
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCoarse-grained molecular dynamics simulations of immobilized micelle systems and their interactions with hydrophobic molecules†
Devashish
Gokhale
a,
Ian
Chen
b and
Patrick S.
Doyle
 *ac
*ac
aDepartment of Chemical Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: pdoyle@mit.edu
bDepartment of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, MA 02139, USA
cHarvard Medical School Initiative for RNA Medicine, Boston, MA 02215, USA
First published on 9th June 2022
Abstract
Micelles immobilized in polymer materials are of emerging interest in drug delivery, water treatment and other applications. Immobilization removes the need for membrane-based separation to eliminate micelles from the medium, enabling facile extraction and delivery in diverse industries. This work lays out a coarse-grained molecular dynamics simulations framework for the rapid identification of surfactants for use in immobilized micelle systems. Micelles are immobilized by constraining one end of the constituent surfactants in space, mimicking what would occur in a copolymer system. We demonstrate that constraints affect how the micelles interact with small hydrophobic molecules, making it important to account for their effects in various drug-micelle and pollutant-micelle simulations. Our results show that in several systems there is stronger interaction between hydrophobic small molecules and micelles in immobilized systems compared to unconstrained systems. These strengthened interactions can have important implications for the design of new micelle-based extraction and delivery processes.
1 Introduction
Surfactants have been used for chemical separations for several decades.1,2 They find applications in several industries and areas, including drug delivery, emulsification, biologics purification, and oil recovery.3–7 These applications are enabled by the self-assembly of surfactants into micelles when above the critical micelle concentration (CMC). Micelles are aggregates with hydrophobic cores and hydrophilic shells. Hydrophobic molecules preferentially partition into the hydrophobic cores, resulting in the formation of large agglomerates. Micelles are therefore useful in both extraction, where the goal is to remove a hydrophobic substance from water, and delivery, where the goal is to deliver a hydrophobic substance into water. In extraction applications, micelles collect hydrophobic molecules in their cores and can then be separated from water by processes like ultrafiltration.8,9 On the other hand, in delivery applications, micelles are reservoirs for the storage and slow release of hydrophobic molecules into an aqueous medium.5 However, studies examining the kinetics of micelle formation and how they are affected by the presence of small molecules and by solvent effects remain limited.Most surfactants cannot be allowed to enter the bulk medium in environmental or physiological applications, imposing extremely restrictive design constraints on the kinds of surfactants which can be used in these areas. While the use of a membrane is feasible in some applications to retain micelles while allowing the passage of small molecules,9 it is not suited to certain applications in drug delivery where it may not be possible to insert or remove a membrane enclosure or in environmental remediation (such as the recovery of oil from a spill) where the scale of the separation is too large to allow the use of a membrane.
Driven by problems where membrane enclosures cannot be used to retain micelles, and by the desire to develop lower cost and more efficient processes that do not require a membrane-based separation step, there has been a growing interest in developing materials (slabs, powders, particles) in which micelles are immobilized.10–13 Immobilization, which can be effected by several methods such as chemically cross-linking micelles into a polymer matrix or by thermally gelling micelles in place, allows retention within the material itself, and no membrane is necessary. The porosity and mesh size of the matrix can be controlled to ensure high mass-transfer rates.13
In the context of delivery, a large body of prior work models micelles for drug delivery applications, focusing on the loading and release of small hydrophobic drug molecules from micelles.14 Apart from the engineering advantages associated with retaining micelles within a macroscopic structure, micelle immobilization has been explored in the context of drug delivery as a way of improving micelle stability by preventing micelle bursting when diluted to below the CMC in biological fluids like blood.15 Prior simulation work generally does not account for the effect of immobilization on micelle–hydrophobic molecule interactions, treating immobilized micelles in the same way as free micelles in solution.14
In the context of extraction, immobilized micelles have been used in prior experimental work in water purification,13 in which context computational studies to study the effects of immobilization are useful and can help optimize absorbent composition. Simulations of micellar systems are uncommon in water purification literature.
As we show in this study, the interactions between immobilized micelles and hydrophobic molecules are fundamentally different from the interactions between free micelles and hydrophobic molecules, which has important implications for the simulation and study of any system comprising immobilized micelles. This difference is primarily attributed to the restrictions on micelle motion conferred by the immobilization process, preventing the aggregation of smaller micelles to form larger agglomerates. From a thermodynamics perspective, constraints prevent the micelle–hydrophobic molecule–solvent system from reaching the global minimum on the potential energy landscape, forcing it into a different, local, minimum. As such, there is a need for scalable simulation studies that quantify the effect of constraints on immobilized micelle systems in the context of diverse chemistries and applications.
The MARTINI coarse-graining framework and force field,16 which has been used in a large body of prior work with great success to study surfactant self-assembly and to study interactions between large and small molecules,17,18 is used in this work to examine the interactions between five hydrophobic molecules and three classes of surfactants to demonstrate the versatility of such a framework while examining the effect of diverse chemistries on micelle–small molecule interactions in conjunction with constraint forces. Coarse-grained simulations are necessary, as opposed to all-atom simulations, for computational accessibility of large domain sizes containing several micelles while allowing rapid testing of potentially useful surfactant-small molecule combinations. The hydrophobic molecules studied here are chosen to represent chemically diverse species drawn from a large variety of sources, reflecting the diversity of industries in which micelle-based separations are, or are likely to be used. They include 2,4-dicholorophenol (DCP, from pesticides), 2-naphthol (NOL, from dyes), perfluorooctanoic acid (PFOA, an industrial surfactant), bisphenol A (BPA, from plastics), and ethinyl estradiol (EDOL, a drug). These molecules are also all contaminants of concern,19 of interest in water purification studies using immobilized micelles.13 The surfactants of interest are drawn from three classes – alkyl–PEO (poly(ethylene oxide)) or polyethoxylated alkane surfactants, PEO–PPO (poly(propylene oxide))–PEO surfactants (also called Pluronics or poloxamers), and sorbitan ester-based surfactants (like Span and Tween), all of which are widely used for separations in the chemical industry, in environmental remediation and water treatment, and in drug delivery.
The present study implements and extends existing coarse-graining schemes for the wide range of molecules listed above. We study the self-assembly of the surfactants into micelles, and examine the interactions between micelles and hydrophobic molecules in presence and absence of constraint forces that mimic immobilization, as shown by the schematic in Fig. 1(A). Machine learning tools are used to study the kinetics of micelle formation from molecular dynamics data in the presence and absence of hydrophobic molecules, and in the presence and absence of constraints. This methodology is robust and can easily be extended to the general study of self-assembly or aggregation kinetics, and enables studies that are otherwise impossible because of data set sizes and the need to count self-assembled domains manually.
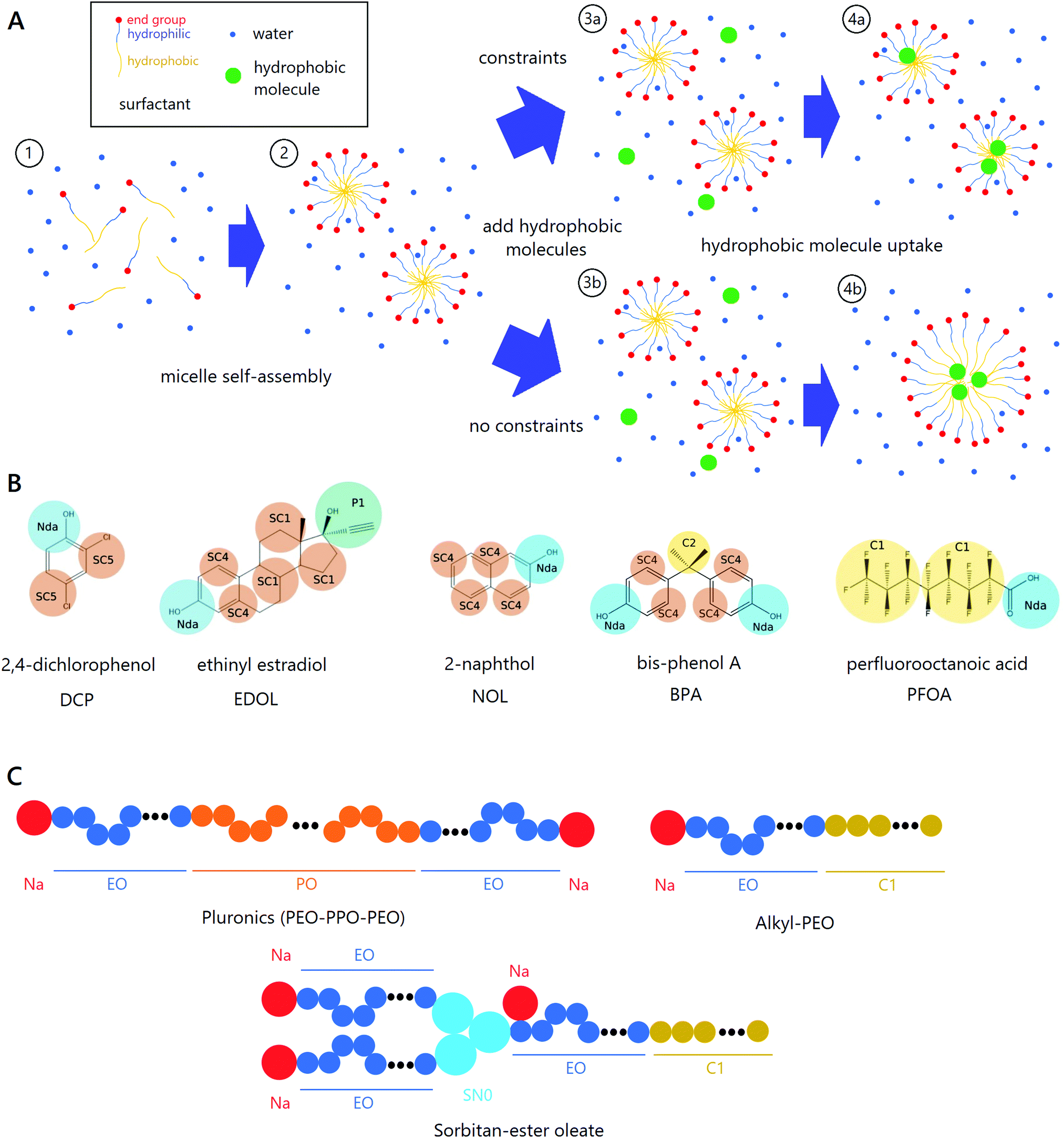 | ||
| Fig. 1 Overview of coarse-grained molecular dynamics simulations. (A) Surfactants are added to a water box (1) and allowed to reach equilibrium in an NPT simulation which produces micelles (2). Hydrophobic small molecules are then added while constraining (3a) or not constraining (3b) the end groups (red circles). Hydrophobic molecules are seen to partition into the micelle cores at equilibrium (4a,4b), while unconstrained micelles coalesce during such partitioning. (B) MARTINI 2.0 coarse-grained structures of the five hydrophobic molecules used in this work. (C) MARTINI 2.0 coarse-grained structures of the three main surfactant classes. | ||
We also study systems wherein the immobilized micelle structures are generated in ethanol–water mixtures. Synthesizing immobilized micelle structures in ethanol–water mixtures is common in both extraction and delivery applications because it often allows higher surfactant loading and helps tune micelle size by changing the polarity of the solvent which controls surfactant self-assembly into micelles.13,20 Adding ethanol to the solvent also allows us to change micelle size independently of the presence of small molecules. We examine the effects of ethanol–water synthesis pathways on micelle growth, sizes, and immobilized micelle–hydrophobic molecule interactions.
2 Methodology
The GROMACS 2019 package was utilized with the MARTINI 2.0 force field (MARTINI FF)16 to carry out CGMD simulations at 293 K with a 5 fs time step. Table 1 lists the surfactants used in our CGMD simulations. 100 molecules were used for each type of surfactant simulated in order to ensure the formation of at least 3 micelles in the system and avoid finite scale effects.21 The resulting system sizes are large and necessitate coarse-graining, implemented using the MARTINI FF that has been used with great popularity to treat similar coarse-grained systems in previous studies.17,18 Using MARTINI enabled us to leverage vast prior literature wherein coarse-grained models for diverse polymers and biomolecules were built and validated, allowing rapid testing of surfactant-small molecule combinations with a long-term perspective. Every simulation utilized a cubic box with periodic boundary conditions. All surfactants studied here are acrylate-terminated, which corresponds to the use of acrylate groups to functionalize surfactants to enable their incorporation into polymer gels.5,13 Acrylate addition in experiments is often accompanied by the introduction of a small amount of methacrylic acid in the solvent to limit acrylate loss by degradation, which we replicate in our simulations but is seen not to affect surfactant self-assembly or micelle behavior. The number of solvent (methacrylic acid (1.25% (v/v)) and water) molecules in simulations was calculated using the basis that the surfactant has a 2.5% (v/v) concentration in the system; this results in concentrations that exceed CMC in all cases. Due to large variation in surfactant molecular weights as seen in Table 1, the number of solvent molecules and box size in each surfactant system was distinct.| Surfactant | # C | # EO | # PO | Molecular weight (g mol−1) |
|---|---|---|---|---|
| Alkyl–PEO surfactants | ||||
| L10 | 12 | 10 | 0 | 627 |
| C10 | 16 | 10 | 0 | 683 |
| L23 | 12 | 23 | 0 | 1268 |
| C20 | 20 | 20 | 0 | 1124 |
| S20 | 18 | 20 | 0 | 1152 |
| B25 | 16 | 24 | 0 | 1496 |
| PEO–PPO–PEO surfactants (pluronics) | ||||
| L31 | 0 | 4 | 16 | 1189 |
| L44 | 0 | 20 | 23 | 2299 |
| L61 | 0 | 6 | 31 | 2147 |
| L64 | 0 | 28 | 31 | 3115 |
| L92 | 0 | 16 | 50 | 3689 |
| P85 | 0 | 52 | 40 | 4693 |
| F68 | 0 | 150 | 30 | 8425 |
| F127 | 0 | 202 | 56 | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 221 221 |
| Sorbitan–ester surfactants | ||||
| Span 20 | 11 | 0 | 0 | 346 |
| Span 80 | 17 | 0 | 0 | 429 |
| Tween 20 | 11 | 20 | 0 | 1228 |
| Tween 80 | 17 | 20 | 0 | 1310 |
Surfactants were modeled using classical MARTINI FF beads utilized in conjunction with the polyethylene glycol (EO) beads modeled in Grunewald, et al.22 and the polypropylene glycol (PO) beads introduced by Nawaz, et al.23 Alkyl groups were represented using the apolar C1 bead, methacrylates and methacrylic acid were modeled using the Na bead, and sugars in sorbitan ester-based surfactants were modeled using the nonpolar SN0 and the polar SP0 beads, as shown in Fig. 1(C), all of which belong to the original MARTINI Force Field.16 As in prior literature, Pluronics, polyethylene/polypropylene glycol block copolymers, are coarse-grained using the EO and PO beads, while sorbitan ester-based surfactants are coarse-grained using EO, SN0, SP0, and C1 beads.24,25 Alkyl–PEO surfactants, also block copolymers, are coarse-grained using polar EO beads and nonpolar C1 beads.16 The coarse-graining for the three types of surfactants are shown in Fig. 1(C). Na beads were used to represent terminal acrylate groups introduced to mimic actual chemistries used in experimental studies. Similar to previous implementations,23 the coarse-grained molecules closely match the geometry of the actual chemical structure to determine bond angles and bond lengths, as shown in the ESI.† The EO beads also incorporated a “restricted bending” potential derived from Grunewald, et al.;22 all other bond lengths and strengths are based on canonical MARTINI literature.16 Coarse-grained models for the small hydrophobic molecules studied here are also based on prior MARTINI literature, which optimizes for thermodynamic parameters that capture self-interactions and interactions with other large molecules including surfactants.16,26–30 Smaller solvent molecules such as water were coarse-grained using a 4:1 coarse-graining ratio and utilized the original MARTINI FF.16
The uptake simulations for each surfactant-hydrophobic molecule pair consist of two subprocesses; the first subprocess simulates the surfactants self-assembling into micelles, as shown in Fig. 1(A)(1). After energy minimization, the system undergoes an NPT production run for 100–150 ns, until the total Lennard-Jones potential energy of interactions in the system and between surfactant molecules, and the surfactant-surfactant radial distribution functions have equilibrated to a stable value. The equilibrium structure corresponds to a state in which surfactants have self-assembled into micelles as shown in Fig. 1(A)(2).
The second subprocess in the uptake simulations then simulates the absorption of hydrophobic molecules by the micelles produced in the previous step. The final system produced by surfactant self-assembly into micelles is first modified by constraining the end-groups on the surfactant molecules, emulating micelle immobilization while retaining the innate mobility of individual surfactant molecules. Acrylate beads, which are the points of attachment between micelles and the polymers within which they are immobilized, are constrained in a steep parabolic potential well to mimic their attachment to a polymer backbone, resulting in the immobilization of micelles. Note that we do not model the polymer backbones themselves in our simulations to limit computational complexity. The introduction of constraints is an approximation that mimics the polymerization of functionalized surfactants into a porous hydrogel,13 and is shown in Fig. 1(A)(3a). Hydrophobic molecules, also coarse-grained using MARTINI FF beads as shown in Fig. 1(B), are then inserted into the constrained system at random locations with an overall concentration of 5 mM and an energy minimization step is again performed. The concentration is of the same order of magnitude as what would be expected in extraction and delivery applications, given the low solubility of the hydrophobic molecules considered in this study. As before, the surfactant-hydrophobic molecule–solvent system now undergoes an NPT production run for 100–150 ns, until the Lennard-Jones potential for the overall system, between surfactants, between surfactant and hydrophobic molecules, and the surfactant-surfactant radial distribution function has equilibrated to a stable value. This process is shown in Fig. 1(A)(4a).
In order to determine the effect of constraining micelles on uptake simulations, a second set of simulations with unconstrained micelles was also run for the representative surfactants B25, L61, L64, P85, Span 20, and Tween 20 on the hydrophobic molecules EDOL, NOL, and PFOA. The process for performing these simulations is very similar to that of the simulations with constrained micelles; however, the constraining step after the self-assembly is ignored, as shown in Fig. 1(A)(3b), and the uptake simulations are run directly on the self-assembled micelles, resulting in the formation of larger agglomerates in the absence of constraints as shown in Fig. 1(A)(4b). The aggregation numbers of the micelles after unconstrained uptake, as well as the Lennard-Jones interaction energies between micelles and the surfactants, were determined. Our aggregation data for the unconstrained micelles of C8E4 compares favorably with prior simulations using a similar MARTINI FF17 (see ESI†). In general, while MARTINI FF simulations tend to underpredict experimentally observed aggregation numbers, trends for a given composition are meaningful and specific aggregation numbers may be quantitatively accurate (also see ESI†).17,31 Trends in structural data (e.g. radial distribution functions) presented in our work are expected to match experiments, and are backed up with quantitative data (e.g. Flory-Huggins χ parameters).
To determine the thermodynamic favorability of the uptake of hydrophobic molecules into the micelles, we performed Gibbs free energy calculations for representative systems. These calculations required two steps: calculating the Gibbs free energy change associated with removing the hydrophobic molecules from the system (i) before, and (ii) after they partition into micelles. The difference between the two values is the Gibbs free energy change associated with the absorption of hydrophobic molecules by the micelles. The computational process for both calculations involves thermodynamic integration and works by utilizing a range of coupling parameters between 0 and 1, as explained in Christ, et al. and associated prior work.32–34 As the interaction parameter incrementally increases from 0 to 1 (usually by a small step, 0.05), the interacting forces between the the hydrophobic molecules and the rest of the system are progressively weakened, emulating the slow removal of the molecules from the system. The change in the Hamiltonian is recorded with every incremental change to the coupling parameter, and can then be converted into Gibbs free energy using a function, developed by Berendsen, et al.35 Surfactants that contained EO beads required modifying the default bond angle potentials due to the Gibbs-free energy simulations being incompatible with restricted-angle potentials developed by Grunewald, et al.22 This modification is implemented by replacing the classical restricted bending potentials by polynomials fitted over the range of bond angles typically sampled through the course of simulations using the restricted bending potentials. Further information is provided in the ESI,† regarding this modification.
Prepolymer surfactant solutions for uptake simulations were also prepared in 10%, 20%, 30% and 40% (v/v) ethanol in water to determine the effect of ethanol on micelle formation and resulting effects on hydrophobic molecule uptake in water. Ethanol–water mixtures are of interest in experimental work because they allow for a greater solubility of certain surfactants such as F127, allowing for more micelles per unit volume of polymer and potentially more hydrophobic molecule uptake as described previously.20 Adding ethanol to the solvent also affects the relative amphiphilicity of surfactants and allows us to change micelle size independently of the presence of small molecules. The process for performing self-assembly simulations in ethanol is almost identical to that of in pure water in Fig. 1(A) (1 → 2); instead, coarse-grained ethanol beads replace water beads so that the desired concentration of ethanol is reached. These ethanol simulations were performed on the representative surfactants Span 20 and P85, utilizing the hydrophobic molecule NOL, which grouping is representative of surfactant-hydrophobic molecule systems utilized in our broader simulations. The aggregation numbers of the micelles in ethanol, as well as the Lennard-Jones interaction energies between micelles and the surfactants, were determined. After micelles assemble in ethanol solutions, they are constrained as before and the solvent is then changed to pure water. Hydrophobic molecules are introduced and uptake studies are performed as before. Since experimental work focuses only on ethanol solutions used for preparing immobilized micelles, we do not perform uptake simulations for unconstrained micelles in ethanol solutions.
We use unsupervised machine learning tools (DBSCAN), which have been used in limited ways in similar contexts recently,36 to cluster the hydrophobic beads in surfactant molecules and automate micelle counting, and enable the determination of average aggregation numbers (and error estimates) over thousands of frames without having to manually count micelles. The DBSCAN algorithm is similar to inbuilt tools in GROMACS (gmx clustsize), but analyzes data frame-by-frame without requiring trajectory data and can be more robust when there is an exchange of surfactant molecules between distinct micelles. DBSCAN is run with an epsilon (the distance at which two beads will be recognized to be parts of the same cluster) of 1 nm and a minimum sample size (the size of a predicted cluster which is a lower threshold separating it from noise) equal to the number of hydrophobic beads per surfactant molecule. We also extend prior work and show that DBSCAN can be used to study the dynamics of micelle formation, which though less studied than aggregation numbers, are also of academic interest and convey useful information about micelle stability. We leverage automatic clustering to investigate the effect of introducing hydrophobic molecules and changing the solvent on the rate of micelle formation as shown in Fig. 2(A). Frames are extracted at periodic intervals from the simulations and hydrophobic beads are isolated within each frame. DBSCAN successfully clusters these beads to identify micelles, as indicated by Fig. 2(B), wherein individual micelles identified by DBSCAN in a box containing 100 B25 molecules are each colored based on the micelle to which DBSCAN predicts them to belong. By applying this technique to a succession of frames, we rapidly count the number of clusters as surfactants assemble into micelles (a process which is impossible to do manually), and enables the study of the kinetics of micelle formation. It is important to note that the MARTINI FF is known to spuriously speed self-assembly, but the relative rates are preserved since the systems being compared contain extremely similar structures and are worth examining.37
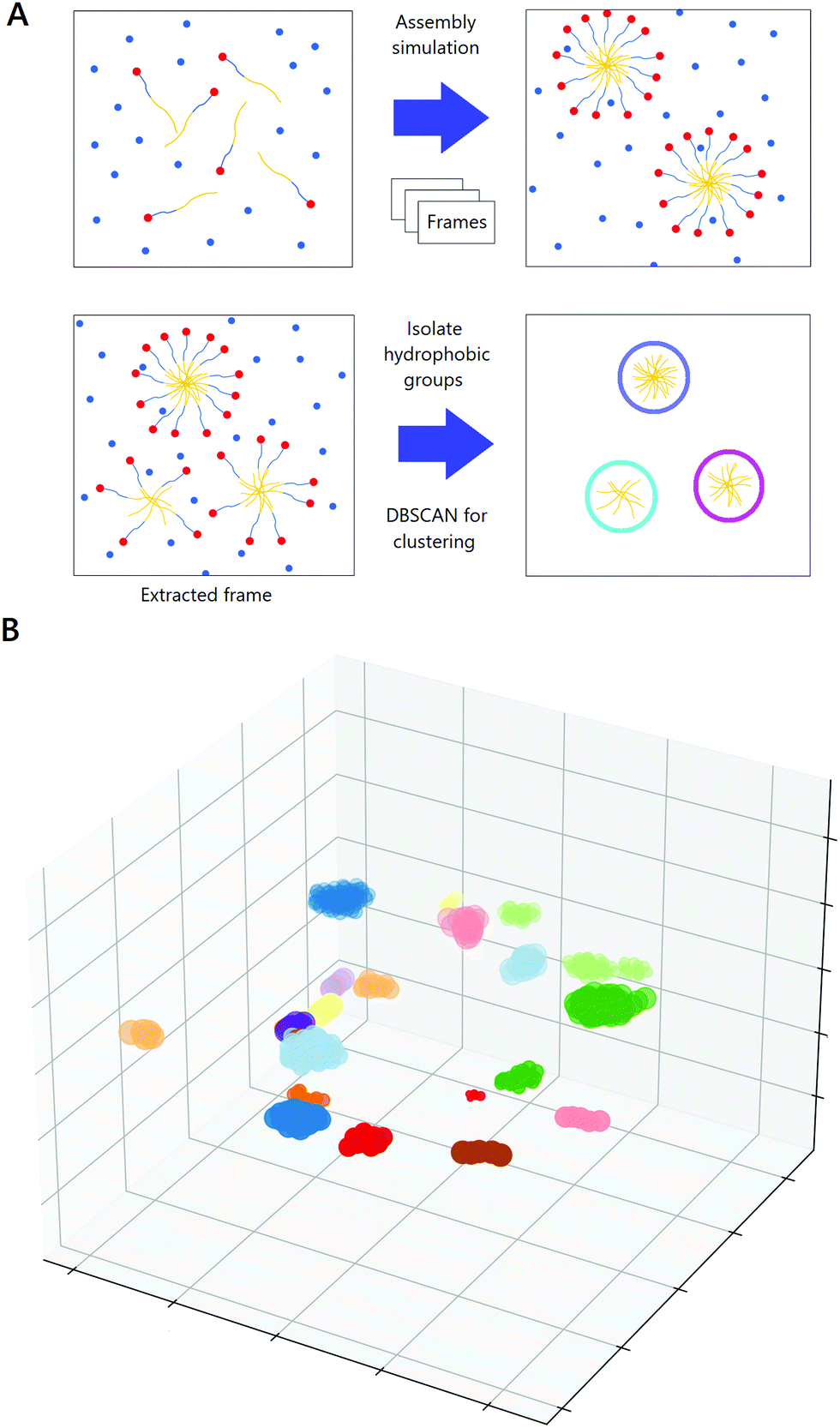 | ||
| Fig. 2 Use of machine learning tools to study the kinetics of micelle formation. (A) Frames are extracted at periodic intervals from the micelle-assembly simulations. Hydrophobic beads are preserved and clustered using DBSCAN, which successfully identifies the numbers and sizes of micelles. (B) B25 molecules (2.5% v/v) in water, colored by DBSCAN based on the cluster they are identified as being a part of. Beads of the same color are seen to belong to the same micelle, indicating successful clustering. Certain colors are repeated when micelles are well-separated visually to limit the total number of colors used. | ||
3 Results and discussion
3.1 Partitioning of hydrophobic molecules into micelles
As described in the methodology, we performed uptake simulations for 17 surfactants and 5 hydrophobic molecules at 293 K, both with and without constraints on the surfactants. Simulation snapshots are useful to validate the scheme shown in Fig. 1(A). Fig. 3(A and B) show constrained surfactant systems at equilibrium after the addition of hydrophobic molecules, indicating the partitioning of hydrophobic molecules into micelles as anticipated. Fig. 3(A) shows a zoomed out view of the entire F68-NOL system, showing that very few hydrophobic molecules exist outside the immobilized micelles at equilibrium, which can be expected at the low hydrophobic molecule concentrations used in our study. Magnified views of single immobilized micelles can be seen in Fig. 3(B) for four representative surfactants, before and after the absorption of hydrophobic small molecules (here, NOL, in green). Note in particular that several hydrophobic molecules sit at the corona–core interface of the micelle. This location is expected based on the presence of small hydrophilic moieties in each of the hydrophobic molecules considered in our study (for example, the hydrophobic molecule shown here, NOL, has a phenolic alcohol moiety). The presence of hydrophilic moieties is common in most hydrophobic molecules that are of interest in drug delivery, water treatment, and other applications. Being located at the interface allows the bulk of the hydrophobic molecule to rest inside the core while hydrophilic moieties can point out into the less hydrophobic corona. Moreover, it is also apparent that interfacial sites are easier for the hydrophobic molecules studied here to access than sites located deep inside the micelle cores. More snapshots demonstrating this effect are shown in the ESI.†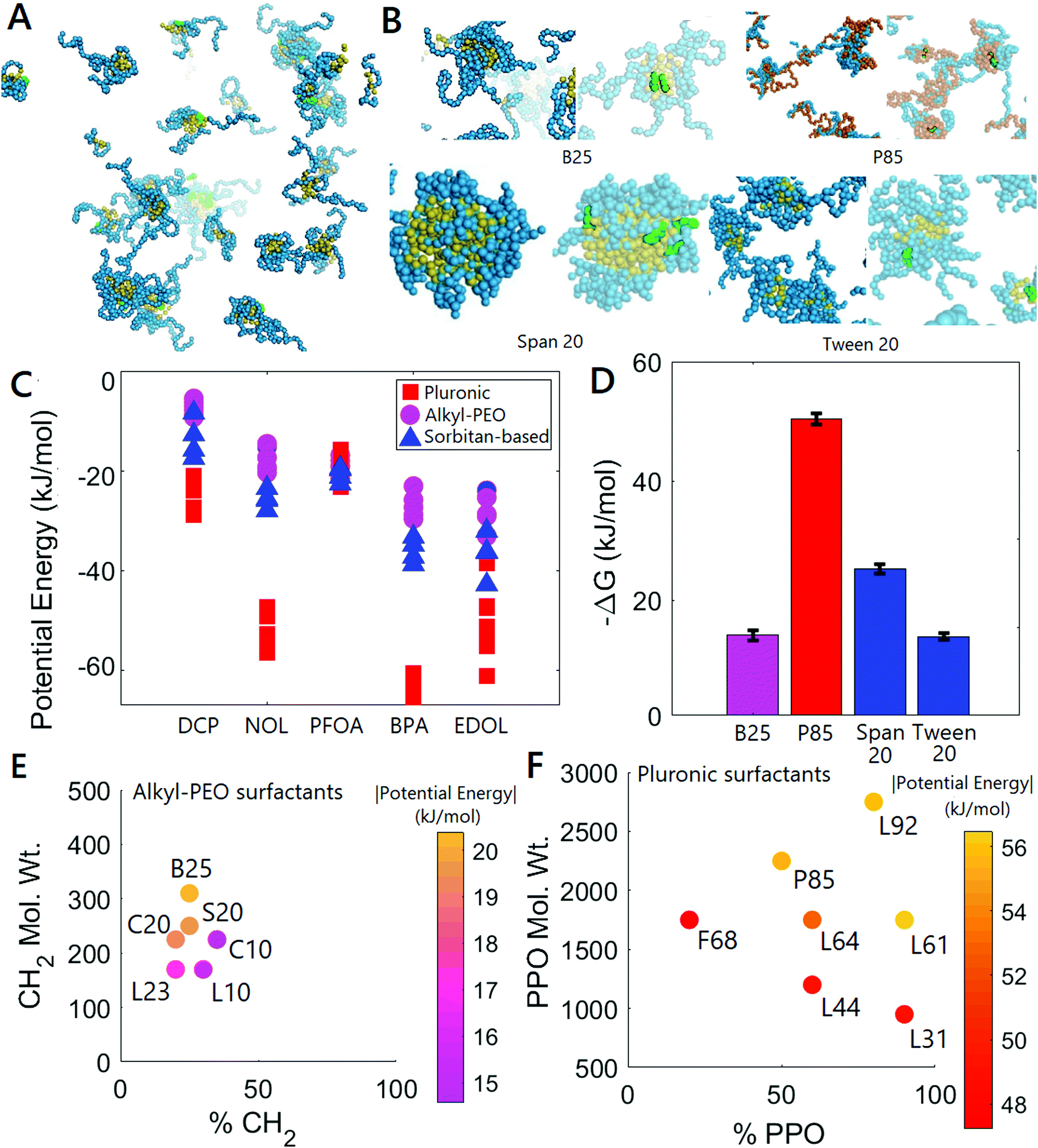 | ||
| Fig. 3 (A and B) Uptake of NOL molecules (green) by immobilized micelles (blue: hydrophilic ethylene oxide groups; orange: hydrophobic propylene oxide groups; yellow: hydrophobic alkyl groups). (A) Shows a zoomed out F68-NOL system at equilibrium, and (B) shows zoomed in images of four surfactant systems containing NOL at equilibrium. (C) Potential energies of interaction between surfactant molecules in immobilized micelles and small hydrophobic molecules. Each point represents one surfactant-hydrophobic molecule pair, with surfactants belonging to the same class having the same marker. (D) Gibbs free energy changes (per mole of small molecules) associated with the partitioning of NOL into immobilized micelles of four representative surfactants. (E and F) Variation of magnitudes of potential energies of surfactants–hydrophobic molecule interactions in (C) as a function of the size of various groups in the surfactant molecule for alkyl–PEO (E) and Pluronic (F) surfactants. Color indicates the magnitude of the potential energy. All potential energy values are reported per mole of surfactant. | ||
Fig. 3(C) shows the potential energies of interactions between surfactants and hydrophobic molecules. This data combined with that for free micelles is shown in Fig. S5 (ESI†). Our observation of the preferential partitioning of hydrophobic molecules into immobilized micelles is consistent with the negative sign of the potential energies. A lower potential energy implies the formation of stronger physical bonds between surfactants and hydrophobic molecules, and can be expected to correlate strongly with the extent of partitioning of hydrophobic molecules at low concentration into immobilized micelles in real systems. Interestingly, all the potential energies determined here are at least several kT (1kT ≈ 2.4 kJ mol−1 at 293 K), which agrees well with our observation that small hydrophobic molecules, after partitioning into micelles, do not escape those micelles into the solvent phase without a change in environmental conditions. It is apparent that the strength of the interaction is a strong function of the surfactant class, and only weakly dependent on variations in sizes of hydrophobic and hydrophilic groups within that class. The Pluronics have the strongest interactions with the hydrophobic molecules, and the alkyl–PEO surfactants have the weakest interactions. Note that the only hydrophobic molecule which does not obey these trends is PFOA, which is itself a strongly-amphiphilic surfactant inherently different from most hydrophobic small molecules.
The potential energies of interactions between the micelles and small hydrophobic molecules are surrogate metrics for the free energy change during the uptake process. The free energy change is computationally expensive to obtain, requiring thermodynamic integration as described in Section 2. Magnitudes of Gibbs free energy changes associated with the partitioning of hydrophobic NOL molecules into immobilized micelles are shown in Fig. 3(D) for four representative surfactants chosen across classes, and can be seen to correlate with the potential energies in Fig. 3(C). For instance, the Pluronic system (P85) shows the largest free energy change, and the alkyl–PEO surfactant (B25) shows the smallest. The absolute values obtained here are similar to values obtained experimentally for other drug-surfactant systems,38–40 and can be expected to be quantitatively accurate since MARTINI is parameterized for accurate free energy determination in self-assembled systems.37 Error bars in Fig. 3(D) reflect the magnitudes of numerical errors produced by discretization in thermodynamic integration, as described in the methodology. Based on the data in Fig. 3(E), we can conclude that the Pluronics, which have the strongest interactions with the hydrophobic small molecules, are also likely to show the fastest uptake or release of those molecules when immobilized in macroscale structures. Recent experimental work using immobilized micelles for water treatment showed the improved performance of Pluronics compared to alkyl–PEO and sorbitan–ester surfactants,13 demonstrating the ability of these simulations to help optimize material compositions.
Fig. 3(E and F) show the dependence of potential energies of interaction between surfactants in immobilized micelles and hydrophobic small molecules on the sizes of surfactant hydrophobic groups (PPO in Pluronics and C in alkyl–PEO surfactants, respectively) and fraction of hydrophobic groups by molecular weight for the Pluronics and alkyl–PEO surfactants respectively. Since the alkyl groups are more hydrophobic than PPO groups, commercial alkyl–PEO surfactants can have a smaller fraction of hydrophobic groups than Pluronics, in order to maintain solubility in water. The data points in Fig. 3(E) are therefore clustered on one side of the plot. Note that this clustering is therefore not an artifact of our surfactant selection process, but arises as a result of the limited range of hydrophobic group sizes and fractions at which commercial alkyl–PEO surfactants are available.
Based on the data in Fig. 3(E and F), it can be inferred that more hydrophobic groups and a larger fraction of hydrophobic groups result in stronger interactions between the surfactants and hydrophobic molecules, as can be expected in free micelles. The same effect is seen in sorbitan–ester surfactants, shown in Fig. S3 (ESI†). In practical applications, improving surfactant performance by increasing the total size and fraction of hydrophobic groups will be limited by solubility, which limits surfactant dissolution in monomer mixtures for immobilization in polymers. The solubility is reduced as the total size and fraction of hydrophobic groups increase.
3.2 Effect of immobilization
We compare the behavior of immobilized micelles as seen in the previous subsection to that of free micelles in water. Videos SV1 and SV2 (ESI†) show the agglomeration of free micelles into larger micelles upon the introduction of hydrophobic molecules. Recent experimental and simulation work on a specific unconstrained surfactant-small molecule system also detected this effect, which was attributed to the disruption in micelle geometry created by the hydrophobic small molecule.41 This geometric disruption is the result of small molecules partitioning either into the micelle core or at the core–corona interface, disrupting interactions between surfactant molecules. To prevent water (or other solvent) from entering the hydrophobic micelle core, an effect which is energetically unfavorable, more surfactant molecules must enter existing micelles to stabilize the geometry. This insertion manifests itself as an increase in the aggregation number, which is the average number of surfactant molecules per micelle. On the other hand, it is apparent that immobilized micelles, being locked in place within a matrix, cannot agglomerate to form these larger structures as seen in videos SV3 and SV4 (ESI†), and their aggregation number remains unchanged after the introduction of hydrophobic molecules. It is interesting to note that the introduction of small molecules into a surfactant solution can slow down micellization, as seen in Fig. S2 (ESI†). Fig. 4(A) shows for four representative surfactants (one from each class) the ratio of the aggregation number of free micelles to the aggregation number of immobilized micelles, after the introduction of hydrophobic molecules of NOL. The ratio is always greater than one, indicating that free micelles always prefer to form larger agglomerates when hydrophobic molecules are added. The ratio can be seen to be weakly dependent on the surfactant type, and is close to 1.5.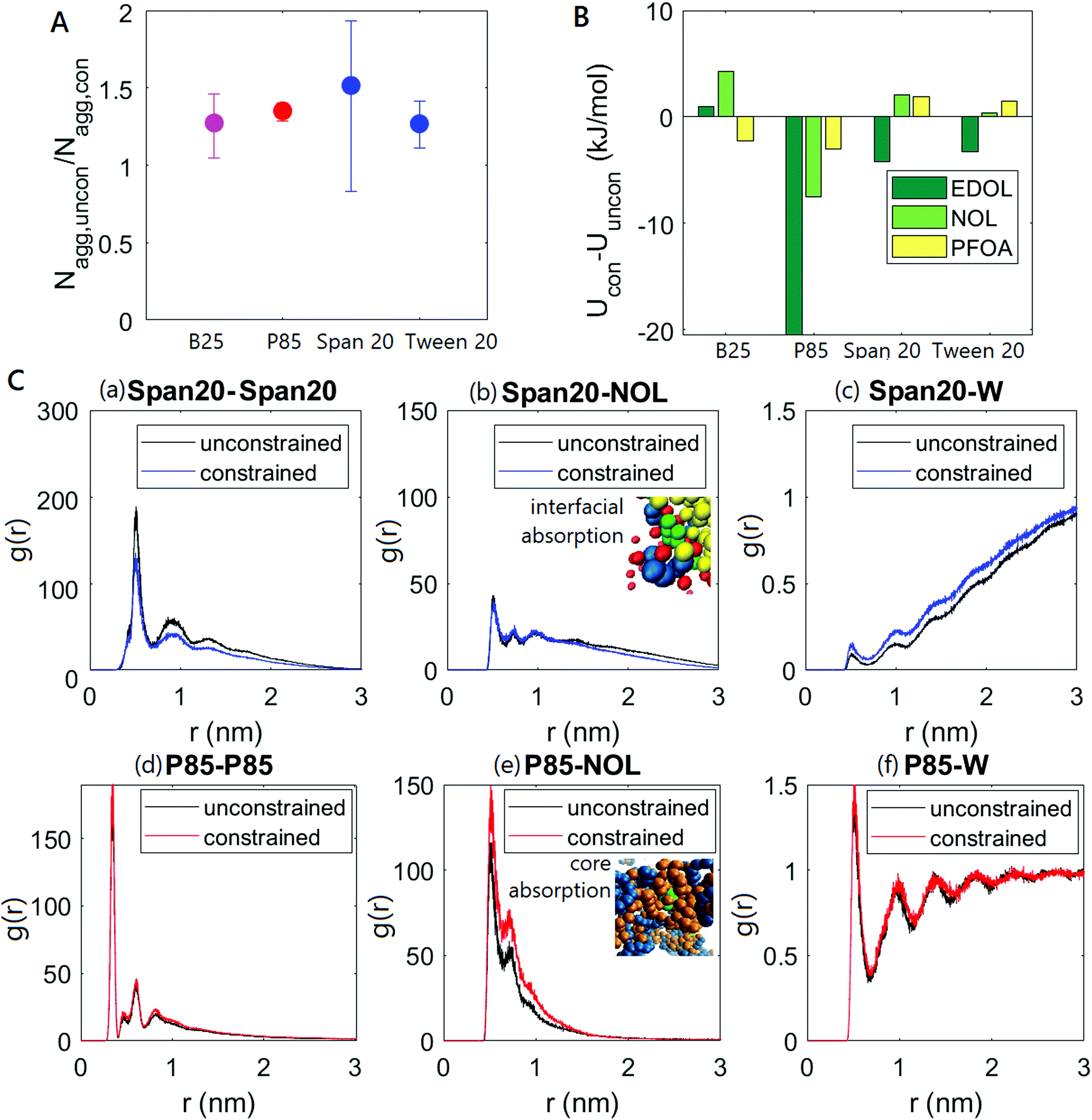 | ||
| Fig. 4 Effect of immobilizing micelles. (A) Ratio of micelle aggregation numbers at equilibrium after the addition of 5 mM NOL without constraints to micelle aggregation numbers at equilibrium after the addition of 5 mM NOL with constraints, for four representative surfactants. (B) Difference between the potential energies of surfactant-hydrophobic molecule interaction with and without constraints. (C) Radial distribution functions for B25 and P85 systems at equilibrium, both containing NOL, with and without immobilizing constraints. These radial distribution functions only account for the hydrophobic beads in B25 (alkyl groups) and P85 (PPO groups), and the origin on the x-axis of these plots corresponds to a point in the interior of the micelle cores. | ||
It is also apparent that a change in micelle size and morphology caused by the introduction of hydrophobic small molecules will affect the interactions between the micelles and those molecules. Fig. 4(B) shows the difference between the interaction energies with constrained and unconstrained micelles for four representative surfactants with three hydrophobic molecules. A negative value here implies that the interaction is stronger when micelles are immobilized, or that the formation of larger micelles inhibits the interactions between the micelles and hydrophobic molecules. It can be seen that the difference is small for most micelle–small molecule systems, but remarkably, is actually negative for specific systems (for example, with surfactant P85 in Fig. 4(B)). These negative values imply that micelle immobilization can strengthen the interactions between hydrophobic small molecules and surfactants. This strengthened interaction can explain the high levels of drug loading seen in prior experimental work, for example with micelle-laden hydrogels used to encapsulate small drug molecules.5
When we immobilize micelles, we constrain the system and prevent certain molecular rearrangments (increased aggregation numbers) which might otherwise lower the free energy of the overall system. In constrained systems, the less efficient packing of the micelles due to the sub-optimal aggregation numbers and other prohibited rearrangements results in a higher overall free energy, but also exposes micelles to more favorable interactions with the small hydrophobic molecules, enhancing the hydrophobic molecule-micelle interactions and lowering the free energy corresponding specifically with these interactions of interest. In general, constraints affect micelle properties in two ways – (1) changed aggregation numbers, and (2) changed space of available conformations that the surfactant molecules can access. To probe the relative importance of these two modes, we studied the P85-NOL system in Fig. 4(B) as an exemplar for systems that show strengthened surfactant-small molecule interactions when constrained. First, we equilibrated the P85 system with NOL in the absence of constraints, which produced micelles with larger aggregation numbers than when produced by constraining after equilibration in the absence of NOL. Then, we measured the potential energy of interaction between P85 and NOL at equilibrium. NOL was then removed from the system, and constraints imposed to immobilize the P85 micelles at this higher aggregation number. Finally, NOL was reinserted and the system allowed to attain a new equilibrium in the presence of constraint forces, and the potential energy of P85-NOL interactions was recorded at equilibrium. The simulation protocol thus gave us two potential energy values, one with constraints, and one without constraints, but both at the same value of aggregation number. We noted a difference of only about 4% in these values, compared to a 16% difference when immobilization is accompanied with an aggregation number change as in real systems. Consequently, it follows that the aggregation number effect is more significant than the constraint force effect.
To probe the mechanisms underlying the effect of aggregation number changes on surfactant-small molecule interactions in greater detail, it is useful to look at the radial distribution functions shown in Fig. 4(C), for the systems constructed using NOL and two surfactants, Span 20 (which does not show enhanced interactions), and P85 (which does). Though MARTINI is not optimized for the quantitative determination of aggregate size and radial distribution functions, qualitative matches with real trends are expected, and the data can be quantitatively accurate for specific systems.17 These radial distribution functions only account for the hydrophobic beads in Span 20 (alkyl groups) and P85 (PPO groups), to separate the core from the corona. The origin on the x-axis of these plots therefore corresponds to a point in the interior of the micelles, close to their centers. B25 is a strongly amphiphilic molecule which forms micelles with strongly hydrophobic cores. On the other hand, P85 is a weak amphiphile and forms micelles with weakly hydrophobic cores. The difference in the breadth and height of peaks seen in Fig. 4(C)(a) and (d) indicate the formation of larger and denser micelles by the strong amphiphile Span 20, compared to the weak amphiphile P85. At the same time, Fig. 4(C)(c and f) indicate the significantly greater presence of water in the cores of the P85 micelles. To further confirm the lower hydrophobicity of the P85 micelle cores relative to Span 20, we performed simulations to measure χ parameters between water and the hydrophobic segments of two surfactants, P85 (a weak amphiphile) and Span 20 (a strong amphiphile), which have been used elsewhere in our work as exemplars for these categories. The beads contained in the hydrophobic segments were kept attached to each other (since bond structure affects overall hydrophobicity), and the hydrophilic portion of the surfactant molecules were removed for the purpose of these simulations. We observed that χP85(hydrophobic)−W = 0.5019 < χSpan20(hydrophobic)−W = 2.1103, demonstrating the difference in surfactant properties.
The small hydrophobic molecules that we examine, and which are representative of those small molecules used in drug delivery, water treatment, and related applications, contain hydrophilic groups even though they are overall hydrophobic. Such molecules are likely to adsorb at the core–corona interface in the Span 20 system, allowing their bulk to reside in the hydrophobic core even as specific hydrophilic groups point out into the corona. In the P85 system, they are much likelier to adsorb into the core of the micelle, which contains hydrophilic oxygen atoms and is still sufficiently hydrophilic to admit several water molecules. A core-site adsorption in Pluronics is supported by prior experimental work.41 This finding is also supported by the radial distribution functions in Fig. 4(C), which show narrower peaks (closer to the core) for P85-NOL (Fig. 4(C)(e)) compared to the longer tails seen for Span 20–NOL (Fig. 4(C)(b)) (also see simulation snapshots in ESI†).
When free micelles are allowed to agglomerate in the presence of hydrophobic small molecules, two different phenomena occur based on the sites into which the small molecules partition. In the Span 20 system, small molecules partition into the core–corona interface, and agglomeration involves the micelle cores becoming denser while driving out water molecules, as seen in Fig. 4(C). The number of interfacial sites does not reduce significantly enough to limit small molecule partitioning at low small molecule concentrations (the Span 20–NOL radial distribution function is unchanged), and micelle–small molecule interactions are not significantly affected by agglomeration. In a system like this, when the surfactants are strongly amphiphilic, micelle immobilization does not affect the micelle–small molecule interactions.
In the P85 system, small molecules prefer to partition into the micelle cores, which are sparse because the micelles are themselves small. The partitioning of small molecules is accompanied by a change in conformation of the micelle cores to admit these molecules, since micelle cores are denser than the corona. The thermodynamic effect of these conformation changes is to improve micelle stability by strengthening surfactant–surfactant interactions, because surfactants are the dominant species in the micelles. This strengthening of surfactant–surfactant interactions is at the expense of surfactant-hydrophobic molecule interactions, and the small hydrophobic molecules are driven out of the micelle cores (Fig. 4(C)(e)). Adding constraints limits these conformation changes upon the addition of small molecules and allows the surfactant–small molecule interactions to remain strong. Therefore, in systems in which the surfactants are weakly amphiphilic, micelle immobilization strengthens the micelle–small molecule interactions compared to systems in which the micelles are not immobilized.
This mechanism is further supported by the variation seen across small molecules in Fig. 4(B). PFOA, the smallest and most amphiphilic molecule which occupies the least volume and always occupies interfaces is least affected by immobilization, while EDOL, the largest and least amphiphilic molecule which strongly prefers core sites that are most sensitive to micelle conformation, is most affected.
To strengthen these conclusions using thermodynamic data, we performed simulations to measure χ parameters for the interactions between two hydrophobic molecules, PFOA (perfluorooctanoic acid, of interest in water treatment) and EDOL (ethinyl estradiol, a drug), and the hydrophobic and hydrophilic segments of P85 and Span 20, taken separately. It must be noted that MARTINI is optimized for the quantitative determination of thermodynamic quantities, and these data can be expected to match experimental values. χ values were determined through free energy simulations using methods similar to those in prior work,42 and are shown in Table 2. The parameter of interest here is the ratio of χ parameters (χhydrophilic/χhydrophobic), which is an indicator of the optimal positioning of the small molecule with respect to the core–corona interface in a hypothetical two-component micelle–small molecule system. Larger values of this ratio indicate that the small molecules would prefer to be closer to the hydrophilic corona, and smaller values indicate that the small molecules would prefer to be closer to the hydrophobic core. From the values in Table 2, we draw two conclusions – (1) the small molecules prefer to be deeper inside the core of weak surfactant micelles like P85 that have less hydrophobic cores, and (2) small molecule chemistry affects the relative position, with EDOL positioned deeper in the core than PFOA in such micelles. Moreover, the location of EDOL deeper inside the core compared to PFOA corresponds well with our prior observation that immobilization strengthens EDOL–P85 interactions more significantly than PFOA–P85 interactions, and that such strengthening is related to core conformations. Of course, it must be noted that the systems studied in our manuscript are three component systems, and also contain water. The effect of water is to push all hydrophobic small molecules deeper into the micelles, and so even when the χ-ratio is greater than one, small molecules still partition into the micelles.
|
χ
P85hydrophobic
|
χ
P85hydrophilic
|
|
χ
Span20hydrophobic
|
χ
Span20hydrophilic
|
|
|
|---|---|---|---|---|---|---|
| PFOA | 0.4593 | 0.5165 | 0.8892 | 0.8950 | 0.6691 | 1.3376 |
| EDOL | 0.1724 | 0.2205 | 0.7818 | 0.4967 | 0.3512 | 1.4144 |


3.3 Effect of ethanol
The study of systems containing immobilized micelles prepared in water–ethanol mixtures is of practical interest in various applications including drug delivery. The addition of ethanol to the synthesis solvent increases surfactant solubility, allowing for higher loading of small molecules.13,20 In the context of examining the effect of aggregation number on micelle–small molecule interactions, preparing micelles in ethanol–water mixtures is a simple way of changing the micelle aggregation number and further investigating the interaction mechanism introduced previously. As previously described in the methodology, we prepared micelles in water–ethanol mixtures, then applied constraints to mimic immobilization inside a polymer, then changed the solvent to pure water and added hydrophobic molecules, and performed uptake simulations.Fig. 5(A) shows the kinetics of micelle formation in water-ethanol mixtures starting with a hundred molecules of Span 20 in the absence of small hydrophobic molecules, for increasing ethanol fractions. Hydrophobic segments in the surfactant molecules interact with ethanol more favorably than with water, and the presence of ethanol in the solvent increases the relative stability of the free surfactant phase (surfactants not incorporated into micelles). At the same time, ethanol–water mixtures have a lower polarity than pure water, and can penetrate deeper into micelle cores, weakening hydrophobic interactions and lowering the stability of the micelle phase. The increased stability of free surfactants and decreases stability of micelles lowers the force driving micellization compared to systems in which the solvent is pure water. This can be seen to slow the formation of micelles significantly at low ethanol fractions. Increasing ethanol fractions continue to marginally lower the micelle formation rate. It is important to note that the MARTINI FF is known to spuriously speed self-assembly, but the relative rates are preserved since the systems being compared contain extremely similar structures.37 The data here are therefore meaningful, though time points are in qualitative time units.
Ethanol also simultaneously reduces the aggregation number, resulting in the production of smaller micelles as seen in Fig. 5(B) for two different surfactants. Note that the aggregation number effect, and consequent effects on micelle properties are solely dependent on the destabilization of micelles due to the reduced solvent polarity, and are independent of the stabilization of free surfactants, as long as surfactant concentrations exceed the CMC. The lowering of aggregation numbers has been well-attested in a large body of previous experimental work,43,44 and the extent of size reduction depends on the surfactant. Strongly amphiphilic surfactants like Span 20 form significantly smaller micelles because the destabilization of the micelle phase due to the lowered polarity of water–ethanol mixtures is large. On the other hand, weakly amphiphilic surfactants like P85 are not significantly affected because the low hydrophobicity of their micelle cores admits polar solvents like pure water, and lowering solvent polarity by adding ethanol does not significantly further destabilize the already less-stable micelle phase. Similarly, we note that the reduction in aggregation number for the more amphiphilic Span 20 is reduced as the ethanol fraction increases beyond 10%. This reduced decrease is a result of the lack of further destabilization of the already destabilized micelle phase.
 | ||
| Fig. 5 Effect of ethanol on micelle formation, structure, and properties. (A) Kinetics of Span 20 micelle formation as ethanol fraction increases (darker to lighter). (B) Aggregation numbers of Span 20 and P85 micelles in the absence of small molecules with increasing ethanol solvent fraction. (C) Potential energies of interaction between constrained Span 20 micelles, produced in varying ethanol fractions, separately for 5 mM of three different hydrophobic molecules. (D) The same interaction energies for P85 systems. | ||
Finally, although micelle aggregation numbers are reduced, the energy of interaction between the surfactants constrained in water and small hydrophobic molecules is not significantly affected as seen in Fig. 5(C and D). This finding is also supported by recent experimental results,13 and again is a result of separate phenomena. Micelle–hydrophobic molecule interactions in systems with strongly amphiphilic surfactants are insensitive to changes in micelle size, as seen previously in Fig. 4(B). For less amphiphilic surfactants, the changes in micelle size with the introduction of ethanol are small as seen in Fig. 5(B), and micelle–small molecule interactions remain unaffected.
4 Conclusions
We have presented a framework that can be used for the scalable and rapid evaluation of immobilized micelle–small molecule systems. The current framework neglects the polymer backbone in which the micelles are immobilized for computational ease while capturing the relevant phenomena. We expect the current framework to be accurate whenever micelles are immobilized using chemical or strong physical bonds, and the formation of immobilizing bonds is on time scales much slower than the time it takes for surfactants to re-equilibrate after perturbation by the formation of these immobilizing bonds, which is true in several experimental systems in literature.5,13 The current framework also predicts relative surfactant performance in experimental studies.13 For a given surfactant class, we showed that micelle–hydrophobic molecule interactions are a stronger function of the chemical nature of the moieties present in each group, and a weaker function of the proportion in which those moieties occur. We have also shown that micelle immobilization can significantly affect interactions between surfactants in micelles and small hydrophobic molecules. Immobilized micelles are constrained to be smaller agglomerates far from equilibrium, and these reduced aggregation numbers are the primary drivers of strengthened surfactant–small molecule interactions. In general, micelle–hydrophobic molecule interactions in systems with highly amphiphilic surfactants are insensitive to changes in aggregation number, but are strengthened in systems with less amphiphilic surfactants as aggregation number is reduced.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the MIT Abdul Latif Jameel Water and Food Systems Lab (J-WAFS).References
- D. W. Armstrong, Sep. Purif. Rev., 1985, 14(2), 213–304 CrossRef CAS.
- W. L. Hinze and E. Pramauro, Crit. Rev. Anal. Chem., 1993, 24(2), 133–177 CrossRef CAS.
- C.-L. Liu, Y. J. Nikas and D. Blankschtein, Bioprocess Technol., 1996, 52(2), 185–192 CAS.
- C.-L. Liu, D. T. Kamei, J. A. King, D. I. C. Wang and D. Blankschtein, J. Chromatogr. B: Biomed. Sci. Appl., 1998, 711(1–2), 127–138 CrossRef CAS.
- P. D. Godfrin, H. Lee, J. H. Lee and P. S. Doyle, Small, 2019, 15, 1803372 CrossRef.
- L.-H. Chen and P. S. Doyle, Adv. Mater., 2021, 2008618 CrossRef CAS PubMed.
- S. Thomas and S. M. F. Ali, J. Can. Pet. Technol., 2001, 40(2), PETSOC-01-02-04 Search PubMed.
- L. Yurlova, A. Kryvoruchko and B. Kornilovich, Desalination, 2002, 144(1–3), 255–260 CrossRef CAS.
- M. Schwarze, Environ. Sci.: Water Res. Technol., 2017, 3, 598–624 RSC.
- T. Polubesova, S. Nir, D. Zadaka, O. Rabinovitz, C. Serban, L. Groisman and B. Rubin, Environ. Sci. Technol., 2005, 39(7), 2343–2348 CrossRef CAS PubMed.
- T. Polubesova, D. Zadaka, L. Groisman and S. Nir, Water Res., 2006, 40(12), 2369–2374 CrossRef CAS PubMed.
- H. Wang, A. A. Keller and K. K. Clark, J. Hazard Mater., 2011, 194, 156–161 CrossRef CAS PubMed.
- D. Gokhale, I. Chen and P. S. Doyle, ACS Appl. Polym. Mater., 2022, 4(1), 746–754 CrossRef CAS.
- N. Thota and J. Jiang, Front. Mater., 2015, 2, 64 Search PubMed.
- Y. Lu, E. Zhang, J. Yang and Z. Cao, Nano Res., 2018, 11(10), 4985–4998 CrossRef.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111(27), 7812–7824 CrossRef CAS PubMed.
- S. A. Sanders and A. Z. Panagiotopoulos, J. Chem. Phys., 2010, 132, 114902 CrossRef.
- E. A. Crespo, L. F. Vega, G. Perez-Sanchez and J. A. P. Coutinho, Soft Matter, 2021, 17, 5183 RSC.
- United States Environmental Protection Agency, Drinking Water Contaminant Candidate List (CCL) and Regulatory Determination, https://www.epa.gov/ccl, Accessed: 2020-09-21.
- C. Chaibundit, N. M. P. S. Ricardo, N. M. P. S. Ricardo, C. A. Muryn, M. Madec, S. G. Yeates and C. Booth, J. Colloid Interface Sci., 2010, 351(1), 190–196 CrossRef CAS PubMed.
- J. J. Harris, G. A. Pantelopulos and J. E. Straub, J. Phys. Chem. B, 2021, 125, 5068–5077 CrossRef CAS.
- F. Grunewald, G. Rossi, A. H. de Vries, S. J. Marrink and L. Monticelli, J. Phys. Chem. B, 2018, 122, 7436–7449 CrossRef CAS PubMed.
- S. Nawaz and P. Carbone, J. Phys. Chem. B, 2014, 118(6), 1648–1659 CrossRef CAS PubMed.
- A. Amani, P. York, H. de Waard and J. Anwar, Soft Matter, 2011, 7, 2900–2908 RSC.
- K. J. Huston and R. G. Larson, Langmuir, 2015, 31, 7503–7511 CrossRef CAS.
- R. Alessandri, J. J. Uusitalo, A. H. de Vries, R. W. A. Havenith and S. J. Marrink, J. Am. Chem. Soc., 2017, 139(10), 3697–3705 CrossRef CAS PubMed.
- S. J. Marrink, A. H. de Vries and A. E. Mark, J. Phys. Chem. B, 2004, 108, 750–760 CrossRef CAS.
- S. J. Marrink, A. H. de Vries, T. A. Harroun, J. Katsaras and S. L. Wassall, J. Am. Chem. Soc., 2008, 130, 10–11 CrossRef CAS.
- M. N. Melo, H. I. Ingolfsson and S. J. Marrink, J. Chem. Phys., 2015, 143, 243152 CrossRef CAS PubMed.
- D. H. de Jong, G. Singh, W. F. D. Bennett, C. Arnarez, T. A. Wassenaar, L. V. Schafer, X. Periole, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2013, 9(1), 687–697 CrossRef CAS.
- S. I. Tuset, D. Malaspina and J. Faraudo, Phys. Chem. Chem. Phys., 2018, 20, 26422–26430 RSC.
- C. D. Christ, A. E. Mark and W. F. van Gunsteren, J. Comput. Chem., 2010, 31, 1569–1582 CAS.
- A. Pohorille, C. Jarzynski and C. Chipot, J. Phys. Chem. B, 2010, 114, 10235–10253 CrossRef CAS PubMed.
- A. Villa and A. E. Mark, J. Comput. Chem., 2002, 23, 548–553 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91(1–3), 43–56 CrossRef CAS.
- H. Chan, M. Cherukara, T. D. Loeffler, B. Narayanan and S. K. R. S. Sankaranarayanan, npj Comput. Mater., 2020, 6, 1 CrossRef.
- S. J. Marrink and D. P. Tieleman, Chem. Soc. Rev., 2013, 42, 6801–6822 RSC.
- V. Bhardwaj, P. Sharma, M. S. Chauhan and S. Chauhan, J. Saudi Chem. Soc., 2016, 20(1), S109–S114 CrossRef CAS.
- M. A. Hoque, M. M. Rahman, S. Mahbub, M. Hossain, M. A. Khan, M. R. Amin, A. S. Alqahtani, M. Z. Ahmed, M. S. Alqahtani and O. M. Almarfadi, Korean J. Chem. Eng., 2021, 38(7), 1487–1498 CrossRef CAS.
- M. Enache, A. M. Toader and M. I. Enache, Molecules, 2016, 21, 1356 CrossRef PubMed.
- H. Luo, K. Jiang, X. Liang, H. Liu and Y. Li, Soft Matter, 2020, 16, 142 RSC.
- A. Chremos, A. Nikoubashman and A. Z. Panagiotopoulos, J. Chem. Phys., 2014, 140, 054909 CrossRef PubMed.
- A. F. H. Ward, Proc. R. Soc. London, Ser. A, 1940, 176, 412–427 CAS.
- B. D. Flockhart, J. Colloid Sci., 1957, 12(6), 557–565 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm00280a |
| This journal is © The Royal Society of Chemistry 2022 |