–83 However, there is no unifying general theory for foam formation as the relevant factors depend on the system and even the foaming procedure used.59,84,85
The slight increase of t1/2 observed for the MG7.5 and MG10.0 at high MG concentrations could be related to the formation of MG aggregates inside the foam films or Plateau borders and subsequent slowdown or blockage of drainage. The overall stabilising effect of aggregate formation or “jamming” inside the liquid “bulk” phase of foams, i.e. the Plateau borders, is well documented.86–89 In addition, MGs with higher cross-linker concentration were reported to form aggregates with close core-core contacts more readily than MGs with lower cross-linker concentrations at a single air/water interface.43,44 Following this argumentation, the increased foam stability at high cross-linker contents and MG concentrations could be rationalised by the presence of MG aggregates in the Plateau borders. However, since there is no direct proof for the existence of these MG aggregates in the Plateau borders, this explanation should not be over interpreted.
Another noteworthy observation regarding the foam stability is the fact that the maximum disjoining pressure before film rupture Πmax is not related to the macroscopic foam stability at a MG concentration of w = 0.3 wt%. Πmax is minimal for MG10.0 (see Fig. 9), whereas t1/2 is nearly constant for all MGs (see Fig. 5, black squares). A discrepancy between stability of individual foam films and macroscopic foam stability is regularly observed.90 The main reason for this is most likely the difference in experimental conditions between comparably large single foam films under static and isolated conditions in a TFPB and a multitude of small foam films in the highly dynamic setting inside a foam, which sometimes makes a direct comparison of foam films in a TFPB and in actual foams difficult.91,92 In addition, further parameters like the MG adsorption speed, size and deformability could influence the foamability and Πmax in different ways.
The temperature responsiveness of PNIPAM MG stabilised foams was already reported in literature46,47 and suggests a link between the collapse of individual MG particles when heated above the VPTT and the macroscopic foam properties. The foam half life time at 50 °C is not influenced by the MG or cross-linker concentration and approximately 15 ± 5 min for every foam. This finding suggests that the MG foams are drastically destabilised once a certain critical internal temperature is reached. This is also underlined by the photograph in Fig. 4(c), in which the foam decay is progressed further at the edge of the foaming vial, which is in contact with the heated water bath, then in the center. Temperature responsiveness was also reported for MG stabilised oil-in-water emulsions, which are stable at temperatures below and rapidly break at temperatures above the VPTT.35–37,93 MGs adsorbed at interfaces do not collapse in the lateral direction when the temperature is increased above the VPTT, which suggests a different destabilisation mechanism than the reduction of surface coverage.38,39 Another explanation for the temperature responsiveness of MG stabilised emulsions is the temperature induced change in interfacial viscoelasticity of MG laden interfaces.94–96 In our opinion, this explanation also appears to be the best candidate for the rationalization of the temperature responsiveness of MG foams, as the interfacial viscoelasticity is also very often related to macroscopic foam stability.97–100
4.2 Foam structure
In the SANS experiments with MG foams the overall scattered intensity decreases slightly with increasing foam height. This was also observed in various other publications investigating foams with SANS and is explained by drainage of the foam.55,101–104 During drainage the liquid films inside the foam become thinner and eventually rupture.5,16,105 As a result, the amount of contrasted structures in the foam decreases, which leads to a decrease in scattered intensity.55 This trend is also visible in the fit parameters B and C (Table 2), which decrease monotonously with increasing foam height.The fact that the theoretical reflectivity curve displays extrema at the same q values as the experimental SANS curves of the MGs suggests the presence of a slightly preferential film thickness of around 30 nm with a pronounced polydispersity. The large polydispersity of the film thickness is reflected by the very high value of the standard deviation of the foam film thickness σ (Table 2), which is one order of magnitude higher than the mean film thickness d0.
In combination with the results obtained for individual foam films in a TFPB the following picture arises: foam films in MG stabilised foams are inhomogeneous in thickness. Only the thinner parts of the foam films (grey areas in Fig. 8, thickness of around 30 nm) are detectable by SANS (detection limit ≈200 nm in the setup used) but the corresponding scattering features are not very well defined as the thin films have a very high polydispersity and only represent a small portion of the total foam film. The foam film's inhomogeneity is also the reason for the rather exact Porod decay of the scattered intensity (I ∝ q−4) as the thicker parts of the foam films are too large to display a distinct form factor and are only detectable as single interfaces by the neutrons.106 In agreement, deviations from strict Porod behaviour were reported for SANS curves from surfactant stabilised foams, which should have thinner foam films with a more homogeneous thickness profile.55,101,103,104,107–109
It is well established that MGs, adsorbed at a single air/water interface, adopt very well defined long-ranged hexagonal structures even at low surface pressures.43–45 The behaviour in foam films however, can be different as the presence of a second air/water interface in close proximity can add further geometrical constraints. Keal et al. were the first to report the inhomogeneous structure of foam films stabilised by PNIPAM MGs.110 This observation is further corroborated here, which suggests that the presence of a second air/water interface changes the interfacial arrangement of MGs as they are gradually confined inside the foam film. Keal et al. were able to verify the depletion of MGs from the thinner regions of the foam film via fluorescence microscopy. The absence of MGs in the thinner and by SANS detectable regions of the foam films also explains the very similar scattering curves irrespective of MG concentration or cross-linker content. The stabilisation mechanism of these thinner regions is still unclear. The dangling ends of MGs might extend into the depleted zones, but since these zones can be 10 μm and even more in diameter this cannot be the sole explanation. Another possibility is that the MGs around the depleted areas are heavily jammed and form a sort of corset structure which stabilises the depleted zones. Finally, small surface active components like PNIPAM oligomers might still be present in small quantities after the dialysis and could stabilise the MG depleted zones, although no impurities could be identified after dialysis. Again, it is possible that the MG structuring inside the foam is slightly different than observed in the TFPB, because of the different geometries. Despite the clear connection between the results obtained with both methods, it is possible that the MG depleted zones in the foam films of the macroscopic foam are for example smaller than the ones observed for single foam films in the TFPB.
The question whether a mono-, bi-, or even multilayer of MGs forms in the foam films was addressed by releasing the pressure inside the TFPB and observing the evolution of the film structure over time. Exemplary photos of a MG5.0 foam film are shown in Fig. S6 in the ESI† as all MGs used in this study show similar behaviour in this regard. Upon pressure release the overall film area decreases gradually. The structuring inside the foam film, i.e. the shape of the thinner regions does not change until the meniscus front reaches them. This also means that the thicker parts of the foam film are “pinned”, which was interpreted by Keal et al. as a monolayer of MGs briding the two air/water interfaces.110 Moreover, the thickness of this monolayer region in the case of a MG with a cross-linker concentration of 5 mol% and a hydrodynamic radius of 330 nm, observed by Keal et al. was 400 nm, which is very similar to the film thickness of 425 nm observed in this study for MG5.0 (Rh(20 °C) = 320 nm).
4.3 Elastic properties of microgels govern their compression in foam films and deformation upon drying
Since the thickness of the colourful areas of the foam films increases with the cross linker concentration of the MGs used for stabilising them (see Fig. 9), a relation between the stiffness or deformability of the individual MGs and this thickness is observed. This correlation is explored in Fig. 12, in which the foam film thickness at the lowest disjoining pressure, where the foam film forms d(Πmin) (black squares, left ordinate) and the elastic modulus extracted from AFM indentation experiments E (red circles, right ordinate), are plotted together as a function of the cross-linker concentration c(BIS). There is a linear relation between these two quantities, which is further highlighted in the inset of this figure, in which E is plotted against d(Πmin). The thickness at the lowest disjoining pressure was used in this evaluation, since the elastic moduli were extracted from the force curves by fitting the first few 10 nm of indentation, which should probe the elastic properties at low compression. The absolute values of E might differ from the values reported here, as the Poisson ratio ν of PNIPAM MGs is still not known exactly. The values reported in literature range from 0.25 to 0.5 for temperatures below the VPTT.29,69,70,111 However, assuming that the Poisson ratio is not strongly influenced by the cross-linker concentration the trend of E is maintained. The foam film thickness should be influenced by more parameters than just the elastic properties. For example the overall MG size might have an impact on the foam film thickness. In fact, the hydrodynamic radius at 50 °C (R50), which can be used to estimate the maximum deformability of MGs, also increases with increasing cross-linker concentration (see Table 1). A possible correlation between R50 and d(Πmin) is explored in Fig. S8 in the ESI.† Despite the absence of a linear correlation between R50 and d(Πmin), a possible influence of the MG size in the foam film thickness cannot be ruled out completely. The MGs in the present study might simply have too similar sizes for this effect becoming notable.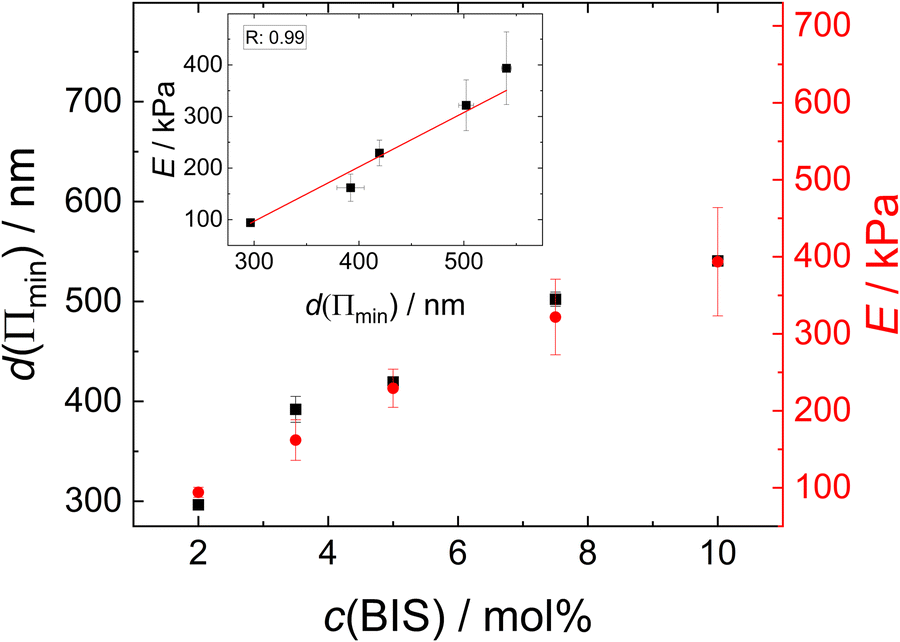 | ||
| Fig. 12 Foam film thickness at the lowest disjoining pressure d(Πmin) (black squares, left ordinate) and elastic modulus E (red circles, right ordinate) as a function the cross-linker concentration c(BIS). The linear correlation between the to parameters is shown in the inset. | ||
The relation observed between d(Πmin) and E suggests that the MGs' resistance to indentation of an AFM tip and to deformation inside a foam film probe the same overall elastic properties. One of the first models for the description of elastic properties of polymer networks is the affine network model.112–114 The main assumption of this model is that the relative macroscopic deformation of a polymer network is the same as the relative deformation of the individual polymer chains. In the following the electrostatic contribution is neglected, since the charges in the MGs are solely caused by the charged initiator fragments, which make up <1% of all monomers. The low charge of the MGs is also reflected by the low ζ potential in the swollen state (see Table 1). The expression for the osmotic pressure arising from the isotropic elastic deformation of such a network is:
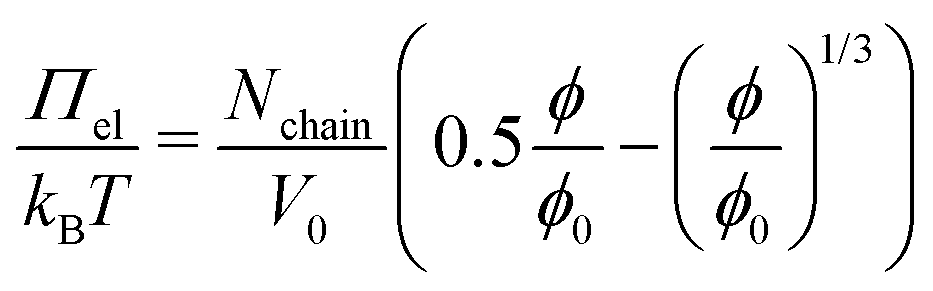 | (4) |
The polymer volume fraction can be estimated by the relation
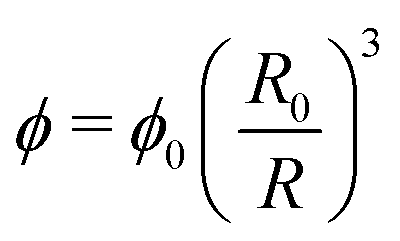 | (5) |
Using this relation and the hydrodynamic radii at 50 °C and 20 °C as reference and experimental state, eqn (4) can be rewritten as:
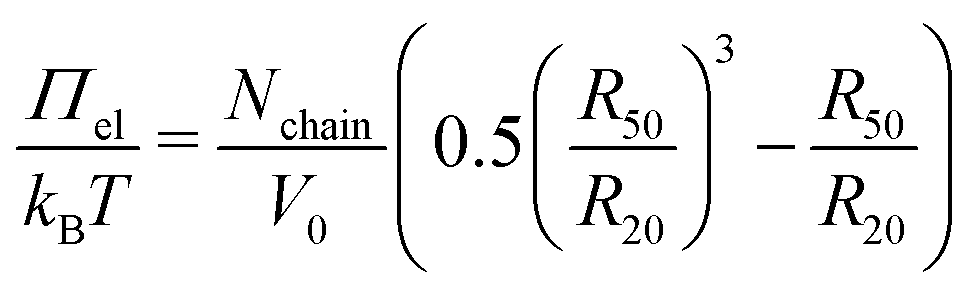 | (6) |
Combining the experimental accessible parameters of the MGs, an affine network factor 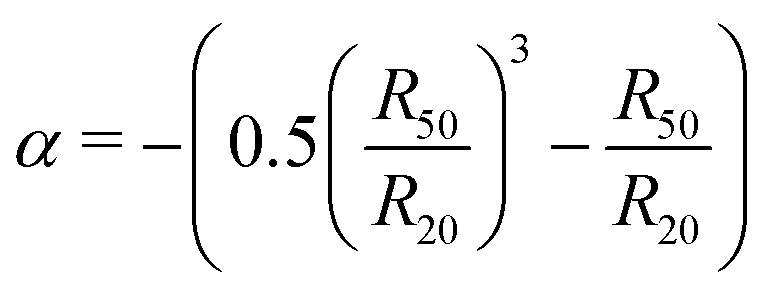 is defined. Possible correlations between α and the elastic moduli E, the foam film thicknesses at the lowest disjoining pressure d(Πmin) and the ratio between the height in the dry state and the hydrodynamic radius at 20 °C h(AFM)/R20(DLS) are explored in Fig. 13. h(AFM)/R20(DLS) can be interpreted as the swelling capacity of MGs when changing from the dry state to the (water-) swollen state.
is defined. Possible correlations between α and the elastic moduli E, the foam film thicknesses at the lowest disjoining pressure d(Πmin) and the ratio between the height in the dry state and the hydrodynamic radius at 20 °C h(AFM)/R20(DLS) are explored in Fig. 13. h(AFM)/R20(DLS) can be interpreted as the swelling capacity of MGs when changing from the dry state to the (water-) swollen state.
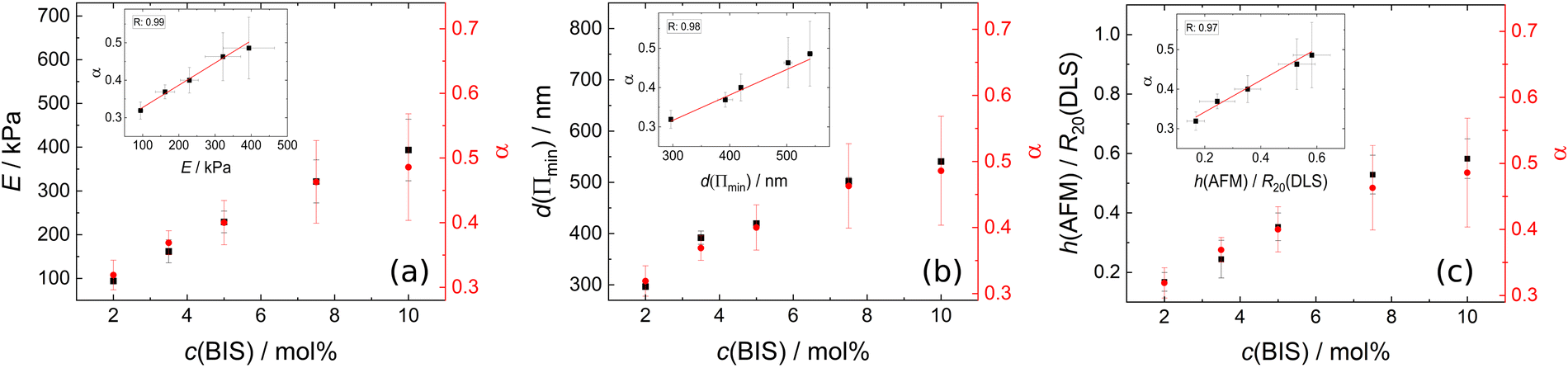 | ||
| Fig. 13 (a) Elastic modulus E (black squares, left ordinate) and affine network factor α (red circles, right ordinate) as a function the cross-linker concentration c(BIS). (b) Foam film thickness at the lowest disjoining pressure d(Πmin) (black squares, left ordinate) and affine network factor α (red circles, right ordinate) as a function the cross-linker concentration c(BIS). (c) Height in the dry state measured by AFM (h(AFM)) related to the hydrodynamic radius at 20 °C measured by DLS (R20(DLS)) (black squares, left ordinate) and affine network factor α (red circles, right ordinate) as a function the cross-linker concentration c(BIS). The linear correlations between the respective parameters is shown in the insets. | ||
All three parameters E, d(Πmin) and h(AFM)/R20(DLS) seem to be strongly linearly correlated with α. These correlations suggest that the deformation of MGs inside a foam film, their swelling capacity and their elastic modulus extracted from AFM indentation measurements can be described by applying the affine network model to MGs. It is worth noting that α in the present case is very similar to the inverse swelling ratio of the MGs, as we used the hydrodynamic radii at 50 °C (R50) and 20 °C (R20) to define it. This means that various pathways of MG deformation (temperature-induced collapse, shrinkage upon drying, deformation in foam films and by an AFM tip), are correlated with each other, which suggests the existence of an underlying concept. The first natural candidate for such a concept is the affine network model, as it is also part of the Flory–Rehner theory, which is widely used for the description of gel swelling. However, some assumptions were made, which have to be kept in mind. The affine network model formalism for isotropic deformation was used, while the deformation of MGs in a foam film is uniaxial and the indentation of a AFM tip into a MG is an even more local deformation. The model for isotropic deformation was used, because the isotropic deformation of MGs upon changing from the reference state (collapsed, T = 50 °C) to the experimental state of interest (here T = 20 °C) can easily be determined using DLS. Despite the difference in deformation symmetry this allows the determination of α with a completely different experimental technique and omits the danger of circular reasoning when correlating α with other parameters. Another point is the question of validity of the affine network model for MGs. Affine swelling was reported for PNIPAM MGs,116 but the exact requirements for the affine network model to be applicable are still unclear.117 In general, there is a variety of models, which try to describe the elastic properties of polymer networks, including the phantom network,118 the constraint-junction,119–121 Edwards tube122 or slip-tube123 model to name a few. The prerequisites for the applicability of each model are still debated, but the polymer concentration and heterogeneity of the polymer network were shown to have an influence on which model describes experimental observations best.124,125 A more detailed understanding of the elastic properties of MGs would be beneficial, especially regarding their possible application as nano-actuators.126–130
5 Conclusion
Aqueous foams stabilised by PNIPAM MGs with varying cross-linker content were studied regarding their macroscopic foam properties and their structure. These foams are thermoresponsive, as they are stable for several hours at temperatures below the VPTT and rapidly collapse when heated above the VPTT. Below the VPTT an increase in foam stability was observed for high cross-linker contents and high MG concentrations, which might be due to the formation of MG aggregates inside the foam and subsequent blockage of drainage. All MGs form heterogeneous foam films inside a TFPB, with small areas (thickness around 30 nm) interspersed in a network structure with a thickness of several 100 nm. This heterogeneity also explains the scattering curves of MG stabilised foams. These SANS curves show a strict Porod behaviour (I ∝ q−4) over a large q-range, which can be attributed to structures larger than 200 nm and therefore includes Plateau borders, nodes and the thicker parts of the foam films. In addition, a strongly damped reflectivity term was used to model the scattering curves. The reflectivity contribution in SANS from foams is typically attributed to foam films with a thickness below 100 nm and was consequently attributed to the thinner regions of the foam films. As these thinner regions only represent a fraction of the complete foam film, the reflectivity contribution to the scattering signal is rather small. A theoretical reflectivity curve of a 30 nm thick D2O layer in air reproduces the position of the extrema in all scattering curves, which leads to the conclusion that the thinner regions of the foam films are around 30 nm thick. The fact that this thickness is comparable for all MGs and much smaller then their hydrodynamic sizes suggests that the MGs are depleted from the thinner areas of the foam film, which might be stabilised by the formation of a corset-like structure, dangling polymer ends of the MGs or residual PNIPAM oligomers. An interesting question for future research is whether other soft particles show similar pattern formation when confined in foam films. The model used for the description of the SANS curves is a restricted version of the model described in a previous publication,55 as the power of the SAS decay was fixed to β = −4 (Porod behaviour). The fact that the model is capable of describing two very different types of foams further corroborates its validity. Finally, a linear correlation between the thickness of the thicker areas of the foam films and the elastic moduli determined by AFM indentation experiments of the individual MGs is observed. This correlation implies that both the deformation in foam films and by an AFM tip are governed by the overall elastic properties of the MGs. In fact, both of these deformations and additionally the MGs' swelling capacity show a strong correlation with the elastic osmotic pressure according to the affine network model describing the elasticity of polymer networks.Author contributions
Conceptualization, M. K.; data curation, M. K., K. G., E. L.; formal analysis, M. K., K. G.; investigation, M. K., K. G., O. S., O. L., H. F.; resources, H. F., R. v. K.; visualization, M. K.; writing – original draft, M. K.; writing – review and editing, M. K., K. G., E. L., O. S., O. L., H. F., R. v. K.; supervision, R. v. K.; funding acquisition, R. v. K.; project administration, R. v. K.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is based upon experiments performed at the KWS-1 instrument operated by JCNS at the Heinz Maier-Leibnitz Zentrum (MLZ), Garching, Germany. This research was funded by the German Federal Ministry for Education and Research (BMBF) within the project “FlexiProb” sample environment grant number 05K2016.References
- R. K. Prud'homme and S. A. Khan, Foams Theory, Measurements, and Applications, Marcel Dekker, New York, 1995 Search PubMed.
- L. L. Schramm, Emulsions, Foams, and Suspensions Fundamentals and Applications, Wiley-VCH, Weinheim, 2005 Search PubMed.
- D. R. Exerowa, G. Gochev, D. Platikanov, L. Liggieri and R. Miller, Foam Films and Foams - Fundamentals and Applications, CRC Press, Boca Raton, FL, 2019 Search PubMed.
- E. Dickinson, Food emulsions and foams, Elsevier, 1987 Search PubMed.
- D. Weaire and S. Hutzler, The Physics of Foams, Oxford University Press, Oxford, New York, 1999 Search PubMed.
- S. A. Koehler, S. Hilgenfeldt and H. A. Stone, Langmuir, 2000, 16, 6327–6341 CrossRef CAS.
- B. P. Binks, Curr. Opin. Colloid Interface Sci., 2002, 7, 21–41 CrossRef CAS.
- T. S. Horozov, Curr. Opin. Colloid Interface Sci., 2008, 13, 134–140 CrossRef CAS.
- B. S. Murray and R. Ettelaie, Curr. Opin. Colloid Interface Sci., 2004, 9, 314–320 CrossRef CAS.
- K. Engelhardt, M. Lexis, G. Gochev, C. Konnerth, R. Miller, N. Willenbacher, W. Peukert and B. Braunschweig, Langmuir, 2013, 29, 11646–11655 CrossRef CAS PubMed.
- P. Wilde, A. Mackie, F. Husband, P. Gunning and V. Morris, Adv. Colloid Interface Sci., 2004, 108, 63–71 CrossRef PubMed.
- A. Bureiko, A. Trybala, N. Kovalchuk and V. Starov, Adv. Colloid Interface Sci., 2015, 222, 670–677 CrossRef CAS PubMed.
- E. Guzmán, S. Llamas, A. Maestro, L. Fernández-Peña, A. Akanno, R. Miller, F. Ortega and R. G. Rubio, Adv. Colloid Interface Sci., 2016, 233, 38–64 CrossRef PubMed.
- R. J. Pugh, Bubble and Foam Chemistry, Cambridge University Press, Cambridge, 2016 Search PubMed.
- Z. Briceño-Ahumada and D. Langevin, Adv. Colloid Interface Sci., 2017, 244, 124–131 CrossRef.
- P. M. Kruglyakov, S. I. Karakashev, A. V. Nguyen and N. G. Vilkova, Curr. Opin. Colloid Interface Sci., 2008, 13, 163–170 CrossRef CAS.
- E. Rio and A. L. Biance, Chem. Phys. Chem., 2014, 15, 3692–3707 CrossRef CAS PubMed.
- S. Fujii and Y. Nakamura, Langmuir, 2017, 33, 7365–7379 CrossRef CAS PubMed.
- A. L. Fameau, A. Carl, A. Saint-Jalmes and R. von Klitzing, ChemPhysChem, 2015, 16, 66–75 CrossRef CAS PubMed.
- A. L. Fameau and S. Fujii, Curr. Opin. Colloid Interface Sci., 2020, 50, 101380 CrossRef CAS.
- M. Schnurbus, M. Hardt, P. Steinforth, J. Carrascosa-Tejedor, S. Winnall, P. Gutfreund, M. Schönhoff, R. A. Campbell and B. Braunschweig, ACS Appl. Mater. Interfaces, 2022, 14, 4656–4667 CrossRef CAS PubMed.
- F. A. Plamper and W. Richtering, Acc. Chem. Res., 2017, 50, 131–140 CrossRef CAS PubMed.
- M. Heskins and J. E. Guillet, J. Macromol. Sci., Part A: Pure Appl. Chem., 1968, 2, 1441–1455 CrossRef CAS.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- M. Füllbrandt, E. Ermilova, A. Asadujjaman, R. Hölzel, F. F. Bier, R. Von Klitzing and A. Schönhals, J. Phys. Chem. B, 2014, 118, 3750–3759 CrossRef PubMed.
- R. Pelton, Adv. Colloid Interface Sci., 2000, 85, 1–33 CrossRef CAS PubMed.
- R. Pelton and P. Chibante, Colloids Surf., 1986, 20, 247–256 CrossRef CAS.
- M. Stieger, W. Richtering, J. S. Pedersen and P. Lindner, J. Chem. Phys., 2004, 120, 6197–6206 CrossRef CAS PubMed.
- N. Boon and P. Schurtenberger, Phys. Chem. Chem. Phys., 2017, 19, 23740–23746 RSC.
- J. Zhang and R. Pelton, Langmuir, 1999, 15, 8032–8036 CrossRef CAS.
- C. Monteux, C. Marliére, P. Paris, N. Pantoustier, N. Sanson and P. Perrin, Langmuir, 2010, 26, 13839–13846 CrossRef CAS PubMed.
- Z. Li, W. Richtering and T. Ngai, Soft Matter, 2014, 10, 6182–6191 RSC.
- T. Ngai, S. H. Behrens and H. Auweter, Chem. Commun., 2005, 331–333 RSC.
- T. Ngai, H. Auweter and S. H. Behrens, Macromolecules, 2006, 39, 8171–8177 CrossRef CAS.
- B. Brugger, B. A. Rosen and W. Richtering, Langmuir, 2008, 24, 12202–12208 CrossRef CAS PubMed.
- W. Richtering, Langmuir, 2012, 28, 17218–17229 CrossRef CAS PubMed.
- V. Schmitt and V. Ravaine, Curr. Opin. Colloid Interface Sci., 2013, 18, 532–541 CrossRef CAS.
- J. Harrer, M. Rey, S. Ciarella, L. M. C. Janssen and N. Vogel, Langmuir, 2019, 10512–10521 CrossRef CAS.
- S. Bochenek, A. Scotti, W. Ogieglo, M. Á. Fernández-Rodríguez, M. F. Schulte, R. A. Gumerov, N. V. Bushuev, I. I. Potemkin, M. Wessling, L. Isa and W. Richtering, Langmuir, 2019, 16780–16792 CrossRef CAS.
- S. Bochenek, A. Scotti and W. Richtering, Soft Matter, 2021, 17, 976–988 RSC.
- K. Geisel, L. Isa and W. Richtering, Langmuir, 2012, 28, 15770–15776 CrossRef CAS.
- K. Geisel, L. Isa and W. Richtering, Angew. Chem., Int. Ed., 2014, 53, 4905–4909 CrossRef CAS.
- C. Picard, P. Garrigue, M.-C. Tatry, V. Lapeyre, S. Ravaine, V. Schmitt and V. Ravaine, Langmuir, 2017, 33, 7968–7981 CrossRef CAS PubMed.
- M. Rey, X. Hou, J. S. J. Tang and N. Vogel, Soft Matter, 2017, 13, 8717–8727 RSC.
- M. Rey, M. A. Fernandez-Rodriguez, M. Karg, L. Isa and N. Vogel, Acc. Chem. Res., 2020, 414–424 CrossRef CAS PubMed.
- Y. Horiguchi, H. Kawakita, K. Ohto and S. Morisada, Adv. Powder Technol., 2018, 29, 266–272 CrossRef CAS.
- A. Maestro, D. Jones, C. Sánchez De Rojas Candela, E. Guzman, M. H. Duits and P. Cicuta, Langmuir, 2018, 34, 7067–7076 CrossRef CAS PubMed.
- J. E. Sader, J. W. Chon and P. Mulvaney, Rev. Sci. Instrum., 1999, 70, 3967–3969 CrossRef CAS.
- O. Bartsch, Kolloidchem. Beih., 1924, 20, 1–49 CrossRef CAS.
- J. Schindelin, I. Arganda-Carreras, E. Frise, V. Kaynig, M. Longair, T. Pietzsch, S. Preibisch, C. Rueden, S. Saalfeld, B. Schmid, J. Y. Tinevez, D. J. White, V. Hartenstein, K. Eliceiri, P. Tomancak and A. Cardona, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- H. Frielinghaus, A. Feoktystov, I. Berts and G. Mangiapia, J. Large-Scale Res. Facil. JLSRF, 2015, 1, 26–29 CrossRef.
- A. V. Feoktystov, H. Frielinghaus, Z. Di, S. Jaksch, V. Pipich, M. S. Appavou, E. Babcock, R. Hanslik, R. Engels, G. Kemmerling, H. Kleines, A. Ioffe, D. Richter and T. Brückel, J. Appl. Crystallogr., 2015, 48, 61–70 CrossRef CAS.
- Vitaliy Pipich, “QtiSAS: user-friendly program for reduction, visualization, analysis and fit of SA(N)S data” (2020), https://www.qtisas.com, accessed on 24th Mar 2022.
- M. Kühnhammer, T. Widmann, L. P. Kreuzer, A. J. Schmid, L. Wiehemeier, H. Frielinghaus, S. Jaksch, T. Bögershausen, P. Barron, H. Schneider, A. Hiess, P. Müller-Buschbaum, T. Hellweg, R. von Klitzing and O. Löhmann, Appl. Sci., 2021, 11, 5116 CrossRef.
- M. Kühnhammer, L. Braun, M. Ludwig, O. Soltwedel, L. Chiappisi and R. von Klitzing, J. Appl. Crystallogr., 2022, 55, 758–768 CrossRef PubMed.
- K. J. Mysels and M. N. Jones, Discuss. Faraday Soc., 1966, 42, 42–50 RSC.
- A. Scheludko and D. Exerowa, Kolloid-Z., 1960, 168, 24–28 CrossRef.
- E. S. Basheva, P. A. Kralchevsky, K. D. Danov, R. D. Stanimirova, N. Shaw and J. T. Petkov, J. Colloid Interface Sci., 2020, 576, 345–355 CrossRef CAS PubMed.
- W. Drenckhan and A. Saint-Jalmes, Adv. Colloid Interface Sci., 2015, 222, 228–259 CrossRef CAS PubMed.
- B. Petkova, S. Tcholakova, M. Chenkova, K. Golemanov, N. Denkov, D. Thorley and S. Stoyanov, Adv. Colloid Interface Sci., 2020, 276, 102084 CrossRef CAS PubMed.
- T. Widmann, L. P. Kreuzer, N. Hohn, L. Bießmann, K. Wang, S. Rinner, J. F. Moulin, A. J. Schmid, Y. Hannappel, O. Wrede, M. Kühnhammer, T. Hellweg, R. von Klitzing and P. Müller-Buschbaum, Langmuir, 2019, 35, 16341–16352 CrossRef CAS PubMed.
- C. Üzüm, J. Hellwig, N. Madaboosi, D. Volodkin and R. von Klitzing, Beilstein J. Nanotechnol., 2012, 3, 778–788 CrossRef PubMed.
- M. F. Schulte, E. Izak-Nau, S. Braun, A. Pich, W. Richtering and R. Göstl, Chem. Soc. Rev., 2022, 51, 2939–2956 RSC.
- A. Burmistrova, M. Richter, C. Uzum and R. von Klitzing, Colloid Polym. Sci., 2011, 289, 613–624 CrossRef CAS.
- A. Burmistrova, M. Richter, M. Eisele, C. Üzüm and R. von Klitzing, Polymers, 2011, 3, 1575–1590 CrossRef CAS.
- M. U. Witt, S. Hinrichs, N. Möller, S. Backes, B. Fischer and R. von Klitzing, J. Phys. Chem. B, 2019, 123, 2405–2413 CrossRef CAS PubMed.
- J. Witte, T. Kyrey, J. Lutzki, A. M. Dahl, M. Kühnhammer, R. von Klitzing, O. Holderer and S. Wellert, ACS Appl. Polym. Mater., 2021, 3, 976–985 CrossRef CAS.
- M. F. Schulte, S. Bochenek, M. Brugnoni, A. Scotti, A. Mourran and W. Richtering, Angew. Chem., Int. Ed., 2021, 60, 2280–2287 CrossRef CAS PubMed.
- S. Schmidt, M. Zeiser, T. Hellweg, C. Duschl, A. Fery and H. Möhwald, Adv. Funct. Mater., 2010, 20, 3235–3243 CrossRef CAS.
- S. M. Hashmi and E. R. Dufresne, Soft Matter, 2009, 5, 3682–3688 RSC.
- S. Backes and R. von Klitzing, Polymers, 2018, 10, 1–23 CrossRef PubMed.
- J. Witte, T. Kyrey, J. Lutzki, A. M. Dahl, J. Houston, A. Radulescu, V. Pipich, L. Stingaciu, M. Kühnhammer, M. U. Witt, R. von Klitzing, O. Holderer and S. Wellert, Soft Matter, 2019, 15, 1053–1064 RSC.
- A. Aufderhorst-Roberts, D. Baker, R. J. Foster, O. Cayre, J. Mattsson and S. D. Connell, Nanoscale, 2018, 10, 16050–16061 RSC.
- I. Lesov, S. Tcholakova and N. Denkov, J. Colloid Interface Sci., 2014, 426, 9–21 CrossRef CAS PubMed.
- N. Politova, S. Tcholakova, Z. Valkova, K. Golemanov and N. D. Denkov, Colloids Surf., A, 2018, 539, 18–28 CrossRef CAS.
- R. Varadaraj, J. Bock, P. Valint, S. Zushma and N. Brons, J. Colloid Interface Sci., 1990, 140, 31–34 CrossRef CAS.
- T. Tamura, Y. Kaneko and M. Ohyama, Dynamic Surface Tension and Foaming Properties of Aqueous Polyoxyethylene n-Dodecyl Ether Solutions, 1995 Search PubMed.
- M. Buzzacchi, P. Schmiedel and W. Von Rybinski, Colloids Surf., A, 2006, 273, 47–54 CrossRef CAS.
- D. Kawale, A. T. van Nimwegen, L. M. Portela, M. A. van Dijk and R. A. Henkes, Colloids Surf., A, 2015, 481, 328–336 CrossRef CAS.
- L. Gupta and D. T. Wasan, Ind. Eng. Chem. Fundam., 1974, 13, 26–33 CrossRef CAS.
- R. Miller, R. Wüstneck, J. Krägel and G. Kretzschmar, Colloids Surf., A, 1996, 111, 75–118 CrossRef CAS.
- M. Bos and T. van Vliet, Adv. Colloid Interface Sci., 2001, 91, 437–471 CrossRef CAS PubMed.
- R. Miller and L. Liggieri, Interfacial rheology, CRC Press, 2009, vol. 1 Search PubMed.
- K. Malysa and K. Lunkenheimer, Curr. Opin. Colloid Interface Sci., 2008, 13, 150–162 CrossRef CAS.
- J. Wang, A. V. Nguyen and S. Farrokhpay, Adv. Colloid Interface Sci., 2016, 228, 55–70 CrossRef CAS PubMed.
- D. Varade, D. Carriere, L. R. Arriaga, A. L. Fameau, E. Rio, D. Langevin and W. Drenckhan, Soft Matter, 2011, 7, 6557–6570 RSC.
- A.-L. Fameau, A. Saint-Jalmes, F. Cousin, B. Houinsou Houssou, B. Novales, L. Navailles, F. Nallet, C. Gaillard, F. Boué and J.-P. Douliez, Angew. Chem., Int. Ed., 2011, 50, 8264–8269 CrossRef CAS PubMed.
- A. L. Fameau and A. Salonen, C. R. Phys., 2014, 15, 748–760 CrossRef CAS.
- B. Haffner, Y. Khidas and O. Pitois, Soft Matter, 2014, 10, 3277–3283 RSC.
- L. Braun, M. Kühnhammer and R. von Klitzing, Curr. Opin. Colloid Interface Sci., 2020, 50, 101379 CrossRef CAS.
- K. Khristov, B. Jachimska, K. Malysa and D. Exerowa, Colloids Surf., A, 2001, 186, 93–101 CrossRef CAS.
- C. Stubenrauch and K. Khristov, J. Colloid Interface Sci., 2005, 286, 710–718 CrossRef CAS PubMed.
- M. Destribats, V. Lapeyre, E. Sellier, F. Leal-Calderon, V. Ravaine and V. Schmitt, Langmuir, 2012, 28, 3744–3755 CrossRef CAS PubMed.
- B. Brugger, S. Rütten, K. H. Phan, M. Moller and W. Richtering, Angew. Chem., Int. Ed., 2009, 48, 3978–3981 CrossRef CAS PubMed.
- B. Brugger, J. Vermant and W. Richtering, Phys. Chem. Chem. Phys., 2010, 12, 14573–14578 RSC.
- M. Destribats, V. Lapeyre, M. Wolfs, E. Sellier, F. Leal-Calderon, V. Ravaine and V. Schmitt, Soft Matter, 2011, 7, 7689 RSC.
- K. Małysa, K. Lunkenheimer, R. Miller and C. Hempt, Colloids Surf., 1985, 16, 9–20 CrossRef.
- H. Fruhner, K. D. Wantke and K. Lunkenheimer, Colloids Surf., A, 2000, 162, 193–202 CrossRef CAS.
- D. Langevin, Adv. Colloid Interface Sci., 2000, 88, 209–222 CrossRef CAS PubMed.
- K. D. Wantke, J. Örtegren, H. Fruhner, A. Andersen and H. Motschmann, Colloids Surf., A, 2005, 261, 75–83 CrossRef CAS.
- M. A. V. Axelos and F. Boué, Langmuir, 2003, 19, 6598–6604 CrossRef CAS.
- M. H. Ropers, B. Novales, F. Boué and M. A. V. Axelos, Langmuir, 2008, 24, 12849–12857 CrossRef CAS PubMed.
- C. Micheau, P. Bauduin, O. Diat and S. Faure, Langmuir, 2013, 29, 8472–8481 CrossRef CAS PubMed.
- S. Yada, H. Shimosegawa, H. Fujita, M. Yamada, Y. Matsue and T. Yoshimura, Langmuir, 2020, 36, 7808–7813 CrossRef CAS PubMed.
- E. Rio, W. Drenckhan, A. Salonen and D. Langevin, Adv. Colloid Interface Sci., 2014, 205, 74–86 CrossRef CAS PubMed.
- C. J. Gommes, S. Jaksch and H. Frielinghaus, J. Appl. Crystallogr., 2021, 54, 1–12 CrossRef PubMed.
- J. Hurcom, A. Paul, R. K. Heenan, A. Davies, N. Woodman, R. Schweins and P. C. Griffiths, Soft Matter, 2014, 10, 3003–3038 RSC.
- S. Perticaroli, J. Herzberger, Y. Sun, J. D. Nickels, R. P. Murphy, K. Weigandt and P. J. Ray, Langmuir, 2020, 36, 14763–14771 CrossRef CAS PubMed.
- M. Hohenschutz, I. Grillo, C. Dewhurst, P. Schmid, L. Girard, A. Jonchère, O. Diat and P. Bauduin, J. Colloid Interface Sci., 2021, 603, 141–147 CrossRef CAS PubMed.
- L. Keal, V. Lapeyre, V. Ravaine, V. Schmitt and C. Monteux, Soft Matter, 2016, 13, 170–180 RSC.
- P. Voudouris, D. Florea, P. Van Der Schoot and H. M. Wyss, Soft Matter, 2013, 9, 7158–7166 RSC.
- W. Kuhn, J. Polym. Sci., 1946, 1, 380–388 CrossRef CAS.
- P. J. Flory, Principles of Polymer Chemistry, Cornell University Press, Ithaca, NY, and London, 1953 Search PubMed.
- R. H. C. M. Rubinstein, Polymer Physics, Oxford University Press, 2003 Search PubMed.
- M. Quesada-Pérez, J. A. Maroto-Centeno, J. Forcada and R. Hidalgo-Alvarez, Soft Matter, 2011, 7, 10536–10547 RSC.
- B. R. Saunders, Langmuir, 2004, 20, 3925–3932 CrossRef CAS PubMed.
- G. Hild, Prog. Polym. Sci., 1998, 23, 1019–1149 CrossRef CAS.
- H. M. James and E. Guth, J. Chem. Phys., 1953, 21, 1039–1049 CrossRef CAS.
- P. J. Flory, J. Chem. Phys., 1977, 66, 5720–5729 CrossRef CAS.
- B. Erman and P. J. Flory, J. Chem. Phys., 1977, 68, 5363–5369 CrossRef.
- P. J. Flory and B. Erman, Macromolecules, 1982, 15, 800–806 CrossRef CAS.
- S. F. Edwards, Proc. Phys. Soc., 1967, 92, 9–16 CrossRef CAS.
- M. Rubinstein and S. Panyukov, Macromolecules, 2002, 35, 6670–6686 CrossRef CAS.
- Y. Akagi, J. P. Gong, U. I. Chung and T. Sakai, Macromolecules, 2013, 46, 1035–1040 CrossRef CAS.
- S. Panyukov, Polymers, 2020, 12, 767–793 CrossRef CAS PubMed.
- D. Suzuki, T. Kobayashi, R. Yoshida and T. Hirai, Soft Matter, 2012, 8, 11447–11449 RSC.
- A. Fernández-Barbero, I. J. Suárez, B. Sierra-Martín, A. Fernández-Nieves, F. J. de las Nieves, M. Marquez, J. Rubio-Retama and E. López-Cabarcos, Adv. Colloid Interface Sci., 2009, 147–148, 88–108 CrossRef PubMed.
- M. Zeiser, I. Freudensprung and T. Hellweg, Polymer, 2012, 53, 6096–6101 CrossRef CAS.
- B. Özkale, J. Lou, E. Ozelci, A. Elosegui-Artola, C. M. Tringides, A. Mao, M. S. Sakar and D. J. Mooney, Lab Chip, 2022, 22, 1962–1970 RSC.
- J. J. Bowen, M. A. Rose and S. A. Morin, MRS Bull., 2021, 46, 337–344 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: AFM scans in air of spin-coated microgels (MGs) and representative height profiles, exemplary photographs used for the determination of macroscopic foam properties, exemplary microscopy images illustrating the influence of pressure release on the foam film structure, the analysis of a possible correlation between foam film thickness and MG size, all force curves used for the determination of the MGs' elasticity and Porod plots of SANS curves of MG stabilised foams at higher MG concentrations. See DOI: https://doi.org/10.1039/d2sm01021f |
| ‡ Current address: Bundesanstalt für Materialforschung und -prüfung (BAM), Unter den Eichen 87, 12205 Berlin, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |