
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Improved alkali metal ion capturing utilizing crown ether-based diblock copolymers in a sandwich-type complexation†
Iklima
Oral
 a and
Volker
Abetz
*ab
a and
Volker
Abetz
*ab
aInstitute of Physical Chemistry, Universität Hamburg, Grindelallee 117, 20146 Hamburg, Germany. E-mail: volker.abetz@hereon.de
bHelmholtz-Zentrum Hereon, Institute of Membrane Research, Max-Planck-Straße 1, 21502 Geesthacht, Germany
First published on 18th January 2022
Abstract
The compexation behavior of metals with free crown ethers (CE) and diblock copolymer-based CE is investigated. The latter shows at least 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times stronger complexation than free CEs. On this basis, a highly stable CE complex within the polymer for efficient extraction of metal ions from low concentrations, e.g. lithium in seawater, is presented.
000 times stronger complexation than free CEs. On this basis, a highly stable CE complex within the polymer for efficient extraction of metal ions from low concentrations, e.g. lithium in seawater, is presented.
Lithium has drawn great interest due to its utilization in the electrical vehicle industry, glass, ceramics, lubricants, catalysis, alloy industries, and pharmaceuticals.1–6 The applications of lithium in the electric vehicle industry have created immense demand, huge price increases, and the consequent shortage of this raw material.7 Currently, lithium is gathered mainly from brines by solar evaporation processes, however, the availability of lithium resources on land is limited.8,9 Around 230 Bt. of lithium can be found in the seawater, therefore, several recovery techniques have been designed, such as adsorption, solvent extraction, and co-precipitation.6,8,10,11 Since many other interfering metal ions, such as Na+, K+, Mg2+, and Ca2+, are present in seawater at much higher concentrations than Li+, a highly selective recovery of the desired Li+ is mandatory.12
A high selectivity towards metal ions is provided by crown ethers (CEs), which form well-known host–guest complexes.13,14 The complexation of metal ions with CEs arises from ion–dipole interactions depending on the cation, stabilizing orbital interactions between charge transfer and polarization, and Pauli repulsive orbital interactions.15 The strength of the interaction, and thus the stability of the complex, can be adjusted by the orientation of the donor atoms in the CE, the cavity size, the choice of donor atom (soft or hard, according to the HSAB principle, e.g. N, S, or O), and substitutions of electron withdrawing or donating groups.16,17 In addition to the strength of the interaction, the stoichiometry of the ligand to metal ion plays a significant role (Fig. 1). Single complexes, sandwich-type complexes as well as club sandwich-type structures can be formed (further higher-ordered complex structures are also possible).14,18–20 This variability of the complexes makes the CEs ligands irreplaceable in the selective complexation of metal ions.
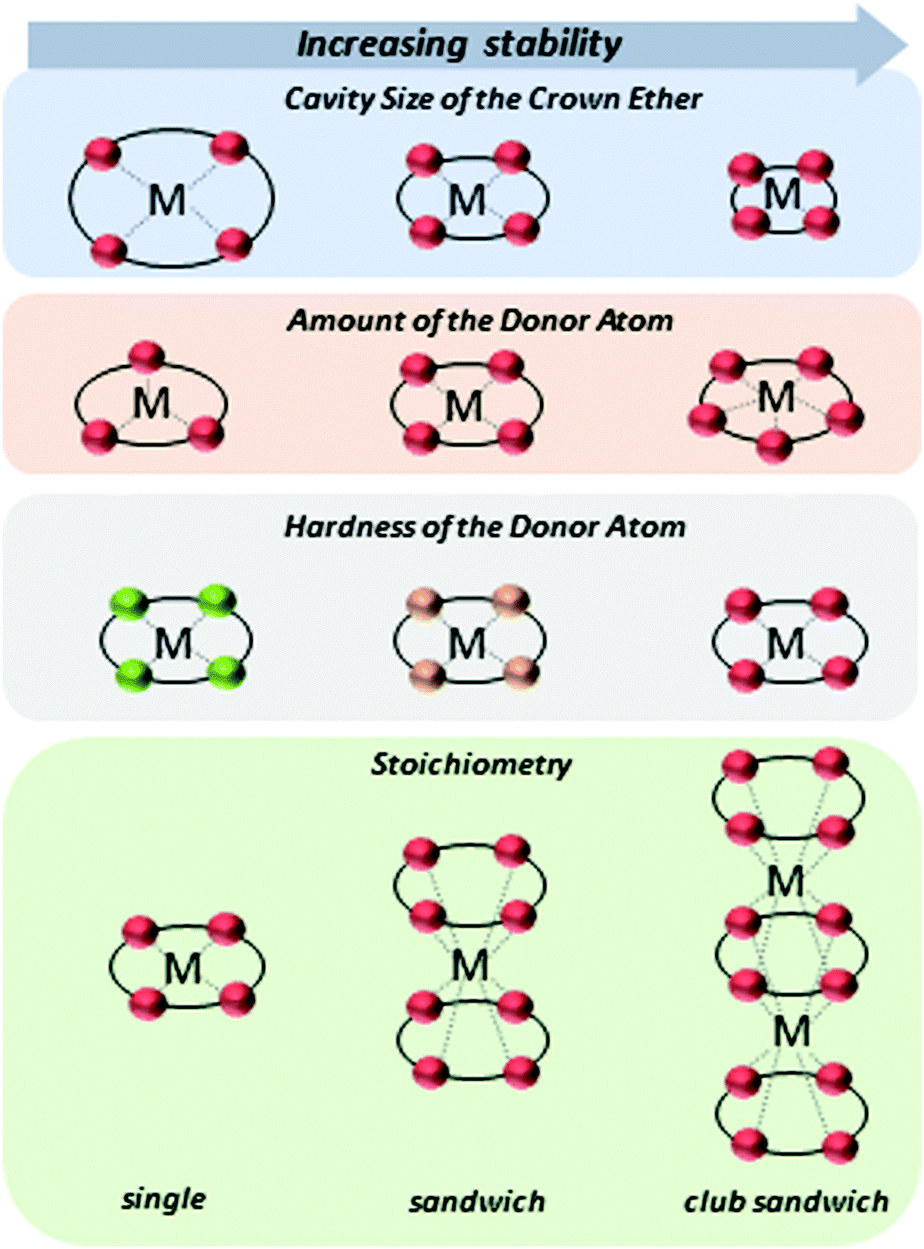 | ||
| Fig. 1 Illustration of the CEs–metal ion complexes. Different factors are illustrated such as the cavity size of the CE, the amount and hardness of the donor atoms as well as the stoichiometry, which affects the interaction between donor and acceptor (metal ion). The red, green, and orange dot represent oxygen, nitrogen, and sulfur atom, respectively. The dashed line represents the interaction between donor atom and metal ion. | ||
In previous work, stronger complexations within the polymer as well as in a sandwich-type formation have been reported.21,22 The aim of this work is to present stronger complexation by sandwich-type structures of the metal ions of the series Li+, Na+ and K+ and CEs of different sizes with 3, 4 and 5 oxygen atoms within the ring. The complex stability in single, as well as sandwich structures is compared and evaluated. A comparison between free CEs and polymer-based CEs is demonstrated. The latter is of particular interest since the polymer can be easily recovered after complexation by simple precipitation.
15-Crown-5 (15C5), 12-crown-4 (12C4), and benzo-9-crown-3 (B9C3) were chosen as CEs to investigate the single and sandwich complexes with Li+, Na+, and K+ within the polymer. Multivalent ions were not considered in this work, since they can easily be separated by electrodialysis using for example monovalent cationic selective membranes.23
A poly(methacrylic acid)-block-polystyrene (PMAA-b-PS) diblock copolymer was chosen (synthesis was reported elsewhere)24 and an overview is shown in Fig. S2 (ESI†). The carboxyl groups make the incorporation of CEs straightforward via Steglich esterification, if the latter are modified with a hydroxyl group (Fig. 2). The Steglich esterification is a simple reaction to esterify long polymer chains and to obtain high yields by adding dicyclohexylcarbodiimide (DCC) and 4-dimethylaminopyridine (DMAP).25,26 The successful incorporation of the CE within the polymer can be analyzed with 1H NMR spectroscopy. The signals of the CE appear broad in the polymer spectrum proving the newly introduced CE group to the PMAA-b-PS diblock copolymer. The average number of CE-esterified repeating units per chain will be called nCE – not “degree of polymerization” which would, by definition, be incorrect in a statistical copolymer and is analyzed by 1H NMR spectroscopy. An example NMR for the 15C5-bonded polymer can be seen in Fig. 3. A detailed determination of the nCE and the degree of functionalization (DF) can be found in the ESI.†
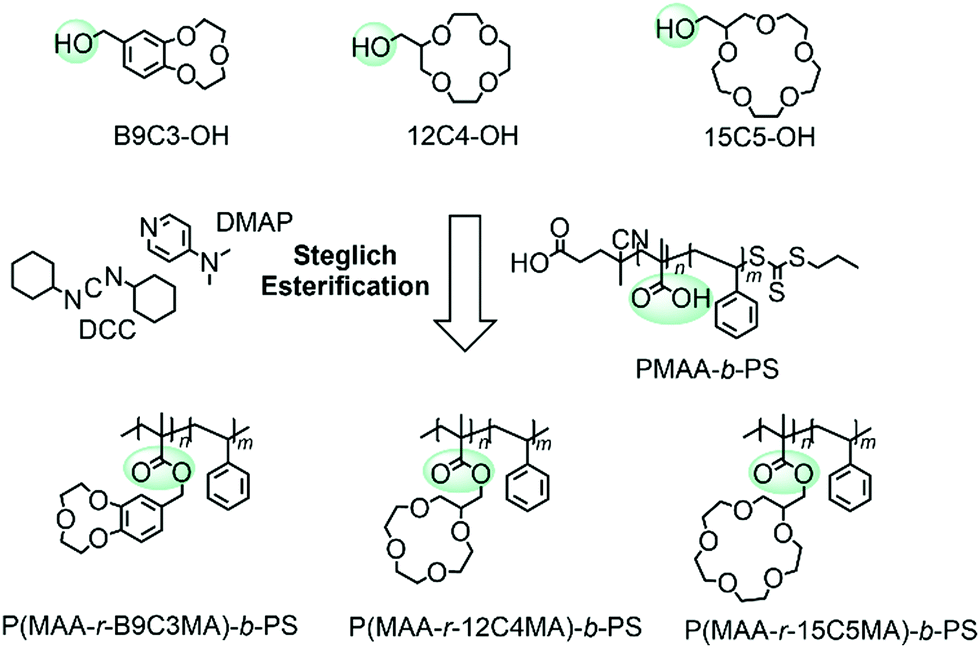 | ||
| Fig. 2 Reaction overview of the Steglich esterification of PMAA-b-PS with hydroxyl functionalized CEs. | ||
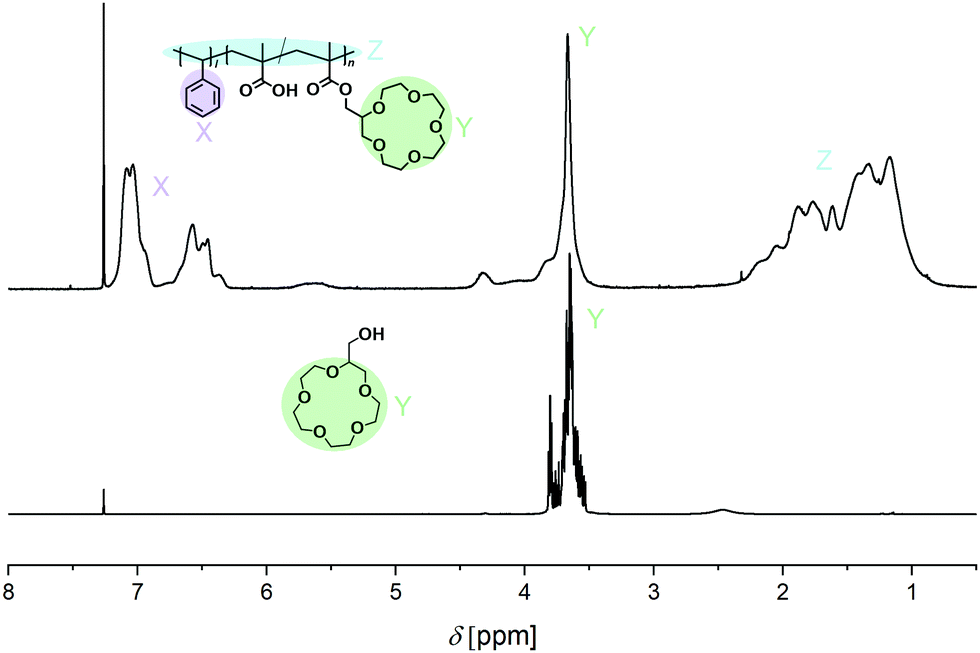 | ||
| Fig. 3 1H NMR spectrum of 15C5–OH (bottom) and P(MAA25-r-15C5MA25)-b-PS5056 diblock copolymer containing randomly distributed CE units (top) in CDCl3. | ||
In the complexation process of metal ions, the dissolved metal ions in water must first be freed from the hydrate shell surrounding them. Among the compared cations, this hydrate shell is most stable for Li+ and least stable for K+.
In a single complex, interactions are established between the donor atoms and the metal ions leading to an enthalpy gain.
In a sandwich complexation, the entropy loss is greater because two CEs participate in the complexation process. However, the enthalpy gain is twice as large, since twice as many interactions take place between donor atoms and metal ions (Fig. 1). In conclusion, the complexation of metals by CEs is a thermodynamic process with an interlude between entropy loss and enthalpy gain. In the thermodynamic process, the complexation constant K play an essential role and provide information about the stability of the complex. The general procedure of the metal ion extraction with CEs follows eqn (1):
| mMz+ + xXv− + nCE ⇌ (MmCEn)mz+ Xxvx | (1) |
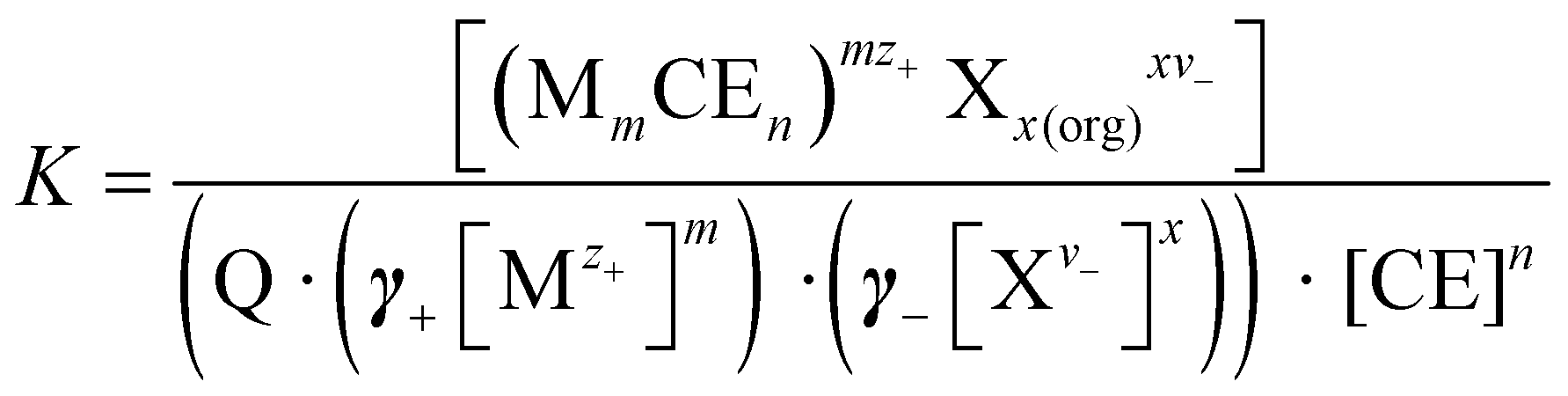 | (2) |
Experimentally, the complex constant K of a CE–metal complex is determined by using a two-phase extraction (Fig. 4). The CE is dissolved in the organic phase and the salt of a dye in the aqueous phase. After mixing the two phases, the cation of the salt dye interacts with the CE at the interface. After successful complexation, the CE–metal complex is in the organic phase (dichloromethane was chosen as the organic phase in these experiments). The aqueous phase (salt dye phase) was investigated by absorption spectroscopy due to spectroscopic limitations of the organic phase. The salt concentrations in the aqueous phase before and after extraction were analyzed and the complex constant K was calculated according to eqn (2). A more detailed description of the two-phase extraction and a detailed calculation of the complex constant K can be found in previous publications.21,22
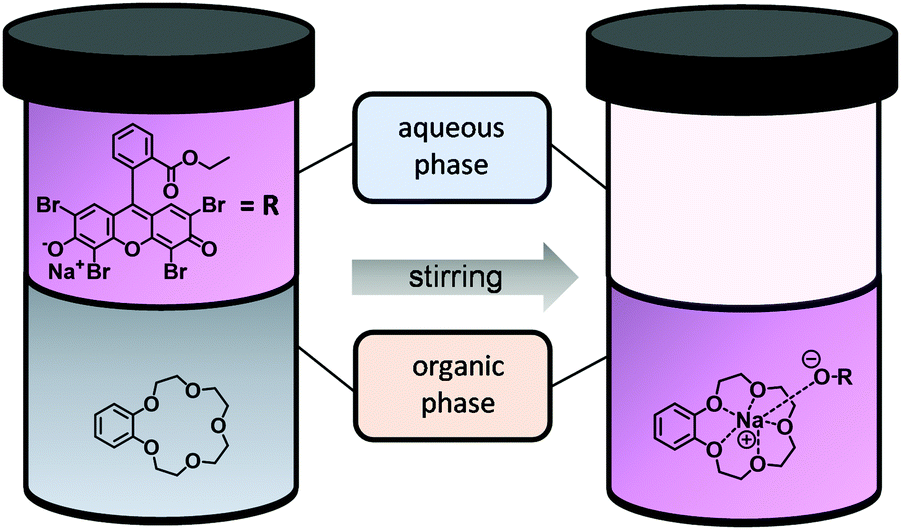 | ||
| Fig. 4 Two-phase extraction of a dichlormethane/water mixture with B15C5 and 2′,4′,5′,7′-tetrabromoeosin ethyl ester sodium salt (NaEE) as sodium dye. The left side shows the phases before the extraction and the right side afterwards. | ||
The free CEs benzo-15-crown-5 (B15C5), benzo-12-crown-4 (B12C4) and B9C3 were extracted as shown in Fig. 3. The 15C5 with a cavity size of 1.70–2.20 Å fits perfectly for Na+ with an ionic diameter (ID) of 2.02 Å and forms well-known “sandwich complexes” with K+ (ID = 2.76 Å).20,27 The values of the complex constant K (pink bar with crosses) can be found in Fig. 5A and B. The B15C5–Na+ and the (B15C5)2–K+ complexes show both stable complexes with values of logK of 5.71 and 7.93, respectively. It is clear that the sandwich complex with K+ is more stable than the single complex with Na+. This may be due to the smaller hydrate shell of K+ compared to Na+, as well as to the increased content of the enthalpy term due to the interaction of twice as many donor atoms with the metal ion. Similar trends are observed for the 12C4 (cavity size of 1.20–1.50 Å), which forms single complexes with Li+ (ID = 1.48 Å) and sandwich complexes with Na+.28,29 Both complexes show high complex stabilities with a log K of 4.71 for the B12C4–Li+ complex and 8.21 for the (B12C4)2–Na+ complex (pink bar with crosses) (Fig. 5C and D). In conclusion, the experiments show a stronger complexation of the sandwich complex compared to the single complex. The B9C3 shows the smallest cavity size among all investigated CEs in this work with a value of approximately 0.25 Å and forms sandwich complexes with Li+.17,21,30 The only ion that could form a single complex with the B9C3 would be beryllium.31 Since this ion is very toxic and only available at extremely low concentrations in aqueous resources (e.g. Be2+ concentration in the Arctic Ocean: 9–15 pmol kg−1i.e. cBe ≈ 80–140 pg L−1), and there is no evidence that Be2+ is found in Li+-relevant aqueous resources such as the Dead Sea (cLi = 18 mg L−1), experiments with beryllium were not further considered and performed.12,32,33 The log K (6.89) of the (B9C3)2–Li+ complex can be seen in Fig. 5E.
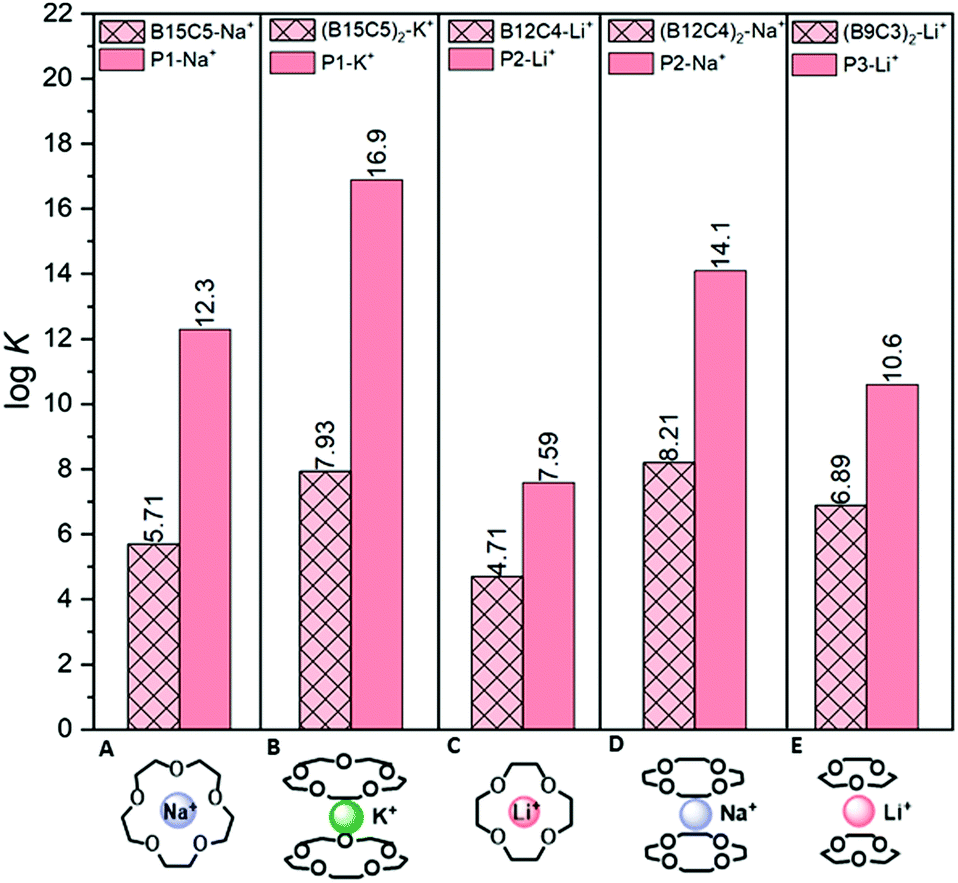 | ||
| Fig. 5 LogK of the CE complexes of the free CEs and the polymer-based CEs. The pink bar with crosses represents the logK of the free CE- and the pink bar of the polymer-based CE complexes. | ||
In the following, the complex stabilities of the free CEs B15C5, B12C4, and B9C3 are compared with those in the polymer. For this purpose, the degrees of functionalization of the CEs in the polymer were synthesized to be as similar as possible.
It is worth mentioning that the polymers P2 and P3 were functionalized with 12C4 and 15C5 (without a benzene group), since these two CEs were commercially available with a hydroxyl functionality.
The B9C3 CE was synthesized and modified with a hydroxyl group via Duff reaction and subsequent reduction with sodium borohydride (NaBH4). An overview of the synthetic procedure can be found in the SI (Fig. S1, ESI†). A detailed report of the post modification of PMAA-b-PS with B9C3 and its extraction with Li+, Na+, and K+ has already been published elsewhere.21 The exact weight fractions of each functionality of the polymers can be taken from Table 1.
| Sample code | Polymer | Metal ion extraction |
|---|---|---|
| P1 | P(MAA25-r-15C5MA25)-b-PS5056.3 | Na+, K+ |
| P2 | P(MAA21-r-12C4MA19)-b-PS6048.6 | Na+, Li+ |
| P3 | P(MAA20-r-B9C3MA20)-b-PS6049.1 | Li+ |
To ensure good comparability of the two-phase extraction of the free CE and polymer-based CEs, the same concentration of CE to metal ion was chosen to be 10:1. The P1–Na+ complex (log K = 12.3) in comparison to the B15C5–Na+complex (log K = 5.71; both forming single complexes with Na+) shows a log K 106.0 times higher than for the free CE (Fig. 5A). The complexation of K+ with polymer P1, shows a particularly high complexation constant of log K = 16.9 and thus has the highest complexation constant of all studied systems and ions (109 times higher than for the free CE). A similar observation can be made for polymer P2. Here, a log K for Li+ (formation of a single complex) of 7.59 is achieved, whereas for the free CE a value of 4.71 was obtained. Thus, the value increases by a factor of 102.9 in the polymer material, although lithium usually must overcome a very strong bounded hydration shell before it can be complexed.
The complexation of polymer P2 with Na+ forms a sandwich complex and reaches a log K of 14.1. It is thus clearly visible that the complexation of the metal ions in the polymer by means of a sandwich complex leads to very stable complexation. The reason for this is most-likely the lower entropy loss within a polymer system, since the CEs are covalently bonded to the polymer and thus have fewer degrees of freedom compared to CEs free in solution.21,22 Sandwich complexation makes this effect even more pronounced.
With respect to the stabilization of lithium complexation, it is clear, that the sandwich complexation of polymer P3 with B9C3 units shows a higher complexation constant of log K 10.6 than polymer P2 with the 12C4 with a logK of 7.59 (formation of a single complex with Li). From the analyzed data, it can be concluded that sandwich complexation of lithium within a polymer system can achieve more stable complexations and thus may be crucial for complexation of lithium from seawater, where it occurs at very low concentrations. First attempts of successful Li+ extraction using the P3 polymer in the presence of much higher Na+ and K+ concentrations were shown in another publication.21 Selective and very stable complexation of lithium within a polymer can thus be a way to isolate the highly desirable raw material.
Author contributions
Iklima Oral and Volker Abetz conceived the experiments, analyzed the data and wrote the manuscript. Iklima Oral designed and performed all experiments.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. R. Kulkarni, H. S. Maiti and A. Paul, Bull. Mater. Sci., 1984, 6, 201–221 CrossRef CAS.
- X. Wu, G. Zhao, X. Wang and W. Liu, Tribol. Lett., 2017, 65, 1–10 CrossRef.
- L. Gupta, A. C. Hoepker, K. J. Singh and D. B. Collum, J. Org. Chem., 2009, 74, 2231–2233 CrossRef CAS PubMed.
- H. Haferkamp, M. Niemeyer, R. Boehm, U. Holzkamp, C. Jaschik and V. Kaese, Mater. Sci. Forum, 2000, 350, 31–42 Search PubMed.
- C. Grosjean, P. Herrera Miranda, M. Perrin and P. Poggi, Renewable Sustainable Energy Rev., 2012, 16, 1735–1744 CrossRef.
- O. A. Avramchik, E. I. Korotkova, E. V. Plotnikov, A. N. Lukina and Y. A. Karbainov, J. Pharm. Biomed. Anal., 2005, 37, 1149–1154 CrossRef CAS PubMed.
- H. Vikström, S. Davidsson and M. Höök, Appl. Energy, 2013, 110, 252–266 CrossRef.
- J. F. Song, L. D. Nghiem, X. M. Li and T. He, Environ. Sci.: Water Res. Technol., 2017, 3, 593–597 RSC.
- A. Kumar, H. Fukuda, T. A. Hatton and J. H. Lienhard, ACS Energy Lett., 2019, 4, 1471–1474 CrossRef CAS.
- B. Swain, Sep. Purif. Technol., 2017, 172, 388–403 CrossRef CAS.
- S. Yang, F. Zhang, H. Ding, P. He and H. Zhou, Joule, 2018, 2, 1648–1651 CrossRef.
- A. Alsabbagh, S. Aljarrah and M. Almahasneh, Miner. Eng., 2021, 170, 107038 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef CAS.
- C. J. Pedersen, J. Am. Chem. Soc., 1970, 92, 386–391 CrossRef CAS.
- A. van der Ham, T. Hansen, G. Lodder, J. D. C. Codée, T. A. Hamlin and D. V. Filippov, ChemPhysChem, 2019, 20, 2103–2109 CrossRef CAS PubMed.
- A. Boda, S. M. Ali, M. R. K. Shenoi, H. Rao and S. K. Ghosh, J. Mol. Model., 2011, 17, 1091–1108 CrossRef CAS PubMed.
- S. De, A. Boda and S. M. Ali, J. Mol. Struct. THEOCHEM, 2010, 941, 90–101 CrossRef CAS.
- J. W. Steed, Coord. Chem. Rev., 2001, 215, 171–221 CrossRef CAS.
- S. P. Gromov, A. I. Vedernikov, N. A. Lobova, L. G. Kuz’Mina, S. S. Basok, Y. A. Strelenko, M. V. Alfimov and J. A. K. Howard, New J. Chem., 2011, 35, 724–737 RSC.
- H. R. Yu, J. Q. Hu, X. H. Lu, X. J. Ju, Z. Liu, R. Xie, W. Wang and L. Y. Chu, J. Phys. Chem. B, 2015, 119, 1696–1705 CrossRef CAS PubMed.
- I. Oral and V. Abetz, Macromol. Rapid Commun., 2021, 42, 2000746 CrossRef CAS PubMed.
- A. Bey, O. Dreyer and V. Abetz, Phys. Chem. Chem. Phys., 2017, 19, 15924–15932 RSC.
- J. Lambert, M. Avila-Rodriguez, G. Durand and M. Rakib, J. Membr. Sci., 2006, 280, 219–225 CrossRef CAS.
- I. Oral, L. Grossmann, E. Fedorenko, J. Struck and V. Abetz, Polymers, 2021, 13, 3675 CrossRef CAS PubMed.
- D. Barker, M. D. McLeod, M. A. Brimble and G. P. Savage, Tetrahedron Lett., 2001, 42, 1785–1788 CrossRef CAS.
- N. Bernhard and S. Wolfgang, Angew. Chem., Int. Ed. Engl., 1978, 17, 522–524 CrossRef.
- J. D. Lamb, R. M. Izatt, C. S. Swain and J. J. Christensen, J. Am. Chem. Soc., 1980, 102, 475–479 CrossRef CAS.
- M. Gjikaj and A. Adam, Z. Anorg. Allg. Chem., 2006, 632, 2475–2480 CrossRef CAS.
- Y. Shibutani, S. Mino, S. S. Long, T. Moriuchi-Kawakami, K. Yakabe and T. Shono, Chem. Lett., 1997, 49–50 CrossRef CAS.
- R. Rencsok, K. A. Jackson, T. A. Kaplan, J. F. Harrison and M. R. Pederson, Chem. Phys. Lett., 1996, 262, 207–212 CrossRef CAS.
- M. R. Ganjali, A. Moghimi and M. Shamsipur, Anal. Chem., 1998, 70, 5259–5263 CrossRef CAS.
- A. N. Shah, M. Tanveer, S. Hussain and G. Yang, Rev. Environ. Sci. Biotechnol., 2016, 15, 549–561 CrossRef CAS.
- M. Frank, D. Porcelli, P. Andersson, M. Baskaran, G. Björk, P. W. Kubik, B. Hattendorf and D. Guenther, Geochim. Cosmochim. Acta, 2009, 73, 6114–6133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01815a |
| This journal is © The Royal Society of Chemistry 2022 |