
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation and characterisation of graphene oxide containing block copolymer worm gels†
Qi
Yue
 ab,
Shang-Pin
Wen
a and
Lee A.
Fielding
*ab
ab,
Shang-Pin
Wen
a and
Lee A.
Fielding
*ab
aDepartment of Materials, School of Natural Sciences, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: lee.fielding@manchester.ac.uk
bHenry Royce Institute, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
First published on 2nd March 2022
Abstract
This paper reports a generic method for preparing reinforced nanocomposite worm-gels. Aqueous poly(glycerol monomethacrylate)-b-poly(2-hydroxypropyl methacrylate) (PGMA–PHPMA) and methanolic poly(glycerol monomethacrylate)-b-poly(benzyl methacrylate) (PGMA–PBzMA) worm gels were prepared by RAFT-mediated polymerisation-induced self-assembly (PISA). The former system undergoes a reversible worm-to-sphere degelation transition upon cooling to 5 °C whilst the latter system undergoes the same transition on heating to 56 °C. This transition allows these copolymer dispersions to be readily mixed with graphene oxide (GO) whilst in a low viscosity state and form nanocomposite gels on returning to room temperature via a sphere-to-worm transition. Various quantities of GO were added to the studied copolymer dispersions at a fixed copolymer content of 15% w/w. A general trend was observed whereby relatively small quantities of GO caused the gel strength of the nanocomposite gel to be higher than that of the pristine worm-gel, as determined by oscillatory rheology. Additional quantities of GO resulted in gel weakening or prevented gel-reformation altogether. For instance, 15% w/w PGMA52–PHPMA130 worm gels had a storage modulus (G′) of approximately 1.5 kPa. The addition of 1.5% w/w GO based on the copolymer caused G′ to increase to approximately 4.0 kPa but >1.5% w/w GO resulted in gel strengths <1.0 kPa. A combination of aqueous electrophoresis and transmission electron microscopy measurements were used to investigate the mechanism of nanocomposite gel formation. It was observed that the PGMA-based copolymers readily absorb onto the surface of GO. Thus, the role of GO is both to strengthen the worm-gels when an optimal concentration of GO is used, but also prevent worm-reformation if too much copolymer becomes absorbed on the surface of the sheets.
Introduction
Composite hydrogels are an interesting class of materials which have become widely investigated in recent years.1–6 For example, in 2002 Haraguchi et al.7 reported a high-strength nanocomposite hydrogel obtained by in situ polymerisation of N-isopropylacrylamide (NIPAm) in a suspension of Hectorite nanosheets. The reported tensile strength of these gels was approximately 10 times that of traditional hydrogels. Subsequent studies have shown that the preparation of nanocomposite gels can be relatively simple, economical and environmentally friendly, and result in hydrogels with significantly improved mechanical properties and swelling behaviour.8–10 At present, numerous types of nanocomposite gel have been reported including graphene oxide-based composite gels,11–16 carbon nanotube composite gels,17–21 nanocellulose composite gels,22–26 inorganic clay composite gels7,27–32 and other inorganic nanoparticle-containing composite gels.33–37Graphene oxide (GO) is a water-dispersible 2D material which has the planar structure of graphene but has a number of oxy-functional groups present.11,38–40 The oxy functional groups are hydroxyl and epoxy groups on the basal plane, with smaller amounts of carboxyl, carbonyl, phenol, lactone, and quinone at the sheet edges.39,41 The presence of these functional groups provides potential advantages for using GO in numerous applications, particularly due to the enhanced water dispersibility of GO in comparison to graphene.42,43 Furthermore, it is known that the incorporation of 2D materials such as graphene oxide into polymeric hydrogels can drastically modify their mechanical,44,45 biocompatibility46–49 and optical properties.50,51 For instance, Liu52 used GO as a cross-linker and acrylamide (AM) as a monomer to prepare PAM-GO nanocomposite gels by in situ polymerisation. Compared with other cross-linked PAM hydrogels, it had improved mechanical properties. For example, breaking elongation reached more than 3000% and the observed tensile strength was nearly 390 kPa. In addition, photothermally sensitive GO-NIPAM composite gels showed optical sensitivity, as the phase transition of the gels could be controlled by an infrared laser.50 Bai et al., reported supramolecular GO containing gels using different copolymers as a crosslinker. In this case, they indicated that hydrogen bonding, π-stacking and hydrophobic effects between copolymer and GO sheets, as well as the lateral dimensions of the GO sheets influenced gel formation. In addition, gel formation could be adjusted by modulating the attraction between the GO sheets and copolymer.53
Block copolymer self-assembly is generally used to prepare nanoparticles and structures in the length scale of 10 nm to 1 μm. This length-scale can greatly affect the physical, chemical and biological properties of these materials.54–56 Block copolymer self-assembly in a dispersion can result in a variety of ordered structures such as spherical micelles (spheres), worm-like micelles (worms), and polymersomes (vesicles).57–61 The self-assembled structures formed can be adjusted by changing e.g., the solvent or relative molar mass of each block of the copolymer. Relatively recently, polymerisation-induced self-assembly (PISA) via reversible addition–fragmentation chain-transfer (RAFT) polymerisation has been demonstrated to be a versatile and efficient route towards the synthesis of various types of block copolymer nano-objects.55,62 The particles formed can have a range of functionalities, both in their core and corona.63 RAFT aqueous dispersion polymerisation can be used to directly prepare block copolymer nanoparticles in water with a range of morphologies. For example, poly(glycerol monomethacrylate)-b-poly(2-hydroxypropyl methacrylate) (PGMA–PHPMA) prepared via RAFT aqueous dispersion polymerisation readily forms so-called ‘worm gels’ which have been widely studied and reported on.58–60,64 These PGMA–PHPMA worm-gels are free-standing at room temperature but become free-flowing liquids when cooled to 4 °C. This reversible de-gelation transition is attributed to worm-to-sphere order-order transitions which cause a loss of worm entanglement.59,65,66 Due to this thermo-responsive property, these gels can be used as sterilisable gels via ultrafiltration at low temperatures. Other systems have been reported which show a similar transition when exposed to a suitable stimulus such as temperature or pH change.59,67,68 However, RAFT aqueous dispersion polymerisation is only suited to a limited range of formulations such as the PGMA–PHPMA system described above and e.g., poly(ethylene glycol)-b-poly(4-hydroxybutyl acrylate).69 Therefore, research into RAFT-mediated dispersion polymerisation in a wider range of media such has proliferated.70–72 Interestingly, reversible gel transitions can also be triggered by heating for some block copolymer worm gel systems. For instance, poly(lauryl methacrylate)–poly(benzyl methacrylate) (PLMA–PBzMA) copolymers showed a worm-to-sphere-to-worm morphological transition when heating above 50 °C and returning to room temperature.66 This behaviour has also been demonstrated for PGMA–PBzMA copolymers prepared in alcoholic media.73 Thus these reversible worm gel transitions provide an opportunity to load these materials with fillers such as GO and study how self-assembly and the resulting properties are affected.
Herein, the preparation of a fascinating class of 3D composite hydrogels comprising 1D block copolymer worm-like micelles and 2D graphene oxide sheets is reported using two previously reported worm-gel formulations, aqueous PGMA–PHPMA and methanolic PGMA–PBzMA.58,59,64,74,75 The reversible de-gelation of the copolymer worm-gels on cooling or heating allows facile mixing of GO with the copolymer dispersions (Fig. 1). On returning to room temperature composite gels are formed and subsequently investigated using a combination of transmission electron microscopy and rheology.
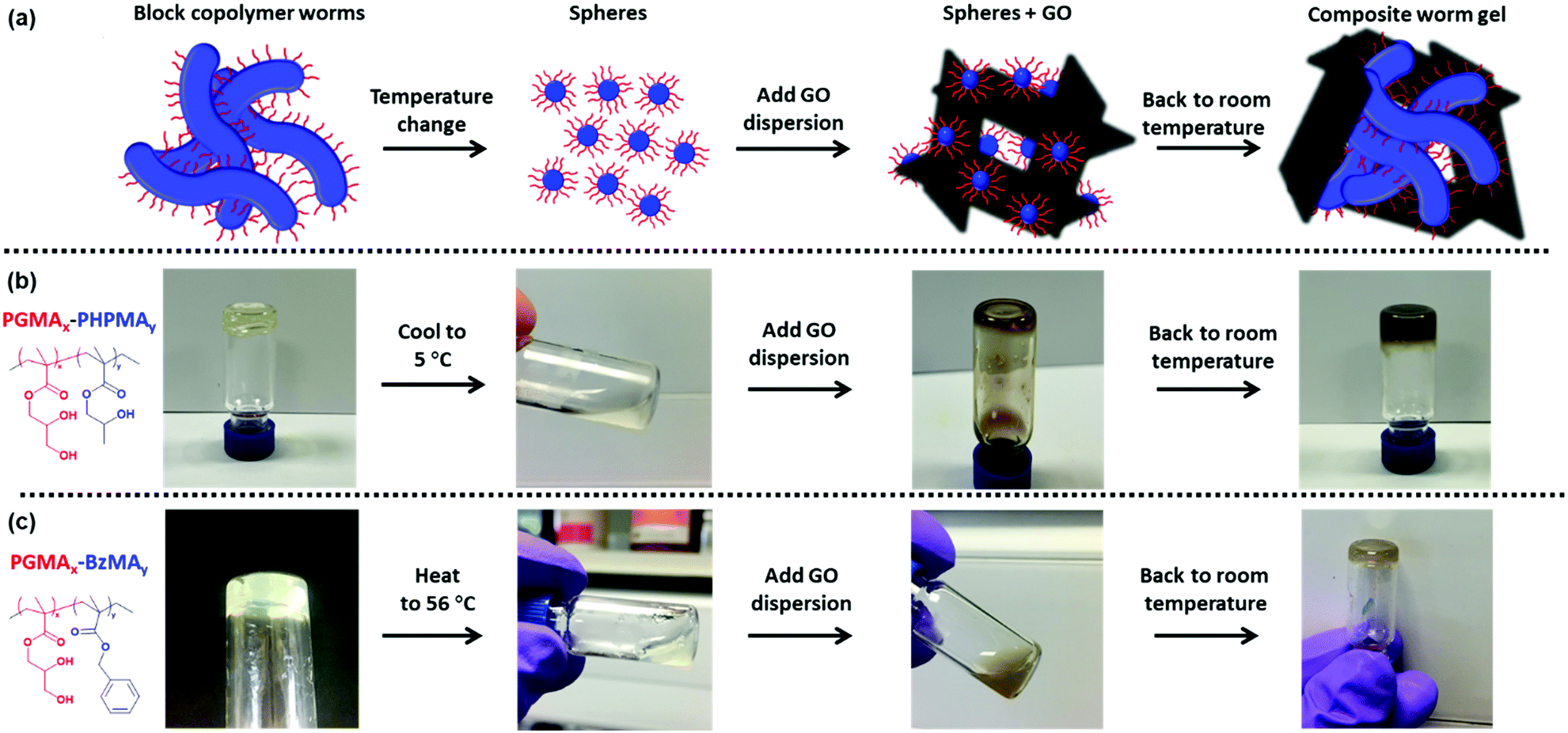 | ||
| Fig. 1 (a) Scheme for preparing graphene oxide containing block copolymer worm gels. (b) PGMAx–PHPMAy copolymer dispersions are cooled to 5 °C, mixed with graphene oxide (GO) and allowed to return to room temperature to form free-standing nanocomposite gels. (c) PGMAx–PBzMAy copolymer dispersions are heated to 56 °C, mixed with graphene oxide and allowed to return to room temperature to form free-standing nanocomposite gels. | ||
Experimental
Materials
All reagents were purchased from Sigma-Aldrich (UK) and used as received, unless otherwise noted. Glycerol monomethacrylate (GMA) was kindly donated from GEO Specialty Chemicals (UK), 2-Hydroxypropyl methacrylate (HPMA, 97%) was purchased from Alfa Aesar (UK). Benzyl methacrylate (BzMA) was purchased from Alfa Aesar (UK) and passed through a column of activated basic alumina to remove inhibitors and impurities before use. Methanol (>99.9%) and ethanol (95%) were purchased from Fisher Scientific (UK) and used as received. Graphene oxide aqueous dispersion was purchased form Graphena (Spain) and purified before use. 4-Cyano-4-(2-phenylethane sulfanylthiocarbonyl) sulfanylpentanoic acid (PETTC) was prepared in-house using previously published methods.76,77 Deionised water was used in all experiments.Synthesis of poly(glycerol monomethacrylate) (PGMA) via RAFT solution polymerisation
The preparation of PGMA by RAFT solution polymerisation has been reported extensively in the literature.59 In this work, polymerisation of GMA was conducted in ethanol at 70 °C. This protocol afforded PGMA macromolecular chain transfer agents (macro-CTAs; denoted Gx) with narrow molecular weight distributions at high yield. A typical protocol is as follows. For a target degree of polymerisation of 50 (G50), GMA monomer (20 g, 124 mmol) and PETTC RAFT agent (0.8476 g, 2.5 mmol) were weighed into a 250 mL round-bottomed flask and purged with N2 for 30 min. 4,4-Azobis(4-cyanovaleric acid) (ACVA) initiator (0.14 g, 0.5 mmol, PETTC/ACVA molar ratio = 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0) and anhydrous ethanol (20 g, previously purged with N2 for 20 min) were then added, and the resulting yellow solution was degassed for a further 10 min while stirring to form a homogeneous solution. The flask was subsequently sealed and immersed in an oil bath set at 70 °C. After 120 min, the polymerisation was quenched by immersion in an ice bath and opening to air. The final degree of polymerisation (DP) was 52, as determined by 1H-NMR analysis (Fig. S3, ESI†) using D2O. The polymer was purified by dialysis (MWCO = 3500 g mol−1) against deionised water and freeze-dried to form a yellow powder. DMF GPC analysis indicated an Mn of 4700 g mol−1 and an Mw/Mn of 1.17 (Fig. S2, ESI†).
:0) and anhydrous ethanol (20 g, previously purged with N2 for 20 min) were then added, and the resulting yellow solution was degassed for a further 10 min while stirring to form a homogeneous solution. The flask was subsequently sealed and immersed in an oil bath set at 70 °C. After 120 min, the polymerisation was quenched by immersion in an ice bath and opening to air. The final degree of polymerisation (DP) was 52, as determined by 1H-NMR analysis (Fig. S3, ESI†) using D2O. The polymer was purified by dialysis (MWCO = 3500 g mol−1) against deionised water and freeze-dried to form a yellow powder. DMF GPC analysis indicated an Mn of 4700 g mol−1 and an Mw/Mn of 1.17 (Fig. S2, ESI†).
Preparation of PGMA–PHPMA worm gels via RAFT aqueous dispersion polymerisation of HPMA
The preparation of PGMA–PHPMA worm-gels by RAFT dispersion polymerisation has been reported extensively in the literature.59 A typical protocol for the synthesis of a PGMA52–PHPMA130 (G52–H130) worm gel is as follows. PGMA52 macro-CTA (5.0 g, 0.566 mmol) and HPMA monomer (11.4 g, 79.274 mmol; target DP = 130) were weighed into a 100 mL round bottomed flask and purged with N2 for 20 min. ACVA was added (31.74 mg, 0.101 mmol, CTA/ACVA molar ratio = 5.0) and purged with N2 for a further 5 min. Deionised water (65.84 mL, producing a 20.0% w/w aqueous solution), which had been previously purged with N2 for 30 min, was then added and the solution was purged for a further 5 min prior to immersion in an oil bath set at 70 °C. The reaction was stirred for 3 h before the polymerisation was quenched by exposure to air. The product was a soft free-standing gel. The absence of signals owing to the vinyl protons of the HPMA monomer in the 1H NMR spectrum indicated that the polymerisation had attained more than 99% conversion (Fig. S3a, ESI†).Preparation of PGMA–PBzMA worm gels via RAFT alcoholic dispersion polymerisation of BzMA
A typical protocol for the synthesis of a PGMA60–PBzMA70 (G60–B70) worm gel is as follows.73 PGMA60 macro-CTA (1.0 g, 0.096 mmol) and BzMA monomer (1.23 g, 7.0 mmol; target DP = 70) were weighed into a 25 mL round bottomed flask and purged with N2 for 15 min. ACVA was added (31.74 mg, 0.101 mmol, CTA/ACVA molar ratio = 5.0) and purged with N2 for a further 5 min. Methanol (8.958 g, producing a 20.0% w/w solution), which had been previously purged with N2 for 30 min, was then added, and the solution was purged for a further 5 min prior to immersion in an oil bath set at 64 °C and reflux for 24 h. The reaction was quenched by exposure to air. The product was a soft free-standing gel. The absence of signals at 6.15 ppm and 5.57 ppm owing to the vinyl protons of the BzMA monomer in the 1H NMR spectrum indicated that the polymerisation had attained high monomer conversion (Fig. S3b, ESI†).Preparation of graphene oxide-containing block copolymer worm gels
All GO dispersions were purified by dialysis against water for 5 days and sonicated in a bath sonicator for 20 min before use. The mean intensity-average particle diameter for the GO dispersion after bath sonication was ∼1900 nm. TEM studies indicated that the dispersion contained a broad distribution of sheet sizes, with a significant population of sub-μm sheets (see Fig. 2 and 3 and Fig. S7, S8, S12 and S13 in the ESI†).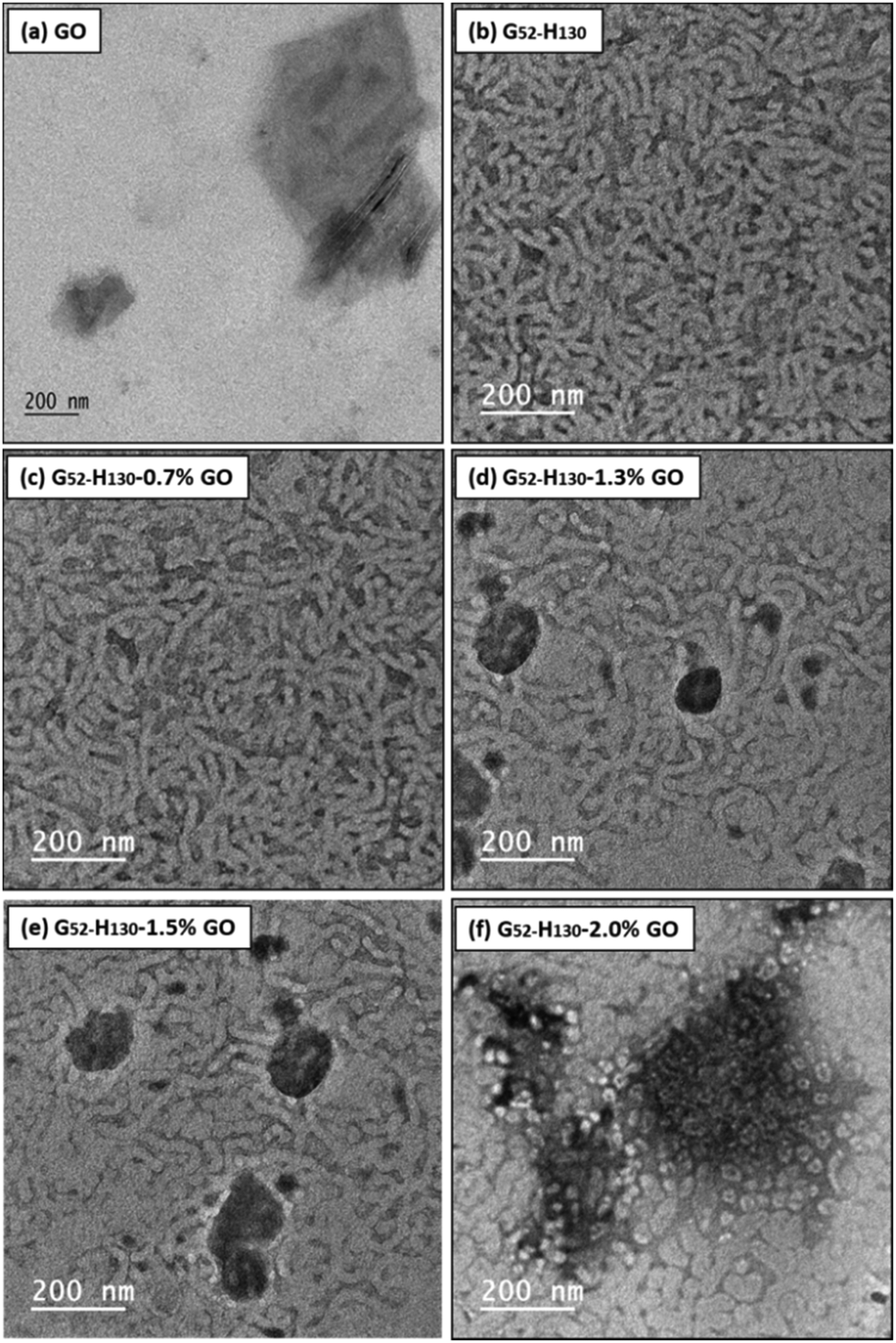 | ||
| Fig. 2 TEM images for (a) GO; (b) 15% w/w G52–H130, (c) G52–H130–0.7% GO, (d) G52–H130–1.3% GO; (e) G52–H130–1.5% GO and (f) G52–H130–2.0% GO. Samples were diluted to 0.01% w/w before being deposited on to carbon-coated TEM grids at room temperature. | ||
 | ||
| Fig. 3 TEM images for (a) 15% w/w G60–B70; (b) G60–B70–2.3% GO, (c) G60–B70–2.5% GO, (d) G60–B70–2.6% GO; (e) G60–B70–2.8% GO and (f) G60–B70–3.1% GO. Samples were diluted to 0.01% w/w before being deposited on to carbon-coated TEM grids at room temperature. | ||
For PGMA52–PHPMA130–x% GO composite worm-gels, GO dispersion (4 mg mL−1, pH 5.1) and 20% w/w G52–H130 copolymer worm gel were cooled to approximately 5 °C until the copolymer dispersion was in a free-flowing state (pH 6.3). Depending on the target GO concentration in the final mixture, appropriate quantities of the pre-cooled GO dispersion and/or deionised water were added to cooled copolymer dispersion to yield a copolymer concentration of 15% w/w (measured dispersion pH 5.9 to 6.2). The samples were mixed thoroughly for 10 s whilst cool using a vortex mixer and subsequently allowed to return to room temperature.
For PGMA60–PBzMA70–x% GO gels, G60–B70 worm gels (pH 6.7) were heated to approximately 56 °C and allowed to equilibrate for ∼10 minutes. On heating, the worm gels transformed into a viscous but free-flowing liquid. Two types of continuous phase were used to prepare G60–B70–x% GO worm gels: methanol and water/methanol mixtures. For water/methanol mixtures, different concentrations of pre-heated aqueous GO dispersion and/or deionised water were added to the methanolic copolymer dispersion. For methanol only formulations, GO was freeze-dried and dispersed in methanol using bath sonication for 40 minutes prior to heating and addition to the heated copolymer dispersion. The samples were mixed thoroughly for 10 s whilst warm using a vortex mixer and subsequently allowed to return to room temperature. In all cases the copolymer content of the final mixture was fixed at 15% w/w (measured dispersion pH 6.4 to 6.6).
Characterisation
Results and discussion
Copolymer worm-gel preparation
PGMA52 (Mw/Mn = 1.17) and PGMA60 (Mw/Mn = 1.26) were synthesised via RAFT solution polymerisation in ethanol at 70 °C. PGMA52 was block-extended with HPMA via RAFT aqueous dispersion polymerisation at 20% w/w and 70 °C (see Fig. S1a, ESI†). The target DP of the core forming PHPMA was 130 to obtain a free-standing worm-gel (Fig. S4d, ESI†).78 GPC analysis confirmed a relatively narrow molecular weight distribution (Mw/Mn = 1.06) (Fig. S2a, ESI†) and the thermal response of the G52–H130 copolymer dispersion was investigated by variable temperature DLS measurements (Fig. S4a, ESI†) on 0.1% w/w aqueous dispersions. The initial particle diameter reported by DLS was approximately 190 nm at 40 °C. This reduced to approximately 30 nm at 2 °C confirming the expected worm-to-sphere morphological transition.59 On heating, a constant diameter of ∼30 nm was recorded, indicating an irreversible transition at low copolymer concentrations. Oscillatory rheology (Fig. S4c, ESI†) conducted on the copolymer at 20% w/w demonstrated fully reversible de-gelation with a critical gelation temperature (CGT), which is defined as crossover temperature for G′ and G′′ curves, of 10 °C on cooling and 11 °C on heating. The gel strength (G′) of the 20% w/w G52–H130 copolymer gel was nearly 1.3 kPa (Fig. S4b, ESI†), as was expected from previous work.59BzMA was polymerised in the presence of PGMA60via RAFT alcoholic dispersion polymerisation at 20% w/w in methanol (Fig. S1b, ESI†).73 The target DP of the core forming PBzMA was 70 to obtain a free standing gel and TEM confirmed that a worm morphology was obtained (Fig. S5c, ESI†). GPC analysis confirmed a relatively narrow molecular weight distribution (Mw/Mn = 1.27) (Fig. S2b, ESI†) and oscillatory rheology studies indicated that G′ of the 20% w/w PGMA60–PBzMA70 gel was approximately 1.1 kPa (Fig. S5b, ESI†).73 Variable temperature DLS measurements examined the thermal response of the G60–B70 dispersions. For 0.1% w/w aqueous dispersions, the Z-average particle diameter was approximately 900 nm at 25 °C (Fig. S5a, ESI†). After heating to 60 °C, the particle diameter reduced to ∼90 nm indicating that the worm-to-sphere morphological transition occurred. On cooling back to 25 °C, DLS confirmed that the transition was irreversible at low copolymer concentration, as observed for G52–H130.66
Preparation of G52–H130–GO composite gels
To enable mixing with GO dispersions, the aqueous G52–H130 diblock copolymer dispersions necessarily required dilution. On reducing the solids content from 20% w/w to 15% w/w, a free-standing gel was retained (Fig. S6a, ESI†) and the copolymer morphology was unchanged (Fig. 2b). Diluting the copolymer reduced the gel strength from ∼1.5 kPa at 20% w/w (Fig. S4b, ESI†) to ∼1.0 kPa at 15% w/w (Fig. 4b). However, when the copolymer was diluted further to 10% w/w, the copolymer was no longer a free-standing gel (Fig. S6a, ESI†). Therefore, all subsequent dilutions of G52–H130 with GO dispersions were conducted at a fixed final copolymer concentration of 15% w/w. | ||
| Fig. 4 (a) Photographs of graphene oxide containing PGMA52–PHPMA130 block copolymer dispersions with graphene oxide concentrations between 2.2 and 0.7% w/w. The upper image shows samples after mixing at 5 °C and the lower image shows samples after equilibration for >2 h at room temperature. Storage modulus (G′) versus % strain for (b) 15% w/w G52–H130 with increasing GO concentration (0–1.5% w/w) and (c) 15% w/w G52–H130 with decreasing GO concentration (1.5–2.2% w/w). (d) Average storage modulus measured between 0.01 and 2% strain for different GO-containing G52–H130 copolymer dispersions. All measurements were conducted at a frequency of 10 Hz, a strain of 1.0% and a controlled temperature of 25 °C. | ||
As shown in Fig. 1b the 15% w/w G52–H130 worm gel dispersion undergoes de-gelation on cooling to form a free-flowing liquid. The low viscosity of this cooled copolymer dispersion (at 5 °C) facilitates mixing with various concentrations of pre-cooled GO dispersion. Therefore, GO was mixed with cooled copolymer to yield a GO content between 0.7 to 2.2% w/w based on copolymer, whilst keeping the total copolymer content fixed at 15% w/w. At low temperature, all samples appeared as brown free-flowing liquids (Fig. 1b). On warming the mixtures to room temperature, most samples returned to being a gel, as judged by tube inversion tests (Fig. 1b). However, gelation did not occur for samples containing relatively high concentrations of GO (≤1.5% w/w based on copolymer, Fig. 4a).
Oscillatory rheology studies of the G52–H130–GO composite gels at room temperature clearly show differences between respective gel strengths with increasing GO loading (Fig. 4). For example, increasing the GO content from 0 to 1.5% w/w based on copolymer increases the measured gel strength from ∼1.5 to ∼4.0 kPa (Fig. 4b). TEM images of the composite gels (Fig. 2c–e) demonstrate that the copolymer worms re-assemble after the cooling-mixing-heating cycle and allow the gels to re-form. Thus, the observed gel strengthening effect is likely a result of attractive polymer–GO interactions and strengthening of the composite/worm network by the incorporated GO flakes. To support the hypothesised interaction between these PGMA-based copolymers and GO, aqueous electrophoresis studies were conducted as function of pH on G52 macro-CTA, diluted G52–H130 worms, GO dispersion (4 mg mL−1), and selected (co)polymer–GO mixtures (0.7%, 1.5% and 2.0% GO) after purification (Fig. S9a and b, ESI†). Both the G52 macro-CTA and the G52–H130 copolymer dispersions exhibited slightly negative Zeta potentials.60 This anionic character is imparted due to the carboxylic acid end-groups from the PETTC RAFT agent.79 The GO dispersion was more anionic across the whole pH range and had a Zeta potential less than −30 mV at pH > 5. On addition of either G52 macro-CTA or G52–H130, the anionic charge from GO is screened and the Zeta potential becomes less negative. For example, with 2.2% GO based on (co)polymer the Zeta potential is highly anionic at pH > 5. However, with 0.7% GO based on (co)polymer the Zeta potential appears more similar to the pristine (co)polymer. Thus, it is likely that on mixing GO with cooled G52–H130 dispersions the copolymer spheres adsorb onto the surface of the GO sheets, become incorporated homogeneously within the reformed composite worm gel and reinforce the gel upon the application of shear.
On increasing the GO content from 1.5 to 2.2% w/w based on copolymer the measured gel strength significantly decreases (Fig. 4c). This is hypothesised to be due to the higher concentration of GO flakes absorbing more copolymer chains on their surface and thus disrupting copolymer worm formation. As such, there is a lower concentration of polymer able to re-form into worms and a weaker gel is formed. Alternatively, it may be that the higher concentration of GO flakes simply prevents sphere–sphere fusion. In either case the presence of a population of spherical micelles can be observed in the TEM images for 1.8% w/w GO based on copolymer and above (Fig. 2f and Fig. S7, ESI†). Thus, there is an optimal concentration of GO for forming composite worm-gels for this particular G52–H130–GO formulation (Fig. 4d).
Influence of graphene oxide concentration on composite gel temperature response
The G52–H130–GO gels retained their thermo-responsive behaviour. Variable temperature rheology studies were conducted on the composite gels between 2 to 40 °C using an applied strain of 1.0% (Fig. 5). The 15% w/w G52–H130 copolymer had a critical gelation temperature (CGT) of 14 °C on cooling and 15 °C on heating, as judged by the cross-over temperature for G′ and G′′.78 The addition of GO initially decreased the CGT of the gels on cooling to 9 °C for 0.7% GO (Fig. 5b) and 6 °C for 1.5% GO (Fig. 5c). For both of these formulations, re-gelation was observed upon heating, with a CGT similar to that on cooling. This observation further supports that the reversible worm-to-sphere transition was retained in the presence of GO. The depression of the CGT can be attributed the association between the GO and copolymer making the gels more temperature stable. | ||
| Fig. 5 Temperature-dependent oscillatory rheology studies obtained for aqueous dispersions of: (a) 15% w/w G52–H130; (b) G52–H130–0.7% GO; (c) G52–H130–1.5% GO; (d) G52–H130–2.2% GO. The temperature was varied from 40 °C to 2 °C to 40 °C in 1 °C steps with 3 minutes equilibration at each step. All measurements were conducted at an angular frequency of 10 Hz and applied strain amplitude of 1.0%. | ||
With GO concentrations >1.5% based on copolymer, the determined gel strengths were significantly decreased (Fig. 4c) and the CGT increased to approximately 20 °C on cooling and 24 °C upon re-heating (Fig. 5d). The higher CGT values and increased hysteresis supports that high GO contents hinder the degree of sphere–sphere fusion on heating but also result in less stable gels on cooling due to the lower worm content of these gels.
In addition, the composite gels exhibited reversible de-gelation behavior over a number of heating-coolong cycles. For instance, the CGT of G52–H130 with 1.5% GO based on copolymer remained relatively consistent over eight cooling/heating cycles (Fig. S10, ESI†).
Preparation of G60–B70–GO composite gels
In contrast to aqueous PGMA–PHPMA copolymer dispersions, which undergo worm-to-sphere de-gelation transitions on cooling, methanolic PGMA–PBzMA copolymer dispersions undergo the same transition on heating.66,75 Thus, this alternative system was investigated as a complimentary route towards the formation of composite GO-containing worm-gels.To facilitate mixing of G–B copolymer dispersions with GO, the dilution of a G60–B70 diblock copolymer dispersion from 20% w/w to 15% w/w was initially studied. When water was added to the methanolic dispersion to reduce the concentration, worm-like micelles were still present (Fig. 3a) and, as expected, the gel-strength was reduced from ∼1.1 kPa (Fig. S5b, ESI†) to ∼0.9 kPa (Fig. 6b) with a free-standing gel being retained (Fig. S6b, ESI†). Interestingly, dilution to 15% w/w with methanol caused the copolymer dispersion to no-longer be a free-standing gel (Fig. S6b, ESI†). Given this observation, and that the GO dispersions obtained were water-based, the initial composite G60–B70–GO gels investigated were therefore methanol/water mixtures.
 | ||
| Fig. 6 (a) 15% w/w PGMAx–PBzMAy block copolymer dispersions after mixing with aqueous graphene oxide between 2.0 and 3.8% w/w based on copolymer. The upper image in (a) shows samples after mixing at 56 °C and the lower image shows samples after equilibration overnight at room temperature. Storage modulus (G′) versus % strain for (b) 15% w/w G60–B70 with increasing GO concentration (0–2.8% w/w) and (c) 15% w/w G60–B70 with decreasing GO concentration (2.8–3.1% w/w). (d) Average storage modulus measured between 0.01 and 2% strain for different GO-containing G60–B70 copolymer dispersions. All measurements were conducted at a frequency of 10 Hz, a strain of 1.0% and a controlled temperature of 25 °C. | ||
To prepare the G60–B70–GO composite gels, the thermo-responsive behaviour of the G60–B70 gels on heating was utilised to facilitate good mixing of the copolymer and GO. As such, 20% w/w methanolic G60–B70 dispersions were heated to approximately 56 °C, where they formed free-flowing liquids (Fig. 1c). The copolymer was then mixed with pre-heated aqueous GO (keeping the copolymer concentration fixed at 15% w/w) and then allowed to cool to room temperature. The range of GO dispersion concentrations studied was initially screened by tube inversion tests. From these simple tests it was determined that the optimal GO concentration for gel re-formation after mixing was between 2.0% w/w and 3.8% w/w based on copolymer (Fig. 6a). As with G52–H130–x% GO, re-gelation did not occur for samples containing relatively high GO concentrations (≤2.8% w/w based on copolymer in this case). Zeta potential measurements were performed on dissolved G60 (Fig. S9c, ESI†) and a G60–B70 copolymer dispersion (Fig. S9d, ESI†). Due to the carboxylic acid end-groups from the PETTC RAFT agent, the G60 and G60–B70 dispersions showed a slightly negative charge. With the addition of GO the measured zeta showed an increase in negative charge with increasing GO concentration. For instance, the zeta potential measured for 3.1% GO based on (co)polymer is more negative than for 2.3% GO at all pH values studied (Fig. S9c and d, ESI†). This again suggests that GO sheets become associated with the (co)polymer during mixing.73
The G60–B70–GO composite gels were investigated using oscillatory rheology. The storage modulus (G′) of the prepared gels was measured as a function of % strain with varying GO contents (Fig. 6b–d). The moduli in the plateau region are strain independent. As the GO concentration increased from 0 to 2.8% w/w based on copolymer, G′ gradually increased to a maximum of approximately 2.7 kPa, which is nearly 3 times greater than for 15% G60–B70 without the addition of GO (Fig. 6b).80 TEM images of the G60–B70–GO composite gels (Fig. 3b–e) show that after the heating-mixing-cooling cycle, copolymer worms are clearly present and that they are associated with the relatively large GO flakes present in the image. As with the G52–H130–GO composites, the association between GO and the G60–B70 copolymer worms is probably providing reinforcement of the re-formed worm-gels.
Increasing the GO content further for led to G′ dramatically decreasing (Fig. 6c). For example, G60–B70–3.1% GO had a gel strength of ∼1.4 kPa compared to 2.7 kPa G60–B70–2.8% w/w GO. TEM images for higher GO loadings (Fig. 3) indicate the formation of shorter worms and a small population of spheres in comparison to lower GO loadings. Thus, the prevention of sphere–sphere fusion with higher GO contents is the proposed reason as to why the gels with relatively high GO loadings are weaker. These observations correlate with those made for G52–H130–GO and thus this seems to be a general mechanism of gel reinforcement/weakening for worm-gel/GO mixtures.
To investigate whether the presence of water was a major factor in the observations made, wholly methanolic copolymer/GO composites were investigated. To facilitate this, freeze-dried GO powder was homogeneously dispersed in methanol using sonication for 40 min. Pre-heated methanolic GO dispersions were then added to G60–B70 at approximately 56 °C whilst keeping the copolymer concentration at 15% w/w.
The addition of GO allowed free-standing gels to form on cooling to room temperature, with an optimal GO concentration between 3.2% w/w and 4.1% w/w based on tube inversion tests (Fig. S11a, ESI†). It is noteworthy that the presence of GO allows gel re-formation on cooling and in fact allows the formation of free-standing gels at 15% w/w copolymer for the dispersion diluted with GO dispersed in methanol (which is not the case in the absence of GO). The measured gel strength (Fig. S11b and c, ESI†) showed a similar trend to the formulations described above, with the expected increase and subsequent decrease in gel strength with increasing GO concentration. However, the recorded values for the wholly methanolic system were less than for G60–B70–GO prepared using aqueous GO. As expected, the recorded TEM images (Fig. S12, ESI†) clearly show an association between the copolymer worms and the GO flakes, indicating the interactions between these particles is not only observed in water. Furthermore, there is clear evidence for the limited amount of worm-reformation with high GO loadings, shown by a high number density of ∼30 nm copolymer spheres and lack of long worms in the TEM image for G60–B70–4.6% GO (Fig. S12d, ESI†).
Conclusions
Block copolymer worm-gels containing GO were prepared. For PGMA52–PHPMA130 worm gels, cooling induces a worm-to-sphere de-gelation transition which allows facile mixing with aqueous GO dispersions. On re-heating to room temperature, GO-containing worm gels are formed for 15% w/w copolymer and GO contents up to ∼1.8% w/w based on copolymer. A maximum gel strength of ∼4 kPa was recorded at a GO content of 1.5% w/w. Aqueous electrophoresis and TEM measurements indicate that there is absorption of the copolymer onto the GO flakes which then allows reinforcement of the gel on worm-reformation. However, too much GO disrupts the worm re-formation process, causing a lower measured gel strength and thus an optimal GO concentration. Similar observations were made for PGMA60–PBzMA70 copolymer worms prepared in methanol. In this case, heating the copolymer dispersions induces the worm-to-sphere transition, with composite gels forming on mixing with either aqueous or methanolic GO and cooling to room temperature. For this system the optimal GO concentration was slightly higher in comparison to the aqueous PGMA52–PHPMA130–GO composite gels. Nevertheless, the same mechanistic process for gel-strengthening and subsequent weakening with increasing GO loadings is observed. Thus, this generic process allows reinforced nanocomposite block copolymer worms gels to be readily prepared by the addition of relatively low concentrations of GO. It is likely that this route is not specific to GO as a re-enforcing nanomaterial and it is expected that this novel class of composite gel has potential applications in many areas of materials science.81–83Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Henry Royce Institute for Advanced Materials, funded through EPSRC grants EP/R00661X/1, EP/S019367/1, EP/P025021/1, and EP/P025498/1 and the Sustainable Materials Innovation Hub, funded through the European Regional Development Fund OC15R19P. The University of Manchester Electron Microscopy Centre is acknowledged for access to electron microscopy facilities.Notes and references
- K. J. Henderson, T. C. Zhou, K. J. Otim and K. R. Shull, Ionically cross-linked triblock copolymer hydrogels with high strength, Macromolecules, 2010, 43(14), 6193–6201 CrossRef CAS.
- Q. Zhu, C. W. Barney and K. A. Erk, Effect of ionic crosslinking on the swelling and mechanical response of model superabsorbent polymer hydrogels for internally cured concrete, Mater. Struct., 2015, 48(7), 2261–2276 CrossRef CAS.
- Y. He, N. Zhang, Q. Gong, H. Qiu, W. Wang, Y. Liu and J. Gao, Alginate/graphene oxide fibers with enhanced mechanical strength prepared by wet spinning, Carbohydr. Polym., 2012, 88(3), 1100–1108 CrossRef CAS.
- Y. He, H.-K. Tsao and S. Jiang, Improved mechanical properties of zwitterionic hydrogels with hydroxyl groups, J. Phys. Chem. B, 2012, 116(19), 5766–5770 CrossRef CAS PubMed.
- Y. Chen, K. Dong, Z. Liu and F. Xu, Double network hydrogel with high mechanical strength: Performance, progress and future perspective, Sci. China: Technol. Sci., 2012, 55(8), 2241–2254 CrossRef CAS.
- S.-a. Riyajan, W. Sukhlaaied and W. Keawmang, Preparation and properties of a hydrogel of maleated poly(vinyl alcohol)(PVAM) grafted with cassava starch, Carbohydr. Polym., 2015, 122, 301–307 CrossRef CAS PubMed.
- K. Haraguchi and T. Takehisa, Nanocomposite hydrogels: A unique organic–inorganic network structure with extraordinary mechanical, optical, and swelling/de-swelling properties, Adv. Mater., 2002, 14(16), 1120–1124 CrossRef CAS.
- S. Hashmi, A. GhavamiNejad, F. O. Obiweluozor, M. Vatankhah-Varnoosfaderani and F. J. Stadler, Supramolecular interaction controlled diffusion mechanism and improved mechanical behavior of hybrid hydrogel systems of zwitterions and CNT, Macromolecules, 2012, 45(24), 9804–9815 CrossRef CAS.
- K. Haraguchi, H.-J. Li, Y. Xu and G. Li, Copolymer nanocomposite hydrogels: Unique tensile mechanical properties and network structures, Polymer, 2016, 96, 94–103 CrossRef CAS.
- D. Wu, M. Yi, H. Duan, J. Xu and Q. Wang, Tough TiO2-rGO-PDMAA nanocomposite hydrogel via one-pot UV polymerization and reduction for photodegradation of methylene blue, Carbon, 2016, 108, 394–403 CrossRef CAS.
- F. Kim, L. J. Cote and J. Huang, Graphene oxide: surface activity and two-dimensional assembly, Adv. Mater., 2010, 22(17), 1954–1958 CrossRef CAS PubMed.
- H. Li, J. Fan, Z. Shi, M. Lian, M. Tian and J. Yin, Preparation and characterization of sulfonated graphene-enhanced poly(vinyl alcohol) composite hydrogel and its application as dye absorbent, Polymer, 2015, 60, 96–106 CrossRef CAS.
- T. Jiao, H. Zhao, J. Zhou, Q. Zhang, X. Luo, J. Hu, Q. Peng and X. Yan, Self-assembly reduced graphene oxide nanosheet hydrogel fabrication by anchorage of chitosan/silver and its potential efficient application toward dye degradation for wastewater treatments, ACS Sustainable Chem. Eng., 2015, 3(12), 3130–3139 CrossRef CAS.
- T. Jiao, H. Guo, Q. Zhang, Q. Peng, Y. Tang, X. Yan and B. Li, Reduced graphene oxide-based silver nanoparticle-containing composite hydrogel as highly efficient dye catalysts for wastewater treatment, Sci. Rep., 2015, 5(1), 1–12 Search PubMed.
- H. Guo, T. Jiao, Q. Zhang, W. Guo, Q. Peng and X. Yan, Preparation of graphene oxide-based hydrogels as efficient dye adsorbents for wastewater treatment, Nanoscale Res. Lett., 2015, 10(1), 1–10 CrossRef CAS PubMed.
- R. Sahraei and M. Ghaemy, Synthesis of modified gum tragacanth/graphene oxide composite hydrogel for heavy metal ions removal and preparation of silver nanocomposite for antibacterial activity, Carbohydr. Polym., 2017, 157, 823–833 CrossRef CAS PubMed.
- Q. Jing, J. Y. Law, L. P. Tan, V. V. Silberschmidt, L. Li and Z. Dong, Preparation, characterization and properties of polycaprolactone diol-functionalized multi-walled carbon nanotube/thermoplastic polyurethane composite, Composites, Part A, 2015, 70, 8–15 CrossRef CAS.
- Y. Zhang, R. Huang, S. Peng and Z. Ma, MWCNTs/cellulose hydrogels prepared from NaOH/urea aqueous solution with improved mechanical properties, J. Chem., 2015, 2015, 413497 Search PubMed.
- Z. Chen, J. W. Wang, C. Lu, Z. Liu, N. Chortos, A. Pan, L. Wei, F. Cui and Y. Bao, Z., A three-dimensionally interconnected carbon nanotube–conducting polymer hydrogel network for high-performance flexible battery electrodes, Adv. Energy Mater., 2014, 4(12), 1400207 CrossRef.
- O. Duman, S. Tunç, T. G. Polat and B. K. Bozoğlan, Synthesis of magnetic oxidized multiwalled carbon nanotube-κ-carrageenan-Fe3O4 nanocomposite adsorbent and its application in cationic Methylene Blue dye adsorption, Carbohydr. Polym., 2016, 147, 79–88 CrossRef CAS PubMed.
- H.-L. Ma, L. Zhang, Y. Zhang, S. Wang, C. Sun, H. Yu, X. Zeng and M. Zhai, Radiation preparation of graphene/carbon nanotubes hybrid fillers for mechanical reinforcement of poly(vinyl alcohol) films, Radiat. Phys. Chem., 2016, 118, 21–26 CrossRef CAS.
- N. Mahfoudhi and S. Boufi, Poly(acrylic acid-co-acrylamide)/cellulose nanofibrils nanocomposite hydrogels: effects of CNFs content on the hydrogel properties, Cellulose, 2016, 23(6), 3691–3701 CrossRef CAS.
- J. Yang, J.-J. Zhao, F. Xu and R.-C. Sun, Revealing strong nanocomposite hydrogels reinforced by cellulose nanocrystals: insight into morphologies and interactions, ACS Appl. Mater. Interfaces, 2013, 5(24), 12960–12967 CrossRef CAS PubMed.
- J. Han, T. Lei and Q. Wu, High-water-content mouldable polyvinyl alcohol-borax hydrogels reinforced by well-dispersed cellulose nanoparticles: Dynamic rheological properties and hydrogel formation mechanism, Carbohydr. Polym., 2014, 102, 306–316 CrossRef CAS PubMed.
- D. Yang, X. Peng, L. Zhong, X. Cao, W. Chen, S. Wang, C. Liu and R. Sun, Fabrication of a highly elastic nanocomposite hydrogel by surface modification of cellulose nanocrystals, RSC Adv., 2015, 5(18), 13878–13885 RSC.
- T. Abitbol, T. Johnstone, T. M. Quinn and D. G. Gray, Reinforcement with cellulose nanocrystals of poly(vinyl alcohol) hydrogels prepared by cyclic freezing and thawing, Soft Matter, 2011, 7(6), 2373–2379 RSC.
- R. Nigmatullin, M. Bencsik and F. Gao, Influence of polymerisation conditions on the properties of polymer/clay nanocomposite hydrogels, Soft Matter, 2014, 10(12), 2035–2046 RSC.
- C. Lian, Y. Yang, T. Wang, W. Sun, X. Liu and Z. Tong, A facile method for reinforcing poly(N-isopropylacrylamide)-hectorite clay nanocomposite hydrogels by heat treatment, Polym. Compos., 2016, 37(5), 1557–1563 CrossRef CAS.
- T. Wang, S. Zheng, W. Sun, X. Liu, S. Fu and Z. Tong, Notch insensitive and self-healing PNIPAm–PAM–clay nanocomposite hydrogels, Soft Matter, 2014, 10(19), 3506–3512 RSC.
- Y. Wan, C. Wu, G. Xiong, G. Zuo, J. Jin, K. Ren, Y. Zhu, Z. Wang and H. Luo, Mechanical properties and cytotoxicity of nanoplate-like hydroxyapatite/polylactide nanocomposites prepared by intercalation technique, J. Mech. Behav. Biomed. Mater., 2015, 47, 29–37 CrossRef CAS PubMed.
- Z. Fang and Q. Feng, Improved mechanical properties of hydroxyapatite whisker-reinforced poly(L-lactic acid) scaffold by surface modification of hydroxyapatite, Mater. Sci. Eng., C, 2014, 35, 190–194 CrossRef CAS PubMed.
- W. L. Hom and S. R. Bhatia, Significant enhancement of elasticity in alginate-clay nanocomposite hydrogels with PEO-PPO-PEO copolymers, Polymer, 2017, 109, 170–175 CrossRef CAS.
- A. Boonmahitthisud, L. Nakajima, K. D. Nguyen and T. Kobayashi, Composite effect of silica nanoparticle on the mechanical properties of cellulose-based hydrogels derived from cottonseed hulls, J. Appl. Polym. Sci., 2016, 134, 44557 Search PubMed.
- J. Yang and J. Zhao, Preparation and mechanical properties of silica nanoparticles reinforced composite hydrogels, Mater. Lett., 2014, 120, 36–38 CrossRef CAS.
- C. Hou, K. Ma, T. Jiao, R. Xing, K. Li, J. Zhou and L. Zhang, Preparation and dye removal capacities of porous silver nanoparticle-containing composite hydrogels via poly(acrylic acid) and silver ions, RSC Adv., 2016, 6(112), 110799–110807 RSC.
- R. Xing, K. Liu, T. Jiao, N. Zhang, K. Ma, R. Zhang, Q. Zou, G. Ma and X. Yan, An injectable self-assembling collagen–gold hybrid hydrogel for combinatorial antitumor photothermal/photodynamic therapy, Adv. Mater., 2016, 28(19), 3669–3676 CrossRef CAS PubMed.
- B. Xu, H. Li, Y. Wang, G. Zhang and Q. Zhang, Nanocomposite hydrogels with high strength cross-linked by titania, RSC Adv., 2013, 3(20), 7233–7236 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, The chemistry of graphene oxide, Chem. Soc. Rev., 2010, 39(1), 228–240 RSC.
- S. C. Thickett and P. B. Zetterlund, Functionalization of graphene oxide for the production of novel graphene-based polymeric and colloidal materials, Curr. Org. Chem., 2013, 17(9), 956–974 CrossRef CAS.
- D. Chen, H. Feng and J. Li, Graphene oxide: preparation, functionalization, and electrochemical applications, Chem. Rev., 2012, 112(11), 6027–6053 CrossRef CAS PubMed.
- N. Lu, Y. Huang, H.-B. Li, Z. Li and J. Yang, First principles nuclear magnetic resonance signatures of graphene oxide, J. Chem. Phys., 2010, 133(3), 034502 CrossRef PubMed.
- K. P. Loh, Q. Bao, G. Eda and M. Chhowalla, Graphene oxide as a chemically tunable platform for optical applications, Nat. Chem., 2010, 2(12), 1015–1024 CrossRef CAS PubMed.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Graphene and graphene oxide: synthesis, properties, and applications, Adv. Mater., 2010, 22(35), 3906–3924 CrossRef CAS PubMed.
- S. Faghihi, M. Gheysour, A. Karimi and R. Salarian, Fabrication and mechanical characterization of graphene oxide-reinforced poly(acrylic acid)/gelatin composite hydrogels, J. Appl. Phys., 2014, 115(8), 083513 CrossRef.
- Y. Huang, M. Zhang and W. Ruan, High-water-content graphene oxide/polyvinyl alcohol hydrogel with excellent mechanical properties, J. Mater. Chem. A, 2014, 2(27), 10508–10515 RSC.
- Q. Fang, X. Zhou, W. Deng, Z. Zheng and Z. Liu, Freestanding bacterial cellulose-graphene oxide composite membranes with high mechanical strength for selective ion permeation, Sci. Rep., 2016, 6(1), 1–11 CrossRef PubMed.
- Y. Piao and B. Chen, Self-assembled graphene oxide–gelatin nanocomposite hydrogels: Characterization, formation mechanisms, and pH-sensitive drug release behavior, J. Polym. Sci., Part B: Polym. Phys., 2015, 53(5), 356–367 CrossRef CAS.
- W. Li, J. Wang, J. Ren and X. Qu, 3D graphene oxide–polymer hydrogel: near-infrared light-triggered active scaffold for reversible cell capture and on-demand release, Adv. Mater., 2013, 25(46), 6737–6743 CrossRef CAS.
- S. R. Shin, B. Aghaei-Ghareh-Bolagh, T. T. Dang, S. N. Topkaya, X. Gao, S. Y. Yang, S. M. Jung, J. H. Oh, M. R. Dokmeci and X. Tang, Cell-laden microengineered and mechanically tunable hybrid hydrogels of gelatin and graphene oxide, Adv. Mater., 2013, 25(44), 6385–6391 CrossRef CAS PubMed.
- C. H. Zhu, Y. Lu, J. Peng, J. F. Chen and S. H. Yu, Photothermally sensitive poly(N-isopropylacrylamide)/graphene oxide nanocomposite hydrogels as remote light-controlled liquid microvalves, Adv. Funct. Mater., 2012, 22(19), 4017–4022 CrossRef CAS.
- W. Liu, X. Zhang, L. Zhou, L. Shang and Z. Su, Reduced graphene oxide (rGO) hybridized hydrogel as a near-infrared (NIR)/pH dual-responsive platform for combined chemo-photothermal therapy, J. Colloid Interface Sci., 2019, 536, 160–170 CrossRef CAS PubMed.
- R. Liu, S. Liang, X.-Z. Tang, D. Yan, X. Li and Z.-Z. Yu, Tough and highly stretchable graphene oxide/polyacrylamide nanocomposite hydrogels, J. Mater. Chem., 2012, 22(28), 14160–14167 RSC.
- H. Bai, C. Li, X. Wang and G. Shi, On the gelation of graphene oxide, J. Phys. Chem. C, 2011, 115(13), 5545–5551 CrossRef CAS.
- S. Darling, Directing the self-assembly of block copolymers, Prog. Polym. Sci., 2007, 32(10), 1152–1204 CrossRef CAS.
- P. De, M. Li, S. R. Gondi and B. S. Sumerlin, Temperature-regulated activity of responsive polymer− protein conjugates prepared by grafting-from via RAFT polymerization, J. Am. Chem. Soc., 2008, 130(34), 11288–11289 CrossRef CAS PubMed.
- P. Farquet, C. Padeste, H. H. Solak, S. A. Gürsel, G. N. G. Scherer and A. Wokaun, Extreme UV radiation grafting of glycidyl methacrylate nanostructures onto fluoropolymer foils by RAFT-mediated polymerization, Macromolecules, 2008, 41(17), 6309–6316 CrossRef CAS.
- C. St. Thomas, H. Maldonado-Textle, A. Rockenbauer, L. Korecz, N. Nagy and R. Guerrero-Santos, Synthesis of NMP/RAFT inifers and preparation of block copolymers, J. Polym. Sci., Part A: Polym. Chem., 2012, 50(14), 2944–2956 CrossRef CAS.
- A. Blanazs, J. Madsen, G. Battaglia, A. J. Ryan and S. P. Armes, Mechanistic insights for block copolymer morphologies: how do worms form vesicles?, J. Am. Chem. Soc., 2011, 133(41), 16581–16587 CrossRef CAS PubMed.
- N. J. Warren, M. J. Derry, O. O. Mykhaylyk, J. R. Lovett, L. P. Ratcliffe, V. Ladmiral, A. Blanazs, L. A. Fielding and S. P. Armes, Critical dependence of molecular weight on thermoresponsive behavior of diblock copolymer worm gels in aqueous solution, Macromolecules, 2018, 51(21), 8357–8371 CrossRef CAS PubMed.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. I. Douglas and S. P. Armes, Sterilizable gels from thermoresponsive block copolymer worms, J. Am. Chem. Soc., 2012, 134(23), 9741–9748 CrossRef CAS PubMed.
- J. R. Lovett, N. J. Warren, L. P. Ratcliffe, M. K. Kocik and S. P. Armes, pH-responsive non-ionic diblock copolymers: Ionization of carboxylic acid end-groups induces an order–order morphological transition, Angew. Chem., Int. Ed., 2015, 54(4), 1279–1283 CrossRef CAS PubMed.
- W. Zhao, H. T. Ta, C. Zhang and A. K. Whittaker, Polymerization-induced self-assembly (PISA)-control over the morphology of 19F-containing polymeric nano-objects for cell uptake and tracking, Biomacromolecules, 2017, 18(4), 1145–1156 CrossRef CAS PubMed.
- S. Sugihara, A. Blanazs, S. P. Armes, A. J. Ryan and A. L. Lewis, Aqueous dispersion polymerization: a new paradigm for in situ block copolymer self-assembly in concentrated solution, J. Am. Chem. Soc., 2011, 133(39), 15707–15713 CrossRef CAS PubMed.
- Y. Li and S. P. Armes, RAFT synthesis of sterically stabilized methacrylic nanolatexes and vesicles by aqueous dispersion polymerization, Angew. Chem., 2010, 122(24), 4136–4140 CrossRef.
- R. Verber, A. Blanazs and S. Armes, Rheological studies of thermo-responsive diblock copolymer worm gels, Soft Matter, 2012, 8(38), 9915–9922 RSC.
- L. A. Fielding, J. A. Lane, M. J. Derry, O. O. Mykhaylyk and S. P. Armes, Thermo-responsive diblock copolymer worm gels in non-polar solvents, J. Am. Chem. Soc., 2014, 136(15), 5790–5798 CrossRef CAS PubMed.
- U. Tritschler, S. Pearce, J. Gwyther, G. R. Whittell and I. Manners, 50th anniversary perspective: Functional nanoparticles from the solution self-assembly of block copolymers, Macromolecules, 2017, 50(9), 3439–3463 CrossRef CAS.
- F. Van Mastrigt, T. Stoffelsma, D. Wever and F. Picchioni, Thermoresponsive comb polymers as thickeners for high temperature aqueous fluids, Mater. Today Commun., 2017, 10, 34–40 CrossRef CAS.
- O. J. Deane, J. Jennings and S. P. Armes, Shape-shifting thermoreversible diblock copolymer nano-objects via RAFT aqueous dispersion polymerization of 4-hydroxybutyl acrylate, Chem. Sci., 2021, 12, 13719–13729 RSC.
- C.-Q. Huang and C.-Y. Pan, Direct preparation of vesicles from one-pot RAFT dispersion polymerization, Polymer, 2010, 51(22), 5115–5121 CrossRef CAS.
- W. Cai, W. Wan, C. Hong, C. Huang and C. Pan, Morphology transitions in RAFT polymerization, Soft Matter, 2010, 6(21), 5554–5561 RSC.
- W.-D. He, X.-L. Sun, W.-M. Wan and C.-Y. Pan, Multiple Morphologies of PAA-b-PSt Assemblies throughout RAFT Dispersion Polymerization of Styrene with PAA Macro-CTA, Macromolecules, 2011, 44(9), 3358–3365 CrossRef CAS.
- M. Semsarilar, E. R. Jones, A. Blanazs and S. P. Armes, Efficient Synthesis of Sterically-Stabilized Nano-Objects via RAFT Dispersion Polymerization of Benzyl Methacrylate in Alcoholic Media, Adv. Mater., 2012, 24(25), 3378–3382 CrossRef CAS PubMed.
- K. L. Thompson, C. J. Mable, A. Cockram, N. J. Warren, V. J. Cunningham, E. R. Jones, R. Verber and S. P. Armes, Are block copolymer worms more effective Pickering emulsifiers than block copolymer spheres?, Soft Matter, 2014, 10(43), 8615–8626 RSC.
- V. J. Cunningham, A. M. Alswieleh, K. L. Thompson, M. Williams, G. J. Leggett, S. P. Armes and O. M. Musa, Poly(glycerol monomethacrylate)–poly(benzyl methacrylate) diblock copolymer nanoparticles via RAFT emulsion polymerization: synthesis, characterization, and interfacial activity, Macromolecules, 2014, 47(16), 5613–5623 CrossRef CAS.
- M. Semsarilar, V. Ladmiral, A. Blanazs and S. Armes, Anionic polyelectrolyte-stabilized nanoparticles via RAFT aqueous dispersion polymerization, Langmuir, 2012, 28(1), 914–922 CrossRef CAS PubMed.
- S.-P. Wen, J. G. Saunders and L. A. Fielding, Investigating the influence of solvent quality on RAFT-mediated PISA of sulfonate-functional diblock copolymer nanoparticles, Polym. Chem., 2020, 11(20), 3416–3426 RSC.
- M. Williams, N. Penfold, J. Lovett, N. Warren, C. Douglas, N. Doroshenko, P. Verstraete, J. Smets and S. Armes, Bespoke cationic nano-objects via RAFT aqueous dispersion polymerisation, Polym. Chem., 2016, 7(23), 3864–3873 RSC.
- J. Lovett, L. Ratcliffe, N. Warren, S. Armes, M. Smallridge, R. Cracknell and B. Saunders, A robust cross-linking strategy for block copolymer worms prepared via polymerization-induced self-assembly, Macromolecules, 2016, 49(8), 2928–2941 CrossRef CAS PubMed.
- H. Bai, C. Li, X. Wang and G. Shi, A pH-sensitive graphene oxide composite hydrogel, Chem. Commun., 2010, 46(14), 2376–2378 RSC.
- T. Chen, X. Qiao, P. Wei, G. Chen, I. T. Mugaanire, K. Hou and M. Zhu, Tough Gel-Fibers as Strain Sensors Based on Strain–Optics Conversion Induced by Anisotropic Structural Evolution, Chem. Mater., 2020, 32(22), 9675–9687 CrossRef CAS.
- B. Wang, J. R. Moon, S. Ryu, K. D. Park and J. H. Kim, Antibacterial 3D graphene composite gel with polyaspartamide and tannic acid containing in situ generated Ag nanoparticle, Polym. Compos., 2020, 41(7), 2578–2587 CrossRef CAS.
- R. Cao, M. Qin, C. Liu, S. Li, P. Guo, G. Han, X. Hu, W. Feng and L. Chen, Photo-and Thermosensitive Polymer Membrane with a Tunable Microstructure Doped with Graphene Oxide Nanosheets and Poly(N-isopropylacrylamide) for the Application of Light-Cleaning, ACS Appl. Mater. Interfaces, 2020, 12(12), 14352–14364 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic schemes; 1H NMR spectra and GPC chromatograms and digital photographs of (co)polymers; variable temperature DLS and rheology studies on copolymers; TEM images of copolymers, GO and composites; aqueous electrophoresis studies of (co)polymer/GO mixtures; critical gelation temperature recorded over several heating–cooling cycles for G52–H130–1.5% GO; rheology data for 15% w/w G60–B70 with various GO concentrations in methanol. See DOI: 10.1039/d2sm00045h |
| This journal is © The Royal Society of Chemistry 2022 |