
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual-reactive nanogels for orthogonal functionalization of hydrophilic shell and amphiphilic network†
Alexandra
Gruber
,
Lucila
Navarro
and
Daniel
Klinger
 *
*
Institute of Pharmacy (Pharmaceutical Chemistry), Freie Universität Berlin, Königin-Luise-Straße 2-4, 14195 Berlin, Germany. E-mail: daniel.klinger@fu-berlin.de
First published on 29th March 2022
Abstract
Amphiphilic nanogels (NGs) combine a soft, water-swollen hydrogel matrix with internal hydrophobic domains. While these domains can encapsulate hydrophobic cargoes, the amphiphilic particle surface can reduce colloidal stability and/or limit biological half-life. Therefore, a functional hydrophilic shell is needed to shield the amphiphilic network and tune interactions with biological systems. To adjust core and shell properties independently, we developed a synthetic strategy that uses preformed dual-reactive nanogels. In a first step, emulsion copolymerization of pentafluorophenyl methacrylate (PFPMA) and a reduction-cleavable crosslinker produced precursor particles for subsequent network modification. Orthogonal shell reactivity was installed by using an amphiphilic block copolymer (BCP) surfactant during this particle preparation step. Here, the hydrophilic block poly(polyethylene glycol methyl ether methacrylate) (PPEGMA) contains a reactive alkyne end group for successive functionalization. The hydrophobic block (P(PFPMA-co-MAPMA) contains random methacryl-amido propyl methacrylamide (MAPMA) units to covalently attach the surfactant to the growing PPFPMA network. In the second step, orthogonal modification of the core and shell was demonstrated. Network functionalization with combinations of hydrophilic (acidic, neutral, or basic) and hydrophobic (cholesterol) groups gave a library of pH- and redox-sensitive amphiphilic NGs. Stimuli-responsive properties were demonstrated by pH-dependent swelling and reduction-induced degradation via dynamic light scattering. Subsequently, copper-catalyzed azide–alkyne cycloaddition was used to attach azide-modified rhodamine as model compound to the shell (followed by UV-Vis). Overall, this strategy provides a versatile platform to develop multi-functional amphiphilic nanogels as carriers for hydrophobic cargoes.
Introduction
Hydrophilic nanogels (NGs) are swollen crosslinked polymer nanoparticles that are promising nanocarriers in next generation therapeutics.1–4 However, due to their overall hydrophilic nature, such NGs are limited in their ability to efficiently encapsulate and deliver hydrophobic drugs. Thus, amphiphilic NGs with a hydrophilic polymer matrix and embedded hydrophobic groups are emerging to expand the therapeutic potential of conventional NGs.5–7To address the challenging synthesis of such amphiphilic colloids (i.e., combine groups of opposing solubility in a single colloidal system) we have recently developed a versatile approach that uses reactive poly(pentafluorophenyl methacrylate) (PPFPMA) precursor nanogels. Transferring the concept of post-polymerization functionalization from linear polymers8–10 to crosslinked NG networks,6,11–13 allows the preparation of NG libraries with varying hydrophobicity but similar colloidal features.6 In this strategy, the internal reactive network of precursor particles is functionalized with different mixtures of hydrophilic and hydrophobic moieties.6 This approach results in amphiphilic NGs containing crosslinked copolymer networks with randomly distributed hydrophilic and hydrophobic side groups. Small angle X-ray scattering (SAXS) on these NGs revealed that the hydrophobic groups phase segregate into hydrophobic nanodomains within a hydrophilic polymer matrix.14 While these domains enable the efficient loading and sustained release of hydrophobic cargoes,6 the amphiphilic character of the network is not restricted to the interior of the NGs but also displays on their surface.15 This can lead to undesired effects such as particle aggregation or the formation of specific protein coronas that determine the nanogels’ interaction with surrounding (biological) systems and can reduce their biological half-life.15–17 Therefore, shielding the amphiphilic structure with a hydrophilic shell is needed to enhance the potential of such colloidal structures for advanced delivery applications. A common strategy to address this need is the coating with hydrophilic macromolecules, e.g., polyethylene glycol (PEG) as the gold standard. This process, also known as PEGylation, has been shown to reduce the uptake by macrophages, thus increasing the circulation time of polymeric nanoparticles.18,19 However, PEGylation can also reduce the desired selective interaction with targeted cells, resulting in the so-called PEGylation dilemma.20,21 To overcome this problem, specific targeting ligands need to be introduced to the hydrophilic polymer shell.
Thus, an ideal amphiphilic NG contains the following elements: (1) it has an amphiphilic interior to enable loading of hydrophobic cargoes. (2) The network needs to be stimuli-responsive and degradable to control the release profile of the cargo. (3) The NGs should contain a hydrophilic shell to cloak the amphiphilic surface, thus preventing aggregation and ultimately increasing biological half-life. (4) The NGs surface should be functionalized with specific moieties that control the interaction with (biological) systems.
To prepare such multi-functional systems, we have developed a detailed synthetic design that takes the following considerations into account (see Scheme 1): to control the interior network functionality, we aim to expand our reactive precursor particle approach. The utilization of cleavable disulfide-based crosslinkers during particle preparation enables reduction-sensitive degradation/swelling of the final nanogels. In addition, network functionalization with a combination of ionic hydrophilic (acidic or basic) and hydrophobic groups can impart pH-responsive swelling properties. In combination with the cleavable crosslinkers, this gives access to a library of double-responsive amphiphilic nanogels, i.e., pH- and reduction sensitive.
 | ||
| Scheme 1 Schematic representation of the synthetic platform for the preparation of functional NGs. Reactive precursor particles with a hydrophilic reactive PPEGMA shell, that contains alkyne end groups, allow orthogonal functionalization of the interior and the surface of the precursor particles. The alkyne groups of the reactive shell can be functionalized with azides, while the interior pentafluorophenyl groups can be reacted with functional amines giving access to a library of NGs with different functionalities. | ||
In contrast to network functionalization, the introduction of an orthogonally reactive hydrophilic shell is much more challenging. To address this challenge, several synthetic strategies are available. These include the physical adsorption of hydrophilic polymers,22,23 the grafting of hydrophilic polymers (e.g., PEG) onto nanoparticles after particle formation,24–26 and the introduction of hydrophilic co-monomers (e.g., polyethylene glycol methyl ether methacrylate (PEGMA)) during particle preparation.27,28 Alternatively, we were drawn to using reactive block copolymer (BCP) surfactants. This versatile approach convinces with a high level of control over the covalent hydrophilic shells since the desired properties can be programmed through the molecular design of the BCP building block as follows (see Scheme 1):
To control the shell properties, we used an amphiphilic block copolymer (hydrophilic-b-hydrophobic) as macromolecular surfactant. In this structure, the hydrophobic block needs to attach to the surfactant to the final nanogels. This is realized already during the preparation of the PPFPMA precursor particles. Here, the BCP enables steric stabilization of the (mini)emulsion droplets consisting of hydrophobic PFPMA monomers and crosslinkers. Consequently, the hydrophobic block of the surfactant also consists of PPFPMA to increase the affinity to the dispersed phase. To anchor the surfactant to the colloidal surface, the hydrophobic PPFPMA block contains reactive groups that enable covalent incorporation into the polymer network during particle synthesis. Such a strategy can be realized by the surfactants acting as macromolecular crosslinker,12,29 macromonomer,30 or macroinitiator.31 In our case, we introduced multiple methacrylate groups to the surfactant's PPFPMA block to enable copolymerization with the network forming monomer. In contrast, the hydrophilic block should be water soluble and show low protein adsorption to provide steric stabilization and increase biological half-life. To provide these properties, we were drawn to PPEGMA500 (poly(polyethylene glycol methyl ether methacrylate); molecular weight of PEG side chains 500 kDa). In such a graft polymer, the PEG side chains are water soluble polymers that are known for the preparation of “stealth”-nanocarriers due to their protein- and cell resistant properties.19,32 In addition, the resulting hydrophilic PPEGMA500 block also contains a reactive end group for further functionalization of the nanogel surface.31 To enable such a modular introduction of functional moieties to the shell, the coupling reaction must not interfere with the modification of the reactive network in the interior of the precursor particles. Thus, shell modification reactions need to be orthogonal to the existing active ester chemistry. Here, we used copper-catalyzed azide–alkyne cycloaddition (CuAAC) as versatile functionalization strategy that allows fast and quantitative reactions in aqueous media (Scheme 1). Moreover, CuAAC is well known to be orthogonal to PFP active ester chemistry.33
Overall, this strategy allows sequential functionalization of the reactive precursor particles’ interior (in organic solvents) and surface (in water). This will be demonstrated by the incorporation of responsive moieties into the network and the determination of pH- and reduction-sensitive swelling and degradation profiles. In addition, the functionalization of the shell with a fluorescent model compound will serve to demonstrate the modular nature of this approach. Focusing on the synthetic aspects of this approach, this work shows a significant improvement of our previously developed synthetic platform.
Results and discussion
Synthesis of reactive block copolymer surfactant
The use of functionalized block copolymer surfactants is a versatile approach to tune colloidal surface properties, e.g., adjusting particle interactions with biological systems. In this context, hydrophilic blocks are of special interest to provide colloidal stability and control protein adsorption on hydrophobic particles. While PEG is still the gold standard, new polymeric building blocks are currently evolving as promising alternatives, e.g., poly(phosphoester)s.34 Combining such hydrophilic blocks with hydrophobic segments enables physical adsorption to the hydrophobic particle surface. While these systems provide versatility, the sole reliance on physical interactions between surfactant and particle limits the stability. Thus, covalent surfactant attachment is required to increase stability.To fulfill the specific surfactant requirements, we synthesized an amphiphilic block copolymer of poly(polyethylene glycol methyl ether methacrylate-b-pentafluorophenyl methacrylate-co-methacrylamido propyl methacrylamide) (P(PEGMA-b-(PFPMA-co-MAPMA)). For the hydrophilic block PPEGMA500 was used as water-soluble polymer that is well-established in biomedical applications. As hydrophobic block PPFPMA was chosen due to its structural similarity to the forming PPFPMA network. Partial functionalization of the PFP esters with N-(3-aminopropyl)-methacrylamide hydrochlorid (APMA) introduces reactive vinyl groups for the covalent attachment of the surfactant to the polymer network during particle formation (Scheme 2).
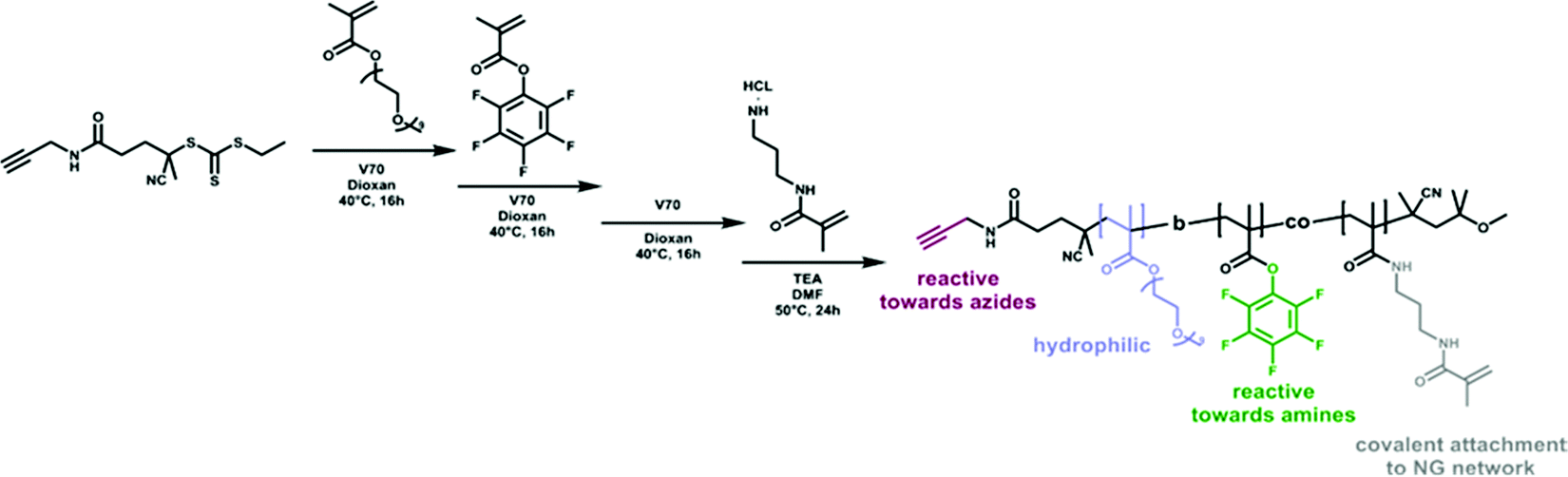 | ||
| Scheme 2 Synthesis of a reactive block copolymer surfactant via RAFT polymerization of PEGMA followed by a second block of PPFPMA using an alkyne-modified CTA. Partial functionalization of the PFP groups with N-(3-aminopropyl)-methacrylamide hydrochlorid (APMA) introduces methacrylamide groups for covalent attachment during particle formation. | ||
The controlled polymerization of the surfactant was realized via reversible addition fragmentation transfer (RAFT) polymerization. This method not only provides control over the polymers’ molecular weight and molecular weight distribution, but also provides access to α,ω-end group-functionalized polymers.35,36 Using an alkyne-modified CTA for the polymerization of PEGMA500 enabled us to install a reactive alkyne in the α-position (Scheme 2). Subsequent chain extension with a second block of PFPMA resulted in the respective surfactant BCP that still contains the α alkyne for modifications of the hydrophilic shell. To avoid side reactions of the alkyne function during the polymerization process, the reaction was performed at a low temperature of 40 °C using 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile) (V70) as initiator. To ensure good stability of (mini)emulsion droplets in an aqueous environment, the volume of the hydrophilic block should be larger than the volume of the hydrophobic block. Therefore, a length of 30 kDa was targeted for the hydrophilic PPEGMA block, corresponding to a degree of polymerization (DP) of 60 monomer units. For the hydrophobic PPFPMA block, a molecular weight of about 10 kDa was aimed for. This was achieved by targeting 20 kDa (DP of 79) and controlling the conversion of the chain extension process. The progress of polymerizations was followed by size exclusion chromatography (SEC) (Fig. S10, ESI†). However, accurate molecular weight determination of block copolymers via SEC can be difficult due to the different solubility behavior of the blocks. Therefore, chain extension of a polymer by a second block can lead to incoherent retention times, resulting in an apparent reduction in molecular weight when, in fact, the opposite occurred.37 These problems became also apparent during molecular weight determination of the PPEGMA500-b-PPFPMA copolymer. Here, SEC showed only a negligible increase of the molecular weight upon chain extension with the second block. Thus, an alternative method was used to examine the molecular weight of the PPEGMA500 polymer and the PPEGMA500-b-PPFPMA copolymer. For this, 1H NMR spectroscopy with DMF as internal standard was used. In this approach, monomer conversions were determined and translated into the respective degrees of polymerizations. For the polymerization of PEGMA500, a conversion of 83% was determined which translates to a 25 kDa PPEGMA500 block (SEC: Mn 28.5 kDa, Đ 1.36). The chain extension with PFPMA showed a conversion of 45%, thus the second hydrophobic PPFPMA block has a length of 9 kDa (SEC BCP: Mn 29.5 kDa, Đ 1.42). After successful polymerization of both blocks, the trithiocarbonate ω-end group was removed by reaction with an excess of V70 before the APMA groups were installed by partial post-functionalization of the PFP groups. For this, a functionalization of 20% of the PFP groups (corresponding to 7 groups) was targeted. 1H NMR evaluation showed an incorporation of 3 APMA groups. Therefore, the final composition of the BCP surfactant is the following: P(PEGMA50-b-(PFPMA32-co-MAPMA3)).
Preparation of reactive precursor particles with a hydrophilic orthogonal reactive shell
To conjugate the functional BCP to the final nanogels, it was used as reactive surfactant during the preparation of the PPFPMA precursor particles. For this, two different methods were compared: miniemulsion and emulsion polymerization of the PFPMA monomer and crosslinker. With this, we aimed to examine the influence of different particle preparation methods on the resulting particle size and size distribution.In our previous work, miniemulsion polymerization was used to prepare the reactive precursor particles: Free radical co-polymerization of PFPMA and EGDMA crosslinker occurred in miniemulsion droplets that are stabilized by sodium dodecyl sulfate (SDS). In this miniemulsion process, the inhibition of net diffusion between droplets (Ostwald ripening) is achieved by the addition of an osmotic pressure agent.38,39 Thus, the droplets serve as stable “nanoreactors”, which bypass nucleation and growth mechanisms. This results in an isotropic distribution of crosslinkers in the particle network.38–40 In this process, we now replaced the small molecule surfactant SDS with the new double-reactive block copolymer surfactant. This installs the hydrophilic shell during particle synthesis. The respective miniemulsion contained the monomer PFPMA, bis(2-methacryloyl)oxyethyl disulfide (DSDMA) (2 mol%) as reduction-cleavable crosslinker, and V70 as oil-soluble initiator in the dispersed phase. The continuous phase consisted of a solution of the BCP surfactant (1 wt%) in water. Polymerization was carried out at 40 °C for 18 hours. Resulting particles were washed with water before their examination via dynamic light scattering (DLS) and transmission electron microscopy (TEM). As can be seen from the TEM images in Fig. 1 (left column), miniemulsion polymerization using the customized surfactant leads to spherical nanoparticles. However, statistical examination of the particle sizes (in TEM) and DLS measurements revealed a broad particle size distribution. Especially in comparison to the particles prepared with the small molecule surfactant SDS (Fig. 1, left column). This phenomenon is a typical drawback of miniemulsions that use non-ionic steric stabilization: realizing a narrow particle size distribution is challenging. Thus, the utilization of tailormade block copolymer surfactants in miniemulsion polymerizations needs to balance an enhanced control over surface properties with a broadening of particle size distribution. For example, size distributions similar to our system are reported for the utilization of bi-functional block copolymer surfactants in the preparation of nanocapsules via miniemulsion polymerization.31
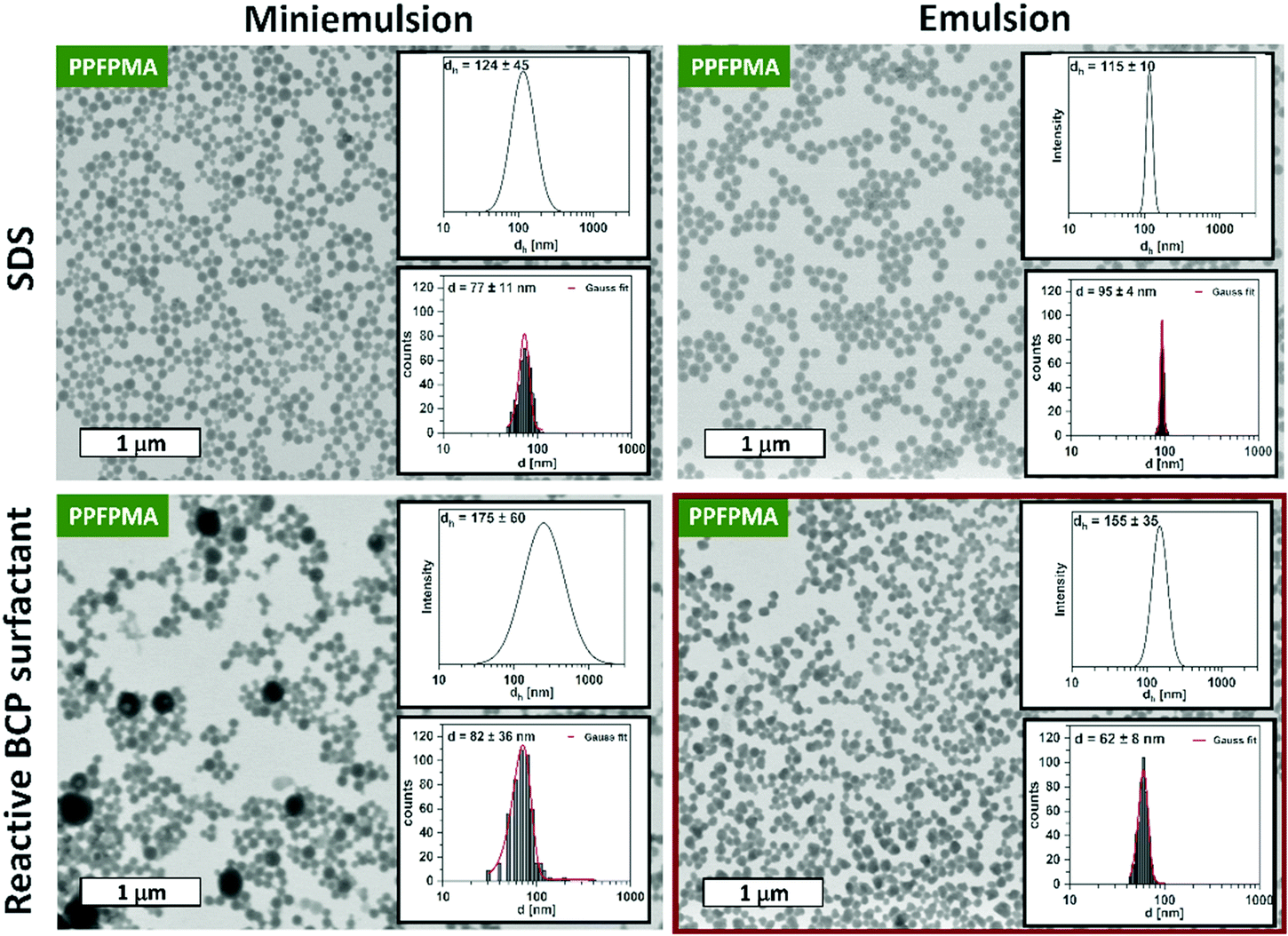 | ||
| Fig. 1 Comparison of particle size and size distribution of reactive precursor particles prepared via miniemulsion (first column) and emulsion polymerization (second column) using SDS (first row) or a customized reactive surfactant (second row). Overall, emulsion polymerization results in more defined precursor particles than miniemulsion polymerization. While the usage of SDS in emulsion polymerization results in very narrow size distributions, also the reactive BCP surfactant during particle preparation results in well-defined particles with narrow particle size distribution as visible by TEM images and their statical evaluation (lower inlet) and DLS curves at 90° (upper inlet). | ||
Therefore, another particle preparation technique is required to improve the size distribution of the precursor particles. Here we were drawn to emulsion polymerization. Emulsion polymerization is a well-known industrially established method for the synthesis of polymeric nanoparticles. It is suitable for the free radical polymerization of a broad range of water-immiscible vinyl monomers and requires water-soluble initiators and surfactants for stabilization. In contrast to the miniemulsion process, statistical monomer diffusion represents a key step in this technique. Therefore, this process is known for the resulting narrow particle size distributions.41,42
To establish a protocol for the emulsion polymerization of PFPMA with DSDMA (2 mol%) as crosslinker, we first used SDS as conventional ionic surfactant that provides high colloidal stability. Using ammonium persulfate (APS) as water soluble initiator, the emulsion polymerization was conducted at 80 °C for 72 hours. As demonstrated by DLS and TEM, this protocol results in monodisperse PFPMA reactive precursor particles (Fig. 1, right column). Thus, in the next step we replaced SDS with the reactive BCP surfactant. However, in first attempts, these conditions resulted in disperse aggregates that show a raspberry-like appearance (Fig. S1, ESI†). It seems like several nanoparticles collapsed into one big aggregate.
It is assumed that this can be explained by the thermo-responsiveness of the hydrophilic PPEGMA block in the BCP surfactant, i.e., PPEGMA500 exhibits a lower critical solution temperature (LCST) of roughly 90 °C.43 Since copolymerization with the hydrophobic PFPMA block can lead to a reduction of the LCST, we suggest that a reaction temperature of 80 °C can cause partial collapse of the PPEGMA chains. The resulting decrease in stabilization can cause aggregation and formation of the observed raspberry-like particles. To circumvent the collapse of the surfactant and the resulting aggregation of the particles, the reaction temperature was lowered to 50 °C. As can be seen in Fig. 1, the reduction of the reaction temperature resulted in well-defined precursor particles with a diameter of 62 nm in their dried state (by TEM) and a hydrodynamic diameter of 155 nm (by DLS). The size distribution of these reactive particles is not as narrow as the one for particles synthesized with SDS by emulsion polymerization. However, it is significantly narrower than the size distributions that are obtained for particles synthesized via miniemulsion polymerization. Thus, emulsion polymerization of PFPMA with the functional BCP surfactant represents a scalable synthetic strategy to prepare well-defined reactive precursor particles with a reactive hydrophilic shell.
Generation of an amphiphilic NG library via post-functionalization of the internal PPFPMA network
Having established the synthesis of dual-reactive precursor particles, we examined the orthogonal and sequential functionalization of core and hydrophilic shell. Regarding the internal functionality, a disulfide-based crosslinker was used to introduce reduction-cleavable network points already during preparation of the hydrophobic precursor particles. The hydrophilic/amphiphilic network properties were then installed in a subsequent functionalization step. Due to the hydrophobic nature of the PFPMA monomer and the DSDMA crosslinker, this two-step approach circumvents the challenging direct incorporation of comparable disulfide-crosslinkers in hydrophilic networks. For example, realizing such systems through inverse (mini)emulsion polymerizations often suffers from limited emulsion stability and can lead to broad particle size distributions.44In addition to the crosslinker-defined network degradability, functionalization of the reactive PPFPMA network enables the introduction of further stimuli-responsive and amphiphilic network properties. Such variations in nanogel properties are traditionally realized by using different monomers in the particle preparation step.45,46 For example, the volume phase transition temperature of thermoresponsive nanogels can be tuned by changing the ratio between N-isopropylacrylamide and N-isopropylmethacrylamide monomers.47 However, this approach involves the synthesis of a new nanogel batch for each new network composition. This can lead to variations in the nanogel features and limits the incorporation of our hydrophobic cholesterol groups. Consequently, our post-polymerization modification allows more synthetic flexibility and ensures comparable colloidal features. For this, different network functionalities (anionic, cationic and/or hydrophobic) can be installed through reaction of functional amines with the reactive PFP ester groups. With this, a library of NGs of varying functionality but similar colloidal features such as size distribution and crosslinking density can be obtained (see Scheme 3). Since these features are determined by the “parent” reactive precursor particles, the resulting high level of comparability enables systematic investigation of structure–property relations.6,11,12,29
 | ||
| Scheme 3 Schematic representation of the synthetic platform for preparation of functional amphiphilic NGs (crosslinker fraction y = 2 mol%) with a hydrophilic, reactive shell. Functionalizing the interior of reactive PPFPMA precursor particles with different hydrophilic moieties gives access to a library of NGs with opposing pH response (cationic, neutral and anionic NGs). Incorporation of additional 20 mol% of hydrophobic amine-functionalized cholesterol (CHOLA) results in amphiphilic NGs with a reactive hydrophilic shell that can be used for further functionalization. | ||
To demonstrate the versatility of this approach, we focused on the preparation of a small library of multi-functional nanogels (Scheme 3 and Table 1). Here, by combining different functionalities, various properties can be programmed into the network. Overall, two sets of nanogels were prepared:
| Sample name | Hydrophilic group | Hydrophobic group | Charge typeb | Sizec [nm] | Degr.d | |||
|---|---|---|---|---|---|---|---|---|
| Type | Amt.a [mol%] | Type | Amt.a [mol%] | |||||
| a Amount with respect to the total number of groups (hydrophilic + hydrophobic). b Charge at pH < pKa for His-containing samples, at pH > pKa for AMPAA-containing samples. c Size in the collapsed state: at pH > pKa for His-containing samples, at pH < pKa for AMPAA-containing samples. d Degradabilty due to incorporation of 2 mol% DSDMA as disulfide-containing crosslinker. | ||||||||
| Set 1 | HIS CHOLA-20 | His | 80 | CHOLA | 20 | Cationic | 150 | Redox |
| PHPMA CHOLA-20 | HPMA | 80 | CHOLA | 20 | Neutral | 170 | Redox | |
| AMPAA CHOLA-20 | AMPAA | 80 | CHOLA | 20 | Anionic | 165 | Redox | |
| Set 2 | HIS | His | 100 | — | — | Cationic | 140 | Redox |
| PHPMA | HPMA | 100 | — | — | Neutral | 250 | Redox | |
| AMPAA | AMPAA | 100 | — | — | Anionic | 180 | Redox | |
Set 1 focuses on amphiphilic nanogels by combining hydrophilic and hydrophobic moieties in the same network. Resulting hydrophobic nanodomains could enhance the loading of hydrophobic drugs.6 For all NGs of this set, amine-functionalized cholesterol (CHOLA) was used to impart the hydrophobic properties in the network. This was combined with various hydrophilic groups. The ratio between these hydrophilic moieties and hydrophobic CHOLA groups was held constant at 20/80 mol-%. In one nanogel, neutral 2-hydroxypropylamine (HPA) was used to provide the NGs with a water swollen, biocompatible matrix, i.e., poly(N-(2-hydroxypropyl) methacrylamide) PHPMA.48,49 In contrast, acidic or basic groups allow the introduction of pH-responsive properties that could be used to tailor release properties. For this, we used a combination of CHOLA and histamine (HIS) or CHOLA and 4-aminomethylphenylacetic acid (AMPAA) to prepare cationic and anionic NGs, respectively. Nanogels are denoted based on their composition as PHPMA CHOLA-20, HIS CHOLA-20, and AMPAA CHOLA-20 (see Scheme 3).
Set 2 focuses on purely hydrophilic nanogels. Here, the networks are functionalized with 100 mol% of the respective hydrophilic groups. By omitting the CHOLA functionality, these NGs serve as control materials to demonstrate the influence of hydrophobic groups on the network swelling. The resulting NGs are denoted as PHPMA, HIS, and AMPAA (see Scheme 3). In all NGs, the successful conversion of the pentafluorophenyl esters to the respective amides was demonstrated by attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) as described previously (see Fig. S2, ESI†).6
Regarding the size and size distribution of the particles, our synthetic strategy suggests that these parameters are determined by the reactive precursor particles and translate to all NGs equally. To test this assumption, TEM images were statistically evaluated with respect to particle sizes. These studies revealed well-defined spherical structures with similar size and size distributions for all NGs (Fig. S3, ESI†). In addition, DLS was used to compare the size of the different NGs to the parent precursor particles in aqueous dispersion. In this case, the pH-responsive particles were measured at pH values that ensure a collapsed state of the networks, thus providing better comparability to the non-swollen hydrophobic PPFPMA precursors. Consequently, DLS measurements were conducted at pH > 10 for the cationic HIS and HIS CHOLA-20, and at pH < 4 for anionic AMPAA and AMPAA CHOLA-20 NGS. The neutral PHPMA-based NGs were measured at pH 7. As shown in Fig. 2, the hydrodynamic diameters of all pH-responsive NGs do not significantly deviate from the size of the precursor particles. For the neutral PHPMA NGs, a significantly increased size is the result of their pH-independent hydrophilicity. Thus, the networks of the PHPMA NGs are measured in their naturally swollen state while the pH-responsive NGs are measured in their collapsed state. Even though the amphiphilic PHPMA CHOLA-20 NGs are also swollen, the difference to the precursor particles is not as pronounced as in their purely hydrophilic PHPMA counterparts. We suggest that the hydrophobic CHOLA groups act as additional physical crosslinks in the network and counteract NG swelling for the amphiphilic system. As a result, the size of the amphiphilic PHPMA CHOLA-20 NGs is comparable to the size of the precursor particles. In general, size differences in dispersion vanish in the dried state. Statistical evaluations of TEM images show very similar diameters in the size range of ca. 70–80 nm for all NGs (see Fig. S3, ESI†). These values are much lower than the DLS diameters in aqueous dispersion (ca. 160–250 nm) due to two reasons: First, DLS provides the hydrodynamic diameters (dh) whereas TEM shows the “hard” geometric diameters. This inherent difference in measuring techniques is enhanced by the collapse of the highly hydrated hydrophilic shell upon drying. Second, an increased size in water also includes swelling of the networks. This effect is most pronounced in the PHPMA samples.
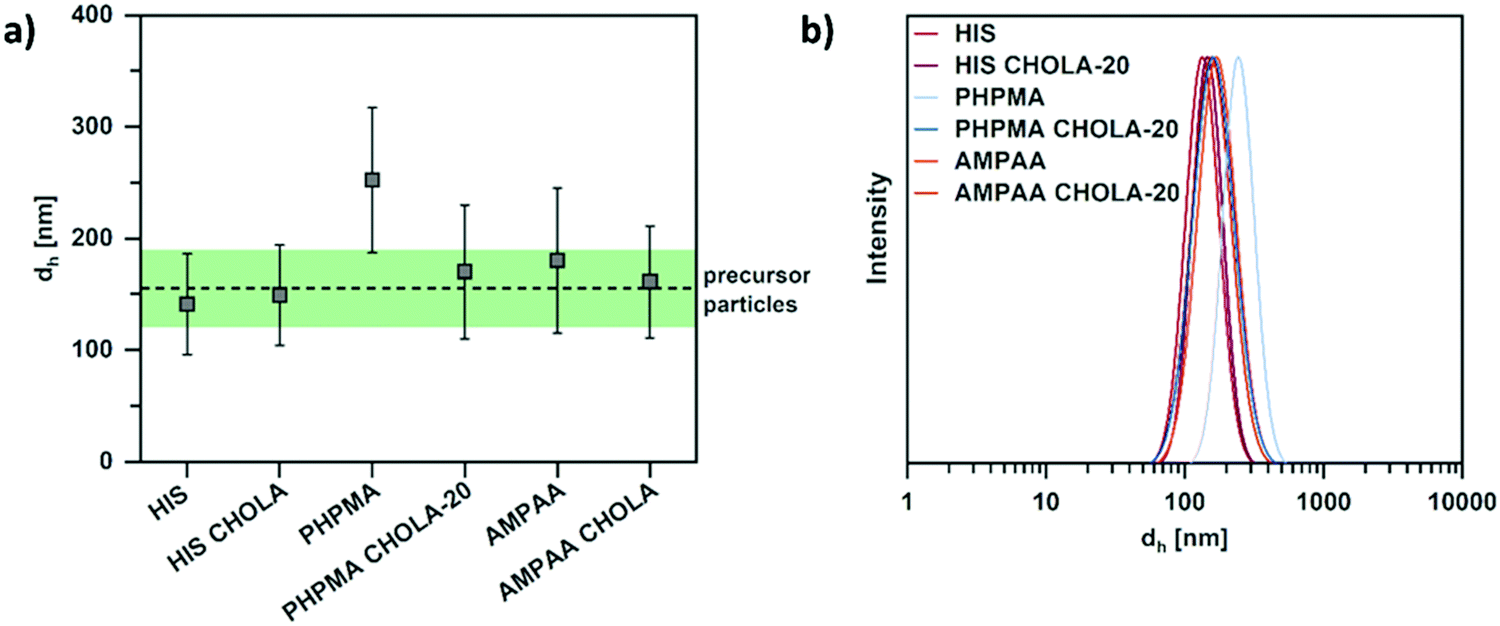 | ||
| Fig. 2 DLS measurements of (collapsed) NGs at an angle of 90° show similar size and size distributions as the reactive precursor particles. (a) Comparison of the size of the NGs. Error bars represent the width of the size distribution as the standard deviation. The dashed line represents the diameter of the reactive precursor particles and the green coloured box the respective width of the size distribution as the standard deviation. (b) DLS curves of the NGs in their collapsed state. | ||
The hydrated properties of the PPPEGMA shell are further demonstrated by the colloidal stability of the particles. Independent from their hydrophobic CHOLA-content, all NGs show no aggregation in aqueous dispersion for at least 3 months (see Fig. S4, ESI†). Thus, PPEGMA shell hydration contributes to a pronounced steric stabilization.
Regarding the morphology of the NGs, TEM images (Fig. S3, ESI†) show a clear difference between the hydrophilic and the amphiphilic NGs. While the purely hydrophilic NGs show a typical deformation of soft particles, the amphiphilic ones are more defined and show less deformation. This supports our previous findings, where phase separated hydrophobic groups form hydrophobic nanodomains that act as additional physical crosslinks in the NG networks. As a result, the rigidity of the NGs increases.14 This concept of combining physical and covalent crosslinks is also well-known from the self-assembly of amphiphilic copolymers in nanoparticles50 and hydrogels51 and bridges the gap between soft hydrophilic NGs and hard hydrophobic latex particles in our system.
Functionalization of the hydrophilic Shell via CuAAC
After functionalization of the NG core, the hydrophilic shell can be modified subsequently. Utilization of end-functionalized block copolymer surfactants is an established versatile strategy to realize this approach. Especially dual-reactive block copolymer surfactants are of interest to combine covalent particle anchoring and orthogonal surface functionalization e.g., demonstrated for the functionalization of nanocapsules.31 Similar to such reported systems, alkyne end groups at the surface of our NG hydrophilic shell enable CuAAC click reactions with functional azides. Since this reaction is orthogonal to the PFP ester functionalization in the core, this platform offers the possibility to attach a broad variety of molecules (dyes or targeting moieties) to the shell. As a proof of concept, the dye rhodamine-azide was attached (Scheme 4). To optimize the reaction conditions, test reactions were performed on the free BCP surfactant, i.e., not bound to a nanogel. This allows monitoring the success of the dye-conjugation via size exclusion chromatography in combination with an UV/VIS absorbance detector. The elugrams in Fig. 3 show SEC traces for the coupled conjugate and the non-coupled control (physical mixture of dye and BCP). Both samples were monitored by a refractive index (RI) and a variable wavelength (VW) detector at 501 nm (the adsorption maximum of rhodamine-azide). In Fig. 3a, the trace for the coupled polymer is clearly visible in both detectors (RI and VW), thus indicating successful attachment of rhodamine to the BCP. To ensure that the dye is covalently bound and not just physically attached, a negative control was performed. Therefore, the reaction was carried out under similar conditions without the addition of copper and ascorbic acid, thus preventing covalent attachment of the dye. While the RI detector shows the signal for the BCP, the VW detector lacks this signal. This supports the covalent conjugation of rhodamine under the optimized CuAAC conditions as seen in Fig. 3a. | ||
| Scheme 4 Post-functionalization of the alkyne groups of the hydrophilic PPEGMA-shell via CuAAC click reaction with rhodamine-azide. | ||
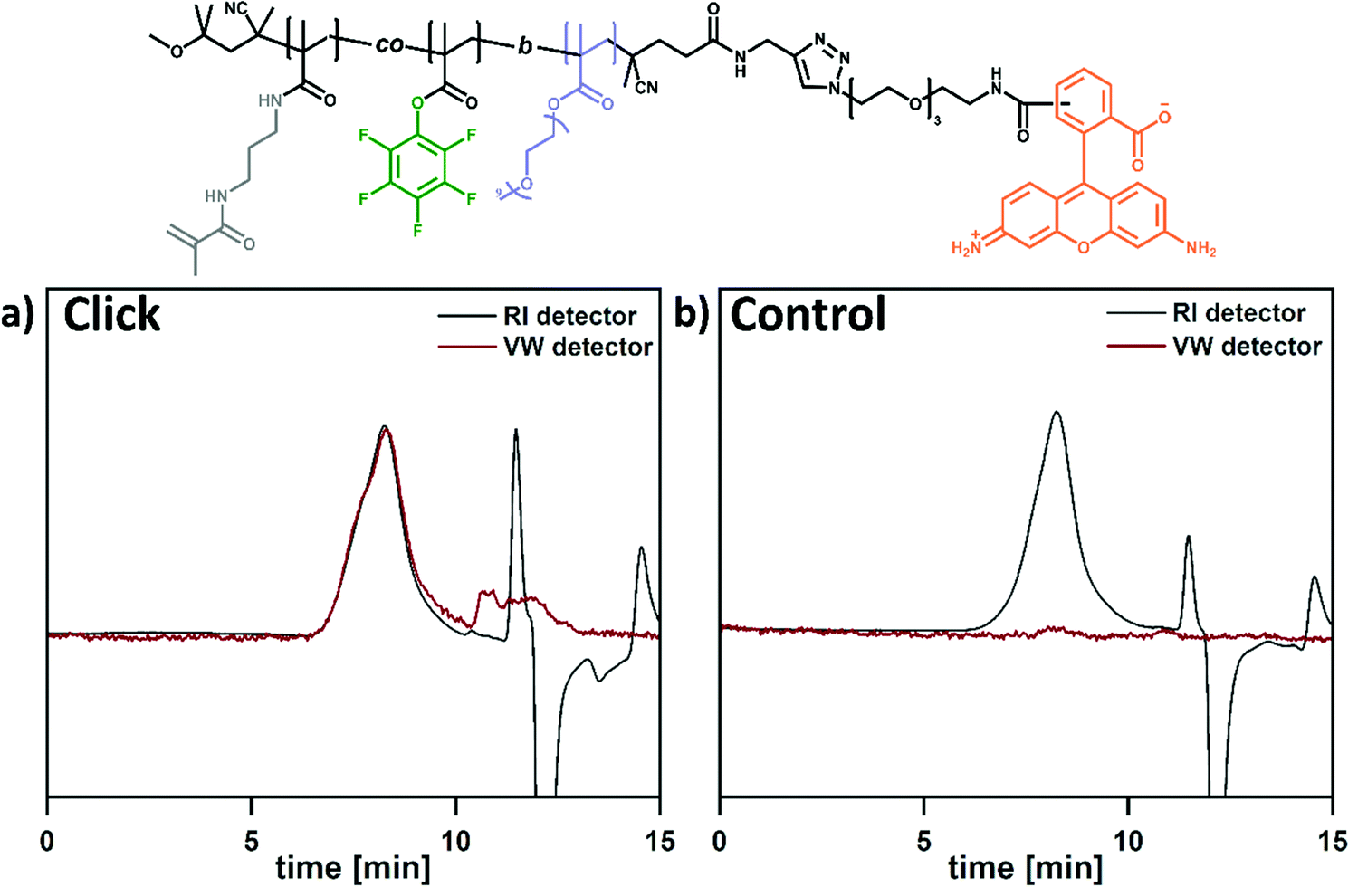 | ||
| Fig. 3 Overlay of the elugram of P(PEGMA-b-(PFPMA-co-MAPMA) detected via refractive index (RI) and variable wavelength at 501 nm (VW) detector proof the successful attachment of rhodamine to the reactive surfactant via CuAAC click reaction. Elugram (a) of click reaction of the reactive surfactant with rhodamine azide an (b) of the negative control (same reaction without the addition of copper and ascorbic acid) after purification. | ||
After having demonstrated the successful click functionalization on a simplified system, we transferred the synthetic procedure to the amphiphilic NGs. For this PHPMA CHOLA-20 NGs were chosen as model system for functionalization with rhodamine. The success of this coupling reaction was demonstrated via fluorescence spectroscopy on the nanogel dispersion. To rule out physical entrapment of the rhodamine, similarly to the BCP test reactions, a negative control was carried out (no coupling reagents). As can be seen in Fig. 4 only negligible amount of dye is entrapped in the control, thus suggesting the covalent conjugation of rhodamine under click conditions. In summary, these experiments provide the proof-of-concept for the orthogonal functionalization of core and shell in the double-reactive NGs. The simplicity and versatility of this approach allows potential functionalization with a variety of different compounds to control the network functionality and tailor the interaction with (biological) materials through respective surface modifications.
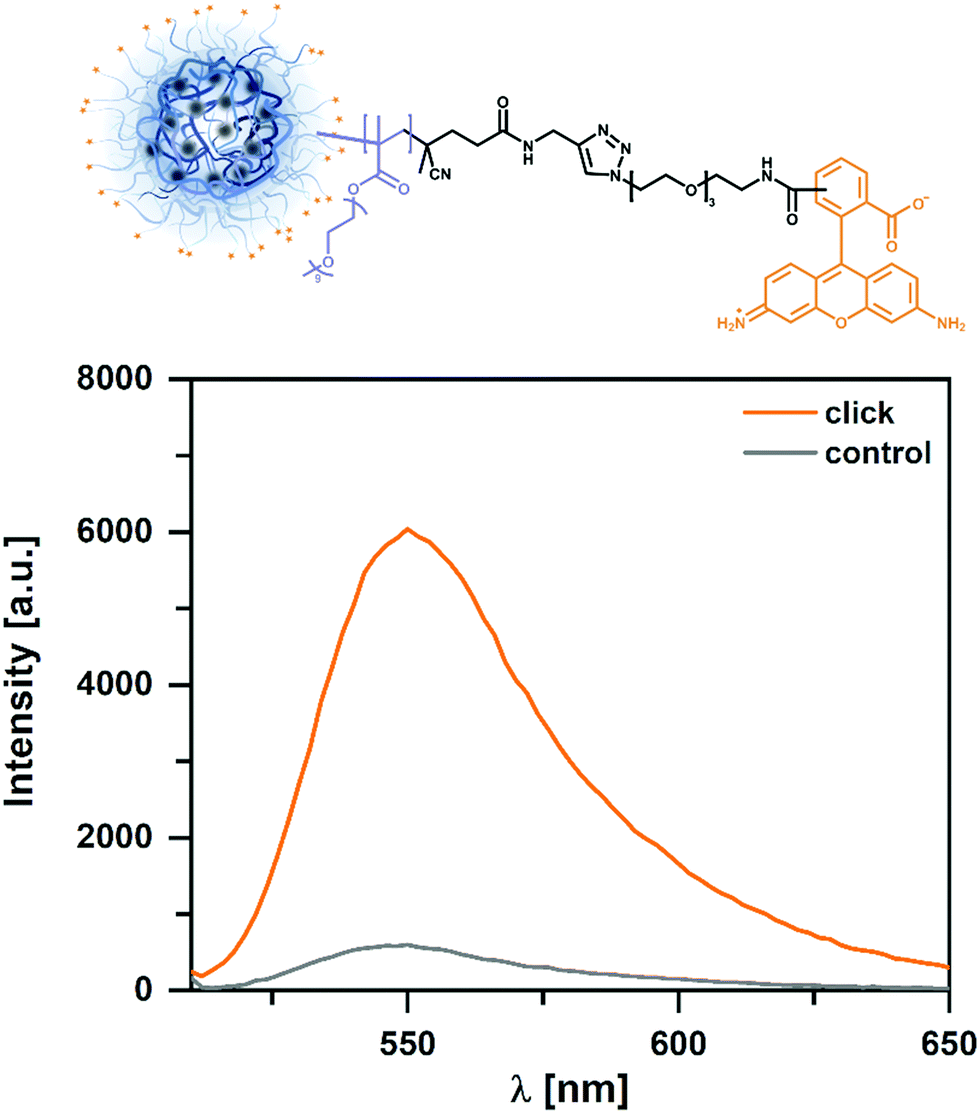 | ||
| Fig. 4 Fluorescence spectra of NGs functionalized with rhodamine via CuAAC demonstrates successful coupling of the dye. To ensure covalent attachment and not only physical entrapment, a control experiment was performed without the addition of copper and ascorbic acid. | ||
Examination of stimuli-responsive network properties
Based on the synthetic modularity of our platform, the multi-functional properties of the nanogel library were investigated. For this, we focused on the stimuli-responsive swelling behavior of the NGs. In aqueous medium, the different ionic functionalities (anionic AMPAA and cationic HIS) are expected to govern the network swelling through their protonation/deprotonation at different pH values. Furthermore, the incorporation of hydrophobic CHOLA is expected to reduce the network swelling due to hydrophobic interactions that act as additional physical crosslinks. To investigate these properties in detail, pH-dependent swelling profiles of cationic and anionic NGs were studied by DLS. Fig. 5 shows a clear pH-dependent volume phase transition of the NGs. The purely anionic AMPAA NGs show the expected transition from a collapsed to a swollen state with increasing pH (Fig. 5a). With increasing pH, the deprotonation of the carboxylic acid groups results in steric repulsion, thus leading to a swelling of the network and a volume phase transition between pH 6 and 7. A similar trend is visible when 20 mol% CHOLA is introduced to the network. However, the change in volume is significantly smaller for AMPAA CHOLA-20 NGs (3 fold) than for purely hydrophilic AMPAA (6 fold). This agrees with a higher structural integrity for the amphiphilic NGs as observed in the TEM images. As mentioned above, the reduced swelling and higher rigidity is suggested to result from the additional physical crosslinks that are formed by self-assembled hydrophobic domains of cholesterol in the NG network.14 In contrast to the anionic AMPAA NGs, the cationic HIS NGs exhibit an opposite swelling profile (Fig. 5b). With decreasing pH, protonation of the basic imidazole groups in the histamine functionalized NGs increases network swelling (5-fold). A volume phase transition is observed between pH 5 and 6. Similar to the anionic NGs, incorporation of 20 mol% CHOLA limits the magnitude of response in swelling significantly (3-fold). Overall, these results demonstrate the successful transfer of properties from the small molecules (acidic AMPAA and basic HIS) to the NG networks via functionalization of the PFP active esters.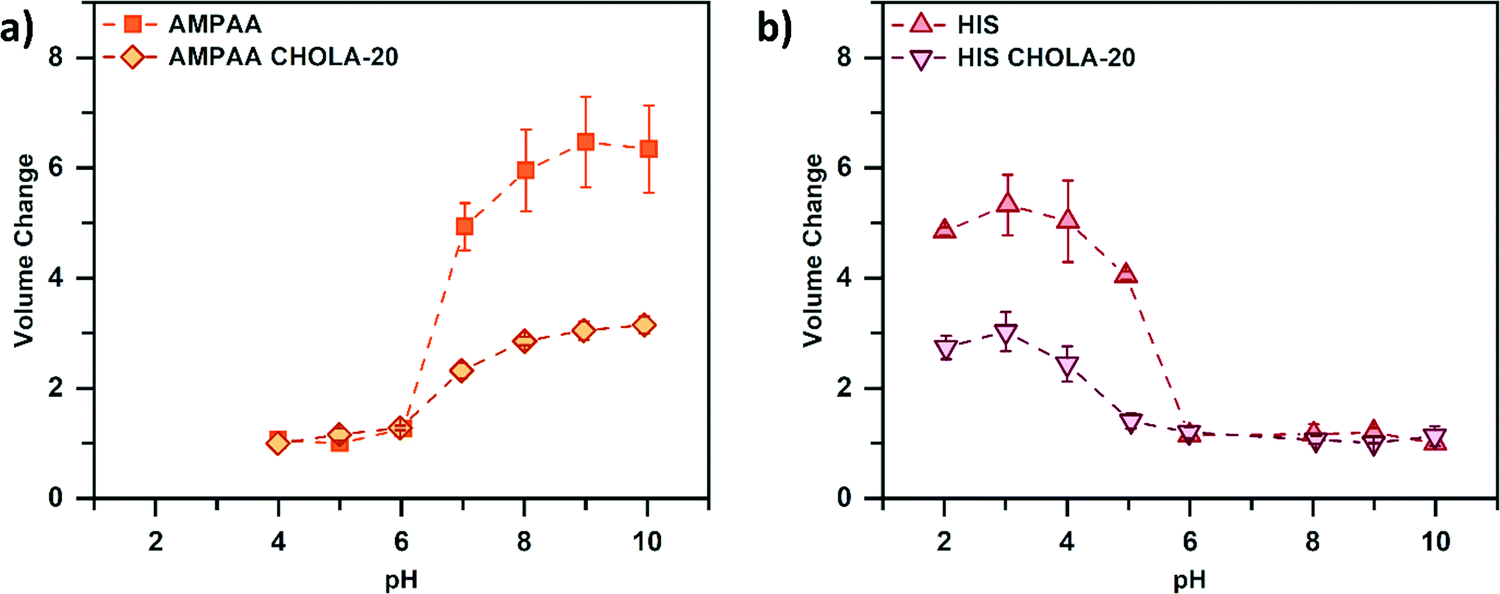 | ||
| Fig. 5 pH-dependent swelling profile of (a) anionic and (b) cationic NGs. Incorporation of 20 mol% of hydrophobic CHOLA groups results in a reduced swelling due to the formation of hydrophobic nanodomains that can act as physical crosslinks in the interior of the NGs. | ||
In addition to the pH-responsive swelling properties, the NG networks are also sensitive to reduction agents due to the incorporated disulfide crosslinker (DSDMA). Cleavage of the –S–S– bonds can be used to control the network integrity.52,53 In amphiphilic nanogel networks, crosslinker cleavage has a complex influence on the NG size. For example, it was shown that degradation of disulfide crosslinked amphiphilic nanogels followed a two-step profile upon incubation with a reducing agent.54 In the first stage, the degree of swelling increased due to the decreased crosslinking density. In the second stage, the size decreased again due to particle fragmentation. Similar profiles are observed in core-crosslinked BCP micelles. Here, reductive cleavage of disulfide crosslinking points needs to be followed by dilution below the critical micellization concentration to trigger complete disintegration.55 In hydrophilic networks, a direct decrease in size can often be observed. In such cases, hydrophilic degradation products/fragments are rapidly released into the medium.56 Thus, to examine the influence of disulfide cleavage on the network properties, turbidity measurements were performed. Such measurements can correlate a decreasing light scattering intensity to an increasing network swelling, i.e., a reduced scattering contrast.57–59 For this, the turbidity of NG dispersions was followed after the addition of 10 mM dithiothreitol (DTT). As can be seen in Fig. 6, the turbidity decreases over time for the overall hydrophilic and anionic NGs when treated with DTT in PBS. In contrast, the turbidity of the controls (no DTT, pure PBS) stays the same. The decrease in turbidity suggests an increasing swelling (increasing mesh size) due to the cleavage of disulfide linkers. However, the turbidity for both NGs reaches a plateau at around 70% of the initial value, thus suggesting an incomplete nanogel disintegration even at long incubation times. It is assumed that this effect can be attributed to the covalent attachment of the BCP surfactant to the NG network. At the surface of the NGs, the reactive hydrophobic anchoring block of the surfactant can act as additional crosslinker. Since these crosslinking points do not contain cleavable disulfide bonds, they prevent complete disintegration of the network. To test this assumption, we prepared PHPMA NGs without the PPEGMA shell, from reactive precursor particles prepared via emulsion polymerization with SDS as surfactant. As can be seen in Fig. S5 (ESI†) the turbidity decreases significantly more than for the NGs with the attached shell, thus indicating a complete degradation. These results support our hypothesis that the NGs with the hydrophilic BCP shell do not disintegrate completely. Instead, the mesh size of the network increases while the surfactant stabilizes the colloidal structure on its surface. Nevertheless, controlling the swelling through reducing agents and pH gives the opportunity to precisely tailor the NGs properties to specific applications and cargoes in future studies.
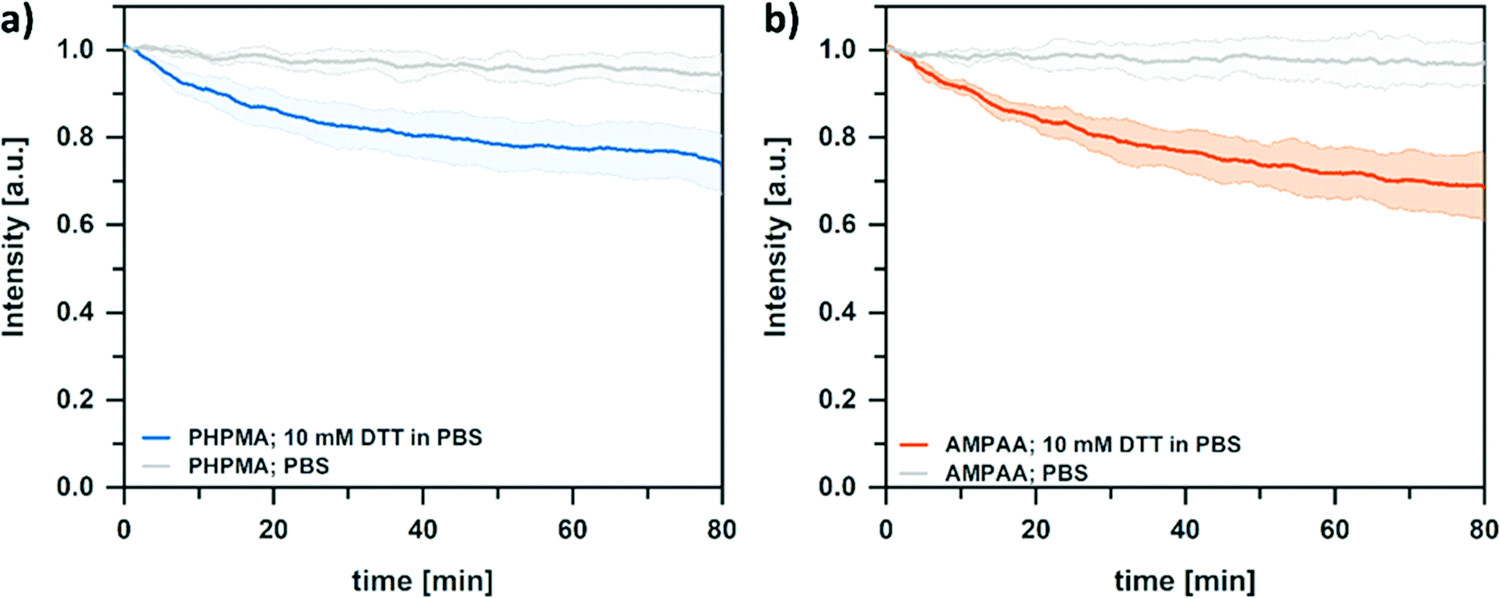 | ||
| Fig. 6 Turbidity measurements of NGs dispersion of (a) PHPMA and (b) AMPAA treated with 10 mM DTT indicate a cleavage of the disulfide bond of the reduction-cleavable DSDMA crosslinker that results in a swelling of the NGs. Solid lines represent the average of 3 turbidity measurements. Colored areas represent the error as standard deviation. | ||
Conclusion
In this work, we demonstrated the development of a versatile synthetic platform for the preparation of multi-functional amphiphilic nanogels. These colloidal particles combine a pH- and reduction-sensitve internal network with a reactive hydrophilic shell that enables further surface modification. Key to this approach is the use of an alkyne end functionalized BCP surfactant for the preparation of reactive PPFPMA precursor particles. As amphiphlic surfactant, alkyne-P(PEGMA-b-(PFPMA-co-MAPMA)) contains a hydrophilic PPEGMA block and a hydrophobic PPFPMA block with reactive APMA vinyl groups. This hydrophobic anchoring block enables covalent incorporation of the surfactant moieties to the PPFPMA nework during free radical emulsion polymerization of PFPMA with a reduction-cleavable crosslinker. Hence, reactive precursor particles were prepared that contain a hydrophilic PPEGMA shell with reactive alkyne groups on the surface. These precursor particles enabled the formation of a small library of multi-functional nanogels. Here, a reduction-sensitive swelling/degradation was realized by the utilization of a disulfide based crosslinker (DSDMA) already during particle synthesis. In addition, more functionalities were introduced in a post-particle preparation approach. Due to the orthogonality of the reactions for modification of interior (active ester chemistry) and surface (copper-catalyzed azide–alkyne cycloaddition), sequential functionalization was enabled. For the interior, acidic AMPAA or basic HIS functionalities were used in combination with hydrophobic groups to prepare pH-sensitive amphiphilic NGs. These nanogels exhibit similar colloidal features such as crosslinking density and size distribution but show opposing swelling profiles as response to pH changes. For the hydrophilic shell, subsequent modification was demonstrated with an azide-containing rhodamine model compound. The successful orthogonal functionalization of core and shell demonstrates the potential of this synthetic platform to tailor nanogel properties to advanced applications very precisely. In this context, it is suggested that functionalizing the surface with specific functionalities such as targeting moieties (e.g. proteins) can be used to accurately tune the interactions of such nanogels with biological systems in future studies. Together with the hydrophilic shell and biodegradable crosslinking points, this modular synthetic platform lays the foundation for the development of novel, highly functional nanocarriers for targeted delivery of (hydrophobic) cargoes in response to external triggers.Experimental
Materials
All starting materials and reagents were purchased from commercial sources and used without further purification, unless otherwise stated. Pentafluorophenyl methacrylate (PFPMA),60,61 CHOLA62,63 and DSDMA64 were prepared according to literature procedures. Dialysis was performed in regenerated cellulose dialysis tubes (Spectra/Por® 4 width: 45 mm, MWCO 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–14000 g mol−1, Repligen, USA).
000–14000 g mol−1, Repligen, USA).
Methods
Synthesis
1H NMR (500 MHz, DMSO-d6): δ = 8.39 (br, J = 7.1 Hz, 2H, –NH–), 3.86 (m, 4H, –CH2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) H), 3.07 (s, 2H, –CH), 2.39–2.04 (m, 8H, –CH2–CH2–C
H), 3.07 (s, 2H, –CH), 2.39–2.04 (m, 8H, –CH2–CH2–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1.68 (s, 3H, –CH3), 1.64 (s, 3H, –CH3).
O), 1.68 (s, 3H, –CH3), 1.64 (s, 3H, –CH3).
Step 2: ethylsulfanyl(ethylsulfanylcarbonylcarbothioyldisulfanyl)methanthione (2) was synthesized according to literature.67 Ethanethiol (10.0 g, 161 mmol) was added dropwise over 30 min to a solution of cold sodium hydroxide (6.44 g, 161 mmol) in anhydrous ether (100 ml). After stirring at room temperature overnight, carbon disulfide (12.25 g, 161 mmol) was added and stirred at room temperature for 2 h. The reaction mixture was washed with hexane and the solvent was evaporated and the residue was dispersed in 150 ml anhydrous ether. Iodine (7.9 g, 31 mmol) was added in portions and stirred at room temperature for 3 h. The reaction mixture was filtered, and the filtrate was washed with an aqueous Na2S2O4 solution (5 wt%), water and brine. The organic phase was dried over Na2SO4 and removed under reduced pressure. The product was used in the following step without further purification.
Step 3: synthesis of 2-cyano-5-oxo-5-(prop-2-yn-1-ylamino)pentan-2-yl ethyl carbonotrithioate (alkyne chain transfer agent) (3) was synthesized according a modified literature procedure for a similar CTA without the click function.67 Under nitrogen atmosphere 1.2 g of crude product of step 2 (4.2 mmol) and dialkyne-V-501 (3 g, 8.4 mmol, 1) were dispersed in 40 mL EtOAc and was heated to 80 °C overnight. The solvent was evaporated, and the residue was purified by automated column chromatography using a gradient solvent mixture (hexane/EtOAc 20% to 100% EtOAc) to yield the product as yellow oil (2 g, 6.6 mmol, 79%).
1H NMR (600 MHz, CDCl3): δ =6.00 (br, 1H, NH), 4.05 (dd, J = 5.3, 2.6 Hz, 2H, NH–CH2–), 3.34 (q, J = 7.4 Hz, 2H, –CH2–CH3), 2.57–2.33 (m, 4H, –CH2–CH2–CO–), 2.25 (t, J = 2.6 Hz, 1H, –CCH), 1.88 (s, 3H, –CH3), 1.35 (t, J = 7.5 Hz, 3H, –CH2–CH3) ppm.
13C NMR (151 MHz, CDCl3): δ = 217.11 (–CS), 170.11 (–CO), 119.29 (–CN), 79.29 (–CCH), 71.97 (–CCH), 47.81 (Cquat), 34.82 (–CH2–CH2–CO–)), 31.63 (–CH2–CO–), 31.49 (–CH2–CH3). 29.49 (–NH–CH2–), 24.29 (–CH3), 12.09 (–CH2–CH3).
HRMS: cal. for C12H16N2OS3 [M + Na]+: 323.0322, found [M + Na]+: 323.0352.
Synthesis of PPEGMA The synthesis was carried out according to a slightly modified literature procedure.68 PEGMA (10 g, 20 mmol), Alkyne CTA (100 mg, 332 μmol) and V70 (13 mg, 42 μmol) were dissolved in 20 ml anhydrous dioxan. 1 ml anhydrous DMF was added as an internal standard. Argon was bubbled through the polymerization solution for 20 min before the reaction vessel was sealed and immersed in an oil bath at 40 °C for 16 hours. The conversion of the reaction was determined via1H NMR spectroscopy of the reaction mixture before and after the reaction. By monitoring the decreasing integral of the vinyl peaks relative to DMF peak showed a monomer conversion of 83%. The resulting polymer was precipitated in cold hexane and centrifugated, redissolved in DCM and precipitated in hexane another 2 times. The precipitated polymer dried on high vacuum and was obtained as yellow oil (8.4 g).
Monomer conversion of 83% (by 1H NMR) corresponds to 25000 Da.
| GPC Mn = 28500 Da, Mw = 39000 Da, Đ = 1.36 |
Monomer Conversion of 45% (by 1H NMR) corresponds to a PPFPMA block of 9000 Da
| GPC Mn = 29500 Da, Mw = 42000 Da, Đ = 1.42 |
000 rpm) and the supernatant was removed and replaced by ultrapure water. The particles were redispersed by vortex and sonication in an ultrasonic bath. The centrifugation-washing-redispersion step was repeated 5 times before the particles were freeze-dried and obtained as white powder (3.7 g).
Because of the low solubility of AMPAA in DMF, the general procedure was modified as follows. AMPAA was dissolved in 30 ml of DMSO and added to dispersed particles in DMF. The reaction was allowed to proceed at 70 °C for 4 days. For AMPAA CHOLA-20 NGs the amines were added sequentially. First, CHOLA (38 mg, 80 μmol) and TEA (120 mg, 1.2 mmol) were added to the particle dispersion in DMF and reacted at 70 °C. After 24 h AMPAA dissolved in 30 mL of DMSO was added and the reaction was allowed to proceed for another 4 days.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially funded by the DFG (Deutsche Forschungsgemeinschaft/German Research Foundation) (KL 3152/2-1, project number: 430915250). It was further supported by the collaborative research center (CRC) 1112 with funding for instrumentation. Furthermore, we would like to acknowledge the assistance of the Core Facility BioSupraMol supported by the Deutsche Forschungsgemeinschaft (DFG). We are thankful for additional financial support for consumables from Verband der Chemischen Industrie eV (VCI) and Focus Area Nanoscale of the Freie Universität Berlin. Furthermore, we would like to thank Ruiguang Cui for assistance with TEM measurements. A. Gruber especially thanks the CRC 1112 and Focus area Nanoscale for scholarships to finance her PhD.References
- D. Klinger and K. Landfester, Polymer, 2012, 53, 5209–5231 CrossRef CAS.
- J. C. Cuggino, E. R. O. Blanco, L. M. Gugliotta, C. I. A. Igarzabal and M. Calderón, J. Controlled Release, 2019, 307, 221–246 CrossRef CAS PubMed.
- M. Karg, A. Pich, T. Hellweg, T. Hoare, L. A. Lyon, J. J. Crassous, D. Suzuki, R. A. Gumerov, S. Schneider, I. I. Potemkin and W. Richtering, Langmuir, 2019, 35, 6231–6255 CrossRef CAS PubMed.
- Q. Zhao, S. Zhang, F. Wu, D. Li, X. Zhang, W. Chen and B. Xing, Angew. Chem., Int. Ed., 2021, 60, 14760–14778 CrossRef CAS PubMed.
- B. Schulte, K. Rahimi, H. Keul, D. E. Demco, A. Walther and M. Moller, Soft Matter, 2015, 11, 943–953 RSC.
- A. Gruber, D. Işık, B. B. Fontanezi, C. Böttcher, M. Schäfer-Korting and D. Klinger, Polym. Chem., 2018, 9, 5572–5584 RSC.
- C. Biglione, T. M. P. Neumann-Tran, S. Kanwal and D. Klinger, J. Polym. Sci., 2021, 59, 2665–2703 CrossRef CAS.
- M. A. Gauthier, M. I. Gibson and H.-A. Klok, Angew. Chem., Int. Ed., 2009, 48, 48–58 CrossRef CAS PubMed.
- M. I. Gibson, E. Fröhlich and H.-A. Klok, J. Polym. Sci., Part A: Polym. Chem, 2009, 47, 4332–4345 CrossRef CAS.
- K. A. Günay, P. Theato and H. A. Klok, J. Polym. Sci., Part A: Polym. Chem, 2013, 51, 1–28 CrossRef.
- A. Gruber, L. Navarro and D. Klinger, Adv. Mater. Interfaces, 2020, 7, 1901676 CrossRef CAS.
- C. Fleischmann, J. Gopez, P. Lundberg, H. Ritter, K. L. Killops, C. J. Hawker and D. Klinger, Polym. Chem., 2015, 6, 2029–2037 RSC.
- X. Wang, J. L. Davis, B. M. Aden, B. S. Lokitz and S. M. Kilbey, Macromolecules, 2018, 51, 3691–3701 CrossRef CAS.
- A. F. Thünemann, A. Gruber and D. Klinger, Langmuir, 2020, 36, 10979–10988 CrossRef PubMed.
- T. Bewersdorff, A. Gruber, M. Eravci, M. Dumbani, D. Klinger and A. Haase, Int. J. Nanomed., 2019, 14, 7861–7878 CrossRef CAS PubMed.
- R. M. Pearson, V. V. Juettner and S. Hong, Front. Chem., 2014, 2, 108 Search PubMed.
- K. Strojan, A. Leonardi, V. B. Bregar, I. Križaj, J. Svete and M. Pavlin, PLoS One, 2017, 12, e0169552 CrossRef PubMed.
- S. Essa, J. M. Rabanel and P. Hildgen, Int. J. Pharm., 2011, 411, 178–187 CrossRef CAS PubMed.
- J. S. Suk, Q. Xu, N. Kim, J. Hanes and L. M. Ensign, Adv. Drug Delivery Rev., 2016, 99, 28–51 CrossRef CAS PubMed.
- H. Hatakeyama, H. Akita and H. Harashima, Biol. Pharm. Bull., 2013, 36, 892–899 CrossRef CAS PubMed.
- Y. Fang, J. Xue, S. Gao, A. Lu, D. Yang, H. Jiang, Y. He and K. Shi, Drug Delivery, 2017, 24, 22–32 CrossRef CAS PubMed.
- M. Mehanny, R. M. Hathout, A. S. Geneidi and S. Mansour, J. Biomed. Mater. Res. A, 2017, 105, 1433–1445 CrossRef CAS PubMed.
- I. Khalin, D. Heimburger, N. Melnychuk, M. Collot, B. Groschup, F. Hellal, A. Reisch, N. Plesnila and A. S. Klymchenko, ACS Nano, 2020, 14, 9755–9770 CrossRef CAS PubMed.
- J. T. Peters, S. Verghese, D. Subramanian and N. A. Peppas, Regen. Biomater., 2017, 4, 281–287 CrossRef CAS PubMed.
- E. Mauri, F. Cappella, M. Masi and F. Rossi, J. Drug Delivery Sci. Technol., 2018, 46, 87–92 CrossRef CAS.
- H. Sui, Z. Gao, J. Guo, Y. Wang, J. Yuan, J. Hao, S. Dong and J. Cui, Langmuir, 2019, 35, 16869–16875 CrossRef CAS PubMed.
- V. F. Motlaq, K. D. Knudsen and B. Nyström, J. Colloid Interface Sci., 2018, 524, 245–255 CrossRef CAS PubMed.
- D. S. Spencer, B. C. Luu, D. W. Beckman and N. A. Peppas, J. Polym. Sci., Part A: Polym. Chem, 2018, 56, 1536–1544 CrossRef CAS PubMed.
- A. Lee, P. Lundberg, D. Klinger, B. F. Lee, C. J. Hawker and N. A. Lynd, Polym. Chem., 2013, 4, 5735–5742 RSC.
- W. H. Lee, J. R. Booth and S. A. F. Bon, Biomacromolecules, 2020, 21, 4599–4614 CrossRef CAS PubMed.
- W. Li, J. A. Yoon and K. Matyjaszewski, J. Am. Chem. Soc., 2010, 132, 7823–7825 CrossRef CAS PubMed.
- D. Y. Joh, Z. Zimmers, M. Avlani, J. T. Heggestad, H. B. Aydin, N. Ganson, S. Kumar, C. M. Fontes, R. K. Achar, M. S. Hershfield, A. M. Hucknall and A. Chilkoti, Adv. Healthcare Mater., 2019, 8, 1801177 CrossRef PubMed.
- A. Das and P. Theato, Chem. Rev., 2016, 116, 1434–1495 CrossRef CAS PubMed.
- J. Müller, K. N. Bauer, D. Prozeller, J. Simon, V. Mailänder, F. R. Wurm, S. Winzen and K. Landfester, Biomaterials, 2017, 115, 1–8 CrossRef PubMed.
- P. J. Roth, F. D. Jochum, R. Zentel and P. Theato, Biomacromolecules, 2010, 11, 238–244 CrossRef CAS PubMed.
- P. J. Roth, M. Haase, T. Basché, P. Theato and R. Zentel, Macromolecules, 2010, 43, 895–902 CrossRef CAS.
- K. Philipps, T. Junkers and J. J. Michels, Polym. Chem., 2021, 12, 2522–2531 RSC.
- M. Antonietti and K. Landfester, Prog. Polym. Sci., 2002, 27, 689–757 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- D. Crespy and K. Landfester, Beilstein J. Org. Chem., 2010, 6, 1132–1148 CrossRef CAS PubMed.
- A. Czajka and S. P. Armes, J. Am. Chem. Soc., 2021, 143, 1474–1484 CrossRef CAS PubMed.
- P. A. Lovell and F. J. Schork, Biomacromolecules, 2020, 21, 4396–4441 CrossRef CAS PubMed.
- J.-F. Lutz, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3459–3470 CrossRef CAS.
- J. K. Oh, D. J. Siegwart, H.-I. Lee, G. Sherwood, L. Peteanu, J. O. Hollinger, K. Kataoka and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 5939–5945 CrossRef CAS PubMed.
- M. A. Macchione, M. F. Sacarelli, A. C. Racca, C. Biglione, G. M. Panzetta-Dutari and M. C. Strumia, Soft Matter, 2019, 15, 9700–9709 RSC.
- C. Biglione, A. Sousa-Herves, M. Menger, S. Wedepohl, M. Calderón and M. C. Strumia, RSC Adv., 2015, 5, 15407–15413 RSC.
- R. Charbaji, M. Kar, L. E. Theune, J. Bergueiro, A. Eichhorst, L. Navarro, P. Graff, F. Stumpff, M. Calderón and S. Hedtrich, Small, 2021, 17, 2007963 CrossRef CAS PubMed.
- J. Kopecek and P. Kopeckova, Adv. Drug Delivery Rev., 2010, 62, 122–149 CrossRef CAS PubMed.
- T. Lammers and K. Ulbrich, Adv. Drug Delivery Rev., 2010, 62, 119–121 CrossRef CAS PubMed.
- N. Morimoto, T. Endo, M. Ohtomi, Y. Iwasaki and K. Akiyoshi, Macromol. Biosci., 2005, 5, 710–716 CrossRef CAS PubMed.
- K. Engberg, D. J. Waters, S. Kelmanovich, R. Parke-Houben, L. Hartmann, M. F. Toney and C. W. Frank, Polymer, 2016, 84, 371–382 CrossRef CAS.
- Y. Zhan, M. Gonçalves, P. Yi, D. Capelo, Y. Zhang, J. Rodrigues, C. Liu, H. Tomás, Y. Li and P. He, J. Mater. Chem. B, 2015, 3, 4221–4230 RSC.
- H. Yang, Q. Wang, S. Huang, A. Xiao, F. Li, L. Gan and X. Yang, ACS Appl. Mater. Interfaces, 2016, 8, 7729–7738 CrossRef CAS PubMed.
- Z.-Y. Qiao, R. Zhang, F.-S. Du, D.-H. Liang and Z.-C. Li, J. Controlled Release, 2011, 152, 57–66 CrossRef CAS PubMed.
- R. Wei, L. Cheng, M. Zheng, R. Cheng, F. Meng, C. Deng and Z. Zhong, Biomacromolecules, 2012, 13, 2429–2438 CrossRef CAS PubMed.
- X. Zhang, K. Achazi, D. Steinhilber, F. Kratz, J. Dernedde and R. Haag, J. Controlled Release, 2014, 174, 209–216 CrossRef CAS PubMed.
- D. Klinger, E. M. Aschenbrenner, C. K. Weiss and K. Landfester, Polym. Chem., 2012, 3, 204–216 RSC.
- N. Al-Manasir, K. Zhu, A.-L. Kjøniksen, K. D. Knudsen, G. Karlsson and B. Nyström, J. Phys. Chem. B, 2009, 113, 11115–11123 CrossRef CAS PubMed.
- M. Lechner, J. Serb. Chem. Soc., 2005, 70, 361–369 CrossRef CAS.
- M. Eberhardt, R. Mruk, R. Zentel and P. Théato, Eur. Polym. J., 2005, 41, 1569–1575 CrossRef CAS.
- J.-C. Blazejewski, J. W. Hofstraat, C. Lequesne, C. Wakselman and U. E. Wiersum, J. Fluorine Chem., 1998, 91, 175–177 CrossRef CAS.
- Y. Nie, M. Günther, Z. Gu and E. Wagner, Biomaterials, 2011, 32, 858–869 CrossRef CAS PubMed.
- M. D. Kearns, A.-M. Donkor and M. Savva, Mol. Pharm., 2008, 5, 128–139 CrossRef CAS PubMed.
- R. H. Utama, Y. Jiang, P. B. Zetterlund and M. H. Stenzel, Biomacromolecules, 2015, 16, 2144–2156 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- K. Luo, J. Yang, P. Kopečková and J. Kopeček, Macromolecules, 2011, 44, 2481–2488 CrossRef CAS PubMed.
- J. Peng, Q. Xu, Y. Ni, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2019, 10, 2064–2072 RSC.
- S. Hartmann, L. Nuhn, B. Palitzsch, M. Glaffig, N. Stergiou, B. Gerlitzki, E. Schmitt, H. Kunz and R. Zentel, Adv. Healthcare Mater., 2015, 4, 522–527 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2sm00116k |
| This journal is © The Royal Society of Chemistry 2022 |