
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A facile method for grafting functional hydrogel films on PTFE, PVDF, and TPX polymers†
Thorsten
Fischer
a,
Jan
Tenbusch
 a,
Martin
Möller
a and
Smriti
Singh
*ab
a,
Martin
Möller
a and
Smriti
Singh
*ab
aDWI—Leibniz-Institute for Interactive Materials e.V., Forckenbeckstr. 50, 52056 Aachen, Germany
bMax-Planck-Institut für medizinische Forschung, Jahnstraße 29, 69120 Heidelberg, Germany. E-mail: smriti.singh@mr.mpg.de
First published on 11th May 2022
Abstract
The use of polymeric materials in biomedical applications requires a judicious control of surface properties as they are directly related to cellular interactions and biocompatibility. The most desired chemical surface properties include hydrophilicity and the presence of functional groups for surface modification. In this work, we describe a method to graft a highly stable, ultra-thin, amine-functional hydrogel layer onto highly inert surfaces of poly(tetrafluoroethylene) (PTFE), poly(vinylidene fluoride) (PVDF), and poly(4-methyl-1-pentene) (PMP or TPX). Covalent grafting is realized with hydrophilic poly(vinylamine-co-acetamide)s by C–H insertion crosslinking (CHic) chemistry initiated by UV light. These polyvinylamides carry tetrafluorophenyl azide groups as photo or thermo activated binding sites and contain further free amine groups, which can be used to bind peptides such as biological ligands, polysaccharides, or other hydrogel layers. The covalently bound surface layers resist intensive Soxhlet extraction confirming the stability of the coating. Fluorescent staining verified the accessibility of free primary amine groups, which can be used for the functionalization of the surface with bioactive molecules. The coating demonstrates hydrophobic wetting behavior when conditioned in air and hydrophilic wetting behavior when conditioned in water showing the presence of loosely crosslinked polymer chains that can re-orient. We believe that the reported application of CHic for the surface modification of fluorinated polymers like PTFE and PVDF as well as TPX can form the basis for advanced biocompatible and biofunctional surface engineering.
Introduction
Chemically inert and biocompatible polymers such as poly(ethylene) (PE) and poly(4-methyl-1-pentene) (PMP or TPX), and their fluorinated derivatives poly(tetrafluoroethylene) (PTFE) and poly(vinylidene fluoride) (PVDF), are extensively used in biomedical devices.1–3 Their application comprises vascular grafts, prostheses, surgical sutures, and extracorporeal membrane oxygenators (ECMOs).3 For instance, PTFE and its expanded form (ePTFE) are used for vascular grafts because of their limited thrombogenic potential. However, protein adsorption can still induce coagulation cascade and thrombus formation, which limits their application in small diameter vascular grafts.4,5 TPX is used for the fabrication of hollow fiber membranes of ECMO devices. Despite great advancements, these membranes also suffer from protein adsorption and thrombus formation, which is one of the main causes of ECMO-related complications.6,7The examples discussed above highlight the imperative need to further modify the surface properties of such biomaterials, in particular for advanced blood compatibility. Ways to achieve this focus on rendering the surface hydrophilic (passive approach) and on endowing the surface with active molecules, which inhibit the coagulative response8–10 and eventually even promote re-endothelization (active approach).11 In order to combine both approaches, an ideal surface modification should consist of a biocompatible hydrogel, to which peptides and proteins do not bind by unspecific adsorption, which, however, can be equipped with specific biofunctional elements like cell-specific ligands, or cytokines and drugs which help to generate a functional natural interface with tissue or blood. Because hydrogels consist mainly of water, they can effectuate a certain stealth character and thus prevent unspecific interaction to enhance the selectivity of specific interactions.
Coating techniques applied so far involve plasma treatments,12 radiation-induced surface grafting,13 and photochemical methods.14 They typically exploit surface oxidation to render the surfaces hydrophilic and to introduce reactive groups to which the coatings can be linked. However, these methods are tedious and require several steps. A powerful alternative is offered, if hydrophilic macromolecules can be grafted directly to the surface of the inert polymer to form a thin or even monomolecular coating. Such grafting of a hydrogel layer to hydrocarbon substrates has been realized based on C–H insertion crosslinking (CHic) described by J. Rühe.15,16 According to this method, a precursor polymer with photoactive groups like aromatic ketones, azides, or diazo groups is coated onto the substrate and irradiated with light. The photoactive groups form highly reactive species such as singlet nitrene or carbene radicals which undergo insertion into C–H bonds. Most photoactive groups require irradiation with short wavelength UV light, which limits the application to rather flat surfaces and thin films. Thioxanthone groups are sensitive to longer wavelengths (λ ≥ 360 nm), but cannot solve the problem for complex surface structures,17 while azides and azo groups can be activated by light as well as by thermal activation.18,19 The latter is advantageous for systems where the coating is not accessible to the light source. The use of CHic has been reported for silicon wafers or glass substrates where a primer layer of functional silanes ensured tight binding to the substrate;16,19–23 direct grafting has been possible to polystyrene24,25 and poly(methylmethacrylate) surfaces.26–28 The reports demonstrate the versatility of the CHic technique to fabricate advanced bioanalytical devices, e.g., immunoassays,17 for anticoagulant surface modifications23 and antimicrobial coatings.22 All examples exploit the introduction of biofunctional molecules or groups within and by the CHic grafting/crosslinking step, i.e., simultaneous grafting, crosslinking, and functionalization. However, CHic has not yet been reported for inert materials like TPX, PVDF or PTFE.
In this report, we demonstrate a facile method to generate thin hydrogel films on extremely inert polymer substrates such as TPX, PVDF and even PTFE binding. As the precursor polymer for the hydrophilic film, we chose poly(vinylacetamide-co-vinylamine) that was partially substituted by 4-azido-2,3,5,6-tetrafluorobenzoic acid by amidation of a fraction of the vinylamine comonomers. Poly(vinylacetamide) is a biocompatible highly hydrophilic thermostable polymer that does not segregate from the aqueous solution even at elevated temperatures up to the boiling point of water.29 The amine functional polymer can be obtained by formamide-selective acid catalyzed hydrolysis of copolymers from vinylacetamide and vinylformamide.30–32 Hence, amidation of only a part of the amine groups with 4-azido-2,3,5,6-tetrafluorobenzoic acid yields a polymer with two chemical functionalities, the CH-inserted nitrene and the primary amine groups, which can be further modified in an aqueous solution, e.g., by peptide chemistry and Michael-type addition reactions. If this polymer is applied as an ultrathin or even monomolecular film, it can serve as a primer to enable wetting and further chemical modification. We chose the fluorinated phenyl azide because it can be safely handled below 100 °C. The azide groups are activated by UV light at 302 nm and are insensitive to polar protic solvents and even oxygen. The nitrenes react only slowly with oxygen compared to their insertion into C–H bonds.33 Furthermore, it allows selective pre-functionalization of the polymers by azide–alkyne cycloaddition or azide–thioacid amidation.34 While the C–H insertion mechanism of nitrenes is broadly studied and successfully employed for C–H insertion crosslinking of polymers, we have been surprised that the precursor polymers could also be attached successfully to PTFE, where the grafting can only occur via attacking the C–C and C–F bonds. We further investigated the wetting behavior of the hydrogel coating using dynamic contact angle measurements and show that the coating resembles a loosely crosslinked hydrogel in which the polymer chains can re-orient once in contact with air or water.
Materials and methods
Materials
N-Vinylformamide (NVF) and N-vinylacetamide (NVA) were purchased from TCI. Methyl pentafluorobenzoate was purchased from abcr. Sodium azide was purchased from Roth. Magnesium sulfate, triethylamine, and N-hydroxysuccinimide (NHS) were purchased from Sigma Aldrich. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) was purchased from Fluorochem. Rhodamine-B-isothiocyanate was purchased from ChemCruz. All solvents, HCl, and NaOH were purchased from VWR. All chemicals were used as received except NVF, which was distilled before usage. Poly(1-methylpenten) (TPX) and poly(vinylidene fluoride) (PVDF) were purchased from Goodfellow. PTFE foil was purchased from RCT. PVDF and PTFE were washed with CHCl3, EtOH and water while TPX was washed with EtOH and water before further modification.Methods
Proton NMR spectra were recorded using a Bruker Avance III-400 FT-NMR spectrometer (Bruker Corporation, Billerica, MA, USA) at 400 MHz. UV-Vis measurements were performed using a Jasco V-780 spectrophotometer (Jasco Corporation, Tokyo, Japan) from 200 to 400 nm at a concentration of 1 mg mL−1 in water. Fourier transformation infrared spectroscopy (FT-IR) was performed using a Nexus 470 spectrometer (Thermo Nicolet, USA). X-ray photoelectron spectroscopy (XPS) was performed using a Kratos Ultra Axis instrument (Kratos, United Kingdom). The samples were excited with monochromatic Al-Kα1,2 radiation (1486.6 eV). The resulting spectra were analyzed using CasaXPS software (Casa Software Ltd, United Kingdom). Confocal laser scanning microscopy (CLSM) images were taken using an SP-8 microscope (Leica GmbH, Wetzlar, Germany). Sessile drop contact angles (CAs) were measured using a DSA 100 analyzer (Krüss, Germany). Drops of 10 μL were placed on the sample surface at a rate of 200 μL s−1. After an equilibration time of 60 s, the CA was determined using a Tangent-2 fitting mode.Advancing and receding CAs, θ, were measured according to the Wilhelmy method35 using a DCAT 25 instrument (DataPhysics, Germany). For the measurement, polymer foil samples were cut into 3 cm × 3 cm squares. PTFE and TPX were pre-modified by static coating, i.e., forming a droplet on the surface of the foil (see below) (2 mL) of the functionalized polymer (2 mg mL−1 in water, 0.1 eq. was carefully spread over the surface and the samples were UV treated afterward at 302 nm for 20 min). The samples were spin-coated (see below); 750 μL of the functionalized polymer (5 mg mL−1 in MeOH, 0.025 eq.) were added slowly to the surface while spinning at 3000 rpm with a subsequent UV treatment at 302 nm for 20 min). Samples were thoroughly washed with water after each UV treatment and coated on both sides. The immersion depth was set to 8 mm with a motor speed of 0.5 mm s−1. Up to 5 cycles were performed in water, to validate the amphiphilic behavior in n-hexadecane. The linear regime was fitted with a linear fit and the contact angle was calculated using the Wilhelmy eqn (1).
 | (1) |
Spin-coating
Spin coating was performed with a Convac 1001s (Convac GmbH, Germany). Unless otherwise noted 300 μL of the functionalized polymer (5 mg mL−1 in MeOH, 0.025 eq.) were added slowly to the surface while spinning at 3000 rpm. Afterward, the samples were UV treated using a CL-1000 Ultraviolet Crosslinker (UVP, Germany) for 20 min at 302 nm. Finally, the samples were washed thoroughly with water and EtOH for at least 30 s and dried in a filtered nitrogen stream.Fluorescent labeling
The samples were incubated with a solution of rhodamine-B-isothiocyanate in water (4 μg mL−1, 1 h). Afterward, they were washed thoroughly with water and EtOH for at least 60 s and dried in a filtered nitrogen stream.Synthesis of P(VAm-co-VAA)
The detailed synthesis is described elsewhere.30 Briefly, N-vinylformamide (NVF) (3.64 g, 51.2 mmol, 1 eq.), N-vinylacetamide (NVA) (6.36 g, 51.2 mmol, 1 eq.), and VA044 (331 mg, 0.51 mmol, 0.01 eq.) were dissolved in water (90 mL) and degassed by at least three freeze–thaw cycles. The reaction mixture was stirred at 60 °C for 20 h. The product was precipitated in acetone, re-dissolved in water and lyophilized. The formamide group was selectively hydrolyzed by dissolving the polymer in NaOH (2 M, 500 mL) and refluxing at 80 °C for 4 h. The reaction mixture was neutralized and dialyzed for one week against water (SpectraPor, MWCO 3.5 kDa). After dialysis, ion exchange was performed to remove residual chloride ions (Ampersep® 900). The product was obtained by lyophilization.1H NMR (400 MHz in D2O, δ in ppm): 1.35–1.75 (–CH2 backbone), 1.75–1.95 (–CH3), 3.0–4.0 (–CH backbone), 7.5–8 (–CHO). SEC: Mn = 5.6 × 104 g mol−1, Mw = 1.2 × 105 g mol−1, PDI = 2.2.
Synthesis of 4-azido-tetrafluorobenzoate
Methyl pentafluorobenzoate (4.02 g, 17.8 mmol, 1 eq.) was dissolved in an acetone/water mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 45 mL). Sodium azide (1.5 g, 23.1 mmol, 1.3 eq.) was added and the mixture was refluxed at 85 °C for 6 h. After the reaction, the reaction mixture was cooled down to room temperature, diluted with water (75 mL), and extracted with Et2O (3 × 75 mL). The combined organic phases were dried with anhydrous MgSO4 and the solvent was removed in a vacuum.
:1, 45 mL). Sodium azide (1.5 g, 23.1 mmol, 1.3 eq.) was added and the mixture was refluxed at 85 °C for 6 h. After the reaction, the reaction mixture was cooled down to room temperature, diluted with water (75 mL), and extracted with Et2O (3 × 75 mL). The combined organic phases were dried with anhydrous MgSO4 and the solvent was removed in a vacuum.
Yield: 3.4 g (=76.7%).
1H NMR (400 MHz in CDCl3): δ 3.95 (s, 3H).
13C NMR (100.6 MHz in CDCl3): δ 53.4, 107.67, 123.5, 139.2, 141.9, 144.2, 146.6, 159.9.
19F NMR (376.5 MHz in CDCl3): δ −151.1, −138.8.
Synthesis of 4-azido-tetrafluorobenzoic acid
4-Azido-tetrafluoromethylbenzoate (2 g, 8.03 mmol) was dissolved in NaOH (3.2 mL, 20 wt%) and methanol (40 mL). The mixture was stirred overnight and subsequently acidified with HCl (2 M) to a pH lower than 1. The mixture was extracted with CHCl3 (3 × 40 mL), the organic phases were dried with anhydrous MgSO4 and the solvent was evaporated in vacuo.Yield: 1.27 g (=67.2%).
13C NMR (100.6 MHz in DMSO-d6): δ 108.5, 122.7, 138.9, 141.6, 143.1, 145.7, 160.2.
19F NMR (376.5 MHz in DMSO-d6): δ −151.7, −141.4.
Synthesis of N-succinimidyl 4-azido-tetrafluorobenzoate
4-Azido-tetrafluorobenzoate (0.25 g, 1.06 mmol, 1 eq.) was dissolved in DCM. NHS (230.15 mg, 1.06 mmol, 1 eq.) and EDC (245 mg, 12.8 mmol, 1.2 eq.) were added and the solution was stirred overnight. This was followed by the addition of 35 mL DCM and 50 mL water and stirred for another 40 min. The organic phase was washed with water (5 × 50 mL) and brine (3 × 10 mL). The mixture was dried with MgSO4 and the solvent was removed in vacuo.Yield: 326.9 mg (92.8%).
1H NMR (400 MHz in CDCl3): δ 2.85 (s, 4H).
19F NMR (376.5 MHz in CDCl3): δ −149.9, −133.6.
Functionalization of p(VAm-co-VAA) with N-succinimidyl 4-azido-tetrafluorobenzoate
P(VAm-co-VAA) (500 mg, 3.94 mmol amino groups) was dissolved in MeOH (25 mL). N-Succinimidyl 4-azido-tetrafluorobenzoate (130.9 mg, 0.394 mmol, 0.1 eq.per -NH2) was dissolved in EtOH and added dropwise to the solution. TEA (250 μL) was added and the solution was stirred overnight. The product was precipitated in cold diethyl ether, re-dissolved in water, and lyophilized. The other prepolymers (azide/amine = 0.075, 0.05, and 0.025) were prepared in a similar manner.Pre-modification of PTFE and TPX
PTFE and TPX foils were cut into a square shape (2 cm × 2 cm). 800 μL of the functionalized polymer (2 mg mL−1 in water, 0.1 eq.) were added to the surface and carefully spread. The samples were UV-treated using a CL-1000 Ultraviolet Crosslinker (UVP, Germany) for 20 min at 302 nm. The liquid phase was removed after UV treatment and the samples were washed with water and EtOH.Crosslinking of PFPA0.025-co-PVAm in Krytox GPL 101
P(VAm-co-VAA) with an azide/amine ratio of 0.025 (PFPA0.025-co-PVAm) was dissolved in MilliQ® water and added to a 10-fold volume of Krytox® GPL101 oil (DuPont, USA). The mixture was subjected to ultrasonication using an ultrasonication finger (Sonifier W-250 D, 10% amplitude, 20 s, G. Heinemann) to form a stable emulsion. The emulsion was subjected to UV irradiation at 302 nm for 20 min as described above. Subsequently, the crosslinked gel-like structure was purified using a 0.1 μm PVDF filter to remove residual Krytox® oil and washed. The residue obtained was isolated after lyophilization as a brown solid, and analyzed via FT-IR spectroscopy.Results and discussion
Scheme 1 shows the polymer synthesis that was used to apply a hydrogel coating to substrates TPX, PVDF and PTFE. The polymer was prepared by free radical co-polymerization of N-vinylacetamide and N-vinylformamide. Subsequent selective acidic hydrolysis of the formamide groups yielded free amine groups. A fraction of those was reacted with N-succinimidyl 4-azido-tetrafluorobenzoate to yield the nitrene functionalized copolymer of vinylacetamide and vinylamine (Scheme 1).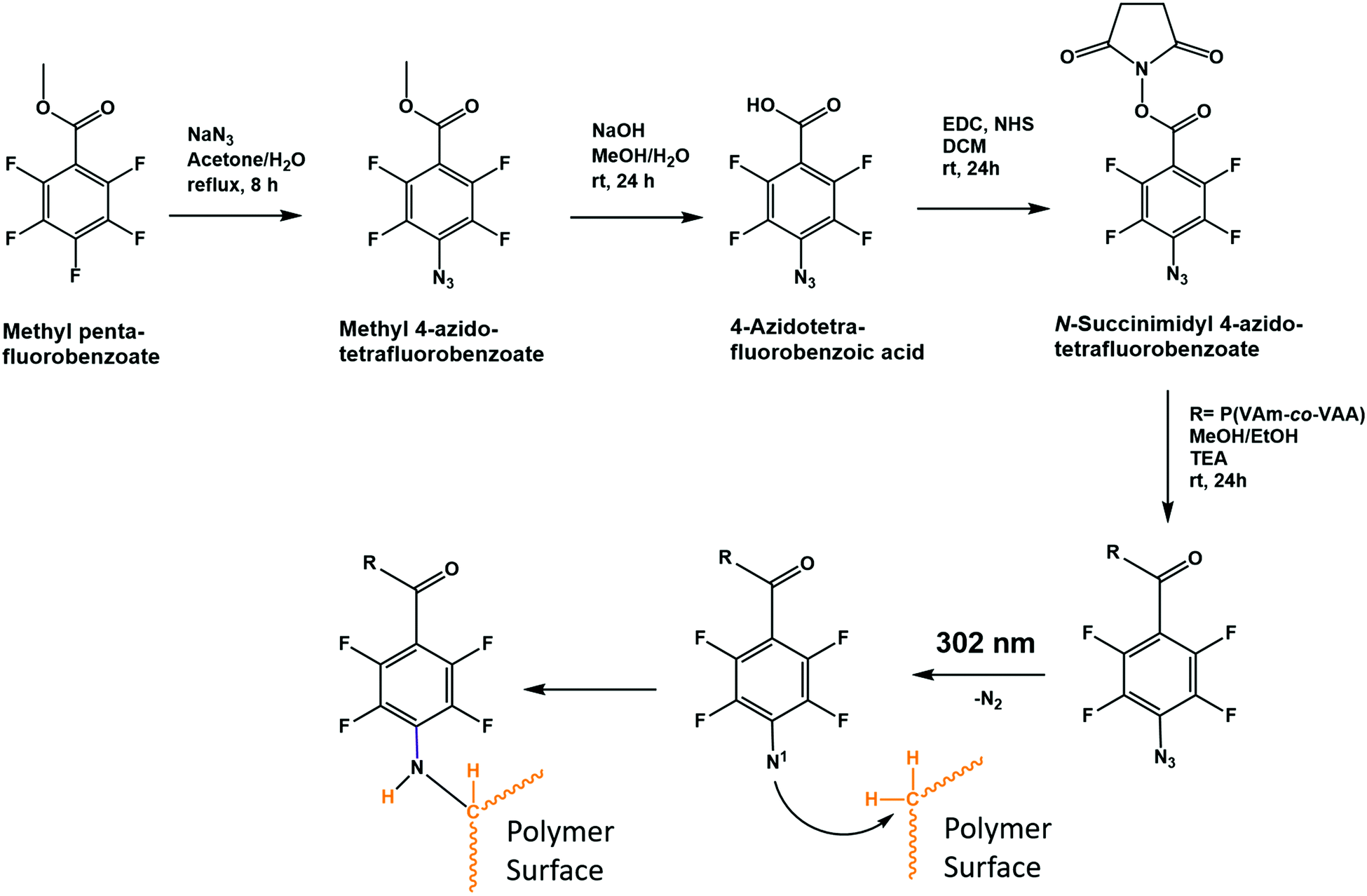 | ||
| Scheme 1 Reaction pathway to synthesize the prepolymer and representation of the proposed CHic reaction. | ||
Table S1 (ESI†) gives an overview of the polymers, which were employed for the preparation of the coatings. They are all based on the same parent P(VAm-co-VAA) sample, which was prepared by free radical polymerization as described before.30 The molecular weight of P(VAm-co-VAA) Mn (SEC) = 5.6 × 104 g mol−1 corresponds to an average degree of polymerization of DP = 670. Because of the nearly ideal co-polymerization kinetics of the two monomers, we assume homogeneous composition for all polymer chains.30 The ratio between N-vinylformamide and N-vinlyacetamide was determined by 1H-NMR in agreement with the monomer ratio employed (1:1). After hydrolysis of the N-vinylformamide units, the analysis of free amine groups corresponded to the original fraction of formamide groups. Substitution by azido tetrafluorobenzamide units was chosen to vary between 2.5 and 10% of the amine groups corresponding to 20–80 monomer units between the crosslinking azide substituents (see Table S1, ESI†). The number of azide groups per chain was a minimum of 10. Experimentally, the azide content was semi-quantitatively verified by the ratio of the IR intensities of the azide vibration (variable) and the amide vibration (Fig. S1 and S2, ESI†).
In the following, we refer to the azide-functionalized poly(vinylamide-co-vinylamine)s as prepolymers. For the preparation of a hydrophilic coating, we spin-coated TPX, PVDF and PTFE substrates with methanolic solutions of the polymer and immediately exposed them to UV irradiation at 302 nm. This leads to the activation of azide to yield the highly reactive nitrenes. For all different substrates, we had to avoid dewetting during the deposition of the polymer when the solvent evaporated. To optimize the surface coating, PVDF was chosen as the substrate in the first instance. With 36 mN m−1 the surface energy of PVDF is the highest of the three polymers compared to γ = 25 mN m−1 and γ = 19 mN m−1 for TPX and PTFE, respectively.36 Spin coating was varied for the solvent, rotation speed, concentration, volume added, and azide to amine ratio of the prepolymer (Fig. S3, ESI†). The coated PVDF samples were subsequently irradiated for 20 min with UV light at 302 nm and thoroughly washed with water and EtOH. Afterward, the hydrophilization was tested by the advancing contact angle against the water with the sessile drop method. A minimum contact angle of 19° was found for the prepolymer with an azide to amine ratio of 0.025 (when mixed with MeOH solution with a polymer concentration of 5 mg mL−1 by a volume of 75 μL cm−2 at a rotation speed of 3000 rpm). Such a low contact angle indicates the formation of a homogeneous, thin hydrogel layer that cannot be washed away by ethanol, which is a good solvent for the prepolymer.
When we used the same procedure to coat TPX and PTFE, we measured rather high contact angles of 108° and 115° against water, indicating either no or only very incomplete coverage by a hydrogel (Fig. S4, ESI†). In order to exclude the poor hydrophilization that was caused predominantly by dewetting, we deposited the prepolymer in two steps. In the first step, a droplet of the prepolymer in water (azide to amine ratio: 0.1; concentration 2 mg mL−1) was applied to the sample and crosslinked by UV to form a pre-layer. Subsequently, a second layer of the prepolymer (azide to amine ratio: 0.025; concentration 5 mg mL−1) was applied by spin coating and irradiated by UV-light. The improvement of the wetting by water with contact angles of 15° and 45° against water for TPX and PTFE, respectively, indicates that the originally bad results must be assigned predominantly to dewetting during spin coating.
Fig. 1(a) and (b) depict the optical micrographs of untreated and hydrophilized tiles of PVDF, TPX and PTFE. In all cases, the coating was colorless and invisible as expected for a very thin film. To visualize the coatings and demonstrate the accessibility of free -NH2 groups, we labeled the coating by treatment with an aqueous solution of rhodamine-B-isothiocyanate. The fluorescent dye attaches readily to the free amine groups in the coating and the coating formation can be observed by confocal laser scanning microscopy (CLSM), with a voxel resolution of 300 nm (Fig. 1c). As can be seen from Fig. 1c all the three substrates show prepolymer coating labeled with rhodamine (red). Of all the three substrates used, TPX which has a tertiary hydrogen atom in the side chain is the most suitable candidate for CHic insertion reaction. Coating PVDF is comparatively difficult because of the electron-withdrawing effect of the CF2 groups which renders the C–H less nucleophilic, still, due to the high reactivity of nitrene, the insertion could be achieved. The image of the coating on PVDF shows lamellar scales of the semicrystalline PVDF which are covered by the red fluorescent hydrogel. The prepolymer attached predominantly to the edges of the lamellae; nevertheless, flat areas between the edges, also show homogeneous coating as indicated by the even fluorescence intensity.
 | ||
| Fig. 1 Photographs of the (a) blank and (b) coated pieces of TPX, PVDF, and PTFE foil respectively, showing that the coating is nearly invisible, and (c) Confocal laser scanning microscopy images of the respective coated samples stained with rhodamine revealing the accessibility of free primary amine groups. The scale bar indicates 100 μm. | ||
However, for PTFE which lacks C–H bonds, C–H insertion is considered not feasible. For the reaction to occur, the electrophilic nitrene must orient to the C–F insertion which, due to the electron-deficient carbon center, is difficult. But in Fig. 1c, we see the rhodamine thio-isocyanate got bound to the surface rather evenly indicating homogeneous coating of the amine group containing prepolymer. The image of the PTFE sample with a fibrous type surface structure is typical of a surface of PTFE that was exposed to some shearing force.
To investigate the binding of the hydrophilic coating to the substrate we subjected the samples to 4 h Soxhlet extraction with methanol. This is a rather rigorous extraction procedure during which non-covalently bound components get extracted about 50 times by hot freshly distilled methanol. For this, the samples were immersed in the hot methanol and rinsed off after about 5 min. After the extraction process, the samples were washed with water and fluorescently labeled with rhodamine-B-isothiocyanate. The confocal laser scanning microscopy images (CLSM) in Fig. 2a–c depict the same staining effect of the surface as is shown in Fig. 1. For comparison, we show in Fig. S6 (ESI†) the images of the polymer surfaces which were exposed to the same extraction and fluorescence labeling processes, but not treated with the prepolymer before.
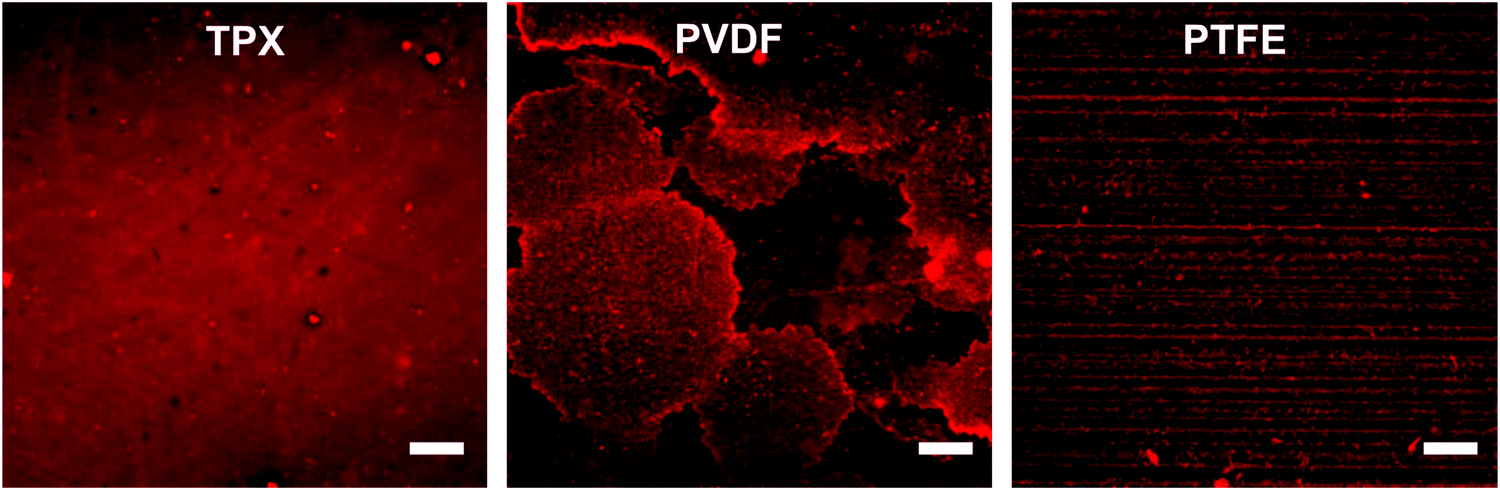 | ||
| Fig. 2 CLSM images of coated TPX, PVDF, and PTFE after treatment in a Soxhlet apparatus for 4 h in MeOH and staining with rhodamine-B-isothiocyanate. The coating is still intact and the amine groups are accessible. The scale bar represents 100 μm. | ||
Since the fluorescent staining only demonstrates that the surfaces treated with the prepolymer can be stained more efficiently than the blank polymer surfaces, it provides rather indirect evidence for the attachment of the hydrogel layer. Thus, we took additional efforts to investigate the grafting of a thin hydrogel layer and to analyze the chemical composition of the surface by X-ray photoelectron spectroscopy (XPS). XPS was performed after Soxhlet extraction in all cases. Fig. 3 depicts the high-resolution C 1s spectra of all the three coated foils that show characteristic C–N and C–O peaks arising from P(VAm-co-VAA). Furthermore, on comparing the high-resolution XPS spectra of TPX, PVDF and PTFE, we observed the presence of the C–F signal at 288 eV arising from the prepolymer backbone in TPX showing the coating stability, while in the case of PVDF and PTFE the C–F signal is reduced by 16% and 48% respectively. The C–F signals in PVDF and PTFE are predominantly from the polymer substrate and the decrease of the signal intensity again proves the presence of a stable hydrogel coating on the substrate ((Fig. 3a–c), Table 1). The N/O ratio of the prepolymer on TPX, PTFE, and PVDF was calculated to be in the range of 0.8 to 1.2 (Table 1). In comparison to the native ‘prepolymer,’ the high oxygen content of the coatings can be due to its reaction with molecular oxygen.37 A detailed discussion of the side reactions can be found in the ESI,† and Fig. S9.
 | ||
| Fig. 3 XPS survey and high-resolution C spectra of (a) TPX, (b) PVDF, and (c) PTFE after Soxhlet extraction. | ||
| Surface | C [atom%] | N [atom%] | O [atom%] | F [atom%] |
|---|---|---|---|---|
| a The values for the prepolymer are derived theoretically from its structure. | ||||
| TPX control | 100 | 0 | 0 | 0 |
| TPX coated | 66.8 | 5.5 | 9.4 | 18.4 |
| PVDF control | 53.1 | 0 | 0 | 46.9 |
| PVDF coated | 69.3 | 7.6 | 15.4 | 7.7 |
| PTFE control | 36.1 | 0 | 0 | 64 |
| PTFE coated | 53.3 | 6.8 | 8.8 | 31.1 |
| Prepolymera | 65.5 | 21.1 | 11.3 | 2.1 |
To further ascertain the grafting of the prepolymer onto PTFE, we conducted a model experiment where a stable w/o emulsion of 5 wt% of PFPA0.025-co-PVAm in water with a perfluorinated oil (Krytox® GPL101) was prepared at a ratio of 1:14 (Fig. S7, ESI†). The rationale behind this experiment was to examine the grafting of the fluorinated oil on the polymer gel upon UV irradiation (Fig. 4a). After UV irradiation, the gel was purified and dried. On comparing the IR spectra of the dried network with those of the prepolymer and the fluorinated oil, we found that the absorption originating from the characteristic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching and N-H stretching at 1640–1550 cm−1 of the N-vinylamides was still present in the emulsion crosslinked gel (Fig. 4b). However, the intensity of C–F stretching absorption at 1200–1000 cm−1 increased significantly, indicating the incorporation of the perfluorinated oil into the network. Due to the overlap with signals of C–F stretching, a distinct verification of the formation of N–F bonds is difficult, especially with the low molar ratio of theoretical N–F to C–F bonds present in both the oil and the prepolymer. Following our observation of the strong adhesion onto PTFE substrates as well as crosslinking within an emulsion with a perfluorinated oil and subsequent analysis, we hypothesize the formation of N–F bonds and, due to the proximity of the electronic environment of the fluorine and the nitrene radical, an attractive electronic interaction cannot be ruled out.38 However, a more detailed molecular analysis will be necessary in the future with more simplistic model compounds.
O stretching and N-H stretching at 1640–1550 cm−1 of the N-vinylamides was still present in the emulsion crosslinked gel (Fig. 4b). However, the intensity of C–F stretching absorption at 1200–1000 cm−1 increased significantly, indicating the incorporation of the perfluorinated oil into the network. Due to the overlap with signals of C–F stretching, a distinct verification of the formation of N–F bonds is difficult, especially with the low molar ratio of theoretical N–F to C–F bonds present in both the oil and the prepolymer. Following our observation of the strong adhesion onto PTFE substrates as well as crosslinking within an emulsion with a perfluorinated oil and subsequent analysis, we hypothesize the formation of N–F bonds and, due to the proximity of the electronic environment of the fluorine and the nitrene radical, an attractive electronic interaction cannot be ruled out.38 However, a more detailed molecular analysis will be necessary in the future with more simplistic model compounds.
 | ||
| Fig. 4 (a) Schematic representation of the investigation of crosslinking of PFPA-co-PVAm within an emulsion with perfluorinated oil. (b) FT-IR spectra of PFPA-co-PVAm, Krytox® GPL101 and crosslinked polymer in a water/perfluorinated oil emulsion indicate the integration of perfluorinated oil within the crosslinked hydrogel network. | ||
The ability of the grafted hydrogel to prevent protein adsorption is associated with the steric repulsion caused by the perturbation that the protein molecules generate when in contact with the hydrogel layer. For effective prevention of protein adsorption surface density of the hydrogel layer is more critical than the thickness. As the surface coverage increases, loosely crosslinked polymeric chains start to repel each other and stretch out to avoid polymer–polymer repulsion and generate an entropic barrier which makes the adsorption of protein thermodynamically unfavorable.39
To investigate the wetting behavior of the coatings the Wilhelmy balance method was used (Fig. 5a). In this method, the force is measured which is needed to immerse (advancing CA) or remove (receding CA) a sample from a liquid (Fig. 5b). This force is related to the CA via the Wilhelmy eqn (1). Unless otherwise stated water was used as the liquid phase.
 | ||
| Fig. 5 (a) Advancing, receding, and sessile drop CA from blank and coated TPX, PVDF, and PTFE. It is visible that the functionalization hydrophilizes the surface. (b) Schematic representation of the principle of the Wilhelmy balance and a graph showing that negative weights refer to hydrophobic CAs, while positive weights refer to hydrophilic CAs. (c) and (d) Exemplary graph of coated PTFE with multiple cycles performed using the Wilhelmy balance with changing the waiting time between the cycles from 0 s to 60 s. The graph shows the wet advancing CA. The arrow in (c) indicates the difference between the dry and the wet advancing CA. (e) The development of the wet advancing CA for the coated samples. With increasing waiting time, a plateau is reached which is in the range of the dry advancing CA. (f) Proving the amphiphilicity of the coating by using n-hexadecane as a solvent for coated and blank PTFE. | ||
As can be seen from Fig. 5a, the coating altered the surface properties of the foils, and all coated samples tend to have the same values for the advancing CA (∼95°) and receding CA (∼15°) resulting in a very high CA hysteresis of ∼80° in comparison to the controls. This was also apparent when static CA measurements were performed using the sessile drop method. Interestingly, when the CAs obtained by the Wilhelmy balance method were compared with those determined by the sessile drop method, it was found that the sessile drop CA for the blank foils was close to the advancing CA and the one for the coated samples was close to the receding CA. Taking the hydrophilicity of P(VAm-co-VAA) into account the advancing CAs of the coated samples are unexpectedly high.30,40 This leads us to conclude that the wetting behavior of the coating was influenced by the swelling and/or re-orientation of the polymer chains. This was ascertained by tracking the CA for the coated samples by the sessile drop method as a function of time (Fig. S8 and Video S1, ESI†). As can be visualized that although a decrease in contact angle was observed, this decrease was not instantaneous but rather in a timeframe of several seconds. To further analyze this, five subsequent cycles of CA measurements were performed with the Wilhelmy balance (as shown in Fig. 5c). It is visible that for coated samples, the advancing CA decreases as soon as the surface is wetted (cycle 2). This means the CA hysteresis has a kinetic factor and the advancing CA depends on the wetting history of the sample. We refer to this as “wet advancing CA”. However, the receding CA remained unaffected by the wetting history and the differences between the 2nd and the following cycles were negligible, which shows that once swollen the wetting behavior of the coating is not affected. In the dry state, because of a low crosslink density (azide to amine ratio, 0.025:1), the hydrophobic backbone of P(VAm-co-VAA) is directed towards the air to reduce the surface tension, while when in contact with water there is a fast re-orientation of the functional hydrophilic groups of polymer chains rendering the surface more hydrophilic indicating the formation of a loosely crosslinked hydrogel layer. This also confirms our previous observation that the lower the azide to amine ratio, the higher is the surface hydrophilicity which means a higher probability of rearrangement in the hydrogel layer, and a higher degree of swelling, indicating a scenario like the highly swollen open structure of a hydrogel film with diffused outer boundary and well solvated inner chain segments. A similar behavior has been reported for PMMA,41–44 where the segmental motion of the polymer chains was observed at the water–polymer interface. Upon contact with water, the initial hydrophobic surfaces turn more hydrophilic due to the re-organization of the carbonyl-groups within seconds.41 Additionally, this behavior was also shown for 2-hydroxy ethyl methacrylate/styrene block copolymers, where the CA hysteresis depended on the wetting history of the surface.45
To quantify this re-orientation time a waiting time between each cycle was introduced ranging from 0–60 s which is in the timescale as previously shown for PMMA.41 The measurement was halted when the sample was completely immersed or removed from the water phase (Fig. 5d). In Fig. 5e the wet advancing CA is plotted against the waiting time. As can be seen from the graph the re-orientation time is related to the film material and decreases from TPX (fast transition) to PVDF and PTFE (slow transition). This can be already seen from the initial wet advancing CA (waiting time 0 s), which also decreases from TPX (81.1° ± 2.1°) to PVDF (72.5° ± 0.8°) and PTFE (39.2° ± 2.0°). The re-orientation is related to the mobility of the polymer chains, which in turn depends on the crosslinking density and the texture of the surface (Fig. 1c).
The hypothesis of re-orientation can be verified by changing the medium from water to non-polar solvents such as n-hexadecane. If the hydrophobic polymer backbone is directed to the air, the sample will show a lipophilic behavior, which was tested for coated PTFE samples. In fact, due to the coating, the sample became more lipophilic than blank PTFE as shown in Fig. 5f (advancing CA 29.4° ± 5.0° and receding CA 7.0° ± 0.7° for the coated sample vs advancing CA 54.8° ± 0.8° and receding CA 16.8° ± 3.4° for the blank sample).
We further investigated the swelling behavior of the coating with respect to changes in the waiting time in the water phase, between the advancing and receding CAs (Fig. 6a). As can be seen from Fig. 6a the weight was increased with a change of the waiting time from 0 to 60 s, indicating the swelling of the hydrogel film. The swelling gives an indirect trend for the thickness of the coating for a given crosslink density and the underlying layer material. In this case, the actual thickness is comparatively difficult to measure due to the surface structure of the foils. To correct differences, arising from the sample geometry the weight was transferred to the CA viaeqn (1) as shown in Fig. 6b. The order of swelling obtained by the Wilhelmy balance shows TPX > PTFE > PVDF.
 | ||
| Fig. 6 (a) Time-dependent plot of the Wilhelmy balance for PTFE with a waiting time of 60 s. When immersed in the water phase the weight increases indicating swelling, while the weight only slightly decreased when removed from the water. (b) The increase of the weight during the waiting time is transferred to the respective CA viaeqn (1). A higher decrease in the CA is correlated with a higher degree of swelling. | ||
Conclusions
In this work, we show a fast and facile method to generate a stable, hydrophilic, and functional hydrogel coating onto PTFE, PVDF, and TPX under ambient conditions. The presence of an accessible primary amine in the hydrogel allows flexible post-modification. While TPX is rich in C–H bonds, PVDF has less, and PTFE has none of it. Moreover, in none of the polymer substrates N–H or CC bonds are present. The absence of C–H bonds in PTFE raises the question of whether the film is chemically or physically bound to the foil. A chemical bonding in PTFE would require the insertion of the nitrene in the C–F bond. In the case of PTFE since the abstraction of the hydrogen atoms is not possible a stable hydrogel coating on the surface of these polymers can only result from N–F bond formation. Non-covalent hydrophobic interactions of the tetrafluoro phenyl group on the PTFE surface are ruled out as it can unlikely form a stable hydrogel coating in the context of our polymer composition. In such a case, the swelling of the hydrogel layer will lead to a strong increase in the volume of this coating, which will induce stress not only normal to the surface but also within the plane of the surface, eventually delaminating the complete layer or parts of it. To the best of our knowledge, there are no studies that explore the C–H insertion for fluorinated polymers.
By using dynamic contact angle measurements at the air–water interface, we showed that the coating resembles a loosely crosslinked hydrogel layer where the polymer chains can re-orient once in contact with air or water. In future studies, the primary amine concentration of the coating will be tailored for bio-functionalization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) via Schwerpunktprogramm “Towards an Implantable Lung” Project Number: SI 2164/2-1. Open Access funding provided by the Max Planck Society.References
- X. Ren, Y. Feng, J. Guo, H. Wang, Q. Li, J. Yang, X. Hao, J. Lv, N. Ma and W. Li, Chem. Soc. Rev., 2015, 44, 5680–5742 RSC.
- S. K. Jaganathan, E. Supriyanto, S. Murugesan, A. Balaji and M. K. Asokan, BioMed Res. Int., 2014, 2014, 459465 Search PubMed.
- A. Subramaniam and S. Sethuraman, Natural and Synthetic Biomedical Polymers, 2014, pp. 301–308 DOI:10.1016/b978-0-12-396983-5.00019-3.
- A. de Mel, B. G. Cousins and A. M. Seifalian, Int. J. Biomater., 2012, 2012, 707863 Search PubMed.
- J. G. Meinhart, M. Deutsch, T. Fischlein, N. Howanietz, A. Fröschl and P. Zilla, Ann. Thorac. Surg., 2001, 71, S327–S331 CrossRef CAS PubMed.
- M. Pflaum, M. Kuhn-Kauffeldt, S. Schmeckebier, D. Dipresa, K. Chauhan, B. Wiegmann, R. J. Haug, J. Schein, A. Haverich and S. Korossis, Acta Biomater., 2017, 50, 510–521 CrossRef CAS PubMed.
- B. C. Keeshan, J. W. Rossano, N. Beck, R. Hammond, J. Kreindler, T. L. Spray, S. Fuller and S. Goldfarb, Pediatr. Transplant, 2015, 19, 294–300 CrossRef PubMed.
- A. Hosseinnejad, T. Fischer, P. Jain, C. Bleilevens, F. Jakob, U. Schwaneberg, R. Rossaint and S. Singh, J. Colloid Interface Sci., 2021, 601, 604–616 CrossRef CAS PubMed.
- A. Hosseinnejad, N. Ludwig, A. K. Wienkamp, R. Rimal, C. Bleilevens, R. Rossaint, J. Rossaint and S. Singh, Biomater. Sci., 2021, 10, 85–99 RSC.
- P. Winnersbach, A. Hosseinnejad, T. Breuer, T. Fechter, F. Jakob, U. Schwaneberg, R. Rossaint, C. Bleilevens and S. Singh, Membranes, 2022, 12 Search PubMed.
- A. I. Cassady, N. M. Hidzir and L. Grøndahl, J. Appl. Polym. Sci., 2014, 131, 40533 CrossRef.
- M. Crombez, P. Chevallier, R. C. Gaudreault, E. Petitclerc, D. Mantovani and G. Laroche, Biomaterials, 2005, 26, 7402–7409 CrossRef CAS PubMed.
- N. Mohd Hidzir, D. J.-T. Hill, E. Taran, D. Martin and L. Grøndahl, Polymer, 2013, 54, 6536–6546 CrossRef CAS.
- S. K. Williams, L. B. Kleinert and V. Patula-Steinbrenner,, J. Biomed. Mater. Res., Part A, 2011, 99, 67–73 CrossRef PubMed.
- O. Prucker, C. A. Naumann, R. Rühe, W. Knoll and C. W. Frank, J. Am. Chem. Soc., 1999, 121, 8766–8770 CrossRef CAS.
- P. F. Kotrade and J. Ruhe, Angew. Chem., Int. Ed., 2017, 56, 14405–14410 CrossRef CAS PubMed.
- S. Zunker and J. Rühe, Macromolecules, 2020, 53, 1752–1759 CrossRef CAS.
- D. S. Breslow, M. F. Sloan, N. R. Newburg and W. B. Renfrow, J. Am. Chem. Soc., 1969, 91, 2273–2279 CrossRef CAS.
- K. Schuh, O. Prucker and J. Rühe, Adv. Funct. Mater., 2013, 23, 6019–6023 CrossRef CAS.
- R. Navarro, M. Perez Perrino, O. Prucker and J. Ruhe, Langmuir, 2013, 29, 10932–10939 CrossRef CAS PubMed.
- C. K. Pandiyarajan, O. Prucker, B. Zieger and J. Ruhe, Macromol. Biosci., 2013, 13, 873–884 CrossRef CAS PubMed.
- P. Zou, D. Laird, E. K. Riga, Z. Deng, F. Dorner, H. R. Perez-Hernandez, D. L. Guevara-Solarte, T. Steinberg, A. Al-Ahmad and K. Lienkamp, J. Mater. Chem. B, 2015, 3, 6224–6238 RSC.
- N. Itagaki, Y. Oda, T. Hirata, H. K. Nguyen, D. Kawaguchi, H. Matsuno and K. Tanaka, Langmuir, 2017, 33, 14332–14339 CrossRef CAS PubMed.
- M. Moschallski, J. Baader, O. Prucker and J. Ruhe, Anal. Chim. Acta, 2010, 671, 92–98 CrossRef CAS PubMed.
- F. D. Scherag, R. Niestroj-Pahl, S. Krusekopf, K. Lucke, T. Brandstetter and J. Ruhe, Anal. Chem., 2017, 89, 1846–1854 CrossRef CAS PubMed.
- T. Brandstetter, S. Bohmer, O. Prucker, E. Bisse, A. zur Hausen, J. Alt-Morbe and J. Ruhe, J. Virol. Methods, 2010, 163, 40–48 CrossRef CAS PubMed.
- M. Moschallski, A. Evers, T. Brandstetter and J. Ruhe, Anal. Chim. Acta, 2013, 781, 72–79 CrossRef CAS PubMed.
- M. Zinggeler, J. N. Schonberg, P. L. Fosso, T. Brandstetter and J. Ruhe, ACS Appl. Mater. Interfaces, 2017, 9, 12165–12170 CrossRef CAS PubMed.
- J. S. Hong, T. Nakahara, H. Maeda, Y. Kikunaga, A. Kishida and M. Akashi, Colloid Polym. Sci., 1996, 274, 1013–1019 CrossRef CAS.
- T. Fischer, J. Köhler, H. Keul, S. Singh and M. Möller, Macromol. Chem. Phys., 2018, 219, 1800399 CrossRef.
- R. K.-J. Pinschmidt, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 2257–2283 CrossRef CAS.
- R. Pelton, Langmuir, 2014, 30, 15373–15382 CrossRef CAS PubMed.
- M. Schock and S. Brase, Molecules, 2020, 25 Search PubMed.
- J.-M. Noy, Y. Li, W. Smolan and P. J. Roth, Macromolecules, 2019, 52, 3083–3091 CrossRef CAS.
- C. D. Volpe and S. Siboni, Surface Innovations, 2018, 6, 120–132 CrossRef.
- https://www.accudynetest.com/ (accessed 14.11.2021, 2021).
- J. Mieres-Perez, E. Mendez-Vega, K. Velappan and W. Sander, J. Org. Chem., 2015, 80, 11926–11931 CrossRef CAS PubMed.
- D. Setiawan, D. Sethio, D. Cremer and E. Kraka, Phys. Chem. Chem. Phys., 2018, 20, 23913–23927 RSC.
- l Szleifer, Curr. Opin. Solid State Mater. Sci., 1997, 2, 337–344 CrossRef.
- J. Illergard, L. Wagberg and M. Ek, Colloids Surf., B, 2011, 88, 115–120 CrossRef CAS PubMed.
- A. Horinouchi, H. Atarashi, Y. Fujii and K. Tanaka, Macromolecules, 2012, 45, 4638–4642 CrossRef CAS.
- B. Zuo, Y. Liu, L. Wang, Y. Zhu, Y. Wang and X. Wang, Soft Matter, 2013, 9, 9376–9384 RSC.
- A. Horinouchi, Y. Fujii, N. L. Yamada and K. Tanaka, Chem. Lett., 2010, 39, 810–811 CrossRef CAS.
- A. Myalitsin, S. Ghosh, S. H. Urashima, S. Nihonyanagi, S. Yamaguchi, T. Aoki and T. Tahara, Phys. Chem. Chem. Phys., 2020, 22, 16527–16531 RSC.
- K. Senshu, S. Yamashita, M. Ito, A. Hirao and S. Nakahama, Langmuir, 1995, 11, 2293–2300 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00313a |
| This journal is © The Royal Society of Chemistry 2022 |