
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double thermoresponsive graft copolymers with different chain ends: feasible precursors for covalently crosslinked hydrogels†
Jingcong
Xu
 a and
Volker
Abetz
*ab
a and
Volker
Abetz
*ab
aInstitute of Physical Chemistry, Universität Hamburg, Grindelallee 117, 20146 Hamburg, Germany
bInstitute of Membrane Research, Helmholtz-Zentrum Hereon, Max-Planck-Straße 1, 21502 Geesthacht, Germany. E-mail: volker.abetz@hereon.de
First published on 17th February 2022
Abstract
The tailored synthesis of graft copolymers from acrylic and methacrylic monomers can be accomplished solely through photoiniferter reversible addition–fragmentation chain transfer (RAFT) polymerization. Samples with poly[oligo(ethylene glycol) methacrylate] (POEGMA) backbones synthesized under green light irradiation and poly(N-isopropylacrylamide) (PNIPAM) side chains growing under blue light irradiation are presented. As monitored by temperature-dependent dynamic light scattering (DLS) measurements and temperature-variable nuclear magnetic resonance (NMR) spectroscopy, the architecture of the graft copolymers allows unique two-step lower critical solution temperature (LCST) transitions in aqueous solutions. Meanwhile, different end-groups introduced by the corresponding RAFT agents affect the detailed thermoresponsive behavior remarkably. This RAFT strategy shows more advantages when the multiple trithiocarbonate groups are converted into thiol reactive pyridyl disulfide (PDS) groups via a facile post-polymerization modification. The PDS-terminated graft copolymer can then be regarded as a usable precursor for various applications, such as thermoresponsive hydrogels.
Introduction
Thermoresponsive polymers can reversibly change their solubility in specific solvents in response to temperature changes. Thermoresponsive homopolymers with a lower critical solution temperature (LCST) in water turn from a hydrophilic into a hydrophobic state once the solutions are heated beyond their cloud points (or phase transition temperatures) (TPT). When two blocks of LCST polymers are coupled together, double thermoresponsiveness can be expected when the concentrations meet the critical aggregation concentration.1–3 To date, a variety of linear block copolymers with two LCST-type TPT have been reported. Their potential applications in biomedical areas were highlighted in recent reviews.4–6 In contrast to linear architectures, multi-thermoresponsive graft copolymers remained sparsely investigated, which is surprising as graft copolymers with stimuli-responsive backbones or side chains usually show unique self-assembling behavior and rheological properties in water due to their confined structures and additional intramolecular interactions.7,8 In this contribution, we present double thermoresponsive graft copolymers with poly[oligo(ethylene glycol) methacrylate] (POEGMA) as their backbones and poly(N-isopropylacrylamide) (PNIPAM) as the side chains. As POEGMA and PNIPAM are both commonly used LCST polymers with wide applications in biomedical areas,9–16 the study of the thermoresponsiveness of their graft copolymers is particularly appealing.The development of different reversible deactivation radical polymerization (RDRP) techniques provides facile and versatile routes for the synthesis of graft copolymers.17,18 However, the polymerization often requires the combination of two RDRP methods, like reversible addition−fragmentation chain transfer (RAFT) polymerization and atom transfer radical polymerization (ATRP).19,20 These methods could lead to laborious synthetic and purification steps. Recently, photoiniferter polymerization was found to be a more efficient route for the preparation of polymers with graft or brush structures.21–24 In 2018, a catalyst-free photoiniferter RAFT polymerization that was selectively initiated by two kinds of visible light irradiation was introduced for the preparation of graft copolymers.22 In that study, methyl methacrylate (backbone) was polymerized via photoiniferter RAFT under green light irradiation, while N,N-dimethylacrylamide (side chains) was polymerized under blue light irradiation. The same two-step synthetic route could also be applied to our work. As shown in Scheme 1, the copolymerization of OEGMA and the RAFT inimer, 2-(2-(n-butyltrithiocarbonate)propionate)ethyl methacrylate (BTPEMA), was performed in the first step. The polymerization was mediated either by 2-cyano-2-propyl dodecyl trithiocarbonate (CPDTC) or 4-cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid (CDTPA). These two RAFT agents with distinct R-groups can both provide good control for the polymerization of methacrylates under green light irradiation (515 nm).22,25 BTPEMA, on the other hand, shows no absorption in the green light region, preventing the photolysis of the trithiocarbonate (TTC) pendant groups (marked in lilac). In addition, the fragmentation to secondary carbon radical R-groups is not favored in the RAFT pre-equilibrium step (i.e., in the chain transfer process).22 BTPEMA thus made no contribution to the radical propagation under green light, enabling the achievement of linear statistical copolymers P(OEGMA-stat-BTPEMA) that bore different functions at α-terminals. In the second synthetic step, all the TTC-groups along the chain could be activated by blue light irradiation (467 nm). They served as branching points from which PNIPAM side chains grew simultaneously under good control. The aim of using two different RAFT agents in the first step was to examine the influence of end-group on the thermoresponsive behavior of polymers in aqueous solutions. Previously, it was reported that the functionality at the chain end of linear LCST polymers sometimes affects the self-assembly of chains to a great extent.26–29 In this work, it was discovered that it plays a crucial role in the synthesized graft copolymers as well. The intriguing self-assembling behaviors of different samples in dilute solutions will be discussed in the following section.
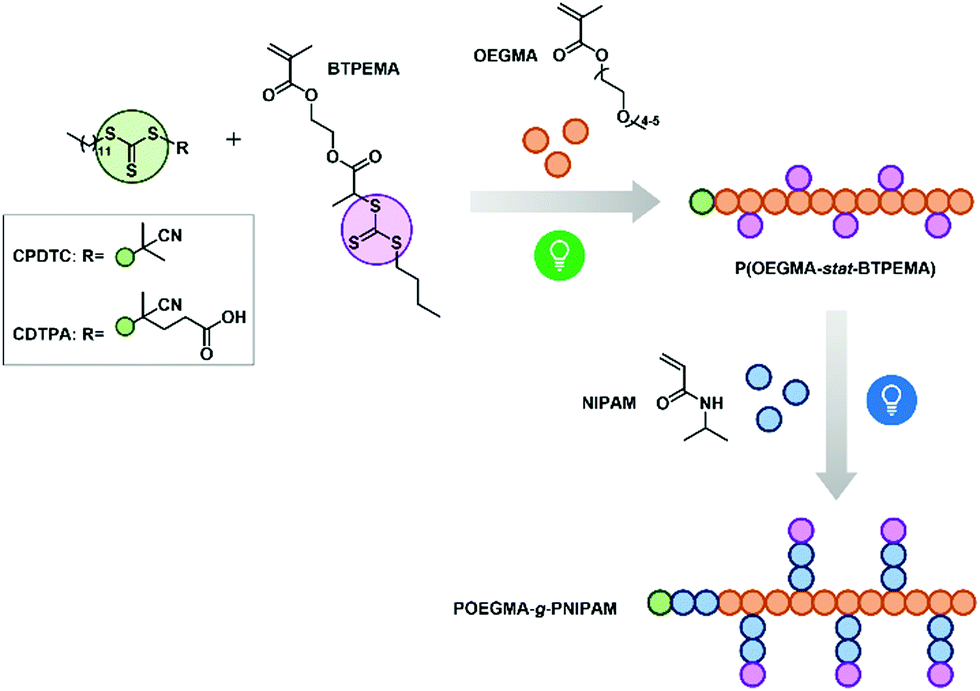 | ||
| Scheme 1 Synthetic route for graft copolymers, and used monomers and RAFT agents in this study. | ||
Besides the convenient accessibility, another compelling advantage of the graft copolymers fabricated solely through the photoiniferter RAFT approach is that all the TTC-groups can undergo various kinds of modifications. In the past decades, abundant processes have been developed to remove the thiocarbonylthio moieties of polymers synthesized by RAFT polymerization for various purposes.30–40 The TTC-group can be converted to thiol functionality when reacting with suitable primary amines.30 By treating the RAFT-synthesized polymers with an amine and 2,2′-dithiodipyridine (DTP) simultaneously, stable pyridyl disulfide (PDS)-terminated polymers can be generated directly. In the field of polymer therapeutics, the PDS-group is known for its thiol reactivity under mild conditions.41–45 Thus, the PDS-functionalized polymers have an enormous potential for further modifications according to specific applications. Herein, we present exemplarily an efficient way to afford a covalently crosslinked hydrogel from PDS-terminated graft polymers.
Experimental
Materials
CPDTC (abcr, 97%), CDTPA (abcr, 97%), DTP (TCI, 98%), 2,2′-(ethylenedioxy)diethanethiol (EDT, Sigma Aldrich, 95%), hexylamine (Sigma Aldrich, 99%). BTPEMA was synthesized according to the literature.22 All the solvents were used as received. OEGMA (Sigma Aldrich,![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 300 Da) was passed through activated basic alumina prior to the polymerizations. NIPAM (TCI, 98%) was recrystallized twice from n-hexane and stored at −30 °C.
n = 300 Da) was passed through activated basic alumina prior to the polymerizations. NIPAM (TCI, 98%) was recrystallized twice from n-hexane and stored at −30 °C.
Procedure for the polymerization of backbones
In a typical photoiniferter RAFT polymerization of P(OEGMA-stat-BTPEMA), OEGMA, BTPEMA and RAFT agent ([OEGMA]/[BTPEMA]/[CPDTC or CDTPA] = 390/10/1) were dissolved in 1,4-dioxane in a polymerization vial. Dimethylformamide (DMF) was added as an internal standard for calculation of conversion. The total solid (RAFT agent plus the monomers) concentration was kept at 20% (w/w). The polymerization vial was purged with nitrogen for 15 min and the reaction was subsequently carried out at 70 °C under green light irradiation (515 nm) of 1.13 mW cm−2. After 3 h, the polymerization was stopped by exposing the mixture to air and cooling with an ice bath. Conversion was determined by nuclear magnetic resonance (NMR) spectroscopy. The polymer was then purified through precipitation in an excess amount of n-hexane three times. The purified product was dried in vacuo at 40 °C for 24 h.Procedure for the polymerization of POEGMA-g-PNIPAM graft copolymers
In a typical photoiniferter RAFT polymerization of POEGMA-g-PNIPAM, NIPAM and the POEGMA backbone ([NIPAM]/[P(OEGMA-stat-BTPEMA)] = 560/1, [NIPAM]/[TTC] = 80/1) were dissolved in 1,4-dioxane in a polymerization vial. DMF was then added as an internal standard for calculation of conversion. The total solid (P(OEGMA-stat-BTPEMA) plus NIPAM) concentration was kept at 15% (w/w). The polymerization vial was purged with nitrogen for 15 min and the reaction was subsequently carried out at 70 °C under blue light irradiation (467 nm) of 3.86 mW cm−2. After 4 h, the polymerization was stopped by exposing the mixture to air and cooling with an ice bath. Conversion was determined by NMR. The polymer was then purified through precipitation in an excess amount of diethyl ether three times. The purified product was dried in vacuo at room temperature for 24 h. For the kinetic study, aliquots of the reaction mixture were taken at specific moments during the polymerization and directly diluted with deuterated chloroform to determine monomer conversions through 1H-NMR spectra.Procedure for the post-polymerization modification of graft copolymers
POEGMA-g-PNIPAM (n,th = 93.1 kDa, 7 TTC-functions per chain) (210 mg, 0.016 mmol TTC-functions) and DTP (173 mg, 50-fold molar excess with respect to TTC) was dissolved in 4 mL 1,4-dioxane. The solution was purged with nitrogen for 15 min to remove oxygen. Afterwards, 0.1 mL of the hexylamine solution (0.1 mL hexylamine dilute in 2 mL 1,4-dioxane) (2.5-fold molar excess with respect to TTC) was added under the protection of nitrogen. The reaction mixture stayed yellow and was stirred overnight at room temperature. After precipitation in diethyl ether (4 times), a white PDS-functionalized polymer, POEGMA-g-PNIPAM-PDS, was recovered.
Procedure for hydrogel preparation
POEGMA-g-PNIPAM-PDS (n,th = 92.8 kDa, 7 PDS-functions per chain) (162 mg, 0.012 mmol PDS-functions) was dissolved in 0.4 mL DMF in a glass vial. Afterwards, 0.02 mL of the EDT solution (0.1 mL EDT dilute in 2 mL DMF) (0.011 mmol thiol-functions) was added quickly. After shaking for seconds, the reaction mixture turned uniformly yellow immediately. In a few minutes, a free-standing gel with yellow color could be observed. The vial was then put aside at room temperature for 30 min to allow further gelation. Afterwards, the gel was immersed in tetrahydrofuran (THF) to remove the crosslinker, uncrosslinked polymer chains, DMF and other small molecules. When the gel became colorless, the residual THF was allowed to evaporate at room temperature. Finally, the gel was dried in vacuo.
Analytics
w) was estimated through the partial Zimm analysis (see eqn (S8), ESI†) in the software ALVStat. The aggregation number (Nagg) of the formed micelles above TPT was estimated as w,agg/w,uni, where w,agg and w,uni are the weight average molecular weights of the micelles and the unimers, respectively.
| Swelling ratio = (Ws − Wd)/Wd, |
Results and discussion
Polymerization of the graft copolymers
As mentioned before, the two RAFT agents, CPDTC and CDTPA (Scheme 1), were used to synthesize the backbones of graft copolymers. After the first synthetic step under the green light, two statistical copolymers, P(O245B6) and P(O234B6)–COOH, were yielded. For the nomenclature of the samples, “O” and “B” stand for OEGMA and BTPEMA, respectively. The subscripts denote the number of the respective monomers in the statistical copolymers based on conversions. The degree of polymerization (DP) is the sum of subscripts. In comparison with the homopolymer POEGMA (PO234–COOH), the statistical copolymers exhibit similar number average molecular weights (n,SEC) and dispersities (Đ) (Table 1). The relatively narrow monomodal molecular weight distributions shown in Fig. 1a suggest successful polymerizations of linear backbones. The similar molar ratios between OEGMA and BTPEMA in the copolymers (OEGMA/BTPEMA [mol]) confirm that the average number of BTPEMA units per chain of the two samples were comparable, i.e., the average number of branching points per backbone were comparable. It can be calculated that both samples own not only one TTC-function at the ω-terminal, but also six pendant ones along every chain.
| Sample namea | OEGMA/BTPEMAb [mol] | OEGMA/NIPAMb [mol] | α [%] |
n,th
[kDa] |
n,SEC
[kDa] |
Đ |
|---|---|---|---|---|---|---|
| a Subscripts following O, B and N denote DP of different monomers based on conversion; for graft copolymers, subscripts following the parentheses denote the number of PNIPAM segments per chain. b Molar ratio between monomers in products calculated from 1H-NMR spectra (see Fig. S4–S7, ESI). c Monomer conversion calculated from 1H-NMR spectra with reference signal of DMF. d Number average molecular weight calculated from monomer conversion. e Determined by SEC in THF or DMAc relative to PMMA standards. | ||||||
| PO234–COOH | — | — | 60 | 70.6 | 38.8 | 1.30 |
| P(O245B6) | 39/1 | — | 63 | 75.9 | 41.3 | 1.37 |
| P(O234B6)–COOH | 41/1 | — | 60 | 72.8 | 41.6 | 1.31 |
| PO245(PN40)7 | 39/1 | 45/55 | 50 | 107.3 | 96.2 | 1.47 |
| PO234(PN43)7–COOH | 41/1 | 45/55 | 54 | 106.9 | 87.6 | 1.42 |
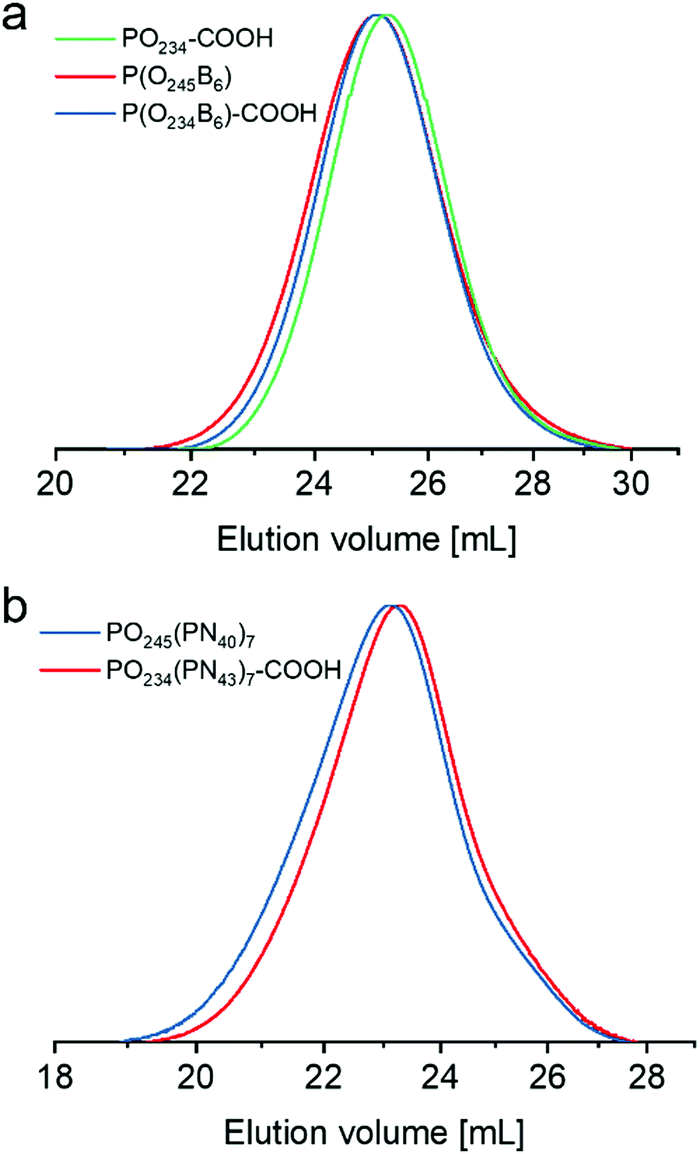 | ||
| Fig. 1 SEC curves of (a) linear polymers measured with THF as the mobile phase and (b) graft copolymers measured with DMAc as the mobile phase. | ||
The PNIPAM side chains, as shown in Scheme 1, were synthesized via blue light irradiation. The DP of PNIPAM per side chain was controlled at about 40 for each polymerization. Kinetic studies were performed during two typical chain extensions (see Fig. S8, ESI†). It is obvious that the rate of chain extension was higher from the backbone P(O234B6)–COOH than from the homopolymer PO234–COOH, which makes sense as more TTC-groups (branching points) could be initiated in the backbone by blue light irradiation, leading to a higher radical concentration. Therefore, graft copolymers instead of linear block copolymers result from the statistical copolymers.
Eventually, two samples with PNIPAM side chains, PO245(PN40)7 and PO234(PN43)7–COOH, were obtained from P(O245B6) and P(O234B6)–COOH, respectively. For the nomenclature of the graft copolymers, “(PNm)7” means that from the 7 TTC-functions (including those from “B”) of every chain, 7 PNIPAM segments with a DP of “m” were synthesized. The 1H-NMR results reveal that the molar ratios between OEGMA and NIPAM (OEGMA/NIPAM [mol]) in these two graft copolymers are nearly identical and agree well with the theoretical values (Table 1). According to the results of SEC analysis, these two graft copolymers also have similar molecular weights (Fig. 1b), enabling further investigations on the influence of the end-group.
Thermoresponsive behavior in dilute aqueous solutions
The dilute aqueous solutions (c = 0.1% [w/w]) of two linear backbone samples were first prepared for temperature-dependent DLS measurements. The results of both samples exhibit reversible LCST transitions with minor hysteresis during heating and cooling (Fig. 2a and b). The LCST-type TPT of P(O245B6) and P(O234B6)–COOH, which are determined as the onset of the increase in the hydrodynamic radius (RH) during heating, are 59 °C and 60 °C, respectively. The ignorable difference between TPT corresponds well to our expectation, considering their similarn,SEC and Đ. However, it is quite surprising that there is a huge difference in their detailed self-assembling behaviors in water. While both backbones could be dissolved in water in the form of unimers with RH < 10 nm below TPT, the sizes of formed aggregates above TPT differed remarkably.
 | ||
| Fig. 2 Evolution of RH of (a) P(O245B6) or (b) P(O234B6)–COOH in dilute aqueous solutions during heating (red dots) and cooling (blue dots) monitored by DLS; (c) assumed self-assembling processes of P(O245B6) and P(O234B6)–COOH chains at different stages (BTPEMA and the oxyethylene brushes are not shown in the illustrations for simplicity). | ||
In the case of P(O245B6) (Fig. 2a), the polymer chains became hydrophobic and started to agglomerate at TPT. The RH of the particles increased to approximately 800 nm at 62 °C and remained relatively constant at higher temperatures. For P(O234B6)–COOH (Fig. 2b), the polymeric aggregates also started growing at TPT, yet the growth ceased already when RH was only around 200 nm. To elucidate this difference reasonably, the mechanism behind the LCST transition of POEGMA must be taken into account. A molecule of homo-POEGMA can basically be divided into two parts: the coiled backbone-like hydrophobic moiety and the pendant oxyethylene brushes. As shown in Fig. 2c, the solubility of the polymers in water below TPT results from the hydration shell formed via the hydrogen bonds between water molecules and the ether oxygen groups of the brushes (brushes are not shown for simplicity). During heating the hydrogen bonds can be disrupted, causing the conformational changes of the oxyethylene brushes and the dehydration of the chains. According to previous research,47 the conformation of the coiled hydrophobic moiety of POEGMA barely changes during the dehydration process. Additionally, the ether oxygen groups of the distorted brushes are still exposed to water molecules to some extent. Therefore, the large aggregates during the LCST transition are mainly formed by the “hydrogen bond bridges” among multiple chains and their structures should be relatively loose. The DLS result of P(O245B6) corresponds well to the mechanism. However, the self-assembling behavior of P(O234B6)–COOH could be disturbed by the carboxyl group which is also supposed to be surrounded by a hydration shell in water.48 This extra hydration shell might not be significantly affected by the LCST transition, and the carboxyl group could stay hydrated during the whole heating process. The bulky hydration shell at the chain end could then hinder the bridging between chains, leading to a smaller size of the aggregates. When heating the P(O234B6)–COOH solution higher than 63 °C, “hydrogen bond bridges” were shortened and water was expelled from the loose aggregates, as reflected by the decrease in RH.47 Moreover, the coiled hydrophobic moiety could start collapsing slowly at higher temperatures due to weak van der Waals interactions.47,49 In principle, the decrease in RH following the rapid phase transition should also be observed in the P(O245B6) solution. The absence of the contraction of P(O245B6) aggregates could be owing to the compensation from more continuously bound free chains.
Through the comparison of these results, it can be found that the end-group plays an important role in the thermoresponsive behaviors of the POEGMA backbones. Subsequently, the aqueous solutions of two different graft copolymers were analyzed by DLS measurements. The end-group again made an immense difference in the sizes of polymeric aggregates at high temperatures.
The DLS result of the PO234(PN43)7–COOH solution (c = 0.1% [w/w]) is easier to interpret (Fig. 3a). All the obtained intensity correlation functions could be fitted by using the standard cumulant approach (see eqn (S9), ESI†). Apparently, a reversible two-step LCST transition occurred during the measurement. It is well-known that homopolymers of PNIPAM possess an LCST-type TPT of about 32 °C.50 Therefore, the PNIPAM side chains are, most likely, responsible for the first increase in RH at 30 °C during heating. Above the first TPT, the side chains turned hydrophobic and are expected to form cores of the micelles. However, the RH of the micelles between 30 °C and 50 °C (region II) was still less than 14 nm. Such a compact dimension can be legitimately explained from two perspectives. First, while the side chains combined together in the core, the hydrophilic backbones consisting of POEGMA were folded on the periphery so that flower-like micelles with thin shells were generated in this temperature region (Fig. 3b).51,52 The strong intramolecular interaction may have limited the Nagg of the macromolecules in a micelle.53 In addition, the hydrated carboxyl groups remaining outside could prevent the formation of larger aggregates. To estimate the Nagg of these compact micelles, SLS measurements were conducted with the same concentration at 25 °C and 40 °C (below and above the first TPT). After fitting the SLS data by the partial Zimm approach (see Fig. S9, ESI†), the Nagg was calculated to be about 2.4, which is indeed quite low and in accordance with the DLS result. Regarding the second LCST transition, the TPT of the POEGMA backbone was lowered to about 45 °C, indicating enhanced polymer–polymer interactions due to the higher density of the chains in the shells. Different from the cases of the statistical copolymers, a slight precontraction (decrease in RH) before the second LCST transition can be observed in region II during heating, corresponding to the dehydration process inside the shells of the amphiphilic micelles. Next, a rapid agglomeration of the micelles occurred based on the mechanism explained in Fig. 2c. Yet, the RH of less than 20 nm at about 55 °C was still relatively small, which can also be attributed to the bulky hydration shells around the carboxyl groups. Above 55 °C (region III), a gradual decrease in RH took place but was less evident than that of P(O234B6)–COOH at high temperatures. The reason could be that the micelles were already relatively dense due to the precontraction before aggregation. The more elaborate CONTIN analysis on the DLS data at three representative temperatures shown in Fig. 4a confirms the two-step LCST transition and the compact sizes of the particles.
 | ||
| Fig. 3 Evolution of RH of (a) PO234(PN43)7–COOH or (c) PO245(PN40)7 in dilute aqueous solutions during heating (red dots) and cooling (blue dots) monitored by DLS; assumed self-assembling process of (b) PO234(PN43)7–COOH or (d) PO245(PN40)7 at different temperatures. | ||
 | ||
| Fig. 4 Intensity-weighted size distributions of the particles formed by (a) PO234(PN43)7–COOH or (b) PO245(PN40)7 at representative temperatures obtained by CONTIN analysis on DLS data. | ||
As for PO245(PN40)7, its DLS result was more complicated to evaluate. The measurement was initially also performed at a concentration of 0.1% (w/w). However, it was difficult to fit the intensity correlation functions reasonably well through the cumulant approach. Excessively large aggregates could be formed in the solution at high temperatures. To avoid such aggregations, the concentration was lowered to 0.05% (w/w). The result of the DLS measurement is shown in Fig. 3c. Below the TPT (region I) from PNIPAM, the DLS data could still be well fitted through the standard cumulant approach, implying that all the polymer chains were dissolved in the form of unimers like PO234(PN43)7–COOH. For the data obtained between 32 °C and 48 °C (region II) (exemplarily shown in Fig. S10, ESI†), however, it was more suitable to fit them by the sum of two exponential decay functions (see eqn (S11), ESI†),54 which means that two particle species emerged when the side chains became hydrophobic. The RH of the smaller species in Fig. 3c and the micelles of PO234(PN43)7–COOH in region II were alike. With the help of the CONTIN analysis, the size distributions of the particles formed at representative temperatures were calculated. As presented in Fig. 4b, a bimodal size distribution was measured in the solution at 40 °C. Considering that the scattering intensity is proportional to RH6, the smaller species still dominated in the solution, which means that the Nagg of most micelles was limited by the intramolecular interaction. The formation of larger aggregates (RH > 1000 nm) could result from the hydrophobic end-group. Unlike that of PO234(PN43)7–COOH, the α-terminal of PO245(PN40)7 could not separate all the particles. The flower-like micelles thus had the chance to link with each other through the hydrophobic interaction between the PNIPAM cores (Fig. 3d). The formation of large aggregates became inevitable despite a lower concentration (0.05% [w/w]). In temperature region II (Fig. 3c), it is also worth noticing that the RH of both particle species started falling off when the solution was heated to 43 °C, indicating the dehydration of the looped POEGMA chains. When the balance between hydrophilic and hydrophobic interactions was disrupted at the second TPT, numerous micelles could agglomerate together to form larger clusters in the absence of carboxyl groups and the small particles disappeared. After reaching its maximum value in region III, the RH of the clusters tended to decrease gradually with the increasing temperature, which resembles the behavior of PO234(PN43)7–COOH. After heating, the solution was cooled to room temperature (25 °C). Although the temperature step was controlled at 1 °C by the thermostat during the whole measurement, the cooling rate was almost twice as fast as the heating rate. Besides, no stirring was allowed for the DLS measurements. It was, therefore, reasonable that the LCST transitions involving the hydrophobic particles with complex structures were not completely reversible. Nevertheless, the chains could still be fully redissolved at room temperature.
Apart from the DLS measurements, the temperature-variable 1H-NMR spectra of the graft copolymers in D2O also confirm the profound influence of the end-group. Fig. 5a and b show the characteristic peaks belonging to POEGMA backbones and PNIPAM side chains at different temperatures. The decline of the signal of PNIPAM at 35 °C was most significant in both samples, revealing the main driving force of the first LCST transition. At 40 °C, the signal of PNIPAM became hardly detectable. Interestingly, from the spectra at 55 °C, it can be noted that the signal of POEGMA in PO245(PN40)7 was much flatter than that in PO234(PN43)7–COOH, suggesting a higher degree of phase separation of PO245(PN40)7 at this temperature. This difference is in good agreement with the DLS results.
 | ||
| Fig. 5 Normalized temperature-variable 1H-NMR spectra of (a) PO234(PN43)7–COOH and (b) PO245(PN40)7 measured in D2O; (c) left: temperature dependence of the transmittance measured for the aqueous solutions of PO234(PN43)7–COOH (0.1% [w/w]) and PO245(PN40)7 (0.05% [w/w]); right: enlarged image of the measurements. | ||
Furthermore, the temperature dependence of the transmittance of the sample solutions is plotted in Fig. 5c. Apparently, the PO245(PN40)7 solution became much more turbid at high temperatures even with a lower concentration. Through the enlarged image of the results, a slight drop of the transmittance can be noticed for both samples at around 35 °C. The second drop of the transmittance of PO245(PN40)7 solution starting at about 43 °C was drastic, while the change of turbidity of PO234(PN43)7–COOH solution in the rest of the heating process was still very small. Although the random error of the measurement caused some fluctuations, the result of PO234(PN43)7–COOH still shows a two-step transition roughly.
All these results above demonstrate that both graft copolymers display a two-step LCST transition originating from POEGMA and PNIPAM. Without the carboxyl groups at the chain end, significantly larger particles arise at high temperatures.
Modification of end-group
In this section, the end-group modification aiming for further chemical crosslinking is discussed. In order to minimize side reactions, a well-defined graft copolymer PO218(PN32)7 without carboxyl groups was prepared. According to the DLS result in Fig. S11 (ESI†) and Fig. 3c, PO218(PN32)7 and PO245(PN40)7 show a similar two-step LCST transition in water. The major difference is that the first LCST transition of PO218(PN32)7 occurred at a higher temperature, which can be ascribed to the shorter PNIPAM side chains, i.e., the weaker polymer−polymer interaction.55 After confirming the reproducibility of the LCST transitions, the polymer was subjected to chemical modification.In the presence of excess DTP and hexylamine, the TTC-groups could be replaced by PDS-groups. The modified sample, PO218(PN32)7-PDS, was first characterized by SEC and UV−vis spectroscopy (Fig. 6). The molecular distribution is slightly broadened after modification, which could be due to the undesired oxidation between thiol groups during the aminolysis.56 Since the oxidation just produces disulfide and the goal of the modification was chemical crosslinking, the impact of this side reaction is trivial in this work. In Fig. 6b, the absorbance of TTC-groups at about 308 nm disappears after modification, manifesting nearly complete removal of TTC-groups.
 | ||
| Fig. 6 (a) SEC curves measured before aminolysis and after aminolysis with DTP; (b) UV−vis spectra measured before and after aminolysis in 1,4-dioxane. | ||
Crosslinking of the graft copolymer
Since the thiol reactive PDS-groups can react with thiol groups at ambient temperature without any catalysts, a dithiol based crosslinker, EDT, was used to obtain a covalently crosslinked hydrogel (Scheme 2). | ||
| Scheme 2 Schematic illustration for chemical crosslinking of POEGMA-g-PNIPAM-PDS with the crosslinker, EDT. | ||
Fig. 7a shows the procedure for the preparation of a polymeric gel briefly. The yellow color of the gel caused by the small molecule, 2-mercaptopyridine, released after crosslinking proved a successful reaction between PDS- and thiol groups. Ultimately, a colorless free-standing gel was formed after purification. After removing the organic solvents, the thermoresponsiveness of the POEGMA–PNIPAM hydrogel was visually tested (Fig. 7b). The hydrogel was fully swollen and transparent at room temperature. At 60 °C, water was squeezed out of the network drastically and the hydrogel became opaque. Once it was allowed to cool down, the hydrogel regained its transparency.
 | ||
| Fig. 7 (a) General procedure for the preparation of POEGMA–PNIPAM hydrogel; (b) macroscopic test on the thermoresponsiveness of the hydrogel. | ||
The swelling ratios of the hydrogel at different temperatures were also determined quantitatively by gravimetry. Because the intensive shrinkage at a high temperature could cause splits in the sample and affect the result, the highest target temperature was lower than 50 °C. As depicted in Fig. 8, the swelling ratio of the hydrogel started to decline below 33 °C, which is lower than the TPT of PNIPAM before crosslinking. This could be correlated with the high local concentration of chains within the network. Moreover, the network had a more confined and complex internal structure than the free polymer chains. After the contraction of PNIPAM chains, the density of POEGMA chains became even higher, which could lead to a huge decrease in the TPT. The phase transitions of PNIPAM and POEGMA could be overlapped. It was thus difficult to distinguish the LCSTs of POEGMA and PNIPAM. Instead, the hydrogel kept shrinking within a relatively broad temperature range.
 | ||
| Fig. 8 Temperature dependence of the swelling ratio of the hydrogel sample. | ||
Conclusions
In conclusion, two well-defined graft copolymers differing merely in the end-group were synthesized by sequentially switching the wavelengths of the light source for photoiniferter RAFT polymerization. Both polymers exhibit a two-step LCST transition in water originating from the backbone and the side chains. However, the sizes of their aggregates formed above each LCST-type TPT are significantly different. Without the carboxyl group at the α-terminal, polymeric aggregates with larger RH were detected by DLS measurements. Furthermore, multiple TTC-groups along the polymer chains were successfully converted into thiol reactive PDS-groups after polymerization by aminolysis in the presence of DTP, enabling simple chemical crosslinking at room temperature without other additives. The obtained thermoresponsive hydrogel may have potential in various fields of applications, like controlled drug release in biomedical applications. Apart from the reaction with a crosslinker, it is also possible to use the reactive thiol groups for attachment of other functional molecules like enzymes which are then shielded or unshielded from substrates as a function of temperature.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The work is financially supported by Universität Hamburg, a collaborative project funded by Helmholtz Association and Russian Science Foundation (grant number HRSF-0075) and by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - GRK 2536. The authors would like to thank Felix Lauterbach and Birgit Hankiewicz for providing the programs attaining convenient fitting of the DLS results. The authors would also like to thank Martin Kehden for his help with SEC measurements.Notes and references
- Q. Li, C. Gao, S. Li, F. Huo and W. Zhang, Polym. Chem., 2014, 5, 2961–2972 RSC.
- Y. Huang, P. Yong, Y. Chen, Y. Gao, W. Xu, Y. Lv, L. Yang, R. L. Reis, R. P. Pirraco and J. Chen, RSC Adv., 2017, 7, 28711–28722 RSC.
- Q. Zhang, J.-D. Hong and R. Hoogenboom, Polym. Chem., 2013, 4, 4322–4325 RSC.
- L. Johnson, D. M. Gray, E. Niezabitowska and T. O. McDonald, Nanoscale, 2021, 13, 7879–7896 RSC.
- Y. Kotsuchibashi, Polym. J., 2020, 52, 681–689 CrossRef CAS.
- Y. Kotsuchibashi, M. Ebara, T. Aoyagi and R. Narain, Polymers, 2016, 8, 380 CrossRef PubMed.
- T. Jiang, V. Aseyev, J. Niskanen, S. Hietala, Q. Zhang and H. Tenhu, Macromolecules, 2020, 53, 8267–8275 CrossRef CAS PubMed.
- L. Lauber, J. Depoorter, T. Nicolai, C. Chassenieux and O. Colombani, Macromolecules, 2017, 50, 8178–8184 CrossRef CAS.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Polymers, 2020, 12, 580 CrossRef CAS PubMed.
- G.-F. Luo, W.-H. Chen and X.-Z. Zhang, ACS Macro Lett., 2020, 9, 872–881 CrossRef CAS.
- L. Tang, L. Wang, X. Yang, Y. Feng, Y. Li and W. Feng, Prog. Mater. Sci., 2021, 115, 100702 CrossRef CAS.
- S. H. Yu, F. Ercole, N. A. Veldhuis, M. R. Whittaker, T. P. Davis and J. F. Quinn, Polym. Chem., 2017, 8, 6362–6367 RSC.
- Y. Ling, J. Ren, T. Li, Y. Zhao and C. Wu, Chem. Commun., 2016, 52, 4533–4536 RSC.
- E. Ellis, K. Zhang, Q. Lin, E. Ye, A. Poma, G. Battaglia, X. J. Loh and T.-C. Lee, J. Mater. Chem. B, 2017, 5, 4421–4425 RSC.
- J. Teo, J. A. McCarroll, C. Boyer, J. Youkhana, S. M. Sagnella, H. T. Duong, J. Liu, G. Sharbeen, D. Goldstein, T. P. Davis, M. Kavallaris and P. A. Phillips, Biomacromolecules, 2016, 17, 2337–2351 CrossRef CAS PubMed.
- M. Bozorg, B. Hankiewicz and V. Abetz, Soft Matter, 2020, 16, 1066–1081 RSC.
- S. Pearson, C. St Thomas, R. Guerrero-Santos and F. D'Agosto, Polym. Chem., 2017, 8, 4916–4946 RSC.
- M. Semsarilar and V. Abetz, Macromol. Chem. Phys., 2021, 222, 2000311 CrossRef CAS.
- D. Shen, B. Xu, X. Huang, Q. Zhuang and S. Lin, Polym. Chem., 2018, 9, 2821–2829 RSC.
- A. Ding, J. Xu, G. Gu, G. Lu and X. Huang, Sci. Rep., 2017, 7, 12601 CrossRef PubMed.
- K. J. Arrington and J. B. Matson, Polym. Chem., 2017, 8, 7452–7456 RSC.
- S. Shanmugam, J. Cuthbert, T. Kowalewski, C. Boyer and K. Matyjaszewski, Macromolecules, 2018, 51, 7776–7784 CrossRef CAS.
- N. Corrigan, F. J. Trujillo, J. Xu, G. Moad, C. J. Hawker and C. Boyer, Macromolecules, 2021, 54, 3430–3446 CrossRef CAS.
- M. Hartlieb, Macromol. Rapid Commun., 2021, 2100514 Search PubMed.
- J. Xu and V. Abetz, Macromol. Rapid Commun., 2021, 42, 2000648 CrossRef CAS PubMed.
- M. L. Ohnsorg, J. M. Ting, S. D. Jones, S. Jung, F. S. Bates and T. M. Reineke, Polym. Chem., 2019, 10, 3469–3479 RSC.
- F. Lauterbach and V. Abetz, Soft Matter, 2020, 16, 2321–2331 RSC.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- P. Kujawa, F. Segui, S. Shaban, C. Diab, Y. Okada, F. Tanaka and F. M. Winnik, Macromolecules, 2006, 39, 341–348 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polym. Int., 2011, 60, 9–25 CrossRef CAS.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- R. N. Carmean, C. A. Figg, G. M. Scheutz, T. Kubo and B. S. Sumerlin, ACS Macro Lett., 2017, 6, 185–189 CrossRef CAS.
- V. H. Dao, N. R. Cameron and K. Saito, Polym. Chem., 2017, 8, 6834–6843 RSC.
- B. A. Abel and C. L. McCormick, Macromolecules, 2016, 49, 6193–6202 CrossRef CAS.
- R. Nicolaÿ, Macromolecules, 2012, 45, 821–827 CrossRef.
- M. R. Newman, S. G. Russell, C. S. Schmitt, I. A. Marozas, T.-J. Sheu, J. E. Puzas and D. S. W. Benoit, Biomacromolecules, 2018, 19, 71–84 CrossRef CAS PubMed.
- P. Chakma, Z. A. Digby, J. Via, M. P. Shulman, J. L. Sparks and D. Konkolewicz, Polym. Chem., 2018, 9, 4744–4756 RSC.
- E. H. Discekici, S. L. Shankel, A. Anastasaki, B. Oschmann, I.-H. Lee, J. Niu, A. J. McGrath, P. G. Clark, D. S. Laitar, J. R. de Alaniz, C. J. Hawker and D. J. Lunn, Chem. Commun., 2017, 53, 1888–1891 RSC.
- A. Hess, B. V. K. J. Schmidt and H. Schlaad, Polym. Chem., 2020, 11, 7677–7684 RSC.
- H. W. Ooi, K. S. Jack, A. K. Whittaker and H. Peng, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4626–4636 CrossRef CAS.
- C. Boyer, V. Bulmus and T. P. Davis, Macromol. Rapid Commun., 2009, 30, 493–497 CrossRef CAS PubMed.
- B. Sui, C. Cheng and P. Xu, Adv. Therap., 2019, 2, 1900062 CrossRef CAS.
- D. Bontempo, K. L. Heredia, B. A. Fish and H. D. Maynard, J. Am. Chem. Soc., 2004, 126, 15372–15373 CrossRef CAS PubMed.
- I. Altinbasak, M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 7603–7624 RSC.
- Q. Song, J. Yang, S. C. L. Hall, P. Gurnani and S. Perrier, ACS Macro Lett., 2019, 8, 1347–1352 CrossRef CAS.
- F. Lauterbach, Advances in RAFT polymerization process design and analysis, Dissertation, Universität Hamburg, 2020 Search PubMed.
- S. Sun and P. Wu, Macromolecules, 2013, 46, 236–246 CrossRef CAS.
- M. Di Gioacchino, F. Bruni, S. Imberti and M. A. Ricci, J. Phys. Chem. B, 2020, 124, 4358–4364 CrossRef CAS PubMed.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J.-F. Lutz, Ö. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- L. I. Atanase, J. Desbrieres and G. Riess, Prog. Polym. Sci., 2017, 73, 32–60 CrossRef CAS.
- Z. Tuzar and P. Kratochvíl, Adv. Colloid Interface Sci., 1976, 6, 201–232 CrossRef CAS.
- F. Bougard, C. Giacomelli, L. Mespouille, R. Borsali, P. Dubois and R. Lazzaroni, Langmuir, 2008, 24, 8272–8279 CrossRef CAS PubMed.
- S. Eggers, B. Fischer and V. Abetz, Macromol. Chem. Phys., 2016, 217, 735–747 CrossRef CAS.
- Y. Xia, X. Yin, N. A. D. Burke and H. D. H. Stöver, Macromolecules, 2005, 38, 5937–5943 CrossRef CAS.
- X.-P. Qiu and F. M. Winnik, Macromolecules, 2007, 40, 872–878 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sm01692j |
| This journal is © The Royal Society of Chemistry 2022 |