
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamically shaping the surface of silica colloids†
Vincent
Niggel
 ,
Chiao-Peng
Hsu‡
and
Lucio
Isa
*
,
Chiao-Peng
Hsu‡
and
Lucio
Isa
*
Laboratory for Soft Materials and Interfaces, Department of Materials, ETH Zurich, CH-8093, Zurich, Switzerland. E-mail: lucio.isa@mat.ethz.ch
First published on 19th September 2022
Abstract
Surface roughness is an important design parameter to influence the processing of particle-based materials. Current methods to synthesize rough particles present some limitations, e.g. low yield, relative methodological complexity, requirements of multiple steps, or poor roughness control. Here, we thoroughly investigate a facile synthesis route where two silanes, tetraethyl orthosilicate (TEOS) and vinyltrimethoxysilane (VTMS), are added in one pot to form silica particles with controlled corrugated surfaces. We first show that the morphology of these particles can be defined by regulating the amount and ratio of the two silane precursors and by adjusting the concentration of ammonia during synthesis. We characterize the surface topography of the particles using atomic force microscopy and show a direct correlation between surface roughness and the synthesis conditions. Furthermore, we carry out an in situ observation of the evolution of surface morphology and propose a mechanism for surface structuring that hinges on the formation of silane droplets, followed by the preferential hydrolysis/condensation reaction of VTMS starting from the droplet surface and evolving towards the center. The exchange of liquid from the droplets through the VTMS shell leads to stress accumulation and wrinkling/buckling of the particles. Moreover, we explicitly show that osmotic imbalances between the inside and the outside of the droplets regulate their shrinking. We therefore demonstrate that exchanging solvents has a comparable impact to adjusting silane and ammonia content in defining the particle shape and that this synthesis route is highly dynamical. Finally, we demonstrate that it is possible to incorporate fluorescent dyes during synthesis to enable future studies on the impact of surface roughness on dynamic processes, including shear, via direct high-resolution imaging. Our findings show that the mechanism for wrinkling and buckling in colloidal silica particles follows a general scheme found in a broad range of systems, from liposomes and polymeric capsules to Pickering emulsion droplets.
1 Introduction
Surface topography is an important design parameter for colloidal systems. In particular, imparting controlled roughness to the surface of particles affects many of their properties, from colloidal stability and dispersibility in solvents,1 through transport,2 dynamics,3 and adsorption4 at fluid interfaces, to colloidal interactions, e.g. depletion5,6 band contact forces such as friction7 and adhesion.8 Consequently, surface roughness plays an essential role in a wide range of processes involving the dispersion of solid particles, the stabilization of foams and emulsions, or the rheology of dense particulate pastes, especially at high deformation rates.9–16 Moreover, small variations in surface roughness may lead to large differences in macroscopic properties. For instance, concerning high-shear rheology, it has been shown that decorating sub-micrometric particles with surface asperities in the range of 10 to 20 nanometers caused a change of fifteen percent in the maximum packing fraction of a suspension, with important consequences in the processing of dense pastes.10 Similarly, in the case of emulsions, changing the height of surface asperities from 20 to 40 nanometers on sub-micrometric particles, allowed or prevented, respectively, the macroscopic mechanical phase inversion of particle-stabilized emulsions.17Systems of applied relevance typically comprise particles with complex shapes displaying roughness over different length scales. Therefore, to disentangle and elucidate the role of roughness at a specific scale in real applications, we need synthetic model systems with a controlled roughness level. Moreover, to map these systems back onto applications, synthetic strategies must not only be controlled but also robust, easy, and scalable. To date, different strategies exist to produce rough colloids exploiting wet chemistry, including heteroaggregation, template confinement, or in situ reactions.18,19 However, these approaches typically suffer from low yields and/or require multiple steps. In contrast, physical methods, such as milling, are simple and applicable to large quantities of material but present a limited control over well-defined roughness scales. Recently, an alternative strategy has emerged, which combines simplicity and scalability with exquisite control over surface topography exploiting surface stress instabilities. Surface stress instabilities have been widely investigated20–22 and exploited for applications in surface patterning,23–25 friction modulation,26 nanoparticle assembly,27 and even fabrication of security features.28 In this mechanism, well-defined out-of-plane corrugations, or wrinkles, of thin stiff films in contact with more compliant substrates develop under a compressive stress to minimize the global elastic energy, up to the extreme case of complete buckling. In the context of shaping colloidal particles, this has been for instance used to yield large amounts of buckled or wrinkled colloids using oil droplets as templates, forming a thin shell around them and then dissolving the oil into the continuous phase, followed by drying.29 More recently, silica microparticles with corrugated surfaces have also been prepared through the combination of two silane precursors. Due to different reaction rates, stresses accumulate within thin films growing inwards from the surface of the particles, eventually triggering a mechanical instability that leads to surface wrinkling and buckling. To date, different pairs of silane precursors have been used to achieve this goal30,31 but interesting questions remain regarding the factors that determine particle morphology. Among the different choices, an interesting synthesis route involves the use of tetraethyl orthosilicate (TEOS) and vinyltrimethoxysilane (VTMS) as silica precursors, which allows forming a broad range of particle morphologies, including ones that have been likened to golf balls and Lilium pollen.32 While previous works demonstrated clear qualitative differences in the particle morphology as a function of reaction conditions,32,33 the specific range of synthesis parameters that allows precise surface modulation, as required in the above-mentioned processes, is currently less defined. Most importantly, a quantification of surface topography in relation to the synthesis parameters, which can be used as a guideline for the design of rough particles with tailored surfaces, remains open. Finally, the evolution of particle morphology involves multiple concurrent processes during the reaction. Current studies have mostly focused on the final result, without providing in situ information on particle evolution. We will indeed show that the formation of these complex silica colloids is a highly dynamical process that can be influenced by many parameters in addition to the initial composition of the reaction mixture.
In our manuscript, we specifically address these open issues. In particular, we first present an extensive scan of a range of synthesis parameters and provide a detailed correlation between synthesis conditions and a careful quantification of the surface topography using atomic force microscopy. We then reflect on the morphogenesis mechanism, presenting a set of in situ observations of the particles in different solvents after the initial stages of synthesis, which clearly demonstrate that shape evolution is highly dependent on the exchange of liquid across the surface of the forming particles. These findings illustrate that the evolution of the particle morphology can be dynamically influenced beyond the initial composition of the reaction mixture. Finally, we show that the particles can be additionally modified to be made fluorescent, as of potential interest for microscopy imaging of different materials and processes.
2 Experimental
2.1 Materials
Tetraethyl orthosilicate (TEOS, ≥99%, 250 mL) and vinyltrimethoxysilane (VTMS, 98%, 100 mL) were acquired from Sigma-Aldrich, an ammonium hydroxyde solution (NH4OH in water, 25% v/v, 1 L) was acquired from VWR Chemicals, 3-Aminopropyltriethoxysilane (APTES, 99%, 100 mL) and Ethanol (EtOH, 99.5%, Extra dry, absolute, Acroseal) were obtained from Acros Organics and ATTO 495 NHS ester (1 mg) was bought from ATTO-TEC GmbH.2.2 Synthesis protocol
The detailed synthesis conditions are given in the ESI,† and presented in the Results section below. A typical synthesis involved magnetically stirring, for 80 minutes and in a closed 15 mL pill glass bottle, given amounts of MilliQ water and of an NH4OH aqueous solution at 25% together with different amounts of VTMS and TEOS. After the 80 minutes had elapsed, the particles were cleaned and collected by centrifuging several times the suspension and replacing the supernatant with MilliQ water. In order to follow the time evolution of the particle synthesis and surface morphology, small aliquots were extracted from the reaction flask and quenched using different solvents, typically MilliQ water. Different washing protocols using different solvents are further discussed in the Results and discussion section.2.3 Particle imaging
The particles were imaged using a combination of scanning electron microscopy (SEM), atomic force microscopy (AFM), and optical microscopy. Standard SEM (LEO 1530 GEMINI, Zeiss) images were collected after coating the samples with a 3 nm layer of platinum (CCU-010, Safematic). Particle size analysis from SEM images was performed manually with Image J (see Fig. S1 and Tables S2–S4, ESI†). The FIB images were obtained with a TFS Helios 5 UX. The particles were coated with 100 nm of tungsten and cut twice with a current of 7 pA to provide a clean cross-section. The AFM imaging was performed with a NanoWizard III from JPK Instruments AG using Olympus OMCL-AC160TS-R3 cantilevers. The image size was 4 μm × 4 μm (256 pixels × 256 pixels) and a scanning rate of 0.2 Hz was used. The analysis process to extract the presented surface parameters is described and explained in the (see Fig. S2, ESI†). The bright-field images used in Section 3.4 were obtained with an Eclipse Ti2 from Nikon with a 100× oil immersion objective. The videos were acquired at 13.3 fps. 5 μL of suspension were placed on a glass slide and sealed with an adhesive spacer and a glass coverslip to avoid evaporation and allow continuous imaging for prolonged times. The bright-field and confocal micrographs of the fluorescent particles were acquired with a Leica TCS SP8. Details of cryo-SEM imaging are given in the ESI.†3 Results and discussion
3.1 Influence of ammonia concentration and the ratio between silanes
We begin our investigation of the synthesis parameters by systematically varying the volumetric ratio between the two silica precursors (VTEOS and VVTMS) while keeping the total volume of the silanes (VSi = VTEOS + VVTMS) constant at 1.36 mL. We express the ratio between silanes as rTEOS = VTEOS/VSi. For each ratio, we also vary the volume of the ammonia catalyst (VNH4OH*), added as a 25% v/v solution, between 12 to 200 μL, while keeping the total volume of water constant at 6.3 mL. All experiments are carried out at room temperature, which is maintained constant in our laboratories at 22 ± 2 °C, by adding the ammonia solution, TEOS, and VTMS to water in a pill glass bottle closed with a lid and stirred for 80 min (see the ESI,† for additional details). Fig. 1 shows an overview of SEM images of the different particles obtained with the different synthesis parameters, where the colored frame indicates the average particle diameter of each batch according to the color bar. Our parameter sweep enables us to recover all of the particle morphologies previously reported in the literature, going from smooth to “bowl-like” particles passing through “golf-ball” and “Lilium-pollen” morphologies.32,33 Often, the highly buckled “bowl-like” particles completely flatten out into disk-like objects, whose dimensions are difficult to evaluate systematically. | ||
| Fig. 1 Overview of SEM images of particles synthesized by varying the volumetric ratio between the two silanes rTEOS = VTEOS/VSi, with VSi = VTEOS + VVTMS, keeping the total silane volume VSi constant at 1.36 mL (columns), and by varying the added volume of ammonia VNH4OH* in a 25% v/v aqueous solution (rows). The total volume of H2O is kept constant at 6.3 mL. The color of the frame around each image indicates the average particle diameter as read from the color bar. A black frame means particle aggregation. The white scale bar represents 1 μm in all SEM images. | ||
Starting from the two extreme cases of using only TEOS or only VTMS, we find that, at the concentration of ammonia we are using, no particles could be detected for a synthesis with only TEOS, i.e. rTEOS = 1. We note that this condition is different from the classical Stöber synthesis using only TEOS,34 because of the absence of ethanol, which is present in the Stöber process to ensure the solubility of TEOS (see Fig. S4, ESI†). In the opposite case, where only VTMS is used, i.e. for rTEOS = 0, particles are formed and display a smooth surface. As we progressively increase the amount of TEOS, for a fixed ammonia content, the presence of the second silane does not seem to affect the size of the particles notably. However, it strongly influences their morphology and surface topography. In particular, for a fixed amount of ammonia, increased surface roughness and an increased propensity to buckle are found with increasing rTEOS, e.g., see rows 3, 4, and 5 in Fig. 1. Lowering VNH4OH* for a fixed rTEOS has a similar effect, as seen in columns 3–6 of Fig. 1. This implies that particles with a similar morphology are formed for lower rTEOS, if the amount of NH4OH is also lowered. As an example, “bowl-like” particles appear at rTEOS = 0.8 for VNH4OH* = 50 μL, while they are already formed at rTEOS = 0.5 for VNH4OH* = 12 μL. Moreover, increasing the amount of NH4OH creates smaller particles, in line with faster reaction kinetics. In the range of ammonia concentrations we used, we obtain particles ranging from around 700 nm at VNH4OH* = 200 μL, to around 3 μm at VNH4OH* = 12 μL. Overall, ammonia plays a key role in the synthesis, as it influences both the shape and the size of the particles. However, going below or above the range reported here leads to uncontrolled synthesis and to the formation of aggregates (see the top-right corner of Fig. 1 and Fig. S6, ESI†).
3.2 Influence of TEOS and VTMS amounts
After examining the influence of the ammonia concentration and ratio between the silane precursors, it is important to investigate the contribution that each silane has on particle morphology. In order to explore these parameters, we change the volume of TEOS (VTEOS) or the volume of VTMS (VVTMS) at constant ammonia concentration and constant total volume of reagents (Fig. 2). | ||
| Fig. 2 Overview of SEM images of particles synthesized by varying the total added volume of TEOS, VTEOS, and the total added volume of VTMS, VVTMS, while keeping the volume of ammonia, VNH4OH*, constant at 50 μL and the total volume of all reagents fixed at 9.1 mL. The color frame around each image indicates the average particle diameter as read from the color bar. The white scale bar represents 1 μm in all SEM images. | ||
In general, adding a greater amount of silica precursors leads to bigger particles. However, adding VTMS has a stronger influence on particle size than adding TEOS. This fact indicates that, at such a low concentration of ammonia, VTMS reacts faster than TEOS and the overall particle size is therefore mainly dictated by the quantity of VTMS in the reaction. Additional considerations on the difference in the reaction rate between VTMS and TEOS are given in the (see Fig. S4 and S5, ESI†). In addition to changing the overall particle dimensions, modulating the quantity of VTMS strongly affects particle morphology. In particular, more buckled particles are formed for lower VVTMS. For example, if VTEOS > 272 μL, the particles we obtain for VVTMS = 816 μL have a “golf-ball-like” shape, while for VVTMS = 204 μL appear “bowl-like”. A higher degree of buckling is also achieved by adding more TEOS for a fixed VVTMS, consistently with the results from Fig. 1. Taken together, these two effects imply that, if rTEOS is kept constant but the total volume of silane is decreased (corresponding to moving diagonally from top right to bottom left in Fig. 2), the produced particles will decrease in size and be more buckled. This trend seems to hold until a certain threshold concentration of TEOS. In fact, for VTEOS = 136 μL, we only obtain smooth particles or particles presenting a spiky surface. This intriguing spiky particle is the result of the grafting of smaller particles to the surface of a bigger nucleus, as it can be seen in Fig. S7, ESI.† The parameter sweep reported in Fig. 2 can also be compared with the one shown in Fig. S8, ESI,† where we also note the formation of smaller and more buckled particles for lower amounts of VTMS. In the cases shown in Fig. S8, ESI,† we added the same amount of silica precursors and ammonia together with a constant amount of MilliQ water. There, changing the volume of the silanes affected both their concentration and the overall ammonia concentration in a more complex but consistent way.
To conclude, ammonia, TEOS, and VTMS all have an influence on particle morphology and size but, more importantly, their roles are all connected and their influence cannot be easily separated. We will come back to a discussion of their contributions in Section 3.4. So far, we have only provided SEM images to qualitatively describe the synthesized particles. SEM images give good and rapid access to qualitative information on the particle morphology, however, from such images it is difficult to extract a quantitative description of particle surface's characteristics, which is at the core of the objective of this work. In the next section, we thus revert to atomic force microscopy to provide a quantification of surface topography.
3.3 Surface characterisation via atomic force microscopy
From the qualitative visualization of the SEM images, we can select the range of synthesis parameters for which the particles present a rough, creased surface but maintain an overall spherical shape before complete buckling takes place. This region is the most interesting one from a synthetic point of view because it allows us to synthesize particles with controlled surface properties and which can be treated as “rough spheres”. This possibility significantly simplifies the definition of both collective and individual properties of suspensions of such particles, e.g., the volume fraction or the particle size, respectively, based on the assumption of an overall spherical shape. It furthermore makes the analysis of their surfaces more reliable, since the surface topography is isotropic on the length scale of the particle, and thus the imaging is independent of the particle orientation.We measure the 3D surface topography of our particles using AFM, after appropriately removing the underlying spherical curvature (see Fig. S2 and S3, ESI†). This three-dimensional reconstruction of the particle surface allows one to extract essentially any topography descriptor. However, from the SEM images, one can see that the surface of these particles consists of valleys delimited by ridges, we decided to characterize the surfaces by evaluating the average valley size via two different parameters: H10 and dvalley. H10 is the difference between the average of the 10% highest values and the average of the 10% lowest values of a surface, which is a good indicator for the depth of the valleys and acts as an effective roughness amplitude parameter. dvalley is the average distance between two valleys and is similar to a roughness spacing parameter.
We imaged different batches of the spherical particles shown in Fig. 1, 2, and Fig. S8, (ESI†). In Fig. 3, we plot the corresponding parameters H10 and dvalley normalized by the radius of the particle Rpart, which we extracted from the images. Taking some of the batches, which correspond to systematic variations of the synthesis parameters, we can, for instance, plot the normalized dvalley/Rpart as a function of VVTMS (Fig. 3A) and VTEOS (Fig. 3B). We observe that by reducing the amount of VTMS in the synthesis or by increasing the amount of TEOS, dvalley increases, confirming the occurrence of stronger surface buckling, as visualized in the SEM images. Moreover, by plotting the two surface parameters against each other (Fig. 3C), we observe that they are positively correlated. An increase in the spacing parameter thus implies a corresponding increase in the amplitude parameter, indicating a strong degree of self-similarity between the different surfaces obtained through different reactions. Moreover, H10 can be associated with the amplitude of surface wrinkling, while dvalley is representative of its wavelength. A linear relationship between these two parameters is predicted by surface elasticity models,35 whose applicability we will discuss later in the text. The measured H10 for the different particles ranges up to 30% of the radius of the particles and dvalley grows to around 70% before the particles lose their isotropy and start to buckle into a “Lilium-pollen-like” shape.
 | ||
| Fig. 3 (A) Evolution of the characteristic size of valleys dvalley normalized by the radius of the particle Rpart as a function of the amount of VTMS, VVTMS. (B) Evolution of the characteristic length of the valley dvalley normalized by Rpart as a function of the amount of TEOS, VTEOS. The numbers next to the data points correspond to the particles whose AFM images are shown in (C). (C) Evolution of normalized H10 as a function of normalized dvalley for different particle batches as indicated by the numbers and the corresponding AFM images. The blue results correspond to particles from Fig. 1 with VNH4OH* = 50 μL. The orange results correspond to particles from Fig. 2. The dashed line indicates particles from the row with VVTMS = 816 μL, while the full line shows particles from the column with VTEOS = 272 μL. The green results correspond to particles from Fig. S8, ESI.† The dashed line indicates particles from the row with VVTMS = 816 μL, while the full line shows particles from the column with VTEOS = 272 μL. The size of the colored ellipses represents the standard errors of the measured quantities computed over at least four particles. The gray bands delimit the accuracy of AFM measurements and image processing. | ||
3.4 Insights on particle morphogenesis
Our observations so far, and the majority of previous studies, have focused on the impact of reaction composition on the final particle morphology.32,33 However, we found interesting differences when following the particle morphology in time for three of the syntheses reported in Fig. 1, i.e., with a fixed amount of ammonia and varying rTEOS. In particular, we first followed the evolution of particle morphology ex-situ, by quenching the reactions at different times by extracting aliquots out of the reaction vessel and diluting them in large amounts of water before drying and imaging via SEM, as shown in Fig. 4. For the first 3 minutes, irrespective of the amount of TEOS, all particles had a smooth spherical shape. However, at 4 minutes, the particles with the highest rTEOS of 0.8, buckled into a bowl-like shape, while the other two remained smooth. Between 5 to 6 minutes, the particles with the intermediate value of rTEOS started to wrinkle, followed 1 or 2 minutes later by the ones with the lowest relative TEOS content (similar experiments have been performed with VNH4OH = 12.5 μL and can be found in Fig. S9, ESI†). The surface morphology continued to evolve over different amounts of time and, the higher the relative concentration of TEOS was, the earlier and the more the particle's surface deformed. When following the particle morphology for even longer times, a more complex behavior was observed (see Fig. S10, ESI†). Moreover, we noticed that the final particle shape varied if the reaction was quenched by washing the particles in different solvents (see Fig. S11, ESI†). The observations we report here hint at the existence of an intricate interplay between concomitant factors, which we further investigated by in situ analysis of the evolution of particle morphology by optical microscopy. | ||
| Fig. 4 SEM images of the time evolution of the particles' morphology. At each time point, 10 μL were removed from the synthesis suspension and added to 1 mL of H2O before imaging. Synthesis conditions: VNH4OH = 50 μL and (A) rTEOS = 0.5; (B) rTEOS = 0.7; (C) rTEOS = 0.8. These conditions are similar the ones in Fig. 1 with corresponding values of rTEOS. The images at 80 min show particles that have been centrifuged in water and then dried on the SEM substrate. The last column shows cross-sections of the particles imaged with FIB-SEM after 80 minutes of reaction and subsequent cleaning. The white and light blue scale bars are 500 nm. | ||
In Fig. 5A, we followed the shape evolution for a synthesis with 12.5 μL of NH4OH* and rTEOS = 0.4, which leads to the formation of highly buckled particles, as seen in Fig. 1. By taking out 5 μL of the reaction mixture and imaging the suspension over time, we saw that, after 20 minutes, the particles were still spherical, and started to buckle at around 40 minutes, before reaching a highly buckled state after 3 hours. If we now took aliquots out of the same reaction vessel at the same times, diluted them into excess MilliQ water (10 μL into 1 mL water), and rapidly observed them under the microscope, we noticed a clear morphological difference. Transferring the particles to water caused a much higher degree of surface buckling (Fig. 5B). The particles in excess water presented a bowl-like shape after 20 minutes and the higher the degree of dilution, the stronger was the buckling (see Fig. S12, ESI†). If we performed the same experiments but diluting the reaction mixture in ethanol instead of water, we got a distinctively different behavior. The presence of excess ethanol outside of the particles induced their swelling, rather than buckling. Particles that were already buckled, e.g., after 40 or 80 minutes of reaction, recovered a smooth spherical shape upon the addition of ethanol (Fig. 5C). If we let the reaction proceed longer, up to 3 hours, a spherical shape was only partially recovered. Similar behavior is also observed if we replace ethanol with methanol (see Fig. S13, ESI†).
 | ||
| Fig. 5 Optical microscopy images of particles as a function of time in different fluids. In row (A), particles are imaged in the reaction mixture (6.2 mL of H2O, 12.5 μL of NH4OH*, 544 μL of TEOS and 816 μL of VTMS − rTEOS = 0.4) as a function of time. In row (B), particles from the same reaction mixture of A are transferred in MilliQ water after 20, 40, 80 and 180 minutes, respectively, (10 μL of the reaction mixture in 1 mL of MilliQ water) and imaged. In row (C), particles from the same reaction mixture of A are transferred to ethanol (10 μL of the reaction mixture in 1 mL of ethanol) and imaged at the corresponding times. The scale is the same for all images and the white bar corresponds to 10 μm. | ||
The occurrence of inward or outward liquid exchange across the particle VTMS shell can be directly visualized by collecting high-frame rate image sequences of the particles during the addition of different solvents. In Fig. 6, we observe that, within less than 1 second, spherical particles taken out of the same reaction mixture of Fig. 5 after a few minutes, completely buckle by adding excess MilliQ water. We then add the same amount of ethanol to the same suspension and, within 2 seconds, the particles re-swell, recovering the initial spherical shape. Notably, we could observe that while re-swelling to a spherical shape, the particles go through an intermediate golf ball-like morphology (see Fig. 6B-5).
 | ||
| Fig. 6 High-frame rate optical image series of the particle morphology upon solvent exchange. 3 μL of the reaction mixture (6.2 mL of H2O, 12.5 μL of NH4OH*, 544 μL of TEOS and 816 μL of VTMS) is placed on a glass slide. While recording with the optical microscope, 70 μL of MilliQ is added. After few minutes, 70 μL of ethanol is also added. The first row (A) shows the evolution of particle morphology when water is added (A-1 just before water addition, A-2 350 ms after, A-3 490 ms after, A-4 560 ms after, A-5 630 ms after, A-6 1 s after and A-7 12 s after). The second row (B) shows the evolution of particle morphology when ethanol is added to the same sample (B-1 3 s before ethanol addition, B-2 300 ms after, B-3 525 ms after, B-4 675 ms after, B-5 1.3 s after, B-6 2 s after, B-7 13 s after). All the images are at the same magnification. The red scale bar in the top left image corresponds to 10 μm. | ||
The combination of all these observations, including the evolution of the particle morphology in response to changes in the initial reaction composition, and to solvent exchange, allows us to shed new light on the mechanisms for particle morphogenesis, as schematically described in Fig. 7.
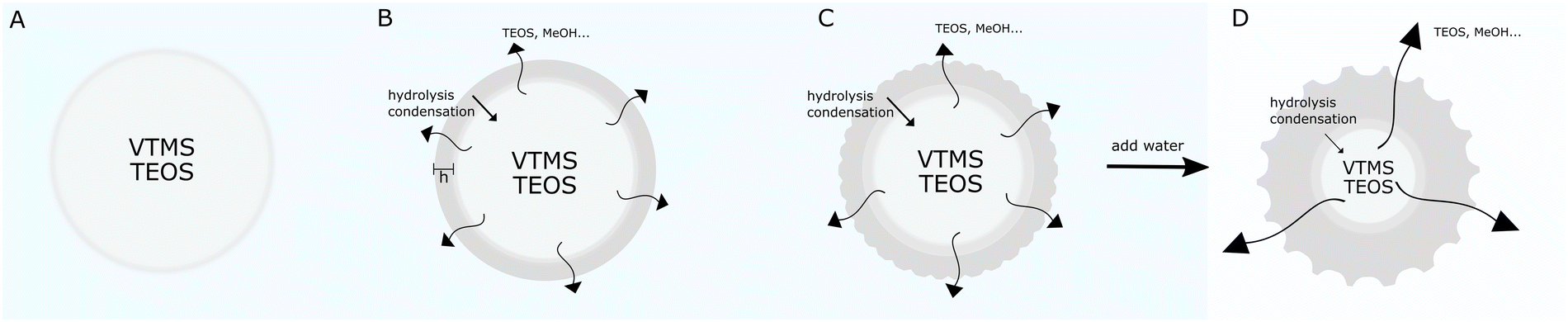 | ||
| Fig. 7 Schematic of the morphogenesis mechanism. (A) In the initial reaction stages, droplets comprising a mixture of VTMS and TEOS are formed in the aqueous phase. (B) The hydrolysis and condensation of the silanes, primarily VTMS, occur at the interface and propagate towards the center of the droplet, while some of the TEOS and the reaction products diffuse out of the droplet. The condensation of the silanes creates a shell with a growing thickness h. (C) The build-up of compressive stress in the droplet, dictated by the osmotic imbalance of reaction products in- and outside the particles, overcomes the critical stress for wrinkling and buckling. (D) Quenching the reaction in water creates greater osmotic stress resulting is the fast expulsion of liquid from the particle's interior towards the outside, leading to a higher degree of wrinkling/buckling. | ||
Our evidence, further supported by the data reported in Fig. S4 (ESI†) indicates that we first form oil (silane) droplets in the aqueous reaction medium. By looking at the individual behavior of the reagents, we could see that in our conditions, i.e. without ethanol, TEOS and VTMS are not soluble in water and form one silane phase when mixed together (see Fig. S4A and B, ESI†). Furthermore, in the presence of the amounts of ammonia we used, and without any ethanol, TEOS does not significantly react in water for many hours and, upon formation of an emulsion, it simply phase separates for the duration of our reactions (see Fig. S4C, ESI†). On the contrary, VTMS (either alone or in the presence of TEOS, see Fig. S4D and E, respectively) spontaneously reacts in the aqueous basic medium even without mixing and the reaction takes place at the interface between the oil and the aqueous phase (see Fig. S4D, ESI†).
The combination of these observations implies that, upon stirring, droplets containing either pure VTMS (see Fig. S14, ESI†) or a mixture of TEOS and VTMS are readily formed and that VTMS starts reacting rapidly at the surface of these droplets creating a shell that grows toward the inside. The faster reaction of VTMS is also confirmed by pH measurements during the synthesis (see Fig. S5, ESI†). The presence of droplets can also explain the influence of ammonia on particle size. Adding more ammonia, within the investigated range, increases the reaction rate of the hydrolysis-condensation process, leaving less time for the droplets to form before the hardening of the shell. It would furthermore explain the influence of the mixing method (see Fig. S15, ESI†) or the injection process of the silanes (see Fig. S16, ESI†) on the reaction. The formation of droplets moreover supports the ability of some particles to buckle to such a degree that they acquire “bowl-like” shapes, as previously reported for capsules with a pure TEOS shell obtained from a silicon oil-in-water emulsion template.29 Finally, from the analysis of the data in Fig. 1, we note that changing the ratio of TEOS rTEOS, while keeping the total volume of the silanes and the amount of ammonia constant, does not significantly change the size of the particles, which implies that the droplets contain a mixture of TEOS and VTMS.
The presence of a rapidly forming VTMS shell is thus essential to enable surface wrinkling and buckling, which we show in Fig. 5 and 6 is caused by the compressive stress on the shell caused by a loss of volume of the droplets. It has been previously shown that silica shells are permeable to small molecules,29e.g., alcohols and water, which can be exchanged with the continuous phase. In particular, the time-dependent experiments reported above show that by diluting the reaction mixture with water, one changes the concentration of the reaction products outside the particles/droplets which causes an osmotic imbalance that drives the rapid expulsion of liquid from the droplets. Conversely, by adding excess ethanol or methanol to the suspension, the osmotic difference is reversed and the particles are rapidly re-swollen. The extent of volume reduction as well as its rate depend both on the initial reaction conditions and on subsequent solvent exchanges. The SEM images in Fig. 1 and 2, in fact, show that a greater degree of buckling is obtained for lower amounts of VTMS and larger amounts of TEOS, corresponding to thinner shells and greater volume losses, respectively. However, differently to the synthesis of pure capsules using only one silane precursor,29 the presence of residual, slowly-reacting TEOS inside the droplets implies that the final particles, after the full reaction has come to completion, are not hollow, but have a solid core as demonstrated in the FIB-SEM images of Fig. 4 and in the freeze-fracture cryo-SEM image of Fig. S17, ESI.† Larger amounts of VTMS enable the formation of thicker shells, which display higher stiffness are resist more to buckling. The exact amount and composition of unreacted silanes expelled during particle formation is difficult to quantify, but by centrifuging the particles after synthesis (e.g. for a reaction with 6.3 mL MilliQ, 0.05 mL NH4OH, 0.680 mL VTMS and 0.680 mL TEOS under stirring for 80 minutes) and collecting the supernatant, residual solid material is found after drying (see Fig. S18, ESI†). These solid films can comprise both unreacted silanes and very small nanoparticles generated by secondary nucleation.36,37 Similar films are found if the particle suspensions are dried without centrifugation and are no longer visible after the repeated centrifugation/redispersion cycles we employed to clean all our particles (see Methods).
The topographies of our particles are highly reminiscent of those obtained during the stress release of core-shell structures38–42 and rely on the onset of a mechanical instability where thin rigid shells release in-plane compressive stresses by out-of-plane deformations. Commonly to what has been observed for liposomes,43 Pickering emulsion droplets,44 silica capsules29 and layer-by-layer polymer capsules45 the volumetric changes are associated to permeability and liquid exchange through the shell, which we now directly capture, providing generality in including osmotic imbalances as a key element to dictate the dynamics of morphologogical evolution. We finally remark that for the thinnest shells, drying may affect the observed particle shape in an electron microscope and combining the latter technique with in situ observation in liquid is essential, e.g. the disk-like particles observed at low ammonia concentration in Fig. 1 have a bowl-like shape in liquid (see Fig. S19, ESI†).
3.5 Synthesis of fluorescent particles
Finally, we proceed to demonstrate that our rough particles with wrinkled/buckled surfaces can also be made fluorescent (see Fig. 8). This is desirable in microscopy studies, where one can investigate both the structure and the dynamics of suspensions of such particles. However, as we have shown so far, this synthesis is highly dependent on the different reagents introduced in the process and on the synthesis route, such that each modification may change the result. We impart fluorescence to the particles by using APTES conjugated to an ATTO-495 NHS ester dye and adding it to the one-pot reaction. As a consequence, for the same amounts of the other reagents (silanes, water, and ammonia) we see a difference in the surface morphology compared to the case without APTES and fluorescent dye (see Fig. S20, ESI†). In general, by adding the fluorescent dye, we increase the buckling of the particles. By examining confocal microscopy images of the corresponding particles, we can confirm that the dye has been incorporated directly in the particles, irrespective of their shape. | ||
| Fig. 8 The first row shows SEM images of the fluorescent particles. The second row displays the corresponding images under a wide-field optical microscope and the third row reports the corresponding confocal micrographs. The blue scale bars represent 1 μm and the white ones represent 10 μm. | ||
4 Conclusions
In this study, we have shown that a detailed control of the initial reaction composition and of the dynamical composition of the solvents can be used to synthesize silica-based particles with controlled surface topography, from “golf-ball-like” to “bowl-like” passing through “Lilium-pollen-like” particles. We show that our particles form via the rapid production of a stiff shell, primarily consisting of VTMS, and that by changing the concentration of ammonia or the volume of silanes, mainly VTMS, we can control the degree of wrinkling and buckling by regulating the ratio between the two silanes. We moreover show that shell formation and deformation are part of a highly dynamical process, where liquid exchange through the forming shell plays a crucial role in determining the particle morphology in analogy with generic observations made for a broad range of core-shell systems.29,43–45 Among all the factors we have investigated, in particular, we have identified a window in the reaction parameters to produce wrinkled spherical particles with finely tuned surfaces. We show that both the height and the distance between the wrinkles formed onto the particle surface can be accurately controlled, giving us access to a broad range of particles with finely tailored surface morphologies.The relative scaling of the roughness parameters, together with the analogy with other systems, strongly supports that buckling and wrinkling occur when the stress build-up originating from the liquid exchange, and subsequent volume loss, overcomes a critical value, which can be in principle predicted by surface elasticity models. Even if this interpretation qualitatively rationalizes our observations, the direct applicability of these models to our system is challenging and a precise quantification of the parameters resulting from the synthesis is extremely complex. In fact, as opposed to model systems where shells of known thickness and elasticity are produced and subjected to controlled stresses, e.g., of osmotic nature,46 in our case, the shell thickness grows as the volume of the interior of the droplet shrinks. Furthermore, as the reaction proceeds for longer times and both VTMS and TEOS have time to react, the different coordination of the organosilica groups (TEOS has four -OR groups, while VTMS only has three) may cause additional internal stresses, as well as a modulation of the shell elasticity across its cross-section. Within the current experimental procedures, we do not yet have direct control over the thickness and the elasticity of the shell, which may be regulated in the future by designing specific reaction-quenching protocols, as we have now elucidated the role they play in the synthesis. We envisage exciting future work along these lines to develop synthetic strategies where the control of elastic instabilities can be used to design silica particles with tailored structures, as model materials to expand our understanding of how surface topography influences a diverse range of processes, from emulsification and emulsion stabilization to the processing of dense particle pastes.
Author contributions
C-P. H., L. I., V. N. Formal analysis: V. N. Funding acquisition: L. I. Investigation: V. N. Methodology: V. N. Project administration: L. I., V. N. Software: V. N. Supervision: L. I. Validation: V. N. Visualization: L. I., V. N. Writing – original draft: L. I., V. N. Writing – review and editing: C-P. H., L. I., V. N.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr. Shivaprakash Ramakrishna for advice on the AFM measurements, Dr. Minghan Hu for assistance with the FIB-SEM imaging, Stephan Handschin for the Cryo-SEM imaging, Prof. André R. Studart for the access to SEM and confocal microscopes and the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zurich for access to their instrumentation. ETH Zurich is acknowledged for financial support.Notes and references
- J. H. Bahng, B. Yeom, Y. Wang, S. O. Tung, J. D. Hoff and N. Kotov, Nature, 2015, 517, 596–599 CrossRef CAS PubMed.
- J. M. Barakat and T. M. Squires, Soft Matter, 2021, 17, 3417–3442 RSC.
- A. Stocco, B. Chollet, X. Wang, C. Blanc and M. Nobili, J. Colloid Interface Sci., 2019, 542, 363–369 CrossRef CAS PubMed.
- C. E. Colosqui, J. F. Morris and J. Koplik, Phys. Rev. Lett., 2013, 111, 028302 CrossRef.
- K. Zhao and T. G. Mason, Phys. Rev. Lett., 2007, 99, 268301 CrossRef PubMed.
- D. J. Kraft, R. Ni, F. Smallenburg, M. Hermes, K. Yoon, D. A. Weitz, A. Van Blaaderen, J. Groenewold, M. Dijkstra and W. K. Kegel, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 10787–10792 CrossRef CAS PubMed.
- S. N. Ramakrishna, P. C. Nalam, L. Y. Clasohm and N. D. Spencer, Langmuir, 2013, 29, 175–182 CrossRef CAS PubMed.
- S. N. Ramakrishna, L. Y. Clasohm, A. Rao and N. D. Spencer, Langmuir, 2011, 27, 9972–9978 CrossRef CAS PubMed.
- M. Zanini, C. Marschelke, S. E. Anachkov, E. Marini, A. Synytska and L. Isa, Nat. Commun., 2017, 8, 15701 CrossRef CAS.
- C.-P. Hsu, S. N. Ramakrishna, M. Zanini, N. D. Spencer and L. Isa, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 5117–5122 CrossRef CAS PubMed.
- P. Bourrianne, V. Niggel, G. Polly, T. Divoux and G. H. McKinley, Phys. Rev. Res., 2022, 4, 033062 CrossRef.
- J. F. Morris, Annu. Rev. Fluid Mech., 2020, 52, 121–144 CrossRef.
- J. M. Monti, P. M. McGuiggan and M. O. Robbins, Langmuir, 2019, 35, 15948–15959 CrossRef CAS PubMed.
- C.-P. Hsu, J. Mandal, S. N. Ramakrishna, N. D. Spencer and L. Isa, Nat. Commun., 2021, 12, 1477 CrossRef CAS.
- E. Akbari Fakhrabadi, C. A. Bullard and M. W. Liberatore, Rheol. Acta, 2021, 60, 251–262 CrossRef CAS.
- C.-P. Hsu, H. E. Baysal, G. Wirenborn, G. Mårtensson, L. Prahl Wittberg and L. Isa, Soft Matter, 2021, 17, 7252–7259 RSC.
- M. Zanini, A. Cingolani, C.-P. Hsu, M. A. Fernandez-Rodriguez, G. Soligno, A. Beltzung, S. Caimi, D. Mitrano, G. Storti and L. Isa, Soft Matter, 2019, 15, 7888–7900 RSC.
- M. Hu, C.-P. Hsu and L. Isa, Langmuir, 2020, 36, 11171–11182 CrossRef CAS.
- L. C. Hsiao and S. Pradeep, Curr. Opin. Colloid Interface Sci., 2019, 43, 94–112 CrossRef CAS.
- S. Nikravesh, D. Ryu and Y. L. Shen, Sci. Rep., 2020, 10, 5728 CrossRef CAS PubMed.
- B. Li, Y. P. Cao, X. Q. Feng and H. Gao, Soft Matter, 2012, 8, 5728–5745 RSC.
- J. Y. Chung, A. J. Nolte and C. M. Stafford, Adv. Mater., 2011, 23, 349–368 CrossRef CAS PubMed.
- M. Pretzl, A. Schweikart, C. Hanske, A. Chiche, U. Zettl, A. Horn, A. Böker and A. Fery, Langmuir, 2008, 24, 12748–12753 CrossRef CAS PubMed.
- H. Jiang, D. Y. Khang, J. Song, Y. Sun, Y. Huang and J. A. Rogers, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15607–15612 CrossRef CAS.
- N. Bowden, W. T. Huck, K. E. Paul and G. M. Whitesides, Appl. Phys. Lett., 1999, 75, 2557–2559 CrossRef CAS.
- H. Yuan, K. Wu, J. Zhang, Y. Wang, G. Liu and J. Sun, Adv. Mater., 2019, 31, 1900933 CrossRef PubMed.
- A. Schweikart, A. Fortini, A. Wittemann, M. Schmidt and A. Fery, Soft Matter, 2010, 6, 5860–5863 RSC.
- H. J. Bae, S. Bae, C. Park, S. Han, J. Kim, L. N. Kim, K. Kim, S. H. Song, W. Park and S. Kwon, Adv. Mater., 2015, 27, 2083–2089 CrossRef CAS PubMed.
- C. I. Zoldesi, C. A. van Walree and A. Imhof, Langmuir, 2006, 22, 4343–4352 CrossRef CAS PubMed.
- Q. Zhou, H. Xiang, H. Fan, X. Yang, N. Zhao and J. Xu, J. Mater. Chem., 2011, 21, 13056–13061 RSC.
- R. Liang, X. Fang, B. Qiu and H. Zou, Soft Matter, 2020, 16, 9113–9120 RSC.
- H. Yang, X. Lu and Z. Xin, Langmuir, 2018, 34, 11723–11728 CrossRef CAS.
- H. Yang, X. Lu and Z. Xin, Langmuir, 2020, 36, 571–575 CrossRef CAS PubMed.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- E. Cerda and L. Mahadevan, Phys. Rev. Lett., 2003, 90, 074302 CrossRef CAS PubMed.
- R. Vacassy, R. Flatt, H. Hofmann, K. Choi and R. Singh, J. Colloid Interface Sci., 2000, 227, 302–315 CrossRef CAS.
- N. Plumeré, A. Ruff, B. Speiser, V. Feldmann and H. A. Mayer, J. Colloid Interface Sci., 2012, 368, 208–219 CrossRef.
- N. Bowden, S. Brittain, A. G. Evans, J. W. Hutchinson and G. M. Whitesides, Nature, 1998, 393, 146–149 CrossRef CAS.
- S. Cai, D. Breid, A. J. Crosby, Z. Suo and J. W. Hutchinson, J. Mech. Phys. Solids, 2011, 59, 1094–1114 CrossRef.
- S. Yang, K. Khare and P. C. Lin, Adv. Funct. Mater., 2010, 20, 2550–2564 CrossRef CAS.
- B. Li, F. Jia, Y. P. Cao, X. Q. Feng and H. Gao, Phys. Rev. Lett., 2011, 106, 234301 CrossRef PubMed.
- Y. Tan, B. Hu, J. Song, Z. Chu and W. Wu, Nanomicro Lett., 2020, 12, 101 CAS.
- R. Kusters, C. Simon, R. Lopes Dos Santos, V. Caorsi, S. Wu, J.-F. Joanny, P. Sens and C. Sykes, Soft Matter, 2019, 15, 9647–9653 RSC.
- S. S. Datta, H. C. Shum and D. A. Weitz, Langmuir, 2010, 26, 18612–18616 CrossRef CAS.
- C. Gao, E. Donath, S. Moya, V. Dudnik and H. Möhwald, Eur. Phys. J. E: Soft Matter Biol. Phys., 2001, 5, 21–27 CrossRef CAS.
- S. Knoche and J. Kierfeld, Soft Matter, 2014, 10, 8358–8369 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details of experimental procedures and report of additional syntheses. See DOI: https://doi.org/10.1039/d2sm00842d |
| ‡ Current address: Center for Protein Assemblies and Lehrstuhl für Zellbiophysik (E27), Physics Department, Technische Universität München, Garching, Germany. |
| This journal is © The Royal Society of Chemistry 2022 |