
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDNA-functionalized colloidal crystals for macromolecular encapsulation†
Maasa
Yokomori
 a,
Hayato
Suzuki
b,
Akiyoshi
Nakamura
b,
Shigeo S.
Sugano
*b and
Miho
Tagawa
*ac
a,
Hayato
Suzuki
b,
Akiyoshi
Nakamura
b,
Shigeo S.
Sugano
*b and
Miho
Tagawa
*ac
aCenter for Integrated Research of Future Electronics (CIRFE), Institute of Materials and Systems for Sustainability (IMaSS), Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8601, Japan. E-mail: tagawa.miho@material.nagoya-u.ac.jp
bPlant Gene Regulation Research Group, Bioproduction Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, 305-8566, Japan. E-mail: shigeo.sugano@aist.go.jp
cDepartment of Materials Process Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya 464-8603, Japan
First published on 5th September 2022
Abstract
Novel DNA-based structures with the ability to encapsulate nanoscale molecules, such as proteins, can be applied to a wide range of areas, including reaction fields and micro/nano drug carriers. DNA-functionalized nanoparticle (DNA-NP) colloidal crystals have emerged as a new class of programmable DNA-based structures harboring metal nanoparticles with improved mechanical properties. The encapsulation of guest molecules into empty spaces in lattice structures is theoretically possible. However, due to the lack of a strategy for versatile encapsulation of guest molecules, the feasibility of nanoscale encapsulation by DNA-NP crystals is unclear. In this study, we developed DNA-functionalized gold nanoparticle (DNA–AuNP) crystals with tunable interparticle spacing for molecular encapsulation. We demonstrated that the modification of DNA–AuNP crystals with functional moieties, that is, biotin molecules, was effective in retaining molecules in the crystals. The crystallinities before and after encapsulation of the molecules were confirmed using small-angle X-ray scattering. We also succeeded in encapsulating CRISPR/Cas9 ribonucleoproteins into DNA–AuNP crystals by harnessing their affinity for target molecules. These findings demonstrated the potential use of metal–DNA hybrid crystals as carriers for direct protein delivery via biolistic bombardment. Thus, this study provides an attractive strategy for creating a new class of DNA-based structures for macromolecular encapsulation, and an alternative research direction toward colloidal crystal engineering using DNA.
Introduction
DNA was proposed as a programmable material for building self-assembled structures several decades ago.1 DNA-based self-assembly opened the field of “structural DNA nanotechnology,” in which DNA-based structures with complex geometrical shapes, biochemical properties, and even spatiotemporal dynamics with 3D architectures have been demonstrated.2,3 In particular, DNA-based structures that encapsulate nanoscale molecules, such as proteins, are expected to be applied in a wide range of areas, including reaction fields and micro/nano drug carriers.4–6 In addition to their excellent programmability and addressability, DNA-based structures provide a suitable environment for enzymes and enhance their stability and/or catalytic activity.7,8 Thus, the interest in exploring novel DNA-based structures with the ability to encapsulate macromolecules has increased significantly.Utilization of DNA-functionalized colloidal crystals is a promising strategy for the development of new classes of DNA-based structures with improved performance and functional properties.9 Gold nanoparticles (AuNPs), which have unique plasmonic features, photothermal properties, and favorable biocompatibility, have been widely used in biological and medical research.10,11 DNA-functionalized AuNPs (DNA–AuNPs) can be assembled into crystalline lattices via DNA hybridization. DNA–AuNP crystals with high programmability have potential as new porous materials for soaking nanoscale molecules inside crystal pores by controlling the distance between nanoparticles and their spatial arrangement in a precise manner.12,13 Moreover, crystals with metal nanoparticles generally exhibit greater mechanical rigidity than that of DNA structures without metal nanoparticles. In addition, there are emergent mechanical properties of colloidal crystals provided by their ordering nature compared to those of amorphous colloidal assemblies.14 In our previous study, we observed that DNA–AuNP crystals tend to maintain their single-crystal polyhedral shapes through the drying process.15,16 However, DNA-functionalized colloidal crystallization for molecular encapsulation has rarely been studied.
In this study, we demonstrated the molecular encapsulation of DNA–AuNP crystals by harnessing their porous structure. Biotin–streptavidin interactions17 were exploited as a versatile approach for retaining molecules in DNA–AuNP crystals. Our confocal microscopic analysis showed that the modification of crystals with biotin enabled the capture of streptavidin and streptavidin-conjugated quantum dots by utilizing its specific and strong binding affinity for streptavidin. Successful encapsulation of CRISPR/Cas9 ribonucleoprotein (RNP)18 into DNA–AuNP crystals utilizing their ability to bind DNA was also demonstrated. Genome editing was achieved by biolistic bombardment using DNA–AuNP crystals harboring RNP, which is a widely used de facto standard method for genome editing in plant molecular biology.19 Our study provides an attractive strategy for creating a new class of DNA-based structures for protein encapsulation, and an alternative research direction toward colloidal crystal engineering with DNA.
Results and discussion
Design, fabrication, and characterization of DNA–AuNP crystals
Fig. 1a shows an overview of the fabrication of the DNA–AuNP crystals and their molecular encapsulation. To construct DNA–AuNP crystals with mesoporous features, we designed binary-component assembly systems in which two types of AuNPs coated with single-stranded DNA were assembled using the corresponding linker DNA strands. One set of DNA–AuNPs was functionalized with ligand DNA strands (A), and the other set with complementary ligand DNA strands (B). The linker DNA consisted of three basic regions: (i) a recognition region that connected the nanoparticle and the linker DNA strand, (ii) a spacer region that controlled interparticle distances, and (iii) a “sticky end” region that drove the assembly of the nanoparticles via specific hybridization between the linker strands (Fig. 1b). For biotin-modified DNA–AuNP crystals, the biotin molecule was conjugated at the end of either the “sticky end” region or spacer region of the linker DNA. By this modification, the capture of streptavidin or streptavidin-conjugated molecules to the crystals was achieved through biotin–streptavidin interactions without harsh conditions such as thermal denaturation. Self-assembly of DNA–AuNPs into colloidal crystals was performed via DNA hybridization during slow continuous annealing. In binary-component assembly systems, a body-centered cubic (bcc) crystal with rhombic dodecahedron morphology is favored to be formed.20 Notably, the incorporation of functional moieties may inhibit the crystallization of DNA–AuNPs because of their charge and steric effects. | ||
| Fig. 1 (a) Schematic illustrations of the fabrication of DNA-functionalized gold nanoparticle (DNA–AuNP) crystals and the molecular encapsulation. Streptavidin (SA) encapsulation for biotin-modified DNA–AuNP crystals is shown as an example. A single interparticle linkage is shown for clarity. (b) DNA design for biotin-modified DNA–AuNP crystal. Illustrations are not drawn to scale. | ||
The crystallinities of DNA–AuNP aggregates were probed with small angle X-ray scattering (SAXS). Fig. 2 shows the structure factors calculated from the 1D SAXS profiles of the DNA–AuNP crystals created from 13 nm diameter AuNPs functionalized with ligand DNAs and linker DNAs of different lengths (designated as n = 0, n = 1, and n = 3). Hereafter, we designated the DNA–AuNP colloidal crystals as DNA–AuNPs(Xnm,n=Y), where “X nm” represents the diameter of the AuNPs and “n = Y” shows the type of linker DNA (Table S1, ESI†). Analysis of the diffraction peak position ratios confirmed that bcc crystals with lattice constants of 42, 55, and 73 nm were obtained. In each unit cell, the surface-to-surface distance between the particles (Dp) is an indicator of the hollow space in the crystal. A comparison of the SAXS data of the non-biotinylated and biotin-modified DNA–AuNP crystals implied that biotin modification minimally affected their crystal structures (Fig. S1, ESI†). We also demonstrated that various derivatives of biotin-modified crystals, such as those modified with a fluorescent molecule (Alexa Fluor 488) and photoreactive 3-cyanovinylcarbazole nucleoside (CNVK), can be fabricated (Fig. S2 and S3, ESI†). The photoreactive chemical group enhances the stability of DNA structures and DNA–AuNP crystals under DNA denaturing conditions by covalent cross-linking of DNA strands.21–25 This feature is especially valuable for the encapsulation of proteins that are sensitive to environmental conditions, including the pH and ionic concentration.
 | ||
| Fig. 2 Small-angle X-ray scattering (SAXS) characterization of biotin-modified DNA-functionalized gold nanoparticle (DNA–AuNP) crystals. SAXS patterns and schematic illustration of the unit cell of biotin-modified DNA–AuNP crystals estimated from the SAXS pattern (inset). A single interparticle linkage is shown for clarity. The SAXS patterns (blue curves; top, n = 3; middle, n = 1; down, n = 0) are shown as plots of the structure factor (S(q)) versus scattering vector (q, in units of nm−1). The black traces are the calculated scattering patterns for a perfect bcc lattice. | ||
Biotin-modified DNA–AuNP crystals were characterized by microscopy. Scanning electron microscopy (SEM) is the most common technique used to observe the morphology of DNA–AuNP crystals. The DNA–AuNP crystals were embedded in silica, which enabled the imaging of DNA–AuNP crystals with the same symmetry and nearly identical interparticle distances as the solution-phase lattice.26 As shown in Fig. 3a, SEM imaging confirmed the formation of DNA–AuNP crystals with a rhombic dodecahedron morphology, which is the equilibrium shape for the bcc system. In contrast, another technique is required to directly observe the crystals and evaluate their molecular encapsulation capacity in a more natural aqueous environment. Accordingly, we performed confocal laser scanning microscopy (CLSM) imaging of the crystals that harbored both biotin and fluorescent dye modifications (Fig. S2, ESI†). Although CLSM is widely employed to evaluate the encapsulation performance of biomaterial-based structures, it has rarely been used to observe DNA–AuNP crystals. Fig. 3b shows microscopy z-stack images of five continuous focal planes from the surface to the interior of the crystals. As shown in Fig. 3c, the 3D image reconstructed from the serial z-stack images of the crystal was consistent with the SEM morphological characterization.
 | ||
| Fig. 3 Microscopic characterization of biotin-modified DNA-functionalized gold nanoparticle (DNA–AuNP)(13nm,n=1) crystals. (a) Scanning electron microscopy image of the shape of a biotin-modified DNA–AuNP embedded in silica and the magnified image of the surface of the crystals where the arrangement of nanoparticles can be observed. Scale bar = 1 μm (100 nm inset). (b) Confocal laser scanning microscopy (CLSM) images of five continuous layers of a biotin-modified DNA–AuNP crystal labeled with Alexa Fluor 488 across 2 μm with an interval of 500 nm. Scale bar = 2 μm. (c) 3D reconstruction of the CLSM images. | ||
Evaluation of molecular encapsulation into biotin-modified DNA–AuNP crystals
To evaluate the encapsulation of macromolecules into biotin-modified DNA–AuNP crystals, the crystals were soaked in fluorescently labeled molecules and observed using CLSM (Fig. 4a). Fig. 4b shows microscopic z-stack images of three continuous focal planes from the surface to the interior of the crystal and the corresponding line profile of fluorescence intensity in the confocal image of fluorescently labeled streptavidin distribution in biotin-modified DNA–AuNP(13nm,n=1) crystals (Dp = 41 nm). The fluorescence intensity, which was more significant than that of the background, was obtained from the interior and surface of the crystal, demonstrating successful streptavidin encapsulation into the crystal. Similar intensity profiles were observed for the DNA–AuNP(13nm,n=0) (Dp = 29 nm) and DNA–AuNP(13nm,n=3) crystals (Dp = 60 nm) (Fig. S4, ESI†). As a control, microscopic observation of the fluorescently labeled streptavidin molecules soaked with no biotinylated crystals is shown in Fig. 4c, indicating the necessity of biotin-dependent interactions for the encapsulation of streptavidin in the crystal. We also confirmed by SAXS that the biotin-modified crystals retained their crystalline nature after encapsulating streptavidin molecules (Fig. 4d and Fig. S5, ESI†). These results suggest that CNVK-modification is an effective approach to maintain the integrity of the crystal structure. In addition to fluorescently labeled streptavidin, streptavidin-conjugated quantum dots (Qdot 585) with an average diameter of 15–20 nm27 were bound to DNA–AuNP(13nm,n=3) crystals (Dp = 60 nm) (Fig. 4e). The fluorescence intensity from the interior of the crystal was found to be larger than that from the background, which decreased significantly compared to the surface, indicating that streptavidin-conjugated quantum dots were also partially adsorbed into the crystal. When the same experiment was conducted using red fluorescent silica particles with a larger diameter (100 nm), the fluorescence intensity from the interior of the crystal was comparable to that of the background (Fig. 4f). These data suggest that DNA–AuNP crystals can encapsulate nanoscale molecules within hollow spaces. Moreover, the encapsulation process involves simple soaking and does not require harsh conditions or complex operations. As streptavidin modification is available for a variety of macromolecules, our approach is especially valuable for biotechnological applications. In addition, quantum dots are semiconductor nanocrystals with unique electronic and optical properties, and increasing their encapsulation capacity is worth investigating.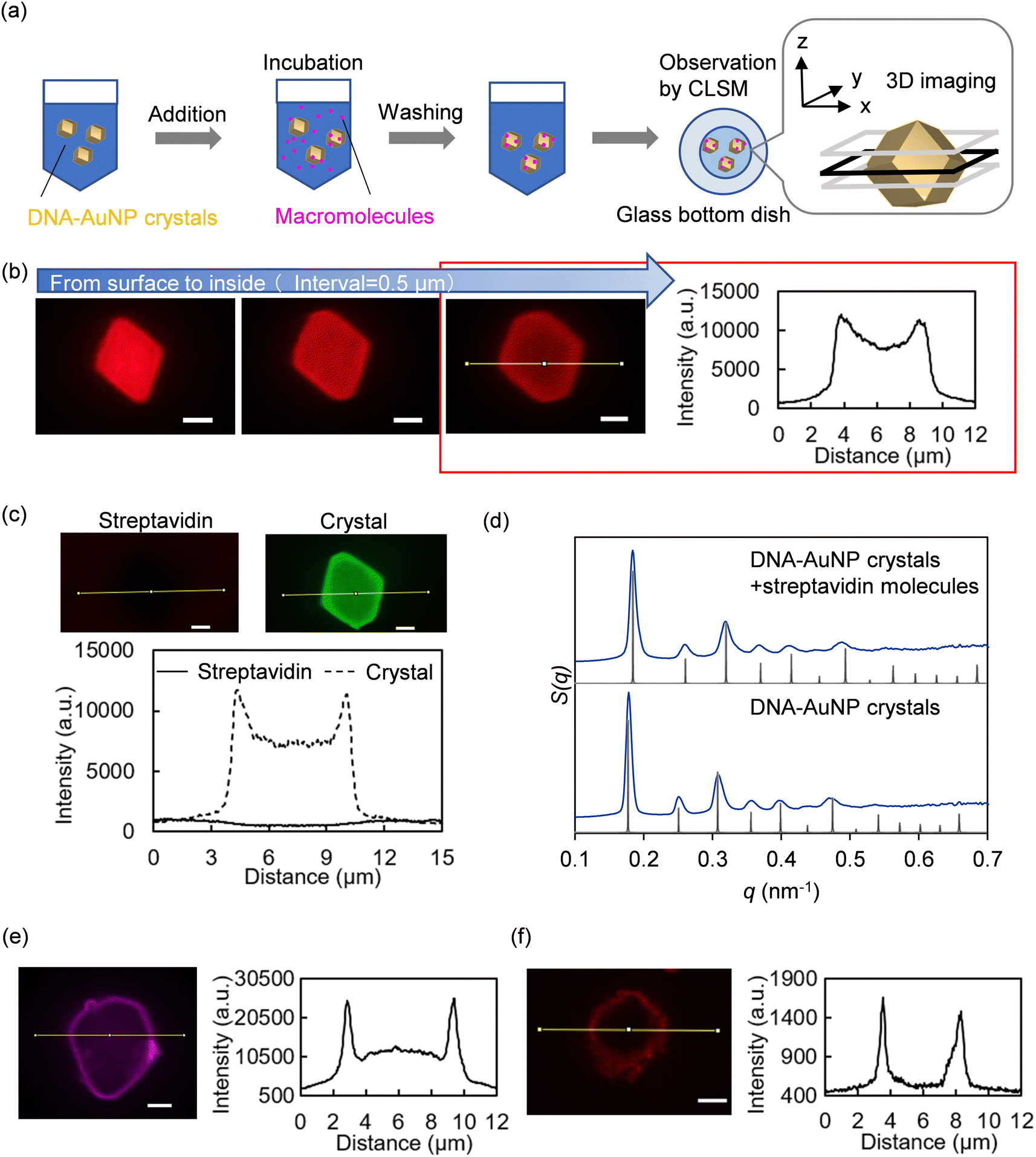 | ||
| Fig. 4 Evaluation of molecular encapsulation into DNA-functionalized gold nanoparticle (DNA–AuNP) crystals. (a) Experimental overview of molecular encapsulation. (b and c) Confocal laser scanning microscopy (CLSM) images and line profiles of the fluorescence intensity of streptavidin molecules labeled with Alexa Fluor 568 (solid lines), which are soaked with (b) biotin-modified and (c) non-biotinylated DNA–AuNP(13nm,n=1) crystals. Non-biotinylated DNA–AuNP crystals were labeled with Alexa Fluor 488 (dashed lines). Fluorescence intensity from the left to the right on the confocal image corresponds to the left to the right in the plot. Scale bar = 2 μm. (d) Comparison of small-angle X-ray scattering (SAXS) measurements of biotin- and CNVK-modified DNA–AuNP(13nm,n=1) crystals before and after streptavidin encapsulation. The SAXS patterns (blue curves) are shown as plots of the structure factor (S(q)) versus scattering vector (q, in units of nm−1). The black traces are the calculated scattering patterns for a perfect body-centered cubic lattice. (e) CLSM image and line profile of the fluorescence intensity of streptavidin-conjugated quantum dots that are soaked with biotin-modified DNA–AuNP(13nm,n=3) crystal at a depth of 1 μm from the crystal surface. Scale bar = 2 μm. (f) CLSM image and line profile of the fluorescence intensity of streptavidin-conjugate silica particles that are soaked with biotin-modified DNA–AuNP(13nm,n=3) crystal at a depth of 1 μm from the crystal surface. Scale bar = 2 μm. | ||
One of the difficulties in studying DNA–AuNP crystals as porous materials is that there is no standard method for characterizing their porous structures and macromolecules. Evaluation of streptavidin encapsulation into DNA–AuNP crystals by CLSM revealed a difference between the fluorescence intensity profiles of streptavidin depending on the dye molecules (Fig. S6, ESI†). The fluorescence intensity ratio of Alexa Fluor 488-streptavidin molecules detected from the interior to the surface of the DNA–AuNP crystal(13nm,n=1) was lower than that of Alexa Fluor 568-streptavidin molecules. One possible explanation for this result is the optical properties of DNA–AuNP crystals. The AuNPs exhibited a UV-vis absorption spectrum with a maximum absorption band at approximately 520 nm, which is the emission wavelength peak of Alexa Fluor 488. The scattering and absorption properties could also influence the fluorescence intensities detected in the interior of DNA–AuNP crystals with different lattice constants (Fig. S4, ESI†). Additionally, the size of the spaces in the DNA–AuNP crystals estimated by SAXS did not directly correlate with the size of molecules capable of diffusing into them because DNA strands are not involved in nanoparticle linkages, especially on the surface of DNA–AuNP crystals. Overall, other assessment methods for molecular encapsulation in DNA–AuNP crystals are important for future research.
The effects of reducing the volume fraction of DNA in the crystals to increase the hollow space of the crystals were also investigated using streptavidin encapsulation experiments. Reducing the volume fraction of DNA in the crystals as much as possible may be an effective approach to increasing the hollow space of the crystals. In contrast, a certain amount of loaded DNA strands on nanoparticles is effective in preventing irreversible aggregation of the nanoparticles in the presence of salt.28 Hence, we prepared AuNPs coated with two different lengths of ligand DNA (Fig. S7a, ESI†). One of these was composed of a recognition region and a flexible spacer region (33-nt DNA, Ligand A or B), which is commonly used for DNA-mediated nanoparticle crystallization. The other was composed of only a flexible spacer region (10-nt DNA, Ligand S) to stabilize the nanoparticles. In this study, 13 nm AuNPs, in which approximately 60 and 15 strands of DNA were coated per nanoparticle, were prepared (Fig. S7b, ESI†). Compared with AuNPs coated with only Ligand A or B, the amounts of Ligand A or B seemed to decrease, especially for Ligand B (Fig. 1a and Fig. S7b, c, ESI†). In addition, the minimum amount of linker DNA strands required to induce crystallization was investigated by changing the ratio of DNA–AuNPs to linker DNA strands used for crystallization. In general, multiple DNA linkages between nanoparticles are involved in crystallization, and the range of linker loading in which crystal formation can occur can vary according to design parameters (particle size, DNA sequences, and lattice parameters). SAXS experiments for a series of samples with different amounts of linker strands revealed that a sample with 25 linker equivalents per AuNP reproducibly formed crystals in biotin-modified DNA–AuNP(13nm,n=1) crystallization (Fig. S8, ESI†). The difference in intensity between the interior and surface of the crystal consisting of nanoparticles coated with two different lengths of ligand DNA with 25 linker equivalents appeared to be slightly narrower than that of nanoparticles coated with long ligand DNA with 100 linker equivalents (Fig. S9, ESI†). These data imply that the design of a hollow space for DNA–AuNP crystals can affect the encapsulation capacity of colloidal crystals.
CRISPR/Cas9 ribonucleoprotein encapsulation and its application to genome editing by a biolistic bombardment
We also demonstrated the encapsulation of CRISPR/Cas9 ribonucleoproteins in DNA–AuNP crystals. The direct delivery of CRISPR/Cas9 effectors into cells is in high demand for effective genome editing.29 The DNA endonuclease Cas9 forms a complex with guide RNA (gRNA), which has a 20-nt programmable RNA sequence that targets the double-stranded DNAs to be cleaved. The Cas9–gRNA complex was designated here as a ribonucleoprotein (RNP). RNPs have an affinity for DNA with a protospacer adjacent motif (PAM) sequence. The PAM sequence is 5′-NGG-3′ in Streptococcus pyogenes Cas9 (SpCas9), which is one of the leading CRISPR/Cas9 systems studied to date.18 Hence, DNA–AuNP crystals that harbor PAM sequences and can be attached to SpCas9-based RNP were designed and synthesized (Table S1, ESI†). Fig. 5a and b show the results of SAXS and SEM measurements for DNA–AuNP crystals using 23 nm diameter AuNPs functionalized with 53-nt ligand DNA and linker DNA (n = 3) (Dp = 73 nm). By utilizing their binding ability to DNA, the successful encapsulation of RNP [SpCas9 fused with a green fluorescent protein (GFP); approximately 7.5 nm hydrodynamic diameter (SpCas9) and approximately 2.5 nm (GFP)]30,31 into DNA–AuNP(23nm,n=3) crystals and the retention of the crystalline nature after encapsulation were confirmed by CLSM and SAXS, respectively (Fig. 5c and d). Furthermore, SEM imaging revealed RNP-encapsulated DNA–AuNP(23nm,n=3) crystals that retained their three-dimensional shape after the drying process (Fig. 5e). These data suggest that DNA–AuNP crystals bound to CRISPR/Cas9 RNP can be handled under dry conditions, maintaining their three-dimensional shape, which is consistent with our previous study on the effects of drying on DNA–AuNP crystals.15 | ||
| Fig. 5 Characterization of DNA-functionalized gold nanoparticle (DNA–AuNP) crystals for ribonucleoprotein (RNP) encapsulation. (a) Small-angle X-ray scattering (SAXS) pattern and schematic illustration of the unit cell of DNA–AuNP(23nm,n=3) crystals estimated from the SAXS pattern (inset). A single interparticle linkage is shown for clarity. The SAXS pattern (blue curve) is shown as a plot of the structure factor (S(q)) versus scattering vector (q, in units of nm−1). The black trace is the calculated scattering pattern for a perfect bcc lattice. (b) Scanning electron microscopy (SEM) image of the shape of a silica-embedded DNA–AuNP(23nm,n=3) crystal and a magnified image of the surface of the crystals where the arrangement of nanoparticles can be observed. Scale bar = 1 μm (200 nm inset). (c) Confocal microscopic images and line profiles of the fluorescence intensity of RNPs (Cas9) labeled with green fluorescent protein (solid lines), which are soaked with the DNA–AuNP crystals. Scale bar = 2 μm. (d) Comparison of SAXS measurements of DNA–AuNP(23nm,n=3) crystals before and after RNP encapsulation. The SAXS patterns (blue curves) are shown as plots of the structure factor (S(q)) versus scattering vector (q, in units of nm−1). The black traces are the calculated scattering patterns for a perfect bcc lattice. (e) SEM image of the shape of RNP-encapsulated DNA–AuNP(23nm,n=3) crystals after the drying process. Scale bar = 1 μm. | ||
Finally, we tried to utilize protein-encapsulated DNA–AuNP crystals with dry resistance as the carriers for biolistic bombardment, because the carriers of this molecule delivery method should be rigid and resistant to drying. DNA–AuNP crystals have potential as alternative carriers of biolistic bombardment with high delivery efficiency by retaining target molecules in the interior as well as the surface of the crystals. However, it is challenging to apply DNA–AuNP crystals to biolistic bombardment because of the lower volume fractions of nanoparticles in the crystals compared to those of conventional gold particles. Fig. 6a and b show the experimental design of the genome-editing experiment using RNP delivery. As plant genome breeding using CRISPR/Cas9 RNP has huge potential, Arabidopsis thaliana, which is widely used as a model flowering plant, was selected as the target organism. The gRNA targeting phytoene desaturase 3 (PDS3) gene was mixed with recombinant Cas9-nucleic localization signal (NLS) protein to form an RNP designated as RNP-PDS3. According to an above-mentioned procedure, DNA–AuNP(23nm,n=3) crystals were soaked in a solvent containing RNP-PDS3. Fully mature A. thaliana leaves were subjected to a biolistic bombardment experiment using either DNA–AuNP(23nm,n=3) crystals harboring RNP-PDS3, or conventional gold microparticles fused with RNP-PDS3. The PDS3 locus was polymerase chain reaction (PCR)-amplified and the PCR products were analyzed by amplicon sequencing (amplicon-seq). Fig. 6c and d show the quantification of genome editing events at the PDS3 locus in the samples. Fig. 6c graphically summarizes the types and positions of the mutations near the gRNA target sites and their frequencies. RNP-introduced samples accumulated significantly more indel mutations compared with those for the non-RNP-introduced control, indicating that these mutations are undoubtedly induced by RNPs delivered via DNA–AuNP crystals and gold particles. To the best of our knowledge, this is the first report of the application of DNA–AuNP crystals for genome editing in plant cells using biolistic bombardment. As different indel mutations should occur in different alleles, more indel variations are likely to indicate higher RNP delivery efficiency. The efficiency of DNA–AuNP crystals was comparable to or slightly lower than that of conventional gold particles (Fig. 6d). However, we could not distinguish between the same indel mutations occurring in different cells in parallel, and the quantitativeness of this amplicon-seq was not perfect. The establishment of a more precise quantification method for genome-editing efficiencies in biolistic-bombarded samples will facilitate improvements in Cas9 RNP delivery via DNA–AuNP crystals.
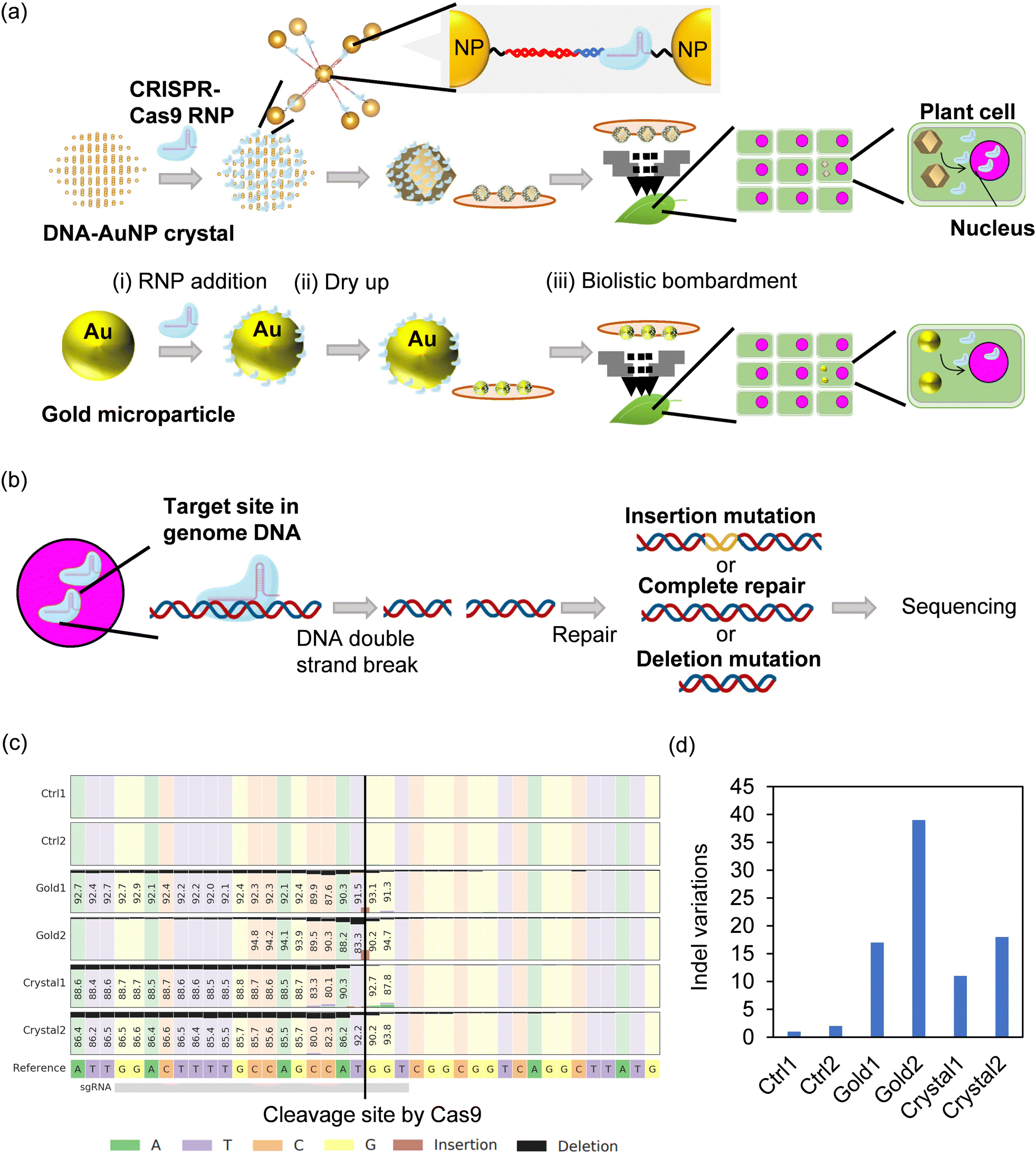 | ||
| Fig. 6 Targeted mutagenesis in Arabidopsis leaves by biolistic delivery of Cas9 ribonucleoproteins (RNPs) using DNA-functionalized gold nanoparticle (DNA–AuNP) crystals. (a) Schematic illustrations of RNP delivery into plant cells using biolistic bombardment methods with DNA–AuNP crystals and conventional gold particles. To prepare the RNP-fused carriers, the crystals or gold particles were soaked into the medium containing RNP (i). The carriers were dried up (ii) for the biolistic bombardment in the vacuum (iii). RNPs were transferred to the nucleus. (b) Schematic illustrations of genome editing events. Cas9 proteins induced double-strand breaks (DSBs) on the DNA targeted by guide RNAs. DSBs are repaired in plant cells and sometimes induce insertion and deletion (indel) mutations. Indel mutations were quantified using high throughput sequencing. (c) Summary of observed indel mutations through amplicon-seq analysis and CRISPResso2 data processing (n = 2). Original sequences surrounding the PDS3 target locus are shown for reference. Black and light brown colors indicate deletion and insertion mutations, respectively. Numbers in boxes represent frequencies of the unedited bases on each position. Ctrl, non-bombarded control samples; Crystal, DNA–AuNP crystal-bombarded samples; Gold, gold particle-bombarded samples. (d) Quantification of the genome editing events at the target site as the number of indel variations. Indel variants with more than 10 reads were regarded as mutational patterns having occurred independently in different alleles but not by PCR errors. | ||
Experimental
Preparation of DNA–AuNPs
AuNP solution, borate buffer, and sodium chloride (NaCl) solutions were purchased from Nanopartz Inc. (Loveland, CO, USA), Thermo Fisher Scientific (Waltham, MA, USA), and Promega Corp. (Madison, WI, USA), respectively. Phosphate buffer powder (pH 7.2) (FUJIFILM Wako Pure Chemical Corp., Tokyo, Japan) was dissolved in ultrapure water using a Direct-Q water purification system. The DNA sequences used to construct DNA–AuNP crystals are listed in Table S1 (ESI†). Thiolated and non-modified DNA were purchased from Nihon Gene Research Laboratories Inc. (Sendai, Japan). Biotin-modified DNA was purchased from Integrated DNA Technologies Inc. (Coralville, IA, USA). DNA modified with 3-cyanovinylcarbazole was purchased from Hokkaido System Science Co. Ltd (Hokkaido, Japan).Two types of DNA–AuNPs (designated as DNA–AuNP A and DNA–AuNP B) were prepared. For the 13 nm AuNP, deprotected thiolated oligonucleotides were added to the aqueous AuNP solution at a molar ratio of 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Ligand A or B:AuNP), or 200:200:1 (Ligand A or B:Ligand S:AuNP). Borate buffer solution was then added, followed by incubation for 20 min with shaking. For the 23 nm AuNP, the deprotected thiolated oligonucleotides were added to the aqueous AuNP solution at a molar ratio of 1600:1 (Ligand A or B:AuNP) and incubated for 30 min with shaking. The NaCl concentration was then gradually increased from 0.05 to 0.5 M using 5 M NaCl. After each NaCl concentration increase, the nanoparticle solutions were sonicated for approximately 10 s, stirred for 15 min, and incubated overnight with shaking. For the 13 nm AuNP, excess oligonucleotides were removed by one cycle of centrifugation for 99 min at 6470 × g and three cycles for 99 min at 8476 × g (Eppendorf Centrifuge 5424; Eppendorf, Hamburg, Germany). For the 23 nm AuNP, excess oligonucleotides were removed by four cycles of centrifugation for 90 min at 5000 × g. After removing the supernatant, the precipitate was resuspended in a phosphate buffer. The final concentrations of the DNA–AuNPs were determined using UV-visible spectroscopy (V-730 BIO; Jasco, Tokyo, Japan).
:1 (Ligand A or B:AuNP), or 200:200:1 (Ligand A or B:Ligand S:AuNP). Borate buffer solution was then added, followed by incubation for 20 min with shaking. For the 23 nm AuNP, the deprotected thiolated oligonucleotides were added to the aqueous AuNP solution at a molar ratio of 1600:1 (Ligand A or B:AuNP) and incubated for 30 min with shaking. The NaCl concentration was then gradually increased from 0.05 to 0.5 M using 5 M NaCl. After each NaCl concentration increase, the nanoparticle solutions were sonicated for approximately 10 s, stirred for 15 min, and incubated overnight with shaking. For the 13 nm AuNP, excess oligonucleotides were removed by one cycle of centrifugation for 99 min at 6470 × g and three cycles for 99 min at 8476 × g (Eppendorf Centrifuge 5424; Eppendorf, Hamburg, Germany). For the 23 nm AuNP, excess oligonucleotides were removed by four cycles of centrifugation for 90 min at 5000 × g. After removing the supernatant, the precipitate was resuspended in a phosphate buffer. The final concentrations of the DNA–AuNPs were determined using UV-visible spectroscopy (V-730 BIO; Jasco, Tokyo, Japan).
DNA–AuNP crystal synthesis
First, an annealing reaction of linker DNA-A (or -B) with the complementary spacer DNA in a phosphate buffer solution containing NaCl was conducted on a Veriti 96-Well Thermal Cycler (Applied Biosystems, Waltham, MA, USA) by incubating the mixture at 95 °C for 5 min and cooling to 25 °C. The linker DNA and DNA–AuNPs were then mixed in a phosphate buffer solution with 0.5 M NaCl (for 13 nm DNA–AuNPs) or with 0.25 M NaCl (for 23 nm DNA–AuNPs). For the 23 nm DNA–AuNPs, 6.5 mM HEPES and 22 mM potassium acetate were also contained in the buffer (final concentration). The amount of linker DNA was 25–100 equivalents per particle (for 13 nm DNA–AuNP) or 200 equivalents per particle (for 23 nm DNA–AuNP). The mixture was slowly cooled over 3 days from 65 °C to 25 °C in an incubator (IS401; Yamato Scientific Co., Ltd, Tokyo, Japan) or thermal cycler (Veriti 96-Well Thermal Cycler (Applied Biosystems) or T100 Thermal Cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA)).Small angle X-ray scattering
Small angle X-ray scattering (SAXS) data were collected using a 2D detector (Pilatus 3S2M; Dectris Ltd, Baden, Switzerland) at the bending-magnet beamline BL40B2 in SPring-8.32 DNA–AuNP aggregates in buffer solution loaded into 1 mm quartz capillary tubes (Charles Supper Company Inc., Westborough, MA, USA) were measured at 25 °C in a temperature controller with a thermoelectric stage (TS62; Instec Inc., Boulder, CO, USA). The wavelength of the incident X-rays, sample-to-detector distance, beam size at the detector position, and X-ray exposure time were 0.11 nm, 1 m, approximately 0.2 mm × 0.2 mm, and 0.3 s or 3 s, respectively.The SAXS analysis was performed as previously described.15 One-dimensional SAXS patterns were obtained by the radial averaging of the 2D scattering patterns. Silver behenate (d = 5.838 nm) was used to calibrate the values of the scattering vector q (q = (4π/λ) sinθ, where θ is half of the scattering angle33). The structure factor was calculated as Iaggr,bg(q)/Idisp,bg(q), where Iaggr,bg(q) and Idisp,bg(q) are the background-corrected 1D scattering intensities for DNA–AuNP aggregates and the dispersed DNA–AuNPs, respectively.
Silica embedding
DNA–AuNP crystals were diluted with a phosphate buffer solution containing 0.5 M NaCl (final concentration of ≈5 nM AuNP). An aliquot of N-trimethoxysilylpropyl-N,N,N-trimethylammonium chloride (Gelest Inc., Morrisville, PA, USA) (∼0.3 μL) was added and stirred at 750 rpm for 15 min using an Eppendorf ThermoMixer C (Eppendorf). Then, triethoxysilane (Sigma-Aldrich, St. Louis, MO, USA) (∼0.6 μL) was added and stirred at 750 rpm for over 70 h. Centrifugation at 20238 × g for 5 min using an Eppendorf Centrifuge 5424 (Eppendorf) and resuspension in water was repeated four times.
Scanning electron microscopy characterization
Silica-encapsulated crystals were dropped onto a silicon substrate, dried for >1 h, and observed by scanning electron microscopy (SEM) (S-4800; Hitachi High-Technologies, Tokyo, Japan). Images were taken using a photodiode-backscattered electron detector at a working distance of approximately 8 mm, accelerating voltage of 15 kV, and probe current between 1 and 5 μA.Encapsulation of macromolecules
First, DNA–AuNP crystals were washed with a phosphate buffer solution containing NaCl, followed by the addition of streptavidin, quantum dot, silica nanoparticles, or RNP solutions. The streptavidin solutions (2 mg mL−1) (Invitrogen, Carlsbad, CA, USA) were prepared by dissolving Alexa Fluor streptavidin conjugate powder into a phosphate buffer with 0.15 M NaCl or commercially available levels. Qdot 585 streptavidin conjugate (Invitrogen) was used as the quantum dot solution (1 μM). In addition, solutions of red fluorescent silica particles coated with streptavidin (10 mg mL−1) (Micromod Partikeltechnologie GmbH, Rostock, Germany) were used. RNP solutions were prepared by mixing Streptococcus pyogenes Cas9 (SpCas9) proteins labeled with green fluorescent protein (GFP) (Sigma-Aldrich) and guide RNA (gRNA) at a molar ratio of 1:1 and incubating the mixture at room temperature for 10 min. The gRNA consisted of crRNA targeting AML5 (target sequence 5′-CGAGACAACTTGAGTATGAG-3′) annealed with tracrRNA (Integrated DNA Technologies). Unlabeled SpCas9 protein (described below) was used for SAXS analysis. The amount of macromolecules added was at least equal to the amount of biotin molecules or protospacer adjacent motif (PAM) sequences in the crystals. After 1 h of incubation, the crystals were washed thrice with a phosphate buffer solution containing NaCl to remove unbound macromolecules.
Confocal laser microscopy experiments
Fluorescence images of the samples were acquired using an Olympus SpinSR10 Ixplore spinning disk confocal microscope with an UplanApo 100×/1.5 oil objective (Olympus Corporation, Tokyo, Japan). Image processing was performed using CellSens Dimensions 1.5 (Olympus) and Fiji ImageJ software (National Institutes of Health, Bethesda, MD, USA).DNA photo-crosslinking between DNA–AuNPs
To improve the stability of the DNA–AuNP crystals, a DNA photocrosslinking reaction was performed by irradiating UV light for 4 min using a ZUV-H10MB lamp with a ZUV-C30H controller (OMRON Corp., Kyoto, Japan; λ = 365 nm).Recombinant Cas9 protein and in vitro synthesis of gRNA
The C-terminal His6-tagged SpCas9 with SV40 NLS proteins cloned into pET28a (Addgene #47327,34 pET-28b-Cas9-His was a gift from Alex Schier (Addgene plasmid # 47327; https://n2t.net/addgene:47327; RRID:Addgene_47327)) were expressed in Escherichia coli Rosetta2 (DE3) (Novagen, Darmstadt, Germany). Cells were grown in lysogeny broth medium containing 25 μg mL−1 kanamycin and 34 μg mL−1 chloramphenicol at 37 °C to an OD600 of 0.6. Isopropyl-β-D-thiogalactopyranoside was then added at a final concentration of 250 μM for induction of overnight protein production at 18 °C. Cells were harvested (4000 × g for 15 min at 4 °C) and disrupted by sonication in buffer A [50 mM Tris–HCl, pH 8.0, 500 mM NaCl, 5 mM MgCl2, 10% (vol/vol) glycerol] with 0.5 mg mL−1 lysozyme and 0.1 mg mL−1 DNase I. Cell debris was removed by centrifugation (12000 × g for 1 h), and clarified supernatant was loaded onto a 1 mL Ni-NTA Superflow (QIAGEN, Hilden, Germany) in a Poly-Prep Column (Bio-Rad Laboratories, Inc.) preequilibrated with buffer A. The column was washed with buffer A containing 10 mM imidazole and the proteins were eluted with 250 mM imidazole. The collected proteins were diluted twice with buffer B [50 mM Tris–HCl (pH 8.0), 5 mM MgCl2, and 10% (v/v) glycerol] and loaded onto a 1 mL HiTrap Heparin HP column (GE Healthcare, Chicago, IL, USA) equilibrated with buffer B. The protein was then eluted using a linear gradient of 0.2–1.5 M NaCl and loaded onto a HiLoad 16/600 Superdex 200 pg column (GE Healthcare) equilibrated with buffer C [20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, and 10% (v/v) glycerol]. Pooled fractions were concentrated by ultrafiltration to a final concentration of 100 μM and stored at −80 °C until use.
The gRNA was transcribed using homemade T7 RNA polymerase. The double-stranded DNA encoding the T7 promoter and gRNA sequence was amplified by polymerase chain reaction (PCR) with four overlapping primers (F1: 5′-TAATACGACTCACTATAGGACTTTTGCCAGCCATGGTGTTTTAGAGCTAGAAATAGCAAGTTAAAATAAG-3′ (for Fig. 6, targeting PDS3 AT4G14210 gene), F2: 5′-GGATCCTAATACGACTCACTATAG-3′, R1: 5′-GCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTC-3′, R2: 5′-AAAAAAGCACCGACTCGGTGCCAC-3′) and purified using a MinElute PCR Purification Kit (QIAGEN). The in vitro transcription was performed at 37 °C for 6 h. The reaction mixture was purified using 8% denaturing urea–polyacrylamide gel electrophoresis. RNA was extracted from gel slices and refolded simultaneously in H2O at 4 °C for 18 h. The extracted RNA samples were precipitated with ethanol, dissolved in H2O, and stored at −80 °C.
Biolistic bombardment
DNA–AuNP crystals consisting of 23 nm diameter DNA–AuNPs and linker DNA (n = 3) were used. Three hundred picomoles of Cas9 protein and 600 pmol of gRNA for Arabidopsis thaliana's PDS3 gene harboring target sequence of 5′-GGACTTTTGCCAGCCATGGT-3′ were mixed in buffer (20 mM Tris–HCl (pH 7.5), 150 mM NaCl, 1 mM MgCl2, 10% glycerol) in a total volume of 30 μL and incubated at 25 °C for 10 min. Five microliters of TransIT-LT (Mirus, Madison, WI, USA; only for conventional gold particle samples) was added and incubated for 5 min. The RNPs were mixed with 15 μL of 180 mg mL−1 gold particles (0.6 μm) and 120 μL of DNA–AuNP(23nm,n=3) crystals (preheated at 40 °C for 10 min), respectively, and incubated on ice for >10 min. The supernatant was replaced with 26 μL of nuclease-free duplex buffer (Integrated DNA Technologies Inc.), followed by centrifugation at 5000 × g for 1 s (gold particles) or 8000 × g for 5 s (DNA–AuNP crystals). The tubes were then subjected to flash sonication and incubated on ice for more than 10 min. Sonication was repeated, and 8 μL portions were spread onto a hydrophilic film (SH2CLHF 1150; 3M, St. Paul, MN, USA) on a carrier of PDS-1000/He Gun (Bio-Rad Laboratories, Inc.). The carriers were air-dried on a clean bench for approximately 20 min.We harvested four leaves per shoot from aged 3–4 weeks old Arabidopsis thaliana accession Col-0 seedlings. Abaxial sides of the leaves were bombarded using a PDS-1000/He Gun (Bio-Rad Laboratories, Inc.) at 1350 psi helium pressure and 6 and 5 cm distances (gold particles and DNA–AuNP crystals, respectively). The evacuation condition for biolistic delivery was set at 28 in Hg. After biolistic bombardment, petioles were embedded in Murasige and Skoog medium (pH 5.7) containing 0.5 g L−1 2-(N-morpholino)ethanesulfonic acid, 5 g L−1 sucrose, and 8 g L−1 agar. They were incubated at 22 °C, 37 °C, and 22 °C for 8, 30, and 2 h, respectively, to induce genome editing events by Cas9.35 Three independent experiments were performed on the gold particles and DNA–AuNP crystals.
Amplicon-sequencing
DNA was extracted from four leaves using a Plant Genomic DNA Extraction Mini Kit (Favorgen Biotech, Ping-Tung, Taiwan) following the manufacturer's instructions. The genome DNA was digested by NcoI-HF (New England Biolabs, Ipswich, MA, USA). The target region was PCR-amplified using a KOD One PCR master Mix (TOYOBO, Osaka, Japan) with the primer set for the first round of PCR (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATATAACTGAACTCCGTTGTAGCATTAG-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTAGCCTACTTGCCTGCTTTTC-3′). PCR cycling was conducted at 98 °C for 2 min, followed by 30 cycles at 98 °C for 10 s, 55 °C for 5 s, and 68 °C for 1 s. The PCR products were digested by NcoI-HF (New England Biolabs) and gel-purified using a FastGene Gel/PCR Extraction Kit (Nippon Genetics, Tokyo, Japan) to reduce unedited PDS3 amplicons. In the second round of PCR, Illumina combinatorial dual index sequences were attached using a primer set (5′-AATGATACGGCGACCACCGAGATCTACACNNNNNNNNTCGTCGGCAGCGTC-3′ and 5′-CAAGCAGAAGACGGCATACGAGATNNNNNNNNGTCTCGTGGGCTCGG-3′). PCR cycling was conducted at 98 °C for 2 min, followed by 30 cycles at 98 °C for 10 s, 55 °C for 5 s, and 68 °C for 3 s. The PCR products were purified using a 1.2 times-volume Sera-Mag Select (Cytiva, Marlborough, MA, USA), whose supernatant was substituted with 20% PEG-8000 and 2.5 M NaCl solution. Equal volumes of purified DNA were pooled, and their concentrations were measured using a Qubit dsDNA HS Assay Kit and a Qubit 3.0 Fluorometer (Thermo Fisher Scientific) and adjusted to 50 pM. The pooled library was used for 2 × 150-bp paired-end sequencing with iSeq100 (Illumina, San Diego, CA, USA) after mixing with 0.25 volume of 50 pM PhiX control v3 (Illumina). The FastQ files obtained were analyzed using CRISPResso2.36 Insertions and deletions (indels) at the Cas9 cleavage site were considered mutations induced by the introduced Cas9-RNPs. The mutation quantification window was set to 10 bp. The number of mutation types in the samples was counted from the results of CRISPResso2. As different sizes of indels should be generated independently in different alleles, the number of indel variations indirectly reflects genome-editing efficiency.Conclusions
We have shown the successful encapsulation of streptavidin, Qdot or CRISPR/Cas9 RNP into DNA–AuNP crystals by harnessing the hollow spaces in the crystal lattices and their affinities for the target molecules. It was also confirmed that DNA–AuNP crystals can be used as carriers for biolistic bombardment, which requires harsh mechanical conditions. The retention of crystallinity after the encapsulation of the molecules, as revealed by SAXS analysis, is a beneficial feature of DNA–AuNP crystals. The ordered structure of the materials contributes to the increased performance of encapsulated molecules such as enzymes.7 The photonic and electronic characteristics of crystalline porous materials will help explore the potential of DNA–AuNP crystals for advanced applications, such as reaction fields, catalysis, and drug carriers.Author contributions
Maasa Yokomori: experimental investigation, data analysis; Hayato Suzuki: experimental investigation, data analysis; Akiyoshi Nakamura: resources; Shigeo S. Sugano: experimental investigation, data analysis, supervision, project administration; Miho Tagawa: supervision, project administration. The manuscript was written with contributions from all the authors. All the authors approved the final version of the manuscript.Conflicts of interest
M.Y., S.S.S., M.T. are co-inventors on patent applications filed by AIST and Nagoya University related to this work.Acknowledgements
We thank R. Miyazaki, C. Sato, H. Onodera, and R. Imai for their helpful discussions and trials of the preliminary experiments. We thank H.-Y. Chou, Y. Takiguchi, A. Kuwazawa, Y. Sugimoto, M. Yamada, M. Taguchi, and Y. Taguchi for their technical assistance. We thank N. Ohta and H. Sekiguchi for technical support and use of the synchrotron radiation experiments performed at BL40B2 of SPring-8, with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal no. 2020A0809, 2021A1518, and 2021B1615). We wish to acknowledge the Division for Medical Research Engineering, Nagoya University Graduate School of Medicine, for technical support and use of the Olympus SpinSR10 Ixplore spinning disk confocal microscope. This work was supported by Core Research for Evolutional Science and Technology (CREST), Japan Science and Technology Agency (JST) (grant number JPMJCR19S6), a Female Researcher Award to M. T. from the Soroptimist Japan Foundation, and a Young Researcher Grant from the Center for Integrated Research of Future Electronics, Institute of Materials and Systems for Sustainability, Nagoya University.References
- N. C. Seeman, J. Theor. Biol., 1982, 99, 237–247 CrossRef CAS PubMed.
- F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed.
- S. Huo, M. Kwak, J. Qin, B. Dittrichn and A. Herrmann, Mater. Today, 2021, 49, 378–390 CrossRef CAS.
- S. Wang, Y. Chen, S. Wang, P. Li, C. A. Mirkin and O. K. Farha, J. Am. Chem. Soc., 2019, 141, 2215–2219 CrossRef CAS PubMed.
- C. Geng and P. J. Paukstelis, J. Am. Chem. Soc., 2014, 136, 7817–7820 CrossRef CAS PubMed.
- L. Chen, J. Zhang, Z. Lin, Z. Zhang, M. Mao, J. Wu, Q. Li, Y. Zhang and C. Fan, Acta Pharm. Sin. B, 2022, 12, 76–91 CrossRef CAS PubMed.
- Y. Tian, J. R. Lhermitte, L. Bai, T. Vo, H. L. Xin, H. Li, R. Li, M. Fukuto, K. G. Yager, J. S. Kahn, Y. Xiong, B. Minevich, S. K. Kumar and O. Gang, Nat. Mater., 2020, 19, 789–796 CrossRef CAS PubMed.
- Z. Zhao, J. Fu, S. Dhakal, A. Johnson-Buck, M. Liu, T. Zhang, N. W. Woodbury, Y. Liu, N. G. Walter and H. Yan, Nat. Commun., 2016, 7, 10619 CrossRef CAS PubMed.
- C. R. Laramy, M. N. O’Brien and C. A. Mirkin, Nat. Rev. Mater., 2019, 4, 201–224 CrossRef CAS.
- X. Cheng, R. Sun, L. Yin, Z. Chai, H. Shi and M. Gao, Adv. Mater., 2017, 29, 1604894 CrossRef PubMed.
- K. Yu, X. Hai, S. Yue, W. Song and S. Bi, Chem. Eng. J., 2021, 419, 129535 CrossRef CAS.
- W. Liu, M. Tagawa, H. L. Xin, T. Wang, H. Emamy, H. Li, K. G. Yager, F. W. Starr, A. V. Tkachenko and O. Gang, Science, 2016, 351, 582–586 CrossRef CAS PubMed.
- R. J. Macfarlane, B. Lee, M. R. Jones, N. Harris, G. C. Schatz and C. A. Mirkin, Science, 2011, 334, 204–208 CrossRef CAS PubMed.
- M. S. Lee, D. W. Yee, M. Ye and R. J. Macfarlane, J. Am. Chem. Soc., 2022, 144, 3330–3346 CrossRef CAS PubMed.
- H. Sumi, N. Ohta, H. Sekiguchi, S. Harada, T. Ujihara, K. Tsukamoto and M. Tagawa, Cryst. Growth Des., 2021, 21, 4506–4515 CrossRef CAS.
- H. Sumi, N. Ohta, H. Sekiguchi, S. Harada, T. Ujihara, K. Tsukamoto and M. Tagawa, Cryst. Growth Des., 2022, 22, 3708–3718 CrossRef CAS.
- N. Michael Green, in Methods in Enzymology, ed. M. Wilchek and E. A. Bayer, Academic Press, 1990, vol. 184, pp. 51–67 Search PubMed.
- M. Jinek, K. Chylinski, I. Fonfara, M. Hauer, J. A. Doudna and E. Charpentier, Science, 2012, 337, 816–821 CrossRef CAS PubMed.
- G. J. Knott and J. A. Doudna, Science, 2018, 361, 866–869 CrossRef CAS PubMed.
- E. Auyeung, T. I. N. G. Li, A. J. Senesi, A. L. Schmucker, B. C. Pals, M. O. de la Cruz and C. A. Mirkin, Nature, 2013, 505, 73–77 CrossRef CAS PubMed.
- Y. Yoshimura and K. Fujimoto, Org. Lett., 2008, 10, 3227–3230 CrossRef CAS PubMed.
- M. Tagawa, K.-I. Shohda, K. Fujimoto and A. Suyama, Soft Matter, 2011, 7, 10931–10934 RSC.
- A. F. De Fazio, A. H. El-Sagheer, J. S. Kahn, I. Nandhakumar, M. R. Burton, T. Brown, O. L. Muskens, O. Gang and A. G. Kanaras, ACS Nano, 2019, 13, 5771–5777 CrossRef CAS PubMed.
- P. K. Harimech, S. R. Gerrard, A. H. El-Sagheer, T. Brown and A. G. Kanaras, J. Am. Chem. Soc., 2015, 137, 9242–9245 CrossRef CAS PubMed.
- A. F. De Fazio, D. Misatziou, Y. R. Baker, O. L. Muskens, T. Brown and A. G. Kanaras, Chem. Soc. Rev., 2021, 50, 13410–13440 RSC.
- E. Auyeung, R. J. Macfarlane, C. H. J. Choi, J. I. Cutler and C. A. Mirkin, Adv. Mater., 2012, 24, 5181–5186 CrossRef CAS PubMed.
- H. Bui, C. Onodera, C. Kidwell, Y. Tan, E. Graugnard, W. Kuang, J. Lee, W. B. Knowlton, B. Yurke and W. L. Hughes, Nano Lett., 2010, 10, 3367–3372 CrossRef CAS PubMed.
- L. Sun, Z. Zhang, S. Wang, J. Zhang, H. Li, L. Ren, J. Weng and Q. Zhang, Nanoscale Res. Lett., 2008, 4, 216–220 CrossRef PubMed.
- S. Svitashev, C. Schwartz, B. Lenderts, J. K. Young and A. Mark Cigan, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- R. Mout, M. Ray, Y.-W. Lee, F. Scaletti and V. M. Rotello, Bioconjugate Chem., 2017, 28, 880–884 CrossRef CAS PubMed.
- M. A. Hink, R. A. Griep, J. W. Borst, A. van Hoek, M. H. M. Eppink, A. Schots and A. J. W. Visser, J. Biol. Chem., 2000, 275, 17556–17560 CrossRef CAS PubMed.
- N. Ohta, H. Sekiguchi, Y. C. Sasaki and N. Yagi, J. Appl. Crystallogr., 2014, 47, 654–658 CrossRef CAS.
- T. C. Huang, H. Toraya, T. N. Blanton and Y. Wu, J. Appl. Crystallogr., 1993, 26, 180–184 CrossRef CAS.
- J. A. Gagnon, E. Valen, S. B. Thyme, P. Huang, L. Akhmetova, A. Pauli, T. G. Montague, S. Zimmerman, C. Richter and A. F. Schier, PLoS One, 2014, 9, e98186 CrossRef PubMed.
- S. Kurokawa, H. Rahman, N. Yamanaka, C. Ishizaki, S. Islam, T. Aiso, S. Hirata, M. Yamamoto, K. Kobayashi and H. Kaya, Plant Cell Physiol., 2021, 62, 1676–1686 CrossRef CAS PubMed.
- K. Clement, H. Rees, M. C. Canver, J. M. Gehrke, R. Farouni, J. Y. Hsu, M. A. Cole, D. R. Liu, J. K. Joung, D. E. Bauer and L. Pinello, Nat. Biotechnol., 2019, 37, 224–226 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00949h |
| This journal is © The Royal Society of Chemistry 2022 |