
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect and indirect effects on molecular mobility in renewable polylactide–poly(propylene adipate) block copolymers as studied via dielectric spectroscopy and calorimetry†
Panagiotis A.
Klonos
 *ab,
Zoi
Terzopoulou
a,
Alexandra
Zamboulis
a,
Miguel Ángel
Valera
c,
Ana
Mangas
c,
Apostolos
Kyritsis
b,
Polycarpos
Pissis
b and
Dimitrios N.
Bikiaris
*a
*ab,
Zoi
Terzopoulou
a,
Alexandra
Zamboulis
a,
Miguel Ángel
Valera
c,
Ana
Mangas
c,
Apostolos
Kyritsis
b,
Polycarpos
Pissis
b and
Dimitrios N.
Bikiaris
*a
aDepartment of Chemistry, Laboratory of Polymer Chemistry and Technology, Aristotle University of Thessaloniki, GR-541 24, Thessaloniki, Greece. E-mail: pklonos@central.ntua.gr; dbic@chem.auth.gr
bDepartment of Physics, National Technical University of Athens (NTUA), Zografou Campus, 15780, Athens, Greece
cAIMPLAS, Asociación de Investigación de Materiales Plásticos Y Conexas, Carrer de Gustave Eiffel, 4, 46980 Valencia, Spain
First published on 14th April 2022
Abstract
In this work, we study a series of sustainable block copolymers based on polylactide, PLA, and poly(propylene adipate), PPAd, both polymers being prepared from renewable resources. Envisaging a wide range of future applications in the frame of a green and circular economy, e.g., packaging materials replacing conventional petrochemicals, the employment of PPAd aims at lowering the glass transition and melting temperatures of PLA and, finally, facilitation of the enzymatic degradation and compostability. The copolymers have been synthesized via ring opening polymerization of lactides in the presence of propylene adipate oligomers (5, 15 and 25%). The direct effects on the molecular mobility by the structure/composition are assessed in the amorphous state employing broadband dielectric spectroscopy (BDS) and calorimetry. BDS allowed the recording of local PLA and PPAd dynamics in all cases. The effects on local relaxations suggest favoring of interchain interactions, both PLA–PPAd and PPAd–PPAd. Regarding the more important segmental dynamics, the presence of PPAd leads to faster polymer chain diffusion, as monitored by the significant lowering of the dielectric and calorimetric glass transition temperature, Tg. This suggests the plasticizing role of PPAd on PLA (majority) in combination with the lowering of the average molar mass, Mn, in the copolymers from ∼75 to ∼30 kg mol−1, which is the actual scope for the synthesis of these materials. Interestingly, a strong suppression in fragility (chain cooperativity) is additionally recorded. In contrast to calorimetry and due to the high resolving power of BDS, for the higher PPAd fraction, the weak segmental relaxation of PPAd was additionally recorded. Overall, the recordings suggest a strong increase in free volume and two individual dynamic states, one for 0 and 5% PPAd and another for 15 and 25% PPAd. Within the latter, we gained indications for partial phase nano-separation of PPAd. Regarding indirect effects, these were followed via crystallization. Independent of the method of crystallization, namely, melt or cold, the presence of PPAd led to the systematic lowering of crystallization and melting temperatures and enthalpies. The effects reflect the decrease of crystalline nuclei, which is confirmed by optical microscopy as in the copolymers fewer although larger crystals are formed.
1. Introduction
Polylactide (PLA) belongs to the class of aliphatic polyesters1–4 prepared mainly by the so-called ring opening polymerization of lactide formed by lactic acid.5 An alternative, although not that efficient, route for the synthesis of PLA is via the condensation of lactic acid. Lactic acid is a product of agricultural compound fermentation,6,7 such as corn starch, potatoes, sugar cane and beet.1,2 This has made PLA a sustainable material as well as a really good candidate for replacing the traditional petrochemicals. PLA has already found use in many different applications, from our everyday life, e.g., 3D-printing8–10 and food packaging,4,11–13 to flame retardancy,14 biotechnology, tissue engineering and drug delivery.12,15–18PLA is a thermoplastic polymer, whereas it can exhibit two characters, i.e., both amorphous, with quite poor mechanical performance,3,19 and semicrystalline.20–22 The presence of crystals induces severe mechanical improvements and thermal stability in PLA. What is actually wanted for processing and modern materials aiming at various applications is the wide range tuning of material performance. These are strongly connected with the polymer chain mobility/rigidity, chain entanglements and interactions as well as semicrystalline morphology.23,24 Within the latter term two main parameters are described: the crystalline fraction, and the size and distribution of crystals throughout the polymer volume. The manipulation of semicrystalline morphology is a valuable tool for tailor-made materials and tuning properties such as the diffusion of small molecules (from oxygen and air to drugs)25,26 or even the heat transfer.27,28 There are many ways to modify crystallization and polymer mobility, for example, by special thermal/processing treatments, by introducing plasticizers29 or inorganic/organic additives (e.g., nanoparticles)3,30,31 or by combining different polymers together, namely, preparing copolymers, mixtures, interpenetrating polymer networks etc.13,32–34 Please note that in our days, there has been an evolving need for material synthesis, application and disposal, respecting the various environmental concerns35–37 and, this way, following the trend for circular and green economic frame.38,39 In connection with the latter comes the poor hydrophilicity of PLA that affects its biodegradation rate, arising from the mechanism of hydrolytic scission of the ester bonds (ref. 40 and references therein). This concerns also many of the known bio-based polyesters.41
A good strategy for the improvement of the properties of polyesters, accounting at the same time for the abovementioned concerns, is proved to be the synthesis of co-polyesters, for example, employing other biobased comonomers, oligomers and polymers.34,42–45 Among the many candidates to prepare PLA-based co-polyesters, poly(n-alkylene adipate)s, PnAAd (with n = 2, 3, 4, 5, 6…) have been recently demonstrated.46 These linear aliphatic polyesters are synthesized from adipic acid, which is listed by the International Energy Agency to be among the most important dicarboxylic acids, whereas it can also be prepared from renewable resources.47 PnAAds are non-toxic and biodegradable as well, while they exhibit a high biodegradation rate and high thermal stability.48 PnAAds are semicrystalline and their crystalline fraction can be easily tuned, by either their structure (molar mass and n-alkylene) or by the targeted thermal treatments.49–53 Compared to PLA, poly(n-alkylene adipate)s demonstrate much easier chain diffusion, equivalently, lower glass transition temperatures (between –63 and –45 °C).34,46,48–51 Thus, PnAAds may act as plasticizers for PLA and, simultaneously, as additional compounds that favor manipulation of crystallization.34,44
A recently synthesized series of block copolymers based on PLA and poly(propylene adipate), PPAd,46 are investigated in the present work. The emphasis is on the direct effects of copolymer composition and chain length on the local and segmental molecular mobility. The said direct effects are assessed mainly in the initially amorphous state of the materials. In the last section of this work, the indirect effects of copolymer composition are studied from the point of view of polymer crystallization. The advanced technique of broadband dielectric spectroscopy (BDS)54 is employed as the main investigation tool for molecular mobility, being a combination of differential scanning calorimetry (DSC) of the conventional as well as the temperature modulation mode (TMDSC) and polarized optical microscopy (POM). The molecular dynamics mapping for these copolymers is presented in this article for the first time, to the best of our knowledge. The results are analyzed and discussed by widely adopted routes and models, whereas we attempt the combination of the overall data and following parameters in order to gain knowledge of the realistic scenarios on the topology of the two polymers and the involvement of interchain associations.
2. Experimental
2.1. Materials
The samples investigated here are block copolymers of PLA and PPAd (i.e., PLA_b_PPAd) with PLA/PPAd % ratios of 95/5, 85/15 and 75/25 (Fig. 1). For comparison we investigate the initial PLA. The synthesis and initial structure characterization of these systems are described in the recent work by Terzopoulou et al.46 Briefly, PPAd oligomers were synthesized by the addition of pre-polymerized adipic acid via two stage polycondensation. The molar mass of PPAd was low and estimated as Mn ∼ 6 kg mol−1. PLA was synthesized via ring opening polymerization, ROP, of lactide at 180 °C. The copolymers were prepared by polymerizing L-lactide in the presence of PPAd oligomers (this diol actually being the initiator for ROP) via a reactive extrusion method in a torque rheometer at 180 °C. This method actually produced the development of PLA blocks on the pre-synthesized PPAd.46 The studied samples are listed Table 1 along with values on the estimated average molar masses (Mn, Mw). | ||
| Fig. 1 Chemical structure of the copolymers under investigation, i.e., consisting of PLA and PPAd blocks. | ||
| Sample | Code name | M n (g mol−1) | M w (g mol−1) |
|---|---|---|---|
| PLA | PLA | 76 k | 120 k |
| PLA(95%)_b_PPAd (5%) | 95/05 | 63 k | 94 k |
| PLA(85%)_b_PPAd (15%) | 85/15 | 41 k | 55 k |
| PLA(75%)_b_PPAd (25%) | 75/25 | 29 k | 39 k |
2.2. Experimental methods
Temperature-modulation DSC measurements were also performed on all samples of about 14 mg in mass, upon melting at 190 °C, fast cooling down to –110 °C and equilibration thereafter for about 20 min. The heat capacity, cp, was recorded upon heating at a heating rate of 2 K min−1 with a modulation period of 60 s and temperature amplitude of 1 K (Fig. S1b in the ESI†), in the temperature range from –110 to 180 °C.
The characteristic temperature of the glass transition step, Tg, was estimated from the heating curve as the point of half cp elevation (TMDSC). From conventional DSC, where heat is measured directly, the half cp (J g K−1) change was nominally estimated upon normalizing the heat flow (mW) to the sample mass (mg) and heating rate (0.167 K s−1). Crystallization and melting events were evaluated in terms of peak temperature maxima and enthalpy changes (ΔH in J g−1).
 | ||
| Fig. 2 Representative BDS sample in the form of electric capacitor (sandwich-like), (left) positioned in the Novocontrol BDS cell. (Right) Dimensionality of the measured sample. | ||
3. Results and discussion
3.1. Direct composition effects on molecular mobility (amorphous state)
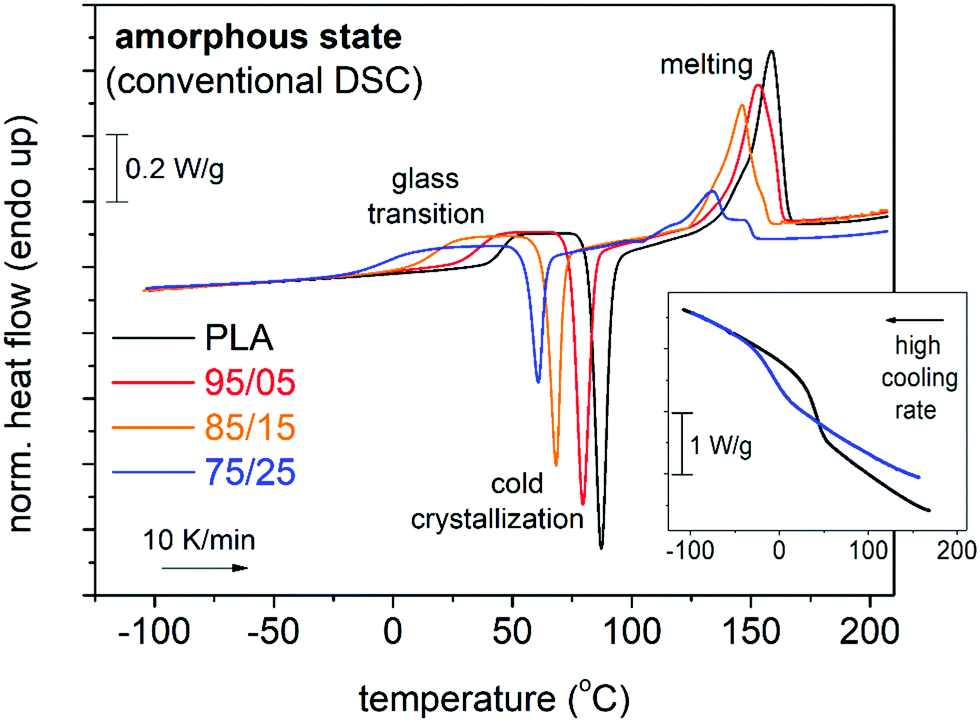 | ||
| Fig. 3 Comparative DSC traces during heating at 10 K min−1 for all initially amorphous samples. The main thermal events are indicated on the plots. The recorded heat flow (in mW) is shown here upon normalization to each sample mass (W g−1). The inset shows examples of prior cooling at a fast rate that led to eliminated crystallization of PLA and 75/25. | ||
In Fig. 3, we present the subsequent DSC heating of the initially amorphous samples at 10 K min−1. Therein, single glass transition steps are recorded in all cases in the range from –25 to 70 °C. For higher temperatures, cold crystallization exothermal peaks are recorded (between 50 and 100 °C), as a result of the vanished melt-crystallization during the prior ‘fast’ cooling. Finally, all samples exhibit melting of crystals via the recorded endothermal peaks in the range from 100 to 170 °C.
It is obvious, already from a glance on the raw data, that there are systematic changes on all thermal transitions. The single glass transition is a first strong indication for the homogeneity (miscibility)13 of the copolymers, namely, no separation of the two polymer phases that individually demonstrate severely different glass transition temperatures. Please note that this result refers to the amorphous state (upon ‘melt-quenching’). In this frame, we may report that Genovese et al.13 studied polylactide-based triblock copolymers and revealed a peculiar mechanism of segregation that depends on the thermal treatment, for example, not detected in the simply quenched samples.13
To further study the glass transition and more accurately evaluate the corresponding heat capacity change, Δcp, TMDSC was employed for initially amorphous samples (melted and fast cooled). The results are shown in Fig. S2 in the ESI.† The reversing part of heat capacity (cp,rev), or else the real cp, is actually exploited for the said evaluation and is shown in Fig. S2b (ESI†), comparatively for all samples.
The characteristic temperatures, Tg (conventional DSC) and Tg,rev (TMDSC), were estimated employing the half Δcp and Δcp,rev changes, respectively. The Tg values have been plotted in Fig. 4a for the four samples against their Mn. Please note that the increase of PPAd content (inset to Fig. 4a) results in a significant decrease of Mn (Table 1). In agreement between the DSC methods, Tg increases monotonically with Mn (and composition) from about –3 °C (75/25) to 46 °C (PLA). The trends suggest much faster chain mobility in the presence of PPAd. This could also suggest easier polymer chain diffusion in the sense of increased free volume and suppression of the chain-chain cooperativity, as expected, when significantly lowering the polymer chain length. However, the origins are not directly clear only by the DSC findings, namely, as to whether the easier chains diffusion arises from the shortening of the chains or the increase in free volume for other reasons. Please note that the two polymer compounds carry significant fraction of active molecular groups, namely, back bone carbonyls (both polymers), side hydroxyls (PLA) and side carboxyls (PPAd)46 that can favor the formation of interchain interactions. These interactions are expected to hinder the chain mobility (diffusion and cooperativity) and be pronounced for shorter chains.55–57 Coming to the strength of glass transition, the Mn dependence of Δcp is shown in Fig. 4b. The trend of Δcp is to increase with Mn, which is in general expected for these relatively low Mns. The trend is, however, not expected when considering the increasing amount of the more mobile PPAd. To more clearly discuss the changes in Δcp it is essential to perform normalization to each polymer phase weight (wt.) content. This is not straightforward for these systems, as both the wt. fractions and the chain lengths change, while at the same time there are no available data for Δcp of PLA and PPAd homopolymers with various low Mns. Therefore, we will attempt to shed more light on molecular mobility, in terms of time scale, cooperativity, and strength of mobilization, by BDS.
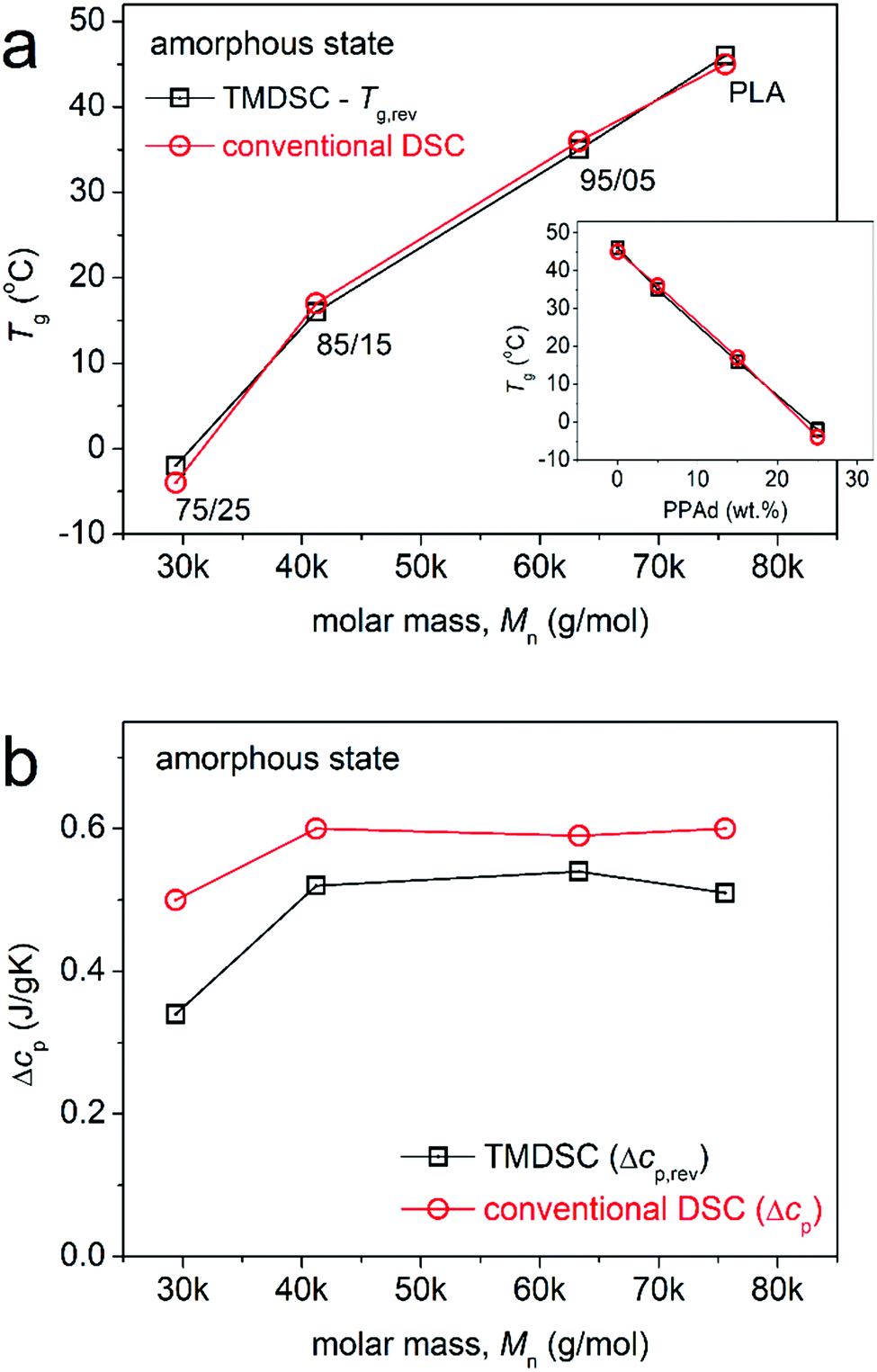 | ||
| Fig. 4 The Mn (sample) dependence of (a) glass transition temperature and (b) heat capacity change during glass transition for the initially amorphous samples. In (a), the sample code names are given along the experimental point, for the sake of clarity, whereas the inset shows the PPAd wt. fraction dependence of Tg. | ||
The effects on cold crystallization and melting, will be briefly discussed in the last section of this article, along with more results by DSC and POM.
 | ||
| Fig. 5 Representative BDS results in terms of the comparative isochronal curves of ε′′ against temperature for all samples. The results are shown for two selected frequencies (a) 3 kHz and (b) 121 Hz. Indicated are the main recorded relaxation processes (peaks of ε′′). | ||
Seeking for more details and better comparisons between the different samples, we present in Fig. S3 (ESI†) the raw BDS isothermal recordings for neat PLA (Fig. S3a, ESI†) and 75/25 (Fig. S3b, ESI†). Therein, the temperature evolutions of the recorded relaxations (peaks) are marked by added arrows. In Fig. S4, (ESI†) we show comparative isothermal ε′′(f) curves for all samples at fixed temperatures, in order to distinguish the direct effects of composition on each type of relaxation.
The complex dielectric data can be summarized and presented in a more efficient manner, namely, in terms of time-scale and dielectric strength in a common dielectric map. To do that, and by adopting widely used methodologies, we analyzed the results of Fig. S3 and S4, by using known mathematical model functions.58,60 Herein, the so called Havriliak–Negami, HN, function (eqn (1)) was fitted to each peak of ε′′(f).61
 | (1) |
The parameters involved within eqn (1) are as follows. ε∝ describes the value of the real part of dielectric permittivity, ε′, for f ≫ f0, Δε is the dielectric strength, f0 is a characteristic frequency related to the frequency of maximum dielectric loss, whereas αHN and βHN are the shape parameters of the relaxation (width and symmetry, respectively).
For the sake of brevity, we do not show examples of this fitting process and proceed directly to the demonstration of the dielectric map of Fig. 6. This map shows for all samples the reciprocal temperature dependences of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) fmax (Fig. 6a, time scale, else called Arrhenius plots) and that of Δε (Fig. 6b). The left part of the figure presents the data at temperatures T ≥ Tg, thus, dealing with segmental dynamics, whereas the right part includes data for T ≤ Tg, focusing on the secondary (local) dynamics. In Fig. 6a, we have added the corresponding points for Tg and Tg,rev at the appropriate equivalent frequencies, logf, –2.8 and –1.8 (logHz), for standard DSC and TMDSC, respectively. We recall that these equivalent frequencies correspond to the slow relaxation time of τ ∼ 100 s at Tg (assumed constant) in the conventional DSC and the 60 s period of temperature modulation in TMDSC. Also, we have marked the range of the expected Tg for various poly(n-alkylene adipate)s according to the literature34,46,48–51,62 and added points on the local and segmental processes of PBAd from ref. 34.
fmax (Fig. 6a, time scale, else called Arrhenius plots) and that of Δε (Fig. 6b). The left part of the figure presents the data at temperatures T ≥ Tg, thus, dealing with segmental dynamics, whereas the right part includes data for T ≤ Tg, focusing on the secondary (local) dynamics. In Fig. 6a, we have added the corresponding points for Tg and Tg,rev at the appropriate equivalent frequencies, logf, –2.8 and –1.8 (logHz), for standard DSC and TMDSC, respectively. We recall that these equivalent frequencies correspond to the slow relaxation time of τ ∼ 100 s at Tg (assumed constant) in the conventional DSC and the 60 s period of temperature modulation in TMDSC. Also, we have marked the range of the expected Tg for various poly(n-alkylene adipate)s according to the literature34,46,48–51,62 and added points on the local and segmental processes of PBAd from ref. 34.
 | ||
Fig. 6 Molecular mobility dielectric/calorimetric map in terms of the reciprocal dependences of (a) the logfmax and (b) dielectric strength, Δε, showing the (left) segmental and (right) local dynamics, for all samples. The symbols for the different samples and relaxations are explained in (a) and (b), respectively. The added red arrows mark the effects imposed by the increasing of PPAd fraction. In (a), the added straight and curved lines connecting the experimental data points are fittings of the Arrhenius and Vogel–Fulcher–Tammann–Hesse equations, respectively, whereas, the ‘star’-points correspond to BDS recordings on poly(butylene adipate), PBAd, from a previous work,34 the calorimetric Tg points have been placed at the corresponding equivalent frequencies and, finally, the added horizontal scale bar addressed as ‘PnAAd’ corresponds to the Tg range of various poly(n-alkylene adipates). In the inset to (right - b), are infra-red spectroscopy data on the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching for all samples (including neat PPAd). The inset of (b) has been adopted from ref. 46, with permission from MPDI, in particular form ESI† Fig S2a, employing different symbols for the different samples and slightly altered scaling. O stretching for all samples (including neat PPAd). The inset of (b) has been adopted from ref. 46, with permission from MPDI, in particular form ESI† Fig S2a, employing different symbols for the different samples and slightly altered scaling. | ||
The local γPPAd seems to screen the most localized motions of PPAd. Obviously, the process is recorded only in the PLA/PPAd copolymers and its strength increases with the fraction of PPAd (Fig. 6b). γPPAd was fitted with a symmetric HN term (i.e., βHN = 1), relatively narrow in 75/25 (αHN ∼ 0.4–0.5) and wider for 85/15 and 95/05 (αHN ∼ 0.2–0.4), suggesting more homogenous relaxation times for increasing the PPAd amount. In general, local relaxations follow the Arrhenius law (eqn (2)).58
 | (2) |
In an Arrhenius behaviour of the relaxation, the activation energy, Eact, is constant and, thus, exhibits a linear time scale, as in our case here in Fig. 6a. Eact is estimated here about 35 kJ mol−1 and barely changes with composition. Interestingly, when increasing the amount of PPAd, γPPAd decelerates, as shown in Fig. 6a by the horizontal migration toward higher temperatures/lower frequencies. Since the literature on molecular mobility for the adipate-based polymers is still limited, we cannot definitely predict the molecular origins of γPPAd. Nevertheless, we are allowed to compare with polymers exhibiting local relaxations of a similar time scale and at the same time carrying similar or same molecular groups. In this context, we have found some resemblances between our γPPAd with a local process of poly(ethylene glycol) (γPEG)63 as well other structurally similar polymers, such as oligo-ethylene glycol methacrylates (OEGMA) and comb-like polymers based on OEGMA64 (and references therein). The said γPEG relaxation is considered to arise from crankshaft motions of methylene sequences at the backbone of the polymer.65,66 Such sequences exist within our PPAd, and therefore, we could propose the same origins for our γPPAd. A similar situation was recently recorded within poly(butylene adipate).34 It is also worth recalling some previous knowledge on the molecular mobility of oligo n-alkylene chains when the latter exist as ‘side chains’, for example of acrylates.67 When the number of carbons is smaller than n ∼ 10–12 their local poly-ethylene-like relaxation is fast, whereas for and n ≥ 12 the PE-like relaxation decelerates due to promoted chain–chain associations for the longer side chains. Coming back to the present findings, the overall effects on homogeneity and timescale provide indications for more associations between methylene sequences of PPAd, for example with neighboring adipate chains, when PPAd increases in the copolymers.
At higher temperatures, namely in the intermediate 1000/T range in Fig. 6, the local βPLA relaxations are recorded. The Eact of βPLA equals ∼50 kJ mol−1. βPLA relaxation originates from fluctuations or local twisting motions of the –CO group at the backbone of PLA.68–70 Schematically, this can be seen in the inset of Fig. 6a. The process is recorded in both neat PLA and all copolymers. Although we have not investigated neat PPAd, a local relaxation of the same molecular origins (–CO) is expected in the copolymers. Still the literature is quite poor on these relatively new polymers. However, our hypothesis should be true, recalling data on PBAd.34 The corresponding points on βPPAd have been included in Fig. 6a and reveal a quite similar timescale to that of βPLA. Regarding its shape characteristics, βPLA was sufficiently fitted with a symmetric HN term of αHN = 0.3 for PLA, 95/05 and 98/15 and slightly narrower for 75/25 (αHN = 0.4). The dielectric strength of the process drops with the addition of PPAd in Fig. 6b. The drop is not monotonic with the PPAd amount. We believe that the strength suppression is due to the involvement of –CO within interchain interactions. The main responsible for that are the terminal hydroxyls (–OH) of both polymer blocks (inset of Fig. 6a). The degree of this interaction (–CO–HO–) precludes the twisting of some ester groups, which become dielectrically less active, reducing Δε. The interaction is favored upon increase of PPAd fraction, as, simultaneously, the average chain length of copolymers drops and, expectedly, the fraction of free chain ends (–OH) increases. More direct evidence on this interaction can be seen from infrared spectroscopy data shown in a recent work.46 These results have been adapted and included in Fig. 6b as an inset. They reveal an increasing contribution of ‘bound’ –CO groups (lower wavenumber band) in the presence of PPAd. Such formalism has been employed within numerous previous cases involving polymer interactions.31,56,57,71,72 In connection to that, Alvarez et al.50 studied poly(butylene isophthalate)/poly(butylene adipate) copolymers by BDS and showed similar changes in the local relaxation of PBAd (βPBAd), and moreover, assigned these changes to the chain–chain associations also related with crystallinity. Despite the said interactions, it is expected that there are still many free end-groups (hydroxyls and carboxyls), as in a previous study, the hydrophilicity of the copolymers was found to increase with PPAd (lowering of Mn) and, subsequently, so did the enzymatic hydrolysis.46
The focus in now turned onto segmental dynamics. In neat PLA, αPLA is recorded and fitted well by an asymmetric HN term with αHN ∼ 0.6 and βHN∼ 0.6. As expected for cooperative dynamics, the activation energy is not constant with T, and moreover, decreases for higher T. Thus, as in most trivial cases, the time scale of αPLA is well described by the so-called Vogel–Fulcher–Tammann–Hesse (VFTH) equation73,74 (eqn (3)). Within VFTH, the pre-exponential factor f0,VFTH is a frequency constant varying in the range 1012–1014 Hz, T0 is the so-called Vogel temperature and D is the so-called fragility strength parameter.74
 | (3) |
Upon the fitting of eqn (3) to the experimental data (curved lines in Fig. 6a) corresponding to the amorphous sample and upon fixing f0,VFTH to the phonon value 1013 Hz,58 we were able to evaluate the dielectric glass transition temperature, Tg,diel, (=50 °C) via the extrapolation of the VFTH fitted curve at the equivalent frequency of DSC (10−2.8 Hz/100 s). Then, the fragility index of α relaxation, mα, was estimated (=178) according to eqn (4).
| mα = 16 + 590/D | (4) |
In Fig. 6b, Δε of αPLA exhibits the expected decreasing trend with T,58 whereas at higher temperatures, Δε exhibits a sharp decrease, due to the evolution of cold crystallization. Subsequently, the process becomes symmetric and wider (βHN = 1 and αHN ∼ 0.4–0.5) and the Δε(T) changes to an increasing trend. The latter is also expected for semicrystalline polymers, due to the gradual loosening of the constraints75–77 imposed by the crystals of the mobile amorphous fraction. Finally, we should report that the calorimetric data on Tg come in quite good agreement with the dielectric ones in Fig. 6a.
Regarding the copolymers, in all cases an α relaxation is recorded. The time scale of the latter shows a systematic acceleration with the addition of PPAd in Fig. 6a (down-pointing triangles). At the same time the curvature of the VFTH fitting decreases. These results are evaluated in terms of Tg,diel and mα, and the overall data are shown in Fig. 7, comparatively for all samples. Tg,diel exhibits a systematic decrease with the addition of PPAd and drop of Mn (acceleration of dynamics), while the fragility drops gradually from 178 to 147 to 107 and to 0. The latter effect suggests the drop of cooperativity (95/05 and 85/15) to the extreme case of vanished cooperativity (75/25). In terms of size/scale, this suggests that either the cooperativity length increases dramatically78,79 or, else, the distances between the neighboring (cooperating) polymer chains increases. This change is accompanied by the fact that the α relaxation in the copolymers has changed to ‘symmetric’ (αHN ∼ 0.5–0.6, βHN = 1). In Fig. 6b, the Δε(T) trends are also different. In particular, for PLA and 95/05 Δε(T) is decreasing, whereas for 85/15 and 75/25 Δε(T) is increasing. The first case is indicative of non-constrained dynamics while the second suggests constraints that are gradually erased or loosened with T. Interestingly, this categorization regarding α relaxation coincides with a similar categorization on the suppression of Δε for β relaxation, noted above and is correlated with constraints due to inter-chain interactions.
 | ||
| Fig. 7 The molar mass (sample) dependence of (left axis) of the dielectric and calorimetric Tg against (right axis) the fragility index of αPLA relaxation, mα. The values correspond to the amorphous state, namely, prior to cold crystallization. | ||
Obviously, the segmental dynamics in the copolymers is strongly modified as compared to PLA. The results indicate that the α relaxation in the copolymers does not arise purely from PLA chains gathered in separate nanodomains and this is an indirect proof for a well-formed copolymeric structure. Therefore, we may conclude the existence of a situation that is schematically described in Scheme 1. The copolymers are quite homogeneous systems, coming from successful synthesis. The increase in the PPAd amount, results in increasing in the inter-chain interactions and, in shorter, thus, more mobile chains which exhibit, however, significantly smaller cooperativity.
 | ||
| Scheme 1 Simplified schematics model for the distribution of PLA and PPAd in the copolymers, considering the overall findings by the present work. | ||
In the extreme case of 75/15, BDS was able to distinguish an additional weak process with αHN ∼ 0.4 and βHN ∼ 1. This is shown in Fig. 6via right-point triangles. The time scale of the process follows the VFTH model better, thus, denoting a level of cooperativity. The extrapolation of this coincides better with previously recorded calorimetric Tg points on various PnAAd (horizontal scale bars in Fig. 6a) and dielectric data on the alpha process of PBAd (stars in Fig. 6a).34 Therefore, it is most likely that the process is the segmental relaxation of the PPAd phase (αPPAd). Please note that in DSC no separate glass transition was recorded. The discrepancy is expected as BDS is a technique with a quite higher resolving power following in principle different modes (dipolar) regarding polymer chain mobility. The results suggest that in 75/25 a part of PPAd is partly formed in domains that are at least a few nanometers in size (Scheme 1). These PPAd domains should not be larger, as in no case of thermal treatment in DSC did we record the individual crystallization of PPAd. Support for that partial nano-organization of PPAd in 75/25 was supplied by scanning electron microscopy in a recent work on the same systems.46
Last but not least, at temperatures well above Tg, an intense signal increase is observed. Another strong ε′′ peak could be resolved therein, i.e., process (1). Such processes are generally related to free electrical charge transport (ions and conductivity relaxation) or phenomena related to charge accumulation at interfaces (Maxwell–Wagner–Sillars phenomenon or electrode polarization).58,59,80 The corresponding time scale and Δε data have been included in Fig. 6, while the fitting shape parameters are βHN = 1 and αHN ∼ 0.5–0.6. In Fig. 6a, the changes in the time scale of process (1) seem to follow those of α relaxation, whereas, interestingly, Δε drops in general on the addition of 15 and 25% PPAd. Thus, the process is dependent on the amount and, possibly, the continuity of the PLA phase. Due to the low amount of available data, we could not safely predict the origins of the said process and, therefore, we will not comment further.
3.2. Composition effects on crystallization
We may discuss now the effects of composition on crystallization. Fig. 8 shows the DSC results for conventional cooling at 10 K min−1 (from the melt, Fig. 8a) and the subsequent heating (Fig. 8b). During cooling, all samples exhibit melt-crystallization peaks, mainly single with the exception of 75/25 that exhibits a double-structured peak. Expectedly, during the subsequent heating, no cold crystallization was recorded, as a result of complete crystallization–nucleation during cooling. The corresponding melting peaks are mainly single and narrow, whereas for 85/15 and 75/25 the peak is double structured. | ||
| Fig. 8 (a) Comparative DSC traces during cooling at 10 K min−1, demonstrating the effects on melt-crystallization for all compositions. The inset shows the results on ‘cold’ crystallization from the previous scan on the initially amorphous samples. (b) Comparative DSC traces during subsequent heating at 10 K min−1. The heat flow is shown upon normalization to the mass of the sample. Details of the glass transition during heating are shown in the inset of (b), within which results of the amorphous samples are included for comparison (dashed–dotted lines, vertically translated for the baseline coincidence at low temperatures). | ||
The results of Fig. 8 have been evaluated in terms of crystallization temperatures and enthalpies, Tc and ΔHc, respectively, and melting temperatures and enthalpies, Tm and ΔHm, respectively. These values are presented in Fig. 9, against Mn, also comparatively with Tcc and ΔHcc, of cold crystallization from the first DSC scan.
 | ||
| Fig. 9 (a) The Mn dependence of crystallization temperatures, Tcc and Tc, the corresponding sample code names being included along the experimental points. (b) The Mn dependence of crystallization, ΔHcc and ΔHc, and melting, ΔHm, enthalpies. The inset of (b) shows the corresponding values of the melting temperature(s). | ||
In Fig. 9a, both the crystallization temperatures and enthalpies increase monotonically with the increase in Mn, or else, with the decrease in PPAd. As discussed above, the effects on Tcc reflect the changes in nucleation more directly, whereas those of Tc (higher values) should be more representative of the rate of crystal growth, as melt-crystallization is developed without prior strong supercooling and at relatively higher temperatures (fast chain diffusion) compared to cold crystallization. In Fig. 9b, the addition of PPAd results to lower ΔHc, ΔHm and ΔHcc. At first thought, this should suggest a drop in the degree of crystallinity (crystalline fraction). To more safely conclude that, the enthalpy change values should be suitably normalized21,51 to the corresponding polymer wt. fraction. This cannot be securely done here, due to the copolymeric structuring of our systems, consisting of two polymers that individually crystallize at very different temperatures; moreover, it is yet not clear as to whether part(s) of the copolymer chains or the overall chains participate in the crystallization process. Finely, the suppression of the enthalpy is also accompanied by a suppression in Tm. This indicates the drop in the quality of crystals. The term bad quality usually reflects lower density of lamellae81 or, in particular cases, much smaller crystallites.31,57 Information on the degree of crystallization as well as on the crystal quality can be better evaluated by suitable experiments via X-ray diffraction and analysis.
In conclusion, the DSC results suggest that the addition of PPAd (or the decrease in Mn) lead to less nucleation, thus, fewer crystals of worse density or/and smaller size. To check this scenario, we employed the more direct method of POM. In Fig. 10 we present POM micrographs for different stages of melt crystallization for all samples. The data show a smaller number of crystals formed in the presence of PPAd, confirming the above mentioned suppressed nucleation. On the other hand, as in PLA the polymer volume is ‘filled’ with spherulites also for the copolymers, which demonstrate larger crystals as compared to neat PLA. Taking into account together the lowering of ΔHc/m and Tm with the addition of PPAd and the larger crystals observed by POM, we may conclude ‘indirectly’ the lowering of the crystal density in the copolymers.
 | ||
| Fig. 10 POM micrographs for all samples suffered melt crystallization, the results being shown for initial, intermediate and final states of crystallization. | ||
Finally, it is worth mentioning another exceptional behavior. In the inset to Fig. 8b, we show a focus on the glass transition region for the melt-crystallized samples (solid lines). Along with these data, we added for comparison the glass transition results of the amorphous samples (dashed–dotted lines) from Fig. 3. Strikingly, upon crystallization, Tg decreases for all samples (Fig. 11a). In general, the presence of crystals is expected to hinder the polymer chain diffusion, thus, to elevate Tg. This is mainly a true fact in homopolymers.82,83 In Fig. 11b, the effect on the glass transition strength, i.e., on the Δcp, is the expected suppression upon crystallization, as a significant fraction of the initially amorphous mobile polymer chains (amorphous Δcp) have become rigid within the crystals. Coming again to the Tg, when directly comparing the change in Tg with the change in ΔH (Fig. 9b), we observe the controversial effect. The maximum change in the Tg occurs for the minimum increase in ΔH and the larger amount of PPAd. This suggests that the role of crystals is most possibly indirect, as actually the formation of crystals leads to further plasticization, or else, an increase in the free volume. This is favoured, again, for the lower Mns and fewer crystals. This point is quite interesting, from the basic research point of view, and is worth being followed in a future study. For the time being, only speculations can be made to rationalize this exceptional behaviour, for example via a scenario involving special organization84,85 of the polymer chains upon crystallization, favoured by the presence of PPAd.
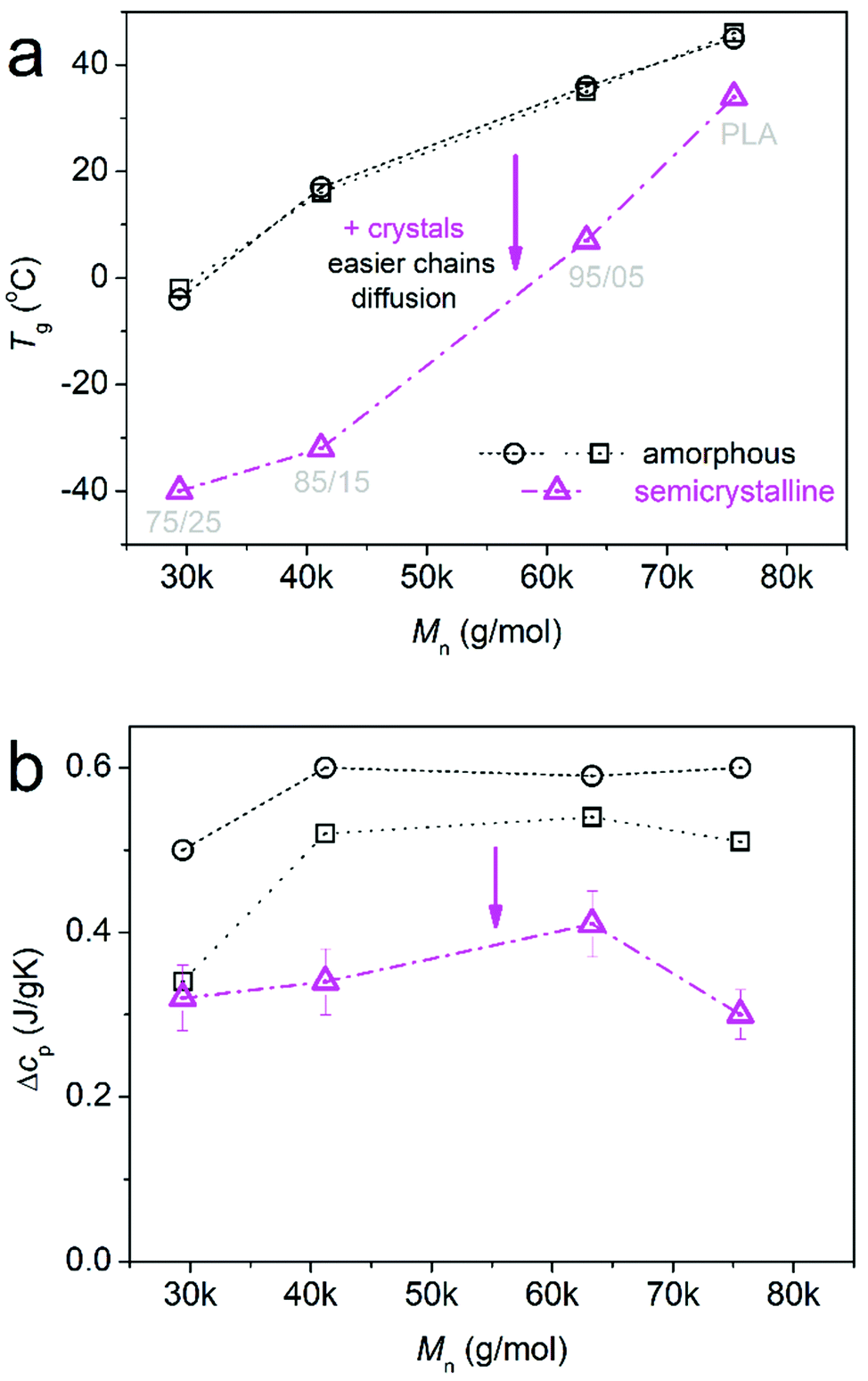 | ||
| Fig. 11 The sample (Mn) dependence of (a) Tg and (b) Δcp, as changed from the amorphous (squares – TMDSC, circles – DSC) to the semicrystalline state (up-triangles – DSC). | ||
Interesting and non-trivial effects were revealed in this work on the molecular mobility of PLA/PPAd copolymers. BDS and the critical analysis employed were proved quite powerful tools on understanding and partly rationalizing the structure–mobility relationship here. Despite these, some questions have opened regarding the structure, namely on partial nano-phase separation, possible self-organization, and, in next level, of the crystal structuring. These points could be further followed in future work, employing, for the amorphous and semicrystalline states small angle X-ray scattering86–88 to illuminate any phase separation, XRD upon targeted thermal treatments to evaluate the degree of crystallinity and investigate alternation on the crystal/lamellar density and to further investigate molecular mobility (by BDS56 and rheology89) on the initially melt- and cold-crystallized systems.
4. Conclusions
In this study we investigated the molecular mobility of new PLA/PPAd block copolymers. Since both compounds are individually semicrystalline, we study the direct composition–structure effects in the amorphous state. Therein, PPAd was found to serve as a good plasticizer for PLA in the copolymers, and also, in connection to its indirect role in the reduction of the copolymer chain length. The local β dynamics arising from the motion of the ester group (CO), a common group in both PLA and PPAd, was found to barely change in terms of time scale. Nevertheless, due to expected interactions between the said group and the terminal polar groups (hydroxyls and carboxyls) the dielectric strength of the local relaxation was significantly reduced, especially for the higher PPAd contents. This is also due to the increase in the fraction of chain ends for the shorter copolymer chains. A faster local mobility was recorded (γ relaxation) and was assigned to the most localized molecular motions of PPAd. The said process was found to accelerate when increasing the PLA content. The most important results refer, as expected, to segmental dynamics. This was followed via the so called α relaxation, the dielectric analogue of glass transition. At the increase of PPAd and lowering of Mn, α relaxation was found to strongly accelerate and exhibiting severely suppressed fragility, denoting increase of cooperativity length or/and dissociation of the relaxing chains. In other words, there seems to be extreme increase in the free volume. The existence of nanodomains of PPAd for 25% PPAd was manifested by the separated weak αPPAd dynamics. Taking together the overall data on dynamics, the temperature of dielectric strength and the shape parameters, we conclude the existence of dynamically distinct behaviors between the copolymers of lower and higher PPAd contents, whereas indirect indications for special morphologies were observed. We also concluded the absence of a continuous phase of PPAd, at any composition, as manifested by the absence of individual PPAd crystallization and ionic conductivity at temperatures below Tg of the copolymers. Coming to the semicrystalline state, nucleation is suppressed in the copolymers, however, larger crystals with presumably lower density are formed. The presence of crystals resulted in the unexpected effect of further lowering of Tg, which is indicative of a non-trivial situation, arising most probably from polymer reorganization upon crystallization. Overall, the results supply evidence for the ability of wide scale manipulation of PLA (semicrystalline morphology, small molecules permeation, mechanical performance etc.), a polymer of high interest regarding applications as well as academia. From the methodological point of view, BDS is once again a quite powerful tool in understanding, among others, the presence of interchain interactions, the degree of chains cooperativity, in addition to indirectly shedding light on the polymers’ topology, which cannot be easily assessed by microscopy.
Author contributions
Panagiotis A. Klonos: conceptualization, methodology, investigation, formal analysis, visualization, writing – original draft; Zoi Terzopoulou: investigation, formal analysis, validation, writing – review & editing; Alexandra Zamboulis: writing – review & editing; Miguel Ángel Valera: resources, validation, writing – review & editing; Ana Mangas: resources, writing – review & editing; Apostolos Kyritsis: resources, validation, writing – review & editing; Polycarpos Pissis: validation, writing – review & editing; Dimitrios Bikiaris: supervision, resources, validation, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Panagiotis Klonos would like to acknowledge Dr Daniel Fragiadakis from the Naval Research Laboratory, Polymer Physics Section, Washington DC, USA, for kindly providing his ‘grafity’ data analysis software (https://grafitylabs.com/).References
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- P. Saini, M. Arora and M. N. V. Kumar, Adv. Drug Delivery Rev., 2016, 107, 47–59 CrossRef CAS PubMed.
- S. Saeidlou, M. A. Huneault, H. Li and C. B. Park, Prog. Polym. Sci., 2012, 37, 1657–1677 CrossRef CAS.
- E. Balla, V. Daniilidis, G. Karlioti, T. Kalamas, M. Stefanidou, N. D. Bikiaris, A. Vlachopoulos, I. Koumentakou and D. N. Bikiaris, Polymers, 2021, 13, 1822 CrossRef CAS PubMed.
- O. Coulembier, J. De Winter, T. Josse, L. Mespouille, P. Gerbaux and P. Dubois, Polym. Chem., 2014, 5, 2103–2108 RSC.
- I. Armentano, N. Bitinis, E. Fortunati, S. Mattioli, N. Rescignano, R. Verdejo, M. A. Lopez-Manchado and J. M. Kenny, Prog. Polym. Sci., 2013, 38, 1720–1747 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed.
- T. D. Ngo, A. Kashani, G. Imbalzano, K. T. Q. Nquyen and D. Hui, Composites, Part B, 2018, 143, 172–196 CrossRef CAS.
- A. Constanzo, R. Spotorno, M. V. Candal, M. M. Fernández, A. J. Müller, R. S. Graham, D. Cavallo and C. McIlroy, Addit. Manuf., 2020, 36, 101415 Search PubMed.
- J. Dominguez-Robles, N. K. Martin, M. L. Fong, S. A. Stewart, N. J. Irwin, M. I. Rial-Hermida, R. F. Donnely and E. Larrenta, Pharmaceutics, 2019, 11, 165 CrossRef CAS PubMed.
- J. Ahmed and S. K. Varshney, Int. J. Food. Prop., 2011, 14, 37–58 CrossRef CAS.
- E. Psochia, L. Papadopoulos, D. J. Gkiliopoulos, A. Francone, M.-E. Grigora, D. Tzetzis, J. Vieira de Castro, N. M. Neves, K. S. Triantafyllidis, C. M. Sotomayor Torres, N. Kehagias and D. N. Bikiaris, Macromol, 2021, 1, 49–63 CAS.
- L. Genovese, M. Soccio, N. Lotti, M. Gazzano, V. Siracusa, E. Salatelli, F. Balestra and A. Munari, Eur. Polym. J., 2017, 95, 289–303 CrossRef CAS.
- S. Bourbigot and G. Fontaine, Polym. Chem., 2010, 1, 1413–1422 RSC.
- T. Casalini, F. Rossi, A. Castrovinci and G. Perale, Front. Bioeng. Biotechnol., 2019, 7, 259 CrossRef PubMed.
- L. Sha, Z. Chen, Z. Chen, A. Zhang and Z. Yang, Int. J. Polym. Sci., 2016, 2016, 6869154 Search PubMed.
- B. Wang, T. Wen, X. Zhang, A. Tercjak, X. Dong, A. J. Müller, D. Wang and D. Cavallo, Macromolecules, 2019, 52, 6274–6284 CrossRef CAS.
- A. Vlachopoulos, G. Karlioti, E. Balla, V. Daniilidis, T. Kalamas, M. Stefanidou, N. D. Bikiaris, E. Christodoulou, I. Koumentakou, E. Karavas and D. N. Bikiaris, Pharmaceutics, 2022, 14, 359 CrossRef CAS PubMed.
- E. Kontou, M. Niaounakis and P. Georgiopoulos, J. Appl. Polym. Sci., 2011, 122, 1519–1529 CrossRef CAS.
- R. Androsch, E. Zhuravlev and C. Schick, Polymer, 2014, 55, 4932–4941 CrossRef CAS.
- M. C. Righetti, M. Gazzano, M. L. Di Lorenzo and R. Androsch, Eur. Polym. J., 2015, 70, 215–220 CrossRef CAS.
- A. J. Müller, M. Ávila, G. Saenz and J. Salazar, in Poly(lactic acid) and technology: processing, properties, additives and applications, ed. A. Jiménez, M. Peltzer and R. Ruseckaite, RSC Publishing, 2015, RSC Polym. Chem. Series No. 12, ch. 3, pp. 66–98 Search PubMed.
- R. Androsch, H. M. Naeem Iqbal and C. Schick, Polymer, 2015, 81, 151–158 CrossRef CAS.
- A. Toda, R. Androsch and C. Schick, Polymer, 2016, 91, 239–263 CrossRef CAS.
- J. Lin, S. Shenogin and S. Nazarenko, Polymer, 2002, 43, 4733–4743 CrossRef CAS.
- A. Karourani, E. G. Andriotis, K. Chachlioudaki, K. N. Kontogiannopoulos, P. A. Klonos, A. Kyritsis, E. Pavlidou, D. N. Bikiaris, D. G. Fatouros and P. Barmpalexis, Mol. Pharmaceutics, 2021, 18, 4393–4414 CrossRef PubMed.
- P. A. Klonos, V. Peoglos, D. N. Bikiaris and A. Kyritsis, J. Phys. Chem. C, 2020, 123, 5469–5479 CrossRef.
- P. A. Klonos, S. N. Tegopoulos, C. S. Koutsiara, E. Kontou, P. Pissis and A. Kyritsis, Soft Matter, 2019, 18, 1813–1824 RSC.
- H. Kang, Y. Li, M. Gong, Y. Guo, Z. Guo, Q. Fang and X. Li, RSC Adv., 2018, 8, 11643–11651 RSC.
- J. M. Raquez, Y. Habibi, M. Murariu and P. Dubois, Prog. Polym. Sci., 2013, 38, 1504 CrossRef CAS.
- Z. Terzopoulou, P. A. Klonos, A. Kyritsis, A. Tziolas, A. Avgeropoulos, G. Z. Papageorgiou and D. N. Bikiaris, Polymer, 2019, 166, 1–12 CrossRef CAS.
- M. J. Wang, S. C. Chen, K. K. Yang and Y. Z. Wang, RSC Adv., 2015, 5, 42162–42173 RSC.
- J. K. Oh, Soft Matter, 2011, 7, 5096–5108 RSC.
- V. Karava, A. Siamidi, M. Vlachou, E. Christodoulou, A. Zamboulis, D. N. Bikiaris, A. Kyritsis and P. A. Klonos, Soft Matter, 2021, 17, 2439–2453 RSC.
- P. Pan and Y. Inoue, Prog. Polym. Sci., 2009, 34, 605–640 CrossRef CAS.
- M. Tanaka, K. Sato, E. Kitakami, S. Kobayashi, T. Hoshiba and K. Fukushima, Polym. J., 2015, 47, 114–121 CrossRef CAS.
- L. Sisti, G. Totaro and P. Marchese, PBS makes its entrance into the family of biobased plastics, John Wiley & Sons, Hobocan, NJ, USA, 2016 Search PubMed.
- M. Hong and E. Y. X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- W. Post, A. Susa, R. Blaauw, K. Molenveld and R. J. I. Knoop, Polym. Rev., 2020, 60, 359–388 CrossRef CAS.
- T. Beslikas, I. Gigis, V. Goulios, J. Christoforides, G. Z. Papageorgiou and D. N. Bikiaris, Int. J. Mol. Sci., 2011, 12, 6597–6618 CrossRef CAS PubMed.
- Q. Zhang, M. Song, Y. Xu, W. Wang, Z. Wang and L. Zhang, Prog. Polym. Sci., 2021, 120, 101430 CrossRef CAS.
- G. Rizis, T. G. van de Ven and A. Eisenberg, Soft Matter, 2014, 10, 2825–2835 RSC.
- J. K. Palacios, L. Zhao, N. Hadjichristidis and A. J. Müller, Macromolecules, 2017, 50, 9683–9695 CrossRef CAS.
- E. Christodoulou, P. A. Klonos, K. Tsachouridis, A. Zamboulis, A. Kyritsis and D. N. Bikiaris, Soft Matter, 2020, 16, 8187–8201 RSC.
- Z. Terzopoulou, L. Papadopoulos, A. Zamboulis, D. G. Papageorgiou, G. Z. Papageorgiou and D. N. Bikiaris, Polymers, 2020, 12, 1209 CrossRef CAS PubMed.
- Z. Terzopoulou, A. Zamboulis, D. N. Bikiaris, M. A. Valera and A. Mangas, Polymers, 2021, 13, 4121 CrossRef CAS PubMed.
- E. Skoog, J. H. Shin, V. Saez-Jimenez, V. Mapelli and L. Olsson, Biotechnol. Adv., 2018, 36, 2248–2263 CrossRef CAS PubMed.
- T. Zorba, K. Chrissafis, K. M. Paraskevopoulos and D. N. Bikiaris, Polym. Degrad. Stab., 2007, 92, 222–230 CrossRef CAS.
- G. Z. Papageorgiou, V. Tsanaktsis and D. N. Bikiaris, CrystEngComm, 2014, 16, 7963–7978 RSC.
- C. Alvarez, M. J. Capitan, N. Lotti, A. Munari and T. A. Ezquerra, Macromolecules, 2003, 36, 3245–3253 CrossRef.
- M. Soccio, N. Lotti, L. Finelli, M. Gazzano and A. Munari, Eur. Polym. J., 2009, 45, 3236–3248 CrossRef CAS.
- J. Lu, Z. Li, L. Zhou, L. Wu and B. G. Li, Polym. Degrad. Stab., 2019, 163, 68–75 CrossRef CAS.
- D. Rohindra, R. Lata, K. Kuboyama and T. Ougizawa, Polym. Cryst., 2019, 2, e10037 Search PubMed.
- A. Schönhals and P. Szymoniak, Dynamics of Composite Materials, Springer, Cham, Switzerland, 2022 Search PubMed.
- M. E. Córdova, A. T. Lorenzo, A. J. Müller, J. N. Hoskins and S. M. Grayson, Macromolecules, 2011, 44, 1742–1746 CrossRef.
- P. A. Klonos, N. D. Bikiaris, E. Christodoulou, A. Zamboulis, G. Z. Papageorgiou and A. Kyritsis, Polymer, 2022, 242, 124603 CrossRef CAS.
- P. A. Klonos, L. Papadopoulos, M. Kasimatis, H. Iatrou, A. Kyritsis and D. N. Bikiaris, Macromolecules, 2021, 54, 1106–1119 CrossRef CAS.
- F. Kremer and F. Schönhals, Broadband Dielectric Spectroscopy, Springer-Verlag, Berlin, 2002 Search PubMed.
- R. Richert, A. Agapov and A. P. Sokolov, J. Chem. Phys., 2011, 134, 104508 CrossRef PubMed.
- L. Genovese, M. Soccio, N. Lotti, A. Munari, A. Szymczyk, S. Paszkiewicz, A. Linares, A. Nogales and T. A. Ezquerra, Phys. Chem. Chem. Phys., 2018, 20, 15696–15706 RSC.
- S. Havriliak and S. Negami, Polymer, 1967, 8, 161–210 CrossRef CAS.
- S. Charlon, L. Delbreilh, E. Dargent, N. Follain, J. Soulestin and S. Marais, Eur. Polym. J., 2016, 84, 366–376 CrossRef CAS.
- P. Klonos, P. Pissis, V. M. Gun’ko, A. Kyritsis, N. V. Guzenko, E. M. Pakhlov, V. I. Zarko, W. Janusz, J. Skubiszewska-Zięba and R. Leboda, Colloids Surf., A, 2010, 360, 220–231 CrossRef CAS.
- O. Vassiliadou, V. Chrysostomou, S. Pispas, P. A. Klonos and A. Kyritsis, Soft Matter, 2021, 17, 1284 RSC.
- S. Kripotou, P. Pissis, P. Sysel, V. Sindelar and V. A. Bershtein, Polymer, 2006, 47, 357–366 CrossRef CAS.
- D. R. Figueroa, J. J. Fontanella, M. C. Wintersgill, J. P. Calame and C. G. Andeen, Solid State Ionics, 1988, 28–30, 1023–1028 CrossRef.
- M. Beiner and H. Huth, Nat. Mater., 2003, 2, 595–599 CrossRef CAS PubMed.
- J. Ren, O. Urakawa and K. Adachi, Polymer, 2002, 44, 847–855 CrossRef.
- E. Laredo, D. Newman, R. Pezzoli, A. J. Müller and A. Bello, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 680–691 CrossRef CAS.
- J. Leng, N. Kang, D. Y. Wang, J. Falkenhagen, A. F. Thünemann and A. Schönhals, Macromol. Chem. Phys., 2017, 218, 1700232 CrossRef.
- M. Füllbrandt, P. J. Purohit and A. Schönhals, Macromolecules, 2013, 46, 4626 CrossRef.
- P. Krisanangkura, A. M. Packard, J. Burgher and F. D. Blum, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1911–1918 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- R. Boehmer, K. Ngai, C. A. Angell and D. J. Plazek, J. Chem. Phys., 1993, 99, 4201–4209 CrossRef CAS.
- C. Alvarez, I. Šics, A. Nogales, Z. Denchev, S. S. Funari and T. A. Ezquerra, Polymer, 2004, 45, 3953–3959 CrossRef CAS.
- L. Yu and P. Cebe, J. Polym. Sci., Part B: Polym. Phys., 2009, 47, 2520–2532 CrossRef CAS.
- P. Klonos, A. Kyritsis and P. Pissis, Eur. Polym. J., 2015, 70, 342–359 CrossRef CAS.
- I. Hodge, J. Res. Natl. Inst. Stand. Technol., 1997, 102, 195–205 CrossRef CAS PubMed.
- N. Delpouve, A. Saiter and E. Dargent, Eur. Polym. J., 2011, 47, 2414–2423 CrossRef CAS.
- G. Georgoussis, A. Kanapitsas, P. Pissis, Y. V. Savelyev, V. Ya Veselov and E. G. Privalko, Eur. Polym. J., 2000, 36, 1113–1126 CrossRef.
- M. Safari, J. Maiz, G. Shi, D. Juanes, G. Liu, D. Wang, C. Mijangos, Á. Alegría and A. J. Müller, Langmuir, 2019, 35, 15168–15179 CrossRef CAS PubMed.
- J. Dobbertin, J. Hannemann, C. Schick, M. Pötter and H. Dehne, J. Chem. Phys., 1998, 108, 9062–9068 CrossRef CAS.
- A. Wurm, M. Ismail, B. Kretzschmar, D. Pospiech and C. Schick, Macromolecules, 2010, 43, 1480 CrossRef CAS.
- M. G. Wessels and A. Jayaraman, Soft Matter, 2019, 15, 3987–3998 RSC.
- E. Mygiakis, E. Glynos and G. Sakellariou, Eur. Polym. J., 2021, 161, 110857 CrossRef CAS.
- P. Szymoniak, B. R. Pauw, X. Qu and A. Schönhals, Soft Matter, 2020, 16, 5406–5421 RSC.
- P. Szymoniak, X. Qu, M. Abbasi, B. R. Pauw, S. Henning, Z. Li, D. Y. Wang, C. Schick, K. Saalwächter and A. Schönhals, Soft Matter, 2021, 17, 2775–2790 RSC.
- N. C. Murillo, P. Szymoniak, G. J. Smales, H. Sturm and A. Schönhals, ACS Appl. Polym. Mater., 2021, 3, 6572–6585 CrossRef CAS.
- Z. C. Yan, E. van Ruymbeke and D. Vlassopoulos, Macromolecules, 2021, 54, 11047–11060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sm00261b |
| This journal is © The Royal Society of Chemistry 2022 |