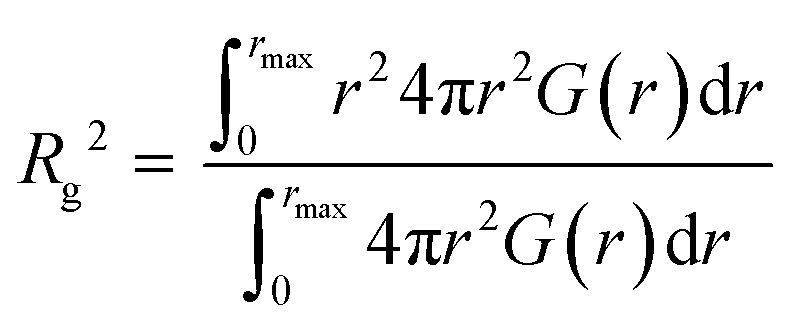DOI:
10.1039/D2SM00853J
(Paper)
Soft Matter, 2022,
18, 8582-8590
Controlling mechanical properties of ultrahigh molecular weight ion gels by chemical structure of ionic liquids and monomers†
Received
27th June 2022
, Accepted 7th October 2022
First published on 11th November 2022
Abstract
A new class of ion gels, termed ultrahigh molecular weight (UHMW) gels, formed by physical entanglement of ultrahigh molecular weight polymers in ionic liquids, are synthesised using facile one step radical polymerisation with significantly low initiator conditions, and exhibit superior mechanical characteristics such as stretchability, recyclability, and room temperature self-healing ability. In this study, UHMW gels are synthesised using various combinations of monomer and IL structures, and the effect of their chemical structures on the physicochemical properties of UHMW gels are thoroughly investigated. UHMW polymers are prepared in situ for all combinations of ILs and monomers used in this study, indicating the wide applicability of this fabrication strategy. The structure–property relationships between chemical structures and mechanical properties of UHMW gels are investigated in detail. Furthermore, the differences in self-healing efficiency of UHMW gels depending on the chemical structure is discussed in terms of individual polymer conformation and polymer–polymer interaction based on molecular dynamics simulations.
Introduction
With the advent of the Internet of things (IoT) era, wearable and flexible electronics have received increasing attention.1–3 In this regard, conductive and stretchable polymeric materials have been intensively investigated in recent years.4–10 Ion-conductive polymeric materials are important components in flexible and wearable devices such as secondary batteries, sensors, electric double layer transistors, supercapacitors, and soft actuators.11–15 Dry ionic conductors such as poly(ethylene oxide) composited with inorganic lithium salts have good mechanical toughness and are free from solvent evaporation.16 However, ionic conductivity is generally low as the ion transport is dominated by the segmental motion of polymer chains. Although hydrogels and organogels that dissolve inorganic salts in swollen polymer networks show relatively high ionic conductivity, they are susceptible to solvent evaporation.17 Whereas, ion gels, defined as a gel-type ionic conductor containing ionic liquids (ILs) as a solvent, are a new class of ionically conductive soft materials that satisfy high ionic conductivity as well as non-volatility.18–21
In general, the poor mechanical properties of polymeric gels due to high liquid content limit their applications in flexible and wearable devices, in which tolerance for repetitive mechanical loading is required.22 So far, various strategies for toughening polymer gels have been proposed.23–28 Recently, introduction of self-healing functionalities into polymer gels has been investigated to achieve significant improvement in material lifetime.29–34 Several studies have reported the fabrication of self-healing and stretchable ion gels using intricately designed reversible bonds.35–41 However, such materials often require time-consuming preparation/purification procedures and complicated synthesis of specially designed functional monomers and polymer networks.
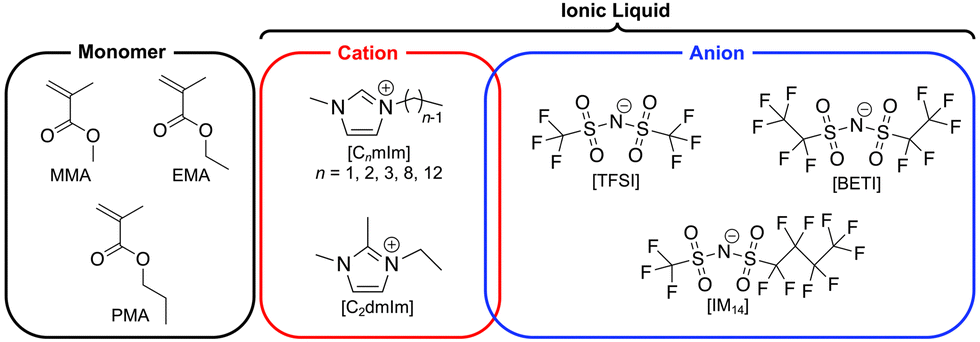
In our previous study, we have reported a facile one-step strategy for fabrication of stretchable and self-healable ion gels based on physical entanglement of ultrahigh molecular weight (UHMW) polymers.42 The radical polymerisation of methacrylate monomers in ILs, for example a typical IL 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([C2mIm][TFSI]), yields a monomer conversion of nearly 100% even at significantly low initiator concentrations, which consequently produced ion gels solely by the physical entanglements of UHMW polymers over 106 Da, which were named as UHMW gels. However, the effect of the chemical structures of ILs and methacrylate monomers on the polymerisation and mechanical properties of UHMW gels have not been understood. In particular, as the possibility to control the mechanical properties of gels through the solvent structure is a unique feature of ion gels, investigation of the effect of IL cation and anion structures has a great importance. Herein, UHMW gels were synthesised from various IL and monomer structures to investigate the effect of their chemical structures on the in situ formed UHMW polymers. The chemical structure of monomers and ILs are shown in Fig. 1. The IL solvents for ion gels were selected based on the compatibility of the methacrylate polymers. Next, the rheological, mechanical properties, and self-healing ability of UHMW gels composed of various IL and polymer structures were investigated in detail to elucidate the structure–property relationship of UHMW gels. Furthermore, molecular dynamics (MD) simulations were performed to decipher the influence of IL and polymer structures on self-healing ability of UHMW gels at microscopic level. It was implied that the individual polymer conformation and interpolymer interactions were adjusted depending on IL/polymer chemical structures, which resulted in the difference in the self-healing ability of UHMW gels.
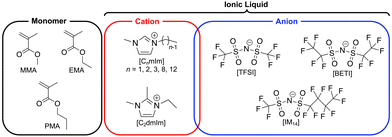 |
| | Fig. 1 The chemical structures of monomer, IL cation, and anions used in this study. | |
Experimental
Materials
1-Alkyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([CnmIm][TFSI], n = 1, 2, 8, 12) and 1-ethyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide ([C2dmIm][TFSI]) were purchased from Kanto Chemical (Japan). [C3mIm][TFSI] was purchased from TCI (Japan). [C2mIm][BETI] (BETI: bis(pentafluoroethanesulfonyl)imide) and [C2mIm][IM14] (IM14: (trifluoromethanesulfonyl)(nonafluorobutanesulfonyl)imide) were synthesised according to a literature reference with slight modifications.43 Lithium salts, Li[BETI] and Li[IM14], which were used for synthesis of these ILs, were purchased from Mitsubishi Materials Electronic Chemical Co., Ltd (Japan). All the ILs were dried under vacuum at 120 °C for 24 h prior to use. Methyl methacrylate (MMA) and ethyl methacrylate (EMA) were purchased from Kanto Chemical (Japan), and propyl methacrylate (PMA) was purchased from TCI (Japan). All monomers were purified by passing them through the basic alumina column to remove polymerisation inhibitor. 2,2′-Azobis(isobutyronitrile) (AIBN) were purchased from FUJIFILM Wako Pure Chemical Corporation (Japan) and used as received.
Synthesis and characterisation of ultrahigh molecular weight gels
For the synthesis of all UHMW gels, the monomer concentration and the AIBN content were set at 40 vol% and 0.02 mol% vs. monomer content, respectively. A representative synthetic procedure for the PMMA/[C2mIm][TFSI] UHMW gel is described as follows. MMA (1.6 mL, 15.0 mmol), [C2mIm][TFSI] (2.4 mL, 9.32 mmol), and AIBN (0.49 mg, 3.0 × 10−3 mmol) were charged in a glass cup and sealed with a rubber septum, through which argon was bubbled for 15 min at room temperature. Subsequently, polymerisation reaction was carried out at 80 °C for 24 h, resulting in the formation of the transparent ion gel. Monomer conversion was determined using 1H nuclear magnetic resonance (NMR) measurements for the as prepared UHMW gels dissolved in deuterated chloroform (CDCl3). For the determination of molecular weight of in situ formed UHMW polymers, UHMW gels were purified by reprecipitation with acetone as good solvent and methanol as poor solvent twice and then vacuum dried at 60 °C, resulting in white powder of UHMW polymers. The molecular weight and polydispersity index (PDI) of the polymers were determined using gel permeation chromatography (GPC), in which 10 mM lithium bromide (LiBr) in N,N-dimethylformamide (DMF) solution was used as the eluent. The GPC columns (Showa Denko, Japan) were calibrated using PMMA standards. Prior to rheological and mechanical measurements, UHMW gels were dried in vacuo at 80 °C overnight to remove residual monomers.
Rheological measurements
Oscillatory shear measurements were performed with an Anton Paar MCR 102 rheometer (Anton Paar, Austria) using the parallel plate geometry with a 12 mm diameter plate. Gap spacing was kept at 0.5 mm for all the measurements. Frequency sweep measurements were performed over a frequency range of 0.1–100 rad s−1 with a strain amplitude of 1% at constant temperatures from 10 to 120 °C at intervals of 10 °C. The time-temperature superposition (tTS) master curves for UHMW gels were constructed from the frequency sweep data at different temperatures.
Tensile tests
Tensile tests of UHMW gels were carried out using a Shimadzu AGS-X tester (Shimadzu, Japan). The gels cut into a dumbbell-shape (JIS K 6251-7 size, width: 2 mm, gauge length: 12 mm) were stretched at a speed of 10 cm min−1 with a 100 N load cell. For the self-healing test, the dumbbell-shaped gels were cut at the centre, subsequently the two cut surfaces were contacted and allowed to heal at room temperature for 24 h, after which tensile tests were performed for the healed samples.
Thermal analyses
Differential scanning calorimetry (DSC) measurements were performed using a DSC250 instrument (TA Instruments, USA) under a nitrogen atmosphere. The samples were placed on an open aluminum pan and heated from 40–200 °C, followed by cooling to −120 °C, and then reheated to 200 °C at a cooling and heating rate of 10 °C min−1. The glass transition temperature (Tg) (the temperature at the peak top of derivative heat flow) were determined from the DSC thermograms during the 2nd heating step.
Molecular dynamics simulations
All-atom molecular dynamics (MD) simulations were carried out using the GROMACS 2020.1 program under the isobaric-isothermal (NTP) ensemble (298 K and 1 atm) in a cubic cell. Procedures similar to that reported in our previous study were followed in this case.42 Number of ILs and polymers in the simulation boxes are listed in Table S1 (ESI†). A 20 mer (approximately 2 kDa) was used as the polymer due to computing performance issues. The total simulation time was set to be 25 ns. From the last 500 ps, the data was collected with 0.1 ps intervals, and analysed to determine the radial distribution function G(r). The density values calculated from the present MD simulations are consistent with the density values estimated from the respective densities of the IL and the polymer, which are also listed in Table S1 (ESI†). CLaP and OPLS-AA force fields, including intermolecular Lennard-Jones (LJ), coulombic interactions, and intramolecular interactions (bond stretching, angle bending, and torsion of dihedral angles) were used for the C2mIm and C12mIm cations, TFSI anion, and PMMA and PEMA polymers.44–46 The radius of gyration (Rg) for the polymer was calculated from the intramolecular component Gintra(r), according to the following equation, where rmax represents the upper limit of the integration, and here we assumed a sufficiently large r (35 Å) that there is no intramolecular correlation of the polymers:
Results and discussion
Synthesis of UHMW gels from various IL and monomer structures
To prepare UHMW gels with different IL structures, methyl methacrylate (MMA) monomer was polymerised in a wide variety of ILs consisting of combinations of different imidazolium-based cations (CnmIm: n = 2–12 and C2dmIm) and sulfonylimide anions with different lengths of perfluoroalkyl groups (TFSI, BETI, and IM14). In addition, ethyl methacrylate (EMA) and propyl methacrylate (PMA) were polymerised in [C2mIm][TFSI] and [C2mIm][BETI], respectively to investigate the effect of monomer structures. Note that when PMA was polymerised in [C2mIm][TFSI], it was too soft to be moulded for mechanical tests probably due to its very low glass transition temperature (Tg). Therefore, polymerisation of PMA monomer was performed in [C2mIm][BETI], which has a higher Tg than that of [C2mIm][TFSI]. The chemical structures of ILs and monomers used in this study are shown in Fig. 1. For the synthesis of UHMW gels, the monomer concentration was fixed at 40 vol% and the initiator (2,2′-azobis(isobutyronitrile), AIBN) content was set at as low as 0.02 mol% vs. monomer content. After argon bubbling of precursor solutions containing ILs, monomers and initiator, polymerisation was allowed to proceed at 80 °C for 24 h. For all combinations of monomers and ILs, transparent ion gels without any macroscopic polymer–IL phase separation were formed in situ as polymerisation proceeded, indicating that the robust physical entanglement of UHMW polymers were formed in ILs. The results of polymerisation (molecular weight, polydispersity index (PDI), and monomer conversion) are summarised in Table 1. GPC traces of synthesised polymers extracted from UHMW gels and 1H NMR spectra of as prepared UHMW gels after polymerisation are shown in Fig. S1 and S2 (ESI†). For all monomer-IL combinations used in this study, UHMW polymers with molecular weights near or above 106 Da were obtained with high monomer conversion of over 95%. These results indicate that the fabrication strategy of UHMW gels can be applicable for wide variety of methacrylate polymer and IL structures, which would render tunability of physicochemical properties of UHMW gels. Previous experimental and computational investigations for the radical polymerisation in ILs suggested that the rate constant of propagation (kp) was enhanced with decrease in activation energy due to the IL-radical interaction.47,48 In addition, our previous study that compared viscosity-tuned toluene and ILs as polymerization media revealed that the effect of solvent viscosity that potentially slow down the termination reaction of growing polymer on the molecular weight was not significant, thus enhancement of kp would be a dominant factor for the formation of UHMW polymers in ILs.42 In the present study, consistent with the previous results, the molecular weight of the synthesised PMMA in various ILs tended to increase with increasing ionic concentration in the precursor solutions (Fig. S3, ESI†), suggesting that the more the ionic moieties around the propagation radicals, the higher the kp due to the IL-radical interaction. Furthermore, the PDIs of obtained UHMW polymers were slightly below 2.0, which were significantly lower than those of high molecular weight polymers produced as a result of decreased termination rate due to high viscosity, which was well-known as the Trommsdorff effect.49 The relatively low PDI in this study could be attributed to the enhanced kp and significantly low concentration of radicals in this polymerization condition that might lower the influence of the termination reaction.
Table 1 Characterisation results of UHMW polymers synthesised in ILs
| Monomer |
Solvent |
M
n
/kDa |
M
w
/kDa |
PDIa |
Conversionb |
|
Calculated by GPC using PMMA standards (eluent: 10 mM LiBr/DMF). For the characterisation by GPC, polymers were extracted from ILs as follows: UHMW gels were dissolved in acetone, subsequently UHMW polymers were reprecipitated into excess amount of methanol.
Calculated from 1H NMR. As prepared UHMW gels were dissolved in CDCl3, and the monomer conversion was calculated by comparing the residual vinyl peaks of monomers with polymer peaks.
|
| MMA |
[C1mIm][TFSI] |
1587 |
2896 |
1.83 |
98.0 |
| [C2mIm][TFSI] |
1280 |
2377 |
1.85 |
98.1 |
| [C3mIm][TFSI] |
1216 |
2400 |
1.97 |
97.3 |
| [C8mIm][TFSI] |
1274 |
2235 |
1.75 |
95.9 |
| [C12mIm][TFSI] |
1169 |
2263 |
1.94 |
97.0 |
| [C2dmIm][TFSI] |
1492 |
2773 |
1.86 |
96.8 |
| [C2mIm][BETI] |
1473 |
2693 |
1.83 |
97.9 |
| [C2mIm][IM14] |
1162 |
2652 |
2.28 |
97.8 |
| EMA |
[C2mIm][TFSI] |
988 |
1700 |
1.72 |
95.8 |
| PMA |
[C2mIm][BETI] |
1019 |
1820 |
1.79 |
95.9 |
Rheological properties of UHMW gels
Rheological properties of UHMW gels with different polymers and IL structures were evaluated by frequency sweep measurements at different temperatures ranging from 10 to 120 °C. Next, time-temperature superposition (tTS) master curves for storage modulus (G′), loss modulus (G′′), and tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ for each UHMW gel was obtained at a reference temperature of 10 °C (Fig. 2 and Fig. S4, ESI†). Note that, although the tTS principle was well applicable, there was a deviation for G′′ and tanδ at high frequency region for all UHMW gels. This phenomenon was observed for other ion gel systems also, and it was originated from the relaxation related to solvent viscosity.50–52
δ for each UHMW gel was obtained at a reference temperature of 10 °C (Fig. 2 and Fig. S4, ESI†). Note that, although the tTS principle was well applicable, there was a deviation for G′′ and tanδ at high frequency region for all UHMW gels. This phenomenon was observed for other ion gel systems also, and it was originated from the relaxation related to solvent viscosity.50–52
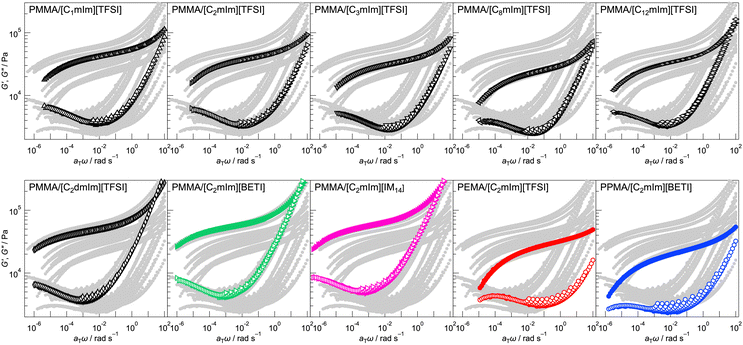 |
| | Fig. 2 Time-temperature superposition (tTS) master curves of G′ (closed symbols) and G′′ (open symbols) for UHMW gels with different combinations of polymers and ILs constructed from the frequency sweep measurements at the temperature range from 10 to 120 °C. The reference temperature = 10 °C. In each graph, only the master curve of interest is highlighted, while the other curves are shown in grey to clarify their relative positions. | |
In all systems, a rubbery plateau region was observed over a wide frequency range; crossover of G′ and G′′ and subsequent terminal relaxation (G′ ∝ ω2 and G′′ ∝ ω) did not appear at least within this measurement condition, which indicates that UHMW gels behave as viscoelastic solids over very long timescales. This is due to the significantly slow relaxation of physical crosslinking owing to the extremely high molecular weight of the polymers, as the reptation theory suggests that the relaxation time of polymer entanglement correlates with the 3rd power of molecular weight. Even at frequency sweep measurement at 120 °C, G′ was higher than G′′ for all frequency region, indicating excellent mechanical robustness against temperature for UHMW gels (Fig. S5, ESI†).
Tensile properties of UHMW gels
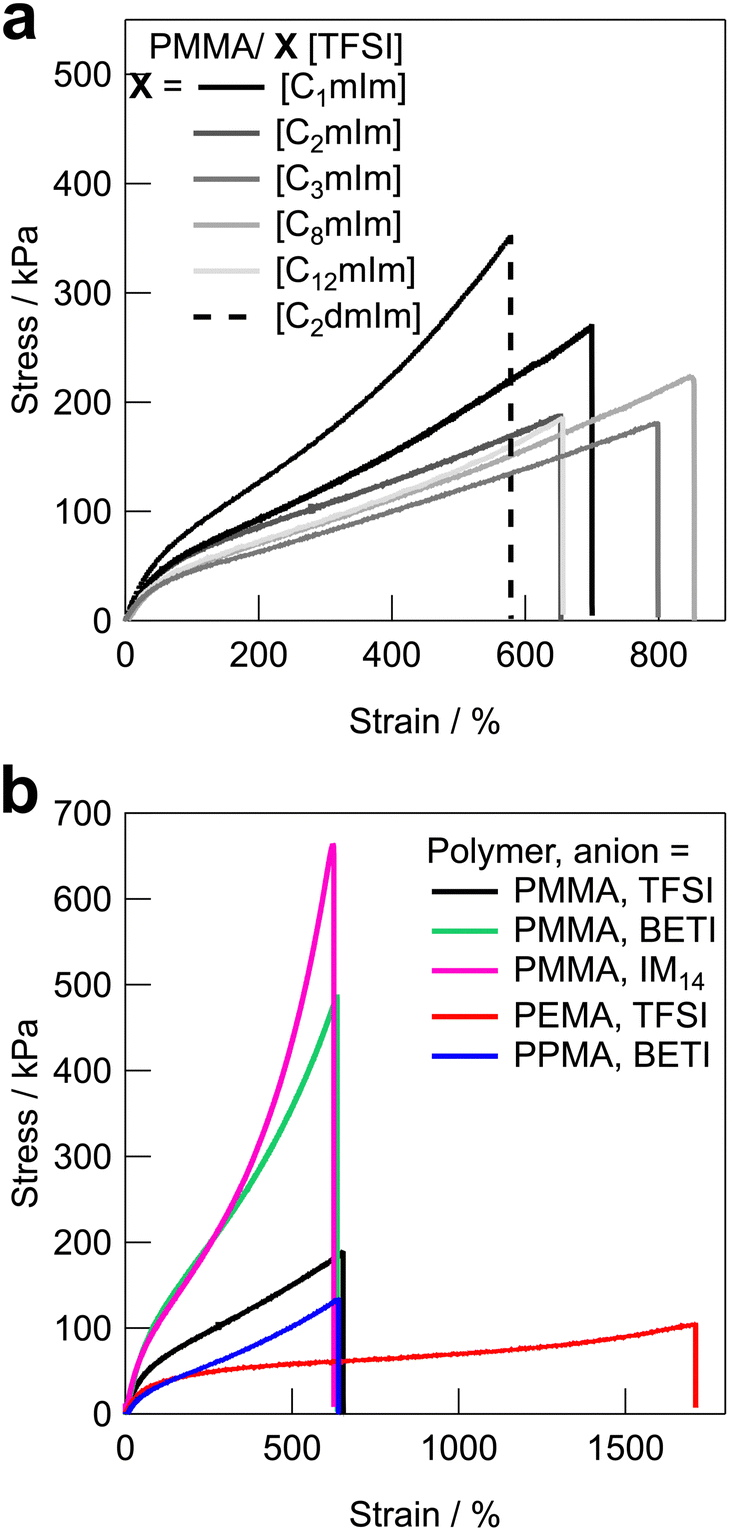
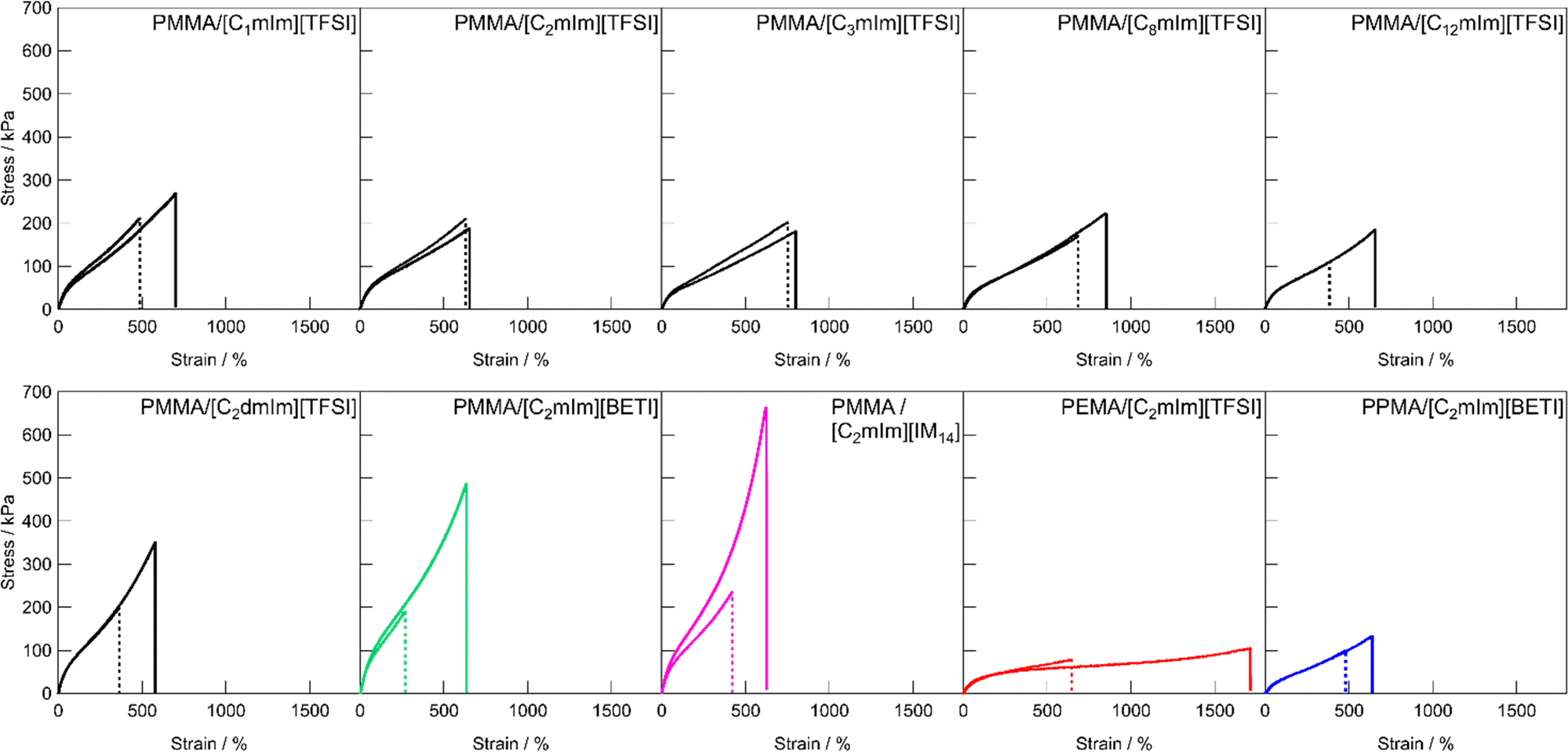
Fig. 3 shows the uniaxial tensile stress–strain curves for UHMW gels obtained with various polymer/IL combinations. The mechanical strength of all the UHMW gels was much better than that of a conventional PMMA ion gel using a chemical crosslinker, which was previously reported as a comparison of the tetra-branched PEO-based stretchable ion gels.53 Tensile properties and glass transition temperatures (Tgs) of UHMW gels are summarised in Table S2 (ESI†). Since the polymer/IL combinations used in this study are all compatible systems, the Tg of the ion gel can be determined by DSC measurements as a single point (Fig. S6, ESI†). When the cation structures of UHMW gels were changed (Fig. 3a), Young's modulus (E) varied from 94 to 167 kPa, and the fracture stress and strain varied from 182 to 353 kPa and 578 to 853%, respectively. On the other hand, when the anion and polymer structures were changed (Fig. 3b), the stress–strain curves changed much significantly compared to changes in the UHMW gels with different cation structures. In particular, UHMW gels from PMMA/[C2mIm][BETI] and PMMA/[C2mIm][IM14] exhibited significantly high fracture stress. One of the possible reasons for high mechanical strength in the PMMA/[C2mIm][BETI] UHMW gel could be attributed to its relatively high Tg of −57.2 °C (Table S2, ESI†), which possibly contributed to stresses associated with micro-Brownian motion of polymer segment. Note that the tensile speed in this study is 10 cm min−1, where the correlation between Tg and E was observed (Fig. S7, ESI†). This could be attributed to the influence of the transition from rubbery plateau to glassy region at this timescale. In contrast, the PMMA/[C2mIm][IM14] UHMW gel showed the upward deviation from the correlation between Tg and E. Furthermore, it exhibited the highest fracture stress among UHMW gels in this study despite possessing relatively low Tg of −69.7 °C. A recent study suggested that nanophase separation occurred in [C2mIm][IM14] due to the aggregation of the long perfluoroalkyl chains of [IM14] anions.54 Therefore, the nanophase separated structure in the UHMW gel might affect the mechanical properties of the UHMW gel. Although the detailed mechanism is still unclear, this result implies that the mechanical strength of UHMW gels could be further enhanced by properly controlling the nanophase separation structure in the gel. On the other hand, although the PMMA/[C12mIm][TFSI] UHMW gel had relatively high Tg of −56.1 °C almost the same as that of PMMA/[C2mIm][BETI], it showed much lower Young's modulus and fracture stress than those of PMMA/[C2mIm][BETI]. The downward deviation from the correlation between Tg and E was also confirmed in Fig. S7 (ESI†). The long alkyl chain of [C12mIm] cation is known to cause polar–apolar nanophase separation,55 suggesting that in this case nanophase separation might have a negative effect on mechanical properties. It is also possible that, although the PMMA/[C12mIm][TFSI] UHMW gel is transparent and thus PMMA and [C12mIm][TFSI] are macroscopically compatible, the low affinity between the IL and the polymer on the microscopic scale may have an influence on the mechanical properties, which will be discussed later in the molecular dynamics (MD) simulations section. When the polymer structure was changed from PMMA, it was observed that the PEMA/[C2mIm][TFSI] UHMW gel had the highest fracture strain and the lowest fracture stress among all UHMW gels. This can be attributed to the lowest Tg of −78.7 °C and slightly lower molecular weight of PEMA/[C2mIm][TFSI] gel compared to other systems (Table 1 and Fig. S2, ESI†), which may lead to low mechanical strength and large elongation at break due to the disentanglement of polymer chains under the timescale of the tensile test.
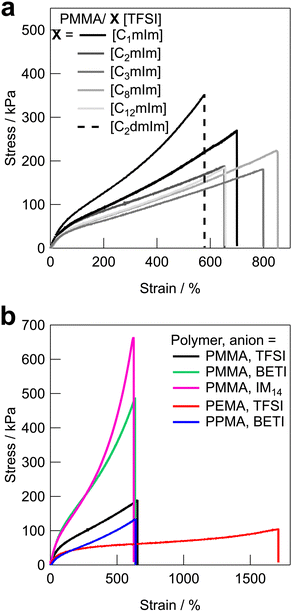 |
| | Fig. 3 Tensile stress–strain curves for UHMW gels with (a) different cation structures with the fixed polymer and anion structures (PMMA and [TFSI] anion), and (b) different polymer and anion structures with the fixed cation structure ([C2mIm] cation). | |
Self-healing ability of UHMW gels
Next, we evaluated the self-healing behaviour of UHMW gels from tensile tests. Our previous study revealed that the PMMA/[C2mIm][TFSI] UHMW gel showed novel self-healing ability at room temperature despite the fact that UHMW gel had a quite long relaxation time owing to the entanglement of UHMW polymers, whereas the PEMA/[C2mIm][TFSI] UHMW gel showed lower healing efficiency than that of PMMA/[C2mIm][TFSI].42 This result implied that the self-healing ability was dependent on the chemical structure of the UHMW gel. However, detailed study to understand the relationship between polymer and IL chemical structures and self-healing ability of the UHMW gels has not been performed so far. Fig. 4 shows the tensile stress–strain curves for the pristine and healed ion gels for each UHMW gel with different polymer and IL structures. For the preparation of healed ion gel samples, dumbbell-shaped ion gels were cut at the centre, after that the two cut surfaces were brought into contact immediately and allowed to heal at room temperature for 24 h. In contrast to conventional chemically cross-linked ion gels with bifunctional chemical crosslinkers that had no self-healing ability, all UHMW gels exhibited self-healing properties, although there were differences in healing efficiencies. For UHMW gels with different cation structures, the healing efficiency reached to 100% for PMMA/[C2mIm][TFSI] and PMMA/[C3mim][TFSI], whereas complete healing was not observed for other gels. When the anion structure was varied from TFSI to BETI and IM14, the healing efficiency was decreased to less than 40%. One of the possible reason of low healing efficiency for the PMMA/[C2mIm][BETI] and PMMA/[C2mIm][IM14] could be attributed to the trade-off relationship between self-healing ability and stiffness. The higher Young's modulus is related to lower dynamic properties of physical cross-linking by entanglement, which might lead to low self-healing properties. The trade-off relationship was also reported in other self-healable polymeric materials.32,56
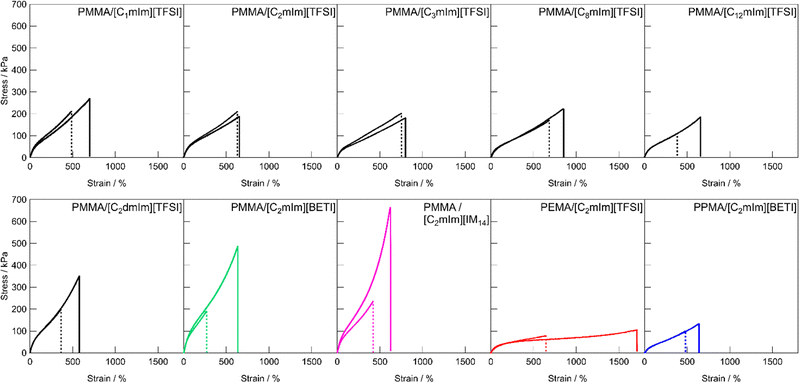 |
| | Fig. 4 Tensile stress–strain curves for pristine and healed UHMW gels with different polymer and IL structures. Solid and dashed lines indicate pristine and healed samples, respectively. | |
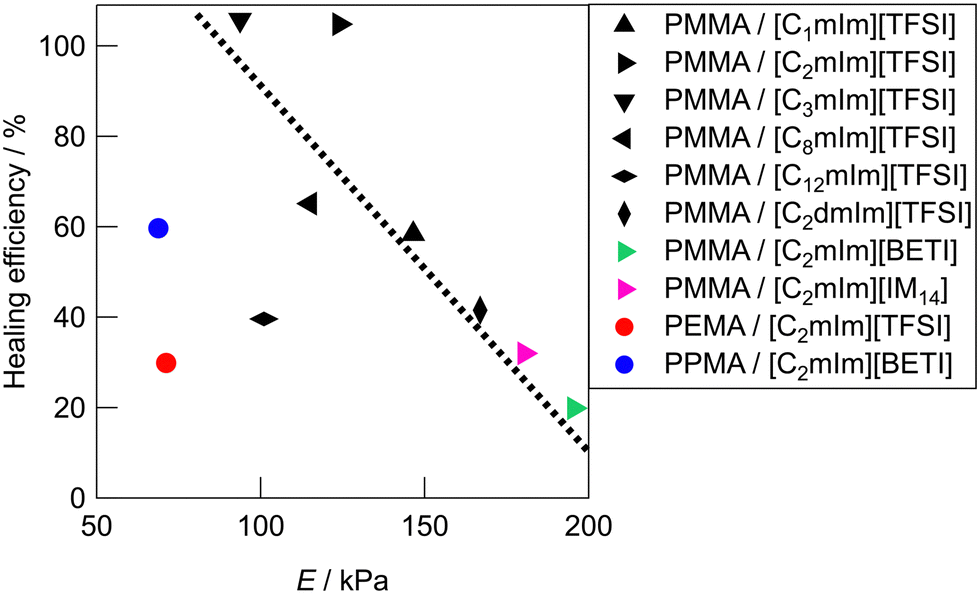
Fig. 5 shows the relationship between Young's modulus and healing efficiency for UHMW gels. While several UHMW gels obeyed the trade-off relationship between Young's modulus and healing efficiency shown by the dotted straight line, some UHMW gels deviated upward and downward from the trade-off line. The PMMA/[C2mIm][TFSI] UHMW gel exhibited upward deviation from the trade-off line, indicating that there exists a driving force which accelerates the reformation of entanglement across the cut surfaces. On the other hand, PMMA/[C12mIm][TFSI], PEMA/[C2mIm][TFSI], and PPMA/[C2mIm][BETI] UHMW gels deviated significantly downward from the trade-off line. Notably, although the tensile stress–strain curves for pristine PMMA/[C12mIm][TFSI] and PMMA/[C2mIm][TFSI] UHMW gels were nearly identical, the healing efficiencies were quite different (Fig. S8, ESI†).
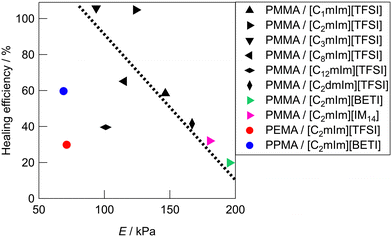 |
| | Fig. 5 Relationship between the Young's modulus (E) and the healing efficiency of UHMW gels. The dotted line is a guide to the eye. The healing efficiency was defined as the ratios of mechanical toughness (the area under the stress–strain curve) between pristine and healed gels. | |
These results suggest that the self-healing behaviour of UHMW gels is not governed only by the trade-off relationship between the stiffness and the healing efficiency, other factors also contribute to the self-healing phenomena. In our previous study, MD simulations for PMMA/[C2mIm][TFSI] and PEMA/[C2mIm][TFSI] systems suggested that PMMA and PEMA showed different solvation structure in [C2mIm][TFSI].42 Therefore, the deviation from the trade-off relationship might have originated from the difference in the microscopic interaction among polymers and IL cations and anions, which would lead to the acceleration or deceleration of reformation of entanglement across cut surfaces.
MD simulations for different polymer/IL systems
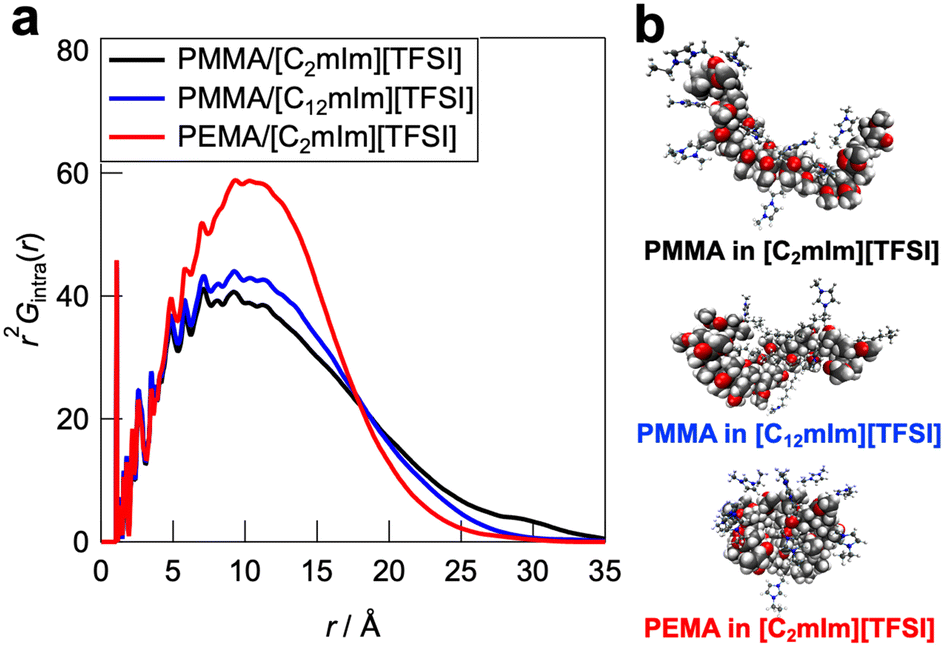
To further understand the polymer–IL and polymer–polymer interactions at the microscopic scale, all-atom MD simulations and detailed statistical analysis were performed on the PMMA/[C2mIm][TFSI], PMMA/[C12mIm][TFSI], and PEMA/[C2mIm][TFSI] systems, in which healing efficiency was quite different. The simulations were carried out in an NTP ensemble. Density values derived from MD simulations are in good agreement with actual systems, indicating the validity of simulated polymer/IL structures (Table S1, ESI†). The radial distribution functions for all atoms (Gtotal(r) as an r-weighted difference form, r2[G(r) − 1], is shown in Fig. S9, ESI†) were calculated from the trajectory data obtained from the 500 ps calculations. The radial distribution function consists of intramolecular and intermolecular correlations for each component. The intramolecular radial distribution function for the polymer (Gintra(r)) is shown in Fig. 6a. The jagged peaks appearing below 5 Å corresponded to correlations between directly bonded atoms or their neighbours in the polymer molecule. On the other hand, the broad peak with a peak top near 10 Å can be attributed to the summation of atomic correlations within non-bonded atoms. Therefore, the broadness of these peaks is an indicator for the degree of expansion of individual polymers solvated by ILs. It was observed that the PMMA/[C2mIm][TFSI] system had a broader peak than those of PMMA/[C12mIm][TFSI] and PEMA/[C2mIm][TFSI] systems, suggesting that the polymer conformation was most extended in the PMMA/[C2mIm][TFSI] system compared to PMMA/[C12mIm][TFSI] and PEMA/[C2mIm][TFSI] systems. In fact, the Rg calculated from the intramolecular radial distribution function was 14.2, 13.1, and 12.3 Å for PMMA/[C2mIm][TFSI], PMMA/[C12mIm][TFSI], and PEMA/[C2mIm][TFSI], respectively. Fig. 6b shows representative polymer conformations for three systems.
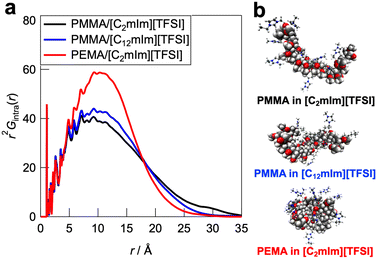 |
| | Fig. 6 (a) Intramolecular contribution of the radial distribution function (Gintra(r)) for the PMMA/[C2mIm][TFSI], PMMA/[C12mIm][TFSI], and PEMA/[C2mIm][TFSI] systems. (b) Representative snapshot of individual polymer conformations in ILs extracted from the MD simulation boxes. | |
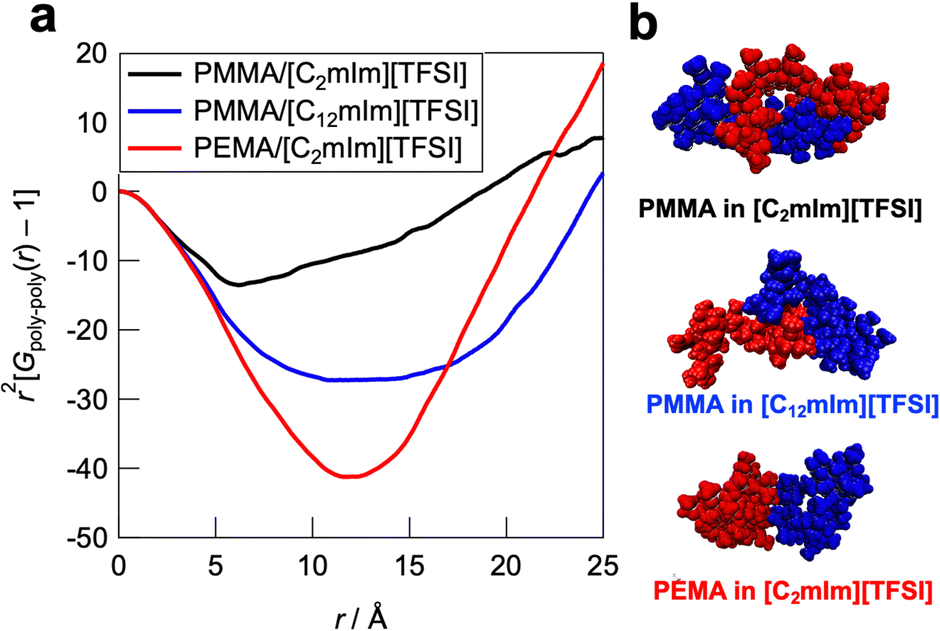
Furthermore, to obtain insight into the polymer–polymer interactions in ILs, we evaluated the intermolecular radial distribution function (Gpoly–poly(r)) that represents the correlation between the different polymer chains (Fig. 7a). In the PMMA/[C2mIm][TFSI] system, the upturn of the intensity was observed around 6 Å, which indicates that the correlation between polymers starts to occur from this distance. On the other hand, in the PMMA/[C12mIm][TFSI] and the PEMA/[C2mIm][TFSI] systems, the upturns were observed from around 9 and 12 Å, respectively, which were farther than that of the PMMA/[C2mIm][TFSI] system. In addition, the depth of the convex curve for the PMMA/[C12mIm][TFSI] and the PEMA/[C2mIm][TFSI] systems in Fig. 7a was much deeper than that of the PMMA/[C2mIm][TFSI] system, suggesting the probability of having different polymer chains in the proximity is low. These results suggest that in the PMMA/[C2mIm][TFSI] system, individual polymer chain has a more extended conformation in the IL, and different polymer chains are more likely to exist in the vicinity (Fig. 7b). Therefore, even at the cut surfaces of the PMMA/[C2mIm][TFSI] UHMW gel, the polymer chains may take an expanded form and different polymer chains are more likely to be present in the vicinity, which facilitates the reformation of entanglement, resulting in the excellent self-healing ability. However, it should be noted that in this study, MD calculations were performed for polymers with a low polymerisation degree of 20 mers, which is quite different from the degree of polymerisation for UHMW polymers (>10000). Therefore, further studies to clarify the polymer conformations and interpolymer interactions in UHMW gels more accurately by combining the large-scale MD calculations at polymerisation degree that exceeds the entanglement molecular weight is under progress along with high-energy X-ray total scattering experiments.57 In addition, the influence of the difference in the polymer conformation calculated from MD simulations on the linear viscoelastic master curves is still elusive. Nonlinear rheological measurements are now underway to investigate the effect of the difference in the polymer conformation in ILs on physical properties.
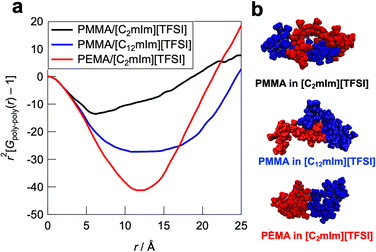 |
| | Fig. 7 (a) Intermolecular contribution of the radial distribution function (Gpoly–poly(r)) for the PMMA/[C2mIm][TFSI], PMMA/[C12mIm][TFSI], and PEMA/[C2mIm][TFSI] systems. (b) Representative snapshot of two adjacent polymers in ILs extracted from the MD simulation boxes. | |
Conclusions
In conclusion, UHMW gels, which were formed solely by physical entanglements of in situ polymerised UHMW polymers in ILs, were fabricated from various IL/monomer structures. In the rheological measurements, all UHMW gels exhibited a wide rubbery plateau without crossover of G′ and G′′ over wide frequency and temperature region, indicating that UHMW gels can maintain solid-like integrity over long timescales. Tensile tests suggested that stress–strain curves of UHMW gels were significantly dependent on the IL/polymer chemical structures. In particular, the PMMA/[C2mIm][IM14] UHMW gel showed the highest fracture stress despite relatively low Tg, implying that the nanophase separation of long perfluoroalkyl chain might affect the tensile property of UHMW gels. Self-healing properties of UHMW gels evaluated by tensile tests revealed the trade-off relationship between the stiffness and the healing efficiency, whereas some UHMW gels deviated from the trade-off line. MD simulations for different polymer/IL structures revealed that the intramolecular polymer conformations as well as interpolymer interactions were dependent of the polymer/IL structures. In the combination of polymer/IL for the UHMW gel that shows high self-healing efficiency, the polymer chain can take an extended conformation in the IL, and that different polymer chains are likely to be present in close proximity to each other. In the field of polymer gel science, “the style of cross-linking”, “polymer network”, and “solvent” are known as the three major components of gels. So far, efforts have been made to realise effects of the style of cross-linking and polymer network towards unprecedented functions and excellent mechanical properties of gels. However, solvents engineering of gels has never received any special attention. The ability to control the mechanical properties of gels by changing the solvent structure is a unique strategy that is not found in other gels such as hydrogels and organogels. Further, our insights into the effects of the chemical structure on the mechanical and self-healing properties of UHMW gels provide a useful basis for the development of a new class of ion gels for wearable and flexible devices with desirable physicochemical properties.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by JST, PRESTO, Japan (Grant Number: JPMJPR2196 to R. T.), and JSPS KAKENHI (Grant Number: 20K15349 to R. T., 20H02804, and 20K21229 to T. U.).
References
- A. Nathan, A. Ahnood, M. T. Cole, S. Lee, Y. Suzuki, P. Hiralal, F. Bonaccorso, T. Hasan, L. Garcia-Gancedo, A. Dyadyusha, S. Haque, P. Andrew, S. Hofmann, J. Moultrie, D. Chu, A. J. Flewitt, A. C. Ferrari, M. J. Kelly, J. Robertson, G. A. J. Amaratunga and W. I. Milne, Proc. IEEE, 2012, 100, 1486–1517 Search PubMed.
- M. Stoppa and A. Chiolerio, Sensors, 2014, 14, 11957–11992 CrossRef CAS PubMed.
- W. Liu, M.-S. Song, B. Kong and Y. Cui, Adv. Mater., 2017, 29, 1603436 CrossRef PubMed.
- T. Sekitani, Y. Noguchi, K. Hata, T. Fukushima, T. Aida and T. Someya, Science, 2008, 321, 1468–1472 CrossRef CAS PubMed.
- Y. Wang, C. Zhu, R. Pfattner, H. Yan, L. Jin, S. Chen, F. Molina-Lopez, F. Lissel, J. Liu, N. I. Rabiah, Z. Chen, J. W. Chung, C. Linder, M. F. Toney, B. Murmann and Z. Bao, Sci. Adv., 2017, 3, e1602076 CrossRef PubMed.
- T. Q. Trung and N.-E. Lee, Adv. Mater., 2017, 29, 1603167 CrossRef PubMed.
- H. Tran, V. R. Feig, K. Liu, Y. Zheng and Z. Bao, Macromolecules, 2019, 52, 3965–3974 CrossRef CAS.
- K. Kim, Y. Park, B. G. Hyun, M. Choi and J. Park, Adv. Mater., 2019, 31, 1804690 CrossRef PubMed.
- B. Wang and A. Facchetti, Adv. Mater., 2019, 31, 1901408 CrossRef PubMed.
- M. L. Hammock, A. Chortos, B. C.-K. Tee, J. B.-H. Tok and Z. Bao, Adv. Mater., 2013, 25, 5997–6038 CrossRef CAS PubMed.
- A. Manuel Stephan, Eur. Polym. J., 2006, 42, 21–42 CrossRef CAS.
- M. Khatib, O. Zohar and H. Haick, Adv. Mater., 2021, 33, 2004190 CrossRef CAS PubMed.
- S. H. Kim, K. Hong, W. Xie, K. H. Lee, S. Zhang, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2013, 25, 1822–1846 CrossRef CAS PubMed.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- C. Jo, D. Pugal, I.-K. Oh, K. J. Kim and K. Asaka, Prog. Polym. Sci., 2013, 38, 1037–1066 CrossRef CAS.
- J. Mindemark, M. J. Lacey, T. Bowden and D. Brandell, Prog. Polym. Sci., 2018, 81, 114–143 CrossRef CAS.
- C. Yang and Z. Suo, Nat. Rev. Mater., 2018, 3, 125–142 CrossRef CAS.
- T. Ueki and M. Watanabe, Macromolecules, 2008, 41, 3739–3749 CrossRef CAS.
- J. Le Bideau, L. Viau and A. Vioux, Chem. Soc. Rev., 2011, 40, 907–925 RSC.
- T. P. Lodge and T. Ueki, Acc. Chem. Res., 2016, 49, 2107–2114 CrossRef CAS PubMed.
- Y. Kitazawa, K. Ueno and M. Watanabe, Chem. Rec., 2018, 18, 391–409 CrossRef CAS PubMed.
- C. Creton, Macromolecules, 2017, 50, 8297–8316 CrossRef CAS.
- C. W. Peak, J. J. Wilker and G. Schmidt, Colloid Polym. Sci., 2013, 291, 2031–2047 CrossRef CAS.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- T. Matsuda, R. Kawakami, R. Namba, T. Nakajima and J. P. Gong, Science, 2019, 363, 504–508 CrossRef CAS PubMed.
- C. Norioka, Y. Inamoto, C. Hajime, A. Kawamura and T. Miyata, NPG Asia Mater., 2021, 13, 34 CrossRef CAS.
- C. Liu, N. Morimoto, L. Jiang, S. Kawahara, T. Noritomi, H. Yokoyama, K. Mayumi and K. Ito, Science, 2021, 372, 1078–1081 CrossRef CAS PubMed.
- T. Fujiyabu, N. Sakumichi, T. Katashima, C. Liu, K. Mayumi, U. Il Chung and T. Sakai, Sci. Adv., 2022, 8, 10 Search PubMed.
- A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128–2140 CrossRef CAS PubMed.
- K. Imato, T. Ohishi, M. Nishihara, A. Takahara and H. Otsuka, J. Am. Chem. Soc., 2014, 136, 11839–11845 CrossRef CAS PubMed.
- A. Campanella, D. Döhler and W. H. Binder, Macromol. Rapid Commun., 2018, 39, 1700739 CrossRef PubMed.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, M. A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- D. L. Taylor and M. in het Panhuis, Adv. Mater., 2016, 28, 9060–9093 CrossRef CAS PubMed.
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 12, 1224–1246 RSC.
- Y. Cao, T. G. Morrissey, E. Acome, S. I. Allec, B. M. Wong, C. Keplinger and C. Wang, Adv. Mater., 2017, 29, 1605099 CrossRef PubMed.
- E. Kamio, T. Yasui, Y. Iida, J. P. Gong and H. Matsuyama, Adv. Mater., 2017, 29, 1704118 CrossRef PubMed.
- R. Tamate, K. Hashimoto, T. Horii, M. Hirasawa, X. Li, M. Shibayama and M. Watanabe, Adv. Mater., 2018, 30, 1802792 CrossRef PubMed.
- R. Tamate and M. Watanabe, Sci. Technol. Adv. Mater., 2020, 21, 388–401 CrossRef CAS PubMed.
- R. Tamate, Polym. J., 2021, 53, 789–798 CrossRef CAS.
- L. Xu, Z. Huang, Z. Deng, Z. Du, T. L. Sun, Z.-H. Guo and K. Yue, Adv. Mater., 2021, 2105306 CrossRef CAS PubMed.
- W. Li, L. Li, S. Zheng, Z. Liu, X. Zou, Z. Sun, J. Guo and F. Yan, Adv. Mater., 2022, 2203049 CrossRef CAS PubMed.
- Y. Kamiyama, R. Tamate, T. Hiroi, S. Samitsu, K. Fujii and T. Ueki, Sci. Adv., 2022, 8, eadd0226 CrossRef PubMed.
- A. Noda, K. Hayamizu and M. Watanabe, J. Phys. Chem. B, 2001, 105, 4603–4610 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- J. N. Canongia Lopes and A. A. H. Pádua, J. Phys. Chem. B, 2006, 110, 19586–19592 CrossRef CAS PubMed.
- S. Harrisson, S. R. Mackenzie and D. M. Haddleton, Chem. Commun., 2002, 2850–2851 RSC.
- K. Low, L. Wylie, D. L. A. Scarborough and E. I. Izgorodina, Chem. Commun., 2018, 54, 11226–11243 RSC.
- Y. Suzuki, Y. Shinagawa, E. Kato, R. Mishima, K. Fukao and A. Matsumoto, Macromolecules, 2021, 54, 3293–3303 CrossRef CAS.
- Y. He, P. G. Boswell, P. Bühlmann and T. P. Lodge, J. Phys. Chem. B, 2007, 111, 4645–4652 CrossRef CAS PubMed.
- A. Noro, Y. Matsushita and T. P. Lodge, Macromolecules, 2008, 41, 5839–5844 CrossRef CAS.
- X. Ma, R. Usui, Y. Kitazawa, R. Tamate, H. Kokubo and M. Watanabe, Macromolecules, 2017, 50, 6788–6795 CrossRef CAS.
- S. Ishii, H. Kokubo, K. Hashimoto, S. Imaizumi and M. Watanabe, Macromolecules, 2017, 50, 2906–2915 CrossRef CAS.
- F. Lo Celso, G. B. Appetecchi, E. Simonetti, M. Zhao, E. W. Castner, U. Keiderling, L. Gontrani, A. Triolo and O. Russina, Front. Chem., 2019, 7, 285 CrossRef PubMed.
- K. Fujii, R. Kanzaki, T. Takamuku, Y. Kameda, S. Kohara, M. Kanakubo, M. Shibayama, S. Ishiguro and Y. Umebayashi, J. Chem. Phys., 2011, 135, 244502 CrossRef PubMed.
- Z. Wang, S. Gangarapu, J. Escorihuela, G. Fei, H. Zuilhof and H. Xia, J. Mater. Chem. A, 2019, 7, 15933–15943 RSC.
- K. Fujii, T. Ueki, K. Hashimoto, Y. Kobayashi, Y. Kitazawa, K. Hirosawa, M. Matsugami, K. Ohara, M. Watanabe and M. Shibayama, Macromolecules, 2017, 50, 4780–4786 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *cd,
Kenta
Fujii
*cd,
Kenta
Fujii