DOI:
10.1039/D1SM01206A
(Paper)
Soft Matter, 2022,
18, 137-145
Tyrosine-based photoluminescent diketopiperazine supramolecular aggregates†
Received
17th August 2021
, Accepted 12th November 2021
First published on 15th November 2021
Abstract
L-Tyrosine diketopiperazine (DKP) derivative 1 was synthesized, and the aggregation and photoluminescence behaviors were examined. A solution of 1 in tetrahydrofuran (THF) gradually became viscous at room temperature, and turned into the gel state 5 hours after preparation, as confirmed by dynamic viscoelasticity measurement. A solution of 1 in THF exhibited photoluminescence. Fibrous patterns were observed by transmission electron, atomic force and fluorescence microscopies. Dynamic light scattering, semiempirical molecular orbital and density functional theory calculations, as well as molecular dynamics simulations, indicated aggregate formation. This was attributed to intermolecular hydrogen bonding, mainly between the DKP moieties and partly between the urethane moieties, resulting in π-orbital overlap of the terminal phenyl groups leading to photoluminescence.
Introduction
The structure of diketopiperazine (DKP), an amino acid dimer and the smallest cyclic peptide, was first determined by single crystal X-ray analysis in the 1930s.1 DKPs are occasionally formed as byproducts or degradation products in peptide synthesis, and are commonly synthesized by intramolecular cyclization of linear dipeptides.2 DKPs are primarily studied from a biological viewpoint due to their physiological activity and enzyme inhibition in a fashion similar to linear peptides.3–7 DKPs consist of two s-cis amide groups capable of forming horizontally tandem hydrogen bonding in a direction parallel to the DKP ring (Fig. 1).8 DKPs form well-defined tapes and two-dimensional layers stabilized by hydrogen-bonding strands.9–11
 |
| | Fig. 1 Hydrogen-bonding network between DKP molecules. | |
DKPs are poorly soluble in organic solvents due to their rigid cyclic amide structure. N-Alkylation of one amide group effectively increases the solubility of DKPs due to the collapse of hydrogen-bonding strands, resulting in a change in aggregation state.12 DKPs having long alkyl groups are aggregated based on van der Waals interaction between the alkyl groups, as well as hydrogen bonding between the amide groups.13D-Hydroxyphenylglycine- and sarcosine-derived DKPs form cyclic supramolecular structures.14 DKPs are regularly arranged based on noncovalent (hydrophobic and electrostatic) interactions, in addition to hydrogen bonding between the amide groups. Thus, DKPs find application in liquid crystalline materials,15 microcapsules16 and oil gelling agents,17–22 and the resulting networks are promising building blocks for molecular assemblies.
Various attempts have been made to utilize DKPs as key components of functional polymers.23,24 We previously synthesized several polymers containing DKP moieties in the main and side chains, and examined the properties based on the interactions between the DKP moieties.25–27 Polyamides and polyesters containing aspartic and glutamic acid DKP moieties in the main chain are associated with each other depending on temperature and concentration.25 The acyclic diene metathesis polycondensation of glutamic acid DKP ω-alkenyl esters gives polymers forming regular crystal structures based on intermolecular interactions between the DKP moieties.26 Poly(phenylacetylene)s substituted with DKPs adopt helical conformations with predominantly one-handed screw sense, which are stabilized by intramolecular hydrogen bonding between the DKP s-cis amide groups on the side chains.27 DKP is also useful as a component which enables polymeric materials as well as supramolecules to adopt regular structures.
Organic photoluminescent materials are widely utilized in chemosensors,28 marking dyes for live cells and tissues,29,30 and solid state laser dyes.31 The most common conventional organic luminescent compounds are polycyclic aromatic hydrocarbons, such as naphthalene, anthracene, pyrene and fluorene derivatives. Recently, several non-conventional organic luminescent compounds have been developed, including benzene with push–pull substituents,32–34 terephthalic acid, dimethyl terephthalate and poly(ethylene terephthalate) in the crystalline and regularly aggregated states,35 as well as polyacrylonitrile clusters36 in a manner similar to aggregation-induced emission.37 Other than these sophisticated examples, benzene derivatives rarely exhibit bright luminescence due to their low quantum yields in solution except at very low temperatures.38,39
In the course of our study on DKP-containing molecules, we coincidentally found that a DKP bearing phenylene and urethane moieties self-assembles to emit photoluminescence, even though the DKP contains neither polycyclic aromatic hydrocarbons nor push–pull substituents on the benzene ring. We herein report the aggregation and photoluminescence of an L-tyrosine-derived DKP, and the mechanistic aspects of the intermolecular interactions of the DKP and urethane moieties by quantum mechanics (QM) calculations and molecular dynamics (MD) simulations.
Results and discussion
Synthesis of compound 1
L-Tyrosine DKP was synthesized by the methyl esterification of L-tyrosine with SOCl2 and MeOH, followed by neutralization and cyclic dimerization of the L-tyrosine methyl ester according to Scheme 1. The structure of L-tyrosine DKP was confirmed by comparing the spectroscopic data with those in the literature.23,40–43 The solvent, temperature and time for the cyclic dimerization reaction were varied to enhance the yield of L-tyrosine DKP. The reaction in toluene/EtOH = 1/1 at 100 °C for 5 days yielded L-tyrosine DKP in 29% yield, the highest among the conditions examined in the present study. It seems that inefficient cyclization is caused by the participation of the phenolic hydroxy group in the transformation from ester to amide. The low yield of L-tyrosine DKP is also attributed to the heterogeneous reaction conditions, which cause a considerable amount of L-tyrosine methyl ester to remain unreacted. L-Tyrosine DKP was soluble in DMF and DMSO, while barely soluble in CHCl3, CH2Cl2, ethyl acetate and hexane, as predicted by formation of a hydrogen-bonded network. Compound 1 was then synthesized by the reaction of L-tyrosine DKP and phenyl isocyanate catalyzed by Bu2Sn(OCOC11H23)2, as illustrated in Scheme 1. Compound 2 and polymer 2p (Mn 1200, Đ 1.7) were also synthesized by the addition of phenol with phenyl isocyanate, and polyaddition of L-tyrosine DKP with 1,4-phenylenediisocyanate, as compounds analogous to 1.
 |
| | Scheme 1 Synthesis of L-tyrosine DKP and 1, and structures of 2 and 2p. | |
Aggregation in the solution state
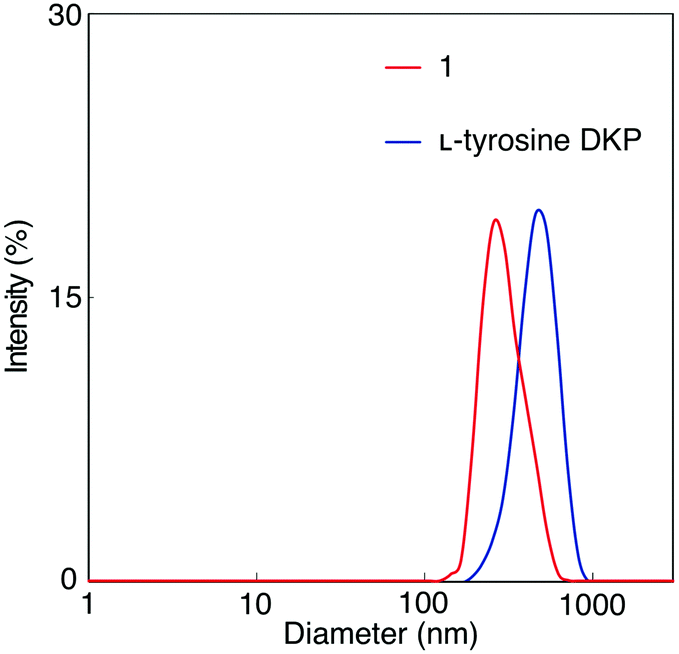
A solution of 1 in DMF was analyzed by dynamic light scattering (DLS) to examine the formation of aggregates, as well as a solution of L-tyrosine DKP in DMF for comparison (Fig. 2). Particles with hydrodynamic diameters around 380 and 300 nm were observed in 1 and L-tyrosine DKP solutions, indicating the formation of aggregates, likely based on hydrogen bonding interaction between the amide and urethane moieties. Observation that the particle sizes of L-tyrosine DKP aggregates are larger than those of 1 is attributable to the presence of hydroxy groups in L-tyrosine DKP, which contribute to the enhancement of intermolecular interaction, resulting in formation of larger aggregates. In fact, the solubility of L-tyrosine DKP was lower than that of 1. The DLS measurements of 1 and L-tyrosine DKP solutions in THF were unsuccessful, presumably due to the formation of aggregates with sizes much larger than those formed in DMF.
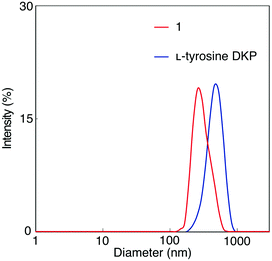 |
| | Fig. 2 DLS diagrams of 1 and L-tyrosine DKP measured in DMF (c = 0.04 mM) at room temperature. | |
Gelation
Compound 1 was dissolved in THF at a concentration of 10 mg mL−1, and the solution was left at room temperature. The solution significantly became viscous after 5 hours, as shown in Fig. 3. It is likely that gelation took place presumably due to gradual growth of network structures driven by intermolecular hydrogen bonding between the DKP and urethane moieties. Hanabusa and coworkers have reported that DKP derivatives substituted with branched long alkyl chains or long alkyl chains with olefin/heteroatoms efficiently serve as oil gelators.17–22 The key importance seems to be the synergistic effects of hydrogen bonding of the DKP and urethane moieties, van der Waals interaction between the alkyl chains, and statistical interactions between olefin/heteroatom moieties. In the present study, it is likely that the urethane moieties of 1 play a key role in gelation, since 1 does not have long alkyl chains. In addition, the π–π interaction between the benzene rings possibly contributes to gel formation, as discussed later in this manuscript. Dynamic viscoelasticity measurements were performed to obtain further information about gel formed from a solution of 1 in THF. The storage modulus was larger than the loss elastic modulus at all frequencies, as shown in Fig. 4, confirming the stability of the gel. In contrast, a DMF solution of 1 did not undergo gelation. It is considered that DMF molecules cause greater weakening of the intermolecular hydrogen bonding interaction between 1 molecules compared to THF molecules, even though 1 forms aggregates in DMF, as shown in Fig. 2. L-Tyrosine DKP was insoluble in THF, and did not form a gel in DMF.
 |
| | Fig. 3 Photographs of a solution of 1 in THF (10 mg mL−1). Left: Sol state just after preparing the solution. Right: Gel state after leaving the solution at room temperature for 5 hours. | |
 |
| | Fig. 4 Dynamic viscoelasticity of a gelated solution 1 in THF (10 mg mL−1). | |
Morphology
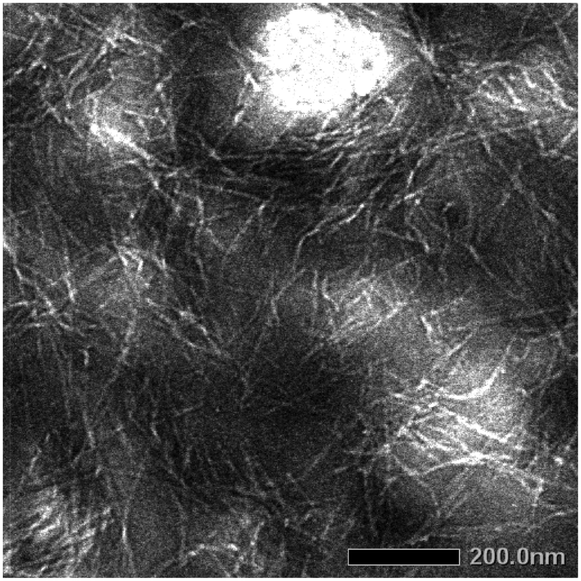
As noted above, compound 1 exhibited aggregation/gelation behavior in the solution state, likely due to intermolecular interaction primarily between the DKP moieties. The sample of 1 was observed by transmission electron microscopy (TEM) and atomic force microscopy (AFM) to determine the morphology in the solid state. Fibrous patterns with ca. 10 nm diameter were observed by TEM, as shown in Fig. 5, and regularly twisted ribbons with 0.7–2.0 nm height were observed by AFM, as shown in Fig. 6. Further AFM images exhibiting assemblies of 1 are shown in Fig. S1.1–S4.2 (ESI†). Such self-assembled structures have often been reported for amide- and aromatic ring-containing systems. Photoelectrically active examples include amide-linked bisporphyrin tethered to trinitrofluorenone to form redox-responsive supramolecules,44 helical conductive nanowires formed by amide-linked tetra(aniline)-amide-ammonium salts,45 and photoluminescent aggregates of triorganophosphinegold(I) 2-mercaptobenzamides.46
 |
| | Fig. 5 TEM image of 1. | |
 |
| | Fig. 6 Height image obtained by AFM showing the twisting of the ribbon-like structure of 1. The graphs on the right are the height profiles of the line sections shown in the image. | |
A THF solution of 1 unexpectedly showed luminescence under black light (λ = 254 nm), even though no remarkable photoluminophore is present. As shown in Fig. 7, 1 emitted light around 300 nm upon excitation at the λmax (280 nm). Fluorescence microscopic observation of a solid sample of 1 was carried out to obtain information on the photoluminescence in conjunction with the solid-state morphology. Fibrous patterns were observed in the sample, as shown in Fig. 8, top, in a fashion similar to the TEM (Fig. 5) and AFM (Fig. 6) images mentioned above. On the contrary, 2p, a compound analogous to 1, did not luminesce under black light and exhibited no fluorescence microscopic image, as shown in Fig. 8, bottom. The photoluminescence quantum yields of 1 were 4–9% in THF, as listed in Table 1. As mentioned above, 1 showed photoluminescence in spite of the absence of typical luminophores. The photoluminescence lifetime was 4 ns (Fig. S5, ESI†). On the other hand, benzene and 2 as the reference compounds of 1, exhibited negligibly small photoluminescence in THF (Fig. S6, ESI†). These results strongly support the importance of aggregation of benzene moieties for photoluminescence.
 |
| | Fig. 7 Absorption (blue) and emission (red, excited at 280 nm) spectra of 1 measured in THF (c = 0.04 mM) at 25 °C. | |
 |
| | Fig. 8 Fluorescence microscopic images of 1 (top) and 2p (bottom). Scale bar size = 100 μm. | |
Table 1 Photoluminescence quantum yields Φ of 1, benzene, 2 and 2pa
| Conc. (mM) |
Φ [%] |
|
1
|
Benzene |
2
|
2p
|
|
Measured in THF using an integration sphere.
Excited at 265 nm.
Excited at 278 nm.
Excited at 277 nm.
Too small to be determined.
Measured in DMF due to the insolubility in THF, excited at 285 nm.
|
| 4 |
4b |
—e |
—e |
—e |
| 0.4 |
9c |
—e |
—e |
0.2 |
| 0.04 |
8d |
—e |
—e |
0.5 |
QM calculations
It was considered that the gelation and photoluminescence of 1 are caused by intermolecular interaction between the molecules. The mechanistic aspects were first examined by QM calculations. Fig. 9 shows the relationships between the relative energy per unit for association of compound 1, and degree of association calculated by the semi-empirical PM747 and DFT methods. The B3LYP-D348,49 and M06-2X50,51 functionals were employed in the DFT calculation, because these functionals consider London dispersion energy, which is necessary for estimating non-covalent interactions such as hydrogen bonding and π-interaction. The relative energy remarkably decreased with increasing degree of association from 1-mer to 8–16-mers, and reached almost constant values around 16–32-mers irrespective of the calculation method. Calculations by the M06-2X and B3LYP-D3 methods of a 64-mer failed due to limitations of available computational resources.
 |
| | Fig. 9 Relationship between the relative energy per unit for the association of 1 and degree of association calculated by the M06-2X/6-31G*, B3LYP-D3/6-31G* and PM7 methods. | |
Fig. 10 illustrates a possible aggregated structure consisting of 32 molecules of 1, whose geometries were optimized by the DFT method (M06-2X/6-31G*). The 32-mer forms a helical supramolecular structure with a diameter of 40 Å and a pitch of 42 Å.52 One turn of the helix consists of 21 molecules of 1. Intermolecular hydrogen bonding strands exist between the DKP-amide moieties, and also between the urethane moieties. The average interatomic distances between the oxygen and hydrogen atoms (>C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N<) of the DKP-amide and urethane moieties were 1.89 Å and 2.25 Å, respectively. Natural bond orbital (NBO) analysis53 of dimeric 1 (Fig. 11) revealed that the average energies of the hydrogen bonds at the DKP-amide and urethane moieties were 27.9 and 29.9 kJ mol−1, respectively. Usually, a larger distance (here in the H-bond with urethane) leads to a weaker bond energy, but in this case the bond energy is greater. This seeming contradiction between the interatomic distance and hydrogen-bonding energy is attributable to the relative angle at the hydrogen-bonding sites. The average angles of CO⋯H– of DKP-amide and urethane moieties are 104.5° and 124.6°, respectively. The average angle of the urethane moiety is closer to 120°, the ideal angle for hydrogen-bond formation by sp2 lone pair electrons. Consequently, the energy is rather large compared with that at the DKP-amide moiety in spite of the longer distance. It is considered that hydrogen bonding strands exist between the DKP-amide moieties and urethane moieties. Such hydrogen-bonding strands were observed in all the 2–64-mers. The terminal phenyl groups were located at a distance of 3.2 Å on average, indicating the presence of π-interactions, possibly leading to photoluminescence. Fig. 11 visualizes the reduced density gradient isosurface representing intermolecular interaction using NCIPLOT,54 which supports the presence of π-interaction between the benzene rings as well as hydrogen bonding.
O⋯H–N<) of the DKP-amide and urethane moieties were 1.89 Å and 2.25 Å, respectively. Natural bond orbital (NBO) analysis53 of dimeric 1 (Fig. 11) revealed that the average energies of the hydrogen bonds at the DKP-amide and urethane moieties were 27.9 and 29.9 kJ mol−1, respectively. Usually, a larger distance (here in the H-bond with urethane) leads to a weaker bond energy, but in this case the bond energy is greater. This seeming contradiction between the interatomic distance and hydrogen-bonding energy is attributable to the relative angle at the hydrogen-bonding sites. The average angles of CO⋯H– of DKP-amide and urethane moieties are 104.5° and 124.6°, respectively. The average angle of the urethane moiety is closer to 120°, the ideal angle for hydrogen-bond formation by sp2 lone pair electrons. Consequently, the energy is rather large compared with that at the DKP-amide moiety in spite of the longer distance. It is considered that hydrogen bonding strands exist between the DKP-amide moieties and urethane moieties. Such hydrogen-bonding strands were observed in all the 2–64-mers. The terminal phenyl groups were located at a distance of 3.2 Å on average, indicating the presence of π-interactions, possibly leading to photoluminescence. Fig. 11 visualizes the reduced density gradient isosurface representing intermolecular interaction using NCIPLOT,54 which supports the presence of π-interaction between the benzene rings as well as hydrogen bonding.
 |
| | Fig. 10 Top and side views of a possible aggregated 32-mer of 1. The geometries were optimized by the DFT method (M06-2X/6-31G*). | |
 |
| | Fig. 11 Reduced density gradient isosurfaces (green) representing the non-covalent intermolecular interactions in dimeric 1, whose geometries were optimized by the DFT method (M06-2X/6-31G*). | |
Vibrational calculations of 1- and 8-mers were carried out in vacuum and THF using the self-consistent reaction field-integral equation formalism polarized continuum model (SCRF-IEFPCM) method in order to estimate the free energy change upon association and the solvent effect. The energies were normalized per monomeric unit (1-mer) for comparison. The ΔH° and ΔG° values of the associated 8-mer were more negative than those of the 1-mer both in vacuum and THF, as listed in Table 2, likely due to the formation of hydrogen-bonding strands between the DKP-amide and urethane moieties, as described above. The relative absolute values of ΔG° were smaller than those of ΔH°, indicating that formation of the associated octamer is entropically unfavorable due to the decrease of mobility. It is considered that the stabilization by hydrogen-bond formation exceeds the entropic loss upon association, resulting in the negative ΔG° in the present system. The absolute ΔH° and ΔG° values of the 8-mer are smaller in THF than those in vacuum, suggesting that the decrease of stabilization effect by hydrogen bonding is due to solvation with THF molecules.
Table 2 Relative enthalpy and free energy of 1-mer and 8-mer of 1aa
| Model |
Relative ΔH° per unit [kJ mol−1] |
Relative ΔG° per unit at 298 K [kJ mol−1] |
| In vacuum |
In THFb |
In vacuum |
In THFb |
|
Calculated by the DFT method (M06-2X/6-31G*).
SCRF-IEFPCM method.
|
| 1-mer |
0.0 |
0.0 |
0.0 |
0.0 |
| 8-mer |
−136.3 |
−98.8 |
−56.7 |
−22.1 |
The phenylene groups of the 8-mer of 1 are present at so-called “slipped parallel positions”, which are favorable for π-stacking.55 The terminal phenyl groups are present in a similar fashion due to the conformation restricted by the intermolecular hydrogen bonding at the DKP-amide moieties. In fact, some sp2 orbitals of the benzene rings overlap, as shown in Fig. 12. The energy of intermolecular interaction of a slipped parallel benzene dimer is estimated to be 2.48 kcal mol−1 (=10.4 kJ mol−1) by the coupled cluster calculations with single and double substitutions with noniterative triple excitations [CCSD(T)] at the basis set,55 in good agreement with the value experimentally obtained (2.4 kcal mol−1).56 It is therefore assumed that the π-stacking at the four benzene rings of 1 contribute to aggregation with a maximum of 40 kJ mol−1 per unit by dispersion interaction, and that hydrogen bonding further promotes aggregation.56 Widely used density functional B3LYP cannot estimate dispersion interaction differently from the CCSD(T), and therefore B3LYP results are unsuitable for investigation of non-covalent interactions such as hydrogen bonding and π-stacking, while B3LYP-D348,49 and M06-2X50,51 functionals are better in this respect.57
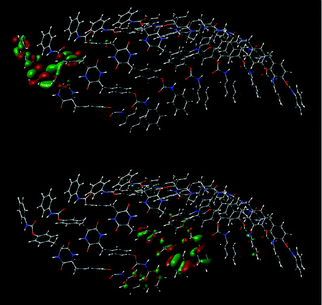 |
| | Fig. 12 Overlap of MOs of an aggregated 8-mer of 1 obtained by the DFT method (M06-2X/6-31G*). Top: LUMO. Bottom: HOMO−9. | |
The photoluminescence behaviors of a single molecule and two molecules of 1 were examined by the TD-DFT method using the M06-2X functional and 6-31+G* basis set at nstates = 3, root = 1, with SCRF-IEFPCM (solvent = THF). The first excited states of single and two molecules of 1 were simulated at 255 and 267 nm with oscillator strengths (f) of 0.0650 and 0.1140, respectively (Fig. S7 and S9, ESI†). The simulated emission wavelength and f of dimeric 1 were longer and larger than those of monomeric 1. Compound 1 emitted light around 300 nm, as shown in Fig. 7. It is likely that many more molecules of 1 interact with each other, resulting in further red shift of emission and increase of f. The transition from MO #298 (LUMO+1) to #296 (HOMO) of the dimer mainly contributes to the emission. The LUMO+1 possesses π-electrons at both the terminal and internal phenyl groups. It should be noted that the atomic orbitals partly overlap between the two molecules, as shown in Fig. S10.4 (ESI†), supporting the proposal that the luminescence of 1 is caused by intermolecular interaction between the molecules of 1. The HOMO of 1 dimer mainly possesses π-electrons at one terminal phenyl group.
MD simulations
MD simulations were carried out in order to obtain further information on aggregation of 1. The DFT-optimized geometries of the aggregated 32-mer (Fig. 10) were employed as the initial geometry for MD simulation. The total and potential energies decreased in the early stage of simulation, and then became smooth and leveled off after ca. 0.15 ns [Fig. S11, (A), ESI†], indicating the completion of relaxation to reach equilibrium. The temperature equilibrated at 300 K, as intended [Fig. S11, (B), ESI†]. Although the pressure largely varied during the simulation, the mean pressure stabilized around 1 atm after 0.15 ns of simulation [Fig. S11, (B), ESI†], sufficiently indicating successful equilibration. The volume decreased in the early stage of simulation as the THF box relaxed, and reached an equilibrium value [376![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Å3, Fig. S11, (C), ESI†]. The density plot showed a mirror image of the volume plot [Fig. S11, (C), ESI†]. The system equilibrated at a density of approximately 0.92 g cm−3. It is likely that the density of the system increases by 4.5% compared to pure THF (0.88 g cm−3 at 300 K) due to the presence a 32-mer of 1. Thus, the smooth transitions in the plots followed by the oscillations about mean values indicate successful equilibration. The mass-weighted root-mean-square deviation (RMSD) fit to the initial structure increased as the 32-mer of 1 relaxed in the THF box, as shown in Fig. 13(A). The representative RMSD for the distance between the DKP ring atoms of units #16 and #17 was almost stable accompanying no large oscillation, as shown in Fig. 13(B). The representative interatomic distances between the oxygen and hydrogen atoms (>CO⋯H–N<) of DKP units #16 and #17 equilibrated at ca. 2.0–2.1 Å, as shown in Fig. 13(C), 0.1–0.2 Å longer than the values obtained by the geometry optimization using the DFT method described above. It is quite reasonable that the MD method estimates the hydrogen-bonding distance longer than the DFT method, because of the difference between the dynamic and static calculation methods.
000 Å3, Fig. S11, (C), ESI†]. The density plot showed a mirror image of the volume plot [Fig. S11, (C), ESI†]. The system equilibrated at a density of approximately 0.92 g cm−3. It is likely that the density of the system increases by 4.5% compared to pure THF (0.88 g cm−3 at 300 K) due to the presence a 32-mer of 1. Thus, the smooth transitions in the plots followed by the oscillations about mean values indicate successful equilibration. The mass-weighted root-mean-square deviation (RMSD) fit to the initial structure increased as the 32-mer of 1 relaxed in the THF box, as shown in Fig. 13(A). The representative RMSD for the distance between the DKP ring atoms of units #16 and #17 was almost stable accompanying no large oscillation, as shown in Fig. 13(B). The representative interatomic distances between the oxygen and hydrogen atoms (>CO⋯H–N<) of DKP units #16 and #17 equilibrated at ca. 2.0–2.1 Å, as shown in Fig. 13(C), 0.1–0.2 Å longer than the values obtained by the geometry optimization using the DFT method described above. It is quite reasonable that the MD method estimates the hydrogen-bonding distance longer than the DFT method, because of the difference between the dynamic and static calculation methods.
 |
| | Fig. 13 (A) Mass-weighted RMSD, (B) RMSD results for the distance between the DKP ring atoms of units #16 and #17, (C) hydrogen atoms (>CO⋯H–N<) of DKP units #16 and #17 in the MD simulation of a 32-mer of 1 in a solvent box consisting of 2662 THF molecules at 300 K for 0–10 ns, AMBER 16 using the ff14SB force field.58 | |
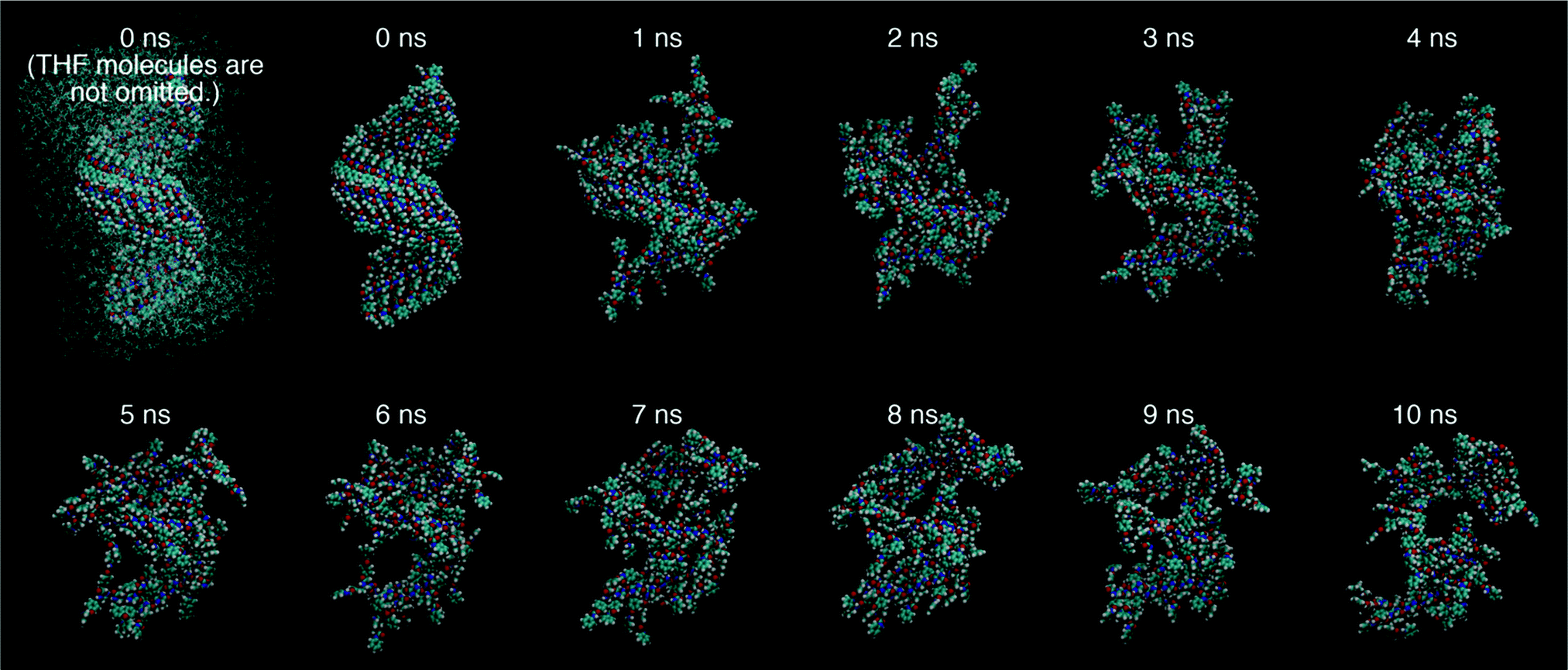
Fig. 14 shows the snapshots of a 32-mer of 1 in the trajectory obtained by the MD simulation in a THF box at 300 K for 0–10 ns, which give information on the dynamical aggregation behavior. The DKP moieties remain aligned because the hydrogen-bonding strands between the DKP-amide moieties are macroscopically intact during dynamic formation/fission. On the other hand, the urethane moieties lose the aligned structures due to the smaller population of hydrogen-bonded urethanes compared with that of non-hydrogen-bonded moieties. The difference of alignment between the DKP and urethane moieties is reasonable, because the mobility of the DKP-amide groups is much smaller compared with that of the urethane groups, due to the motion-constrained ring-structure and location of the DKP moieties (center of molecule). The DFT calculations estimated that the average interatomic distances of oxygen and hydrogen atoms (>CO⋯H–N<) of the DKP moieties were 1.89 Å, closer than those of the urethane moieties (2.25 Å), as mentioned above, coinciding with the trend obtained by the MD simulation.
 |
| | Fig. 14 Snapshots of a 32-mer of 1 in the trajectory obtained by the MD simulation performed in a solvent box consisting of 2662 THF molecules at 300 K for 0–10 ns. For clarity, THF molecules are omitted in the snapshots except the left top (0 ns). A movie is available in ESI.† | |
The diameters of the fibrils observed by TEM and the heights of the patterns observed by AFM are comparable to the diameter of the helical supramolecular structure of 1 optimized by the DFT method. It is suggested that the hydrogen bonds between the DKP moieties and those between the urethane moieties play an important role in the formation of fibrous objects and networks in a manner similar to that in the solution state. Fig. 15 depicts the image of formation of nanoscale fibrils by hierarchical self-assembly of 1. A helical tube with 24 helix turns consisting of 504 molecules of 1 corresponds to a 100 nm fiber according to the DFT-optimized helical tube structure shown in Fig. 10.
 |
| | Fig. 15 Image of formation of fibril structure by hierarchical self-assembly of 1. | |
Conclusions
In this study, we demonstrated the aggregation and photoluminescence behavior of novel L-tyrosine diketopiperazine derivative 1. Compound 1 serves as a gelator for THF by forming supramolecular fibril structures based on hydrogen bonding between the DKP moieties in conjunction with H-bonding between the urethane moieties. The dispersion interaction between the benzene rings also plays a role, as supported by QM calculations and MD simulations, as well as DLS, dynamic viscoelasticity, TEM, AFM, fluorescence microscopy and photoluminescence measurements. We believe that the present study will lead to the development of molecular design of photoluminescent materials accompanying gelation, without the presence of a polycyclic aromatic hydrocarbon moiety as a luminophore.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partly supported by the JSPS KAKENHI grant (Grant Number JP21H05511, JP20H05210, and JP19H02565). The authors are grateful to Dr Motohiro Shizuma at Osaka Research Institute of Industrial Science and Technology for measurement of ESI-TOF-MS, Prof. Takashi Miyata, Prof. Akifumi Kawamura and Mr Shingo Yamashita at Kansai University for measurement of dynamic viscoelasticity, Prof. Yasuhiko Iwasaki and Ms Masako Tanaka for measurement of fluorescence microscopy, Prof. Kenneth B. Wagener and Dr Kathryn R. Williams at the University of Florida for their helpful suggestions and comments.
Notes and references
- R. B. Corey, J. Am. Chem. Soc., 1938, 60, 1598–1604 CrossRef CAS.
- C. J. Dinsmore and D. C. Beshorep, Tetrahedron, 2002, 58, 3297–3312 CrossRef CAS.
- A. Peterkofsky, F. Battaini, Y. Koch, Y. Takahara and P. Dannies, Mol. Cell. Biochem., 1982, 42, 45–63 CrossRef CAS PubMed.
- M. B. Martins and I. Carvalho, Tetrahedron, 2007, 63, 9923–9932 CrossRef CAS.
- Y. Hayashi, Y. Yamazaki-Nakamura and F. Yakushiji, Chem. Pharm. Bull., 2013, 61, 889–901 CrossRef CAS.
- Y.-M. Ma, X.-A. Liang, Y. Kong and B. Jia, J. Agric. Food Chem., 2016, 64, 6659–6671 CrossRef CAS.
- M. Moutiez, P. Belin and M. Gondry, Chem. Rev., 2017, 117, 5578–5618 CrossRef CAS.
- J. C. MacDonald and G. M. Whitesides, Chem. Rev., 1994, 94, 2383–2420 CrossRef CAS.
- G. T. R. Palmore and M. T. McBride, J. Chem. Soc., Chem. Commun., 1998, 145–146 RSC.
- Y. Ishida and T. Aida, J. Am. Chem. Soc., 2002, 124, 14017–14019 CrossRef CAS.
- Y. Du, J. C. Creighton, B. A. Tounge and A. B. Reitz, Org. Lett., 2004, 6, 309–312 CrossRef CAS.
- R. A. Weatherhead-Kloster, H. D. Selby, W. B. Miller III and E. A. Mash, J. Org. Chem., 2005, 70, 8693–8702 CrossRef CAS.
- Y. Ohta, K. Terada, T. Masuda and F. Sanda, Heterocycles, 2009, 78, 1477–1483 CrossRef CAS.
- Y. Ohta, K. Terada, T. Masuda and F. Sanda, Heterocycles, 2009, 78, 2523–2530 CrossRef CAS.
- R. A. Kloster, M. D. Carducci and E. A. Mash, Org. Lett., 2003, 5, 3683–3686 CrossRef CAS.
- R. J. Bergeron, O. Phanstiel IV, G. W. Yao, S. Milstein and W. R. Weimar, J. Am. Chem. Soc., 1994, 116, 8479–8484 CrossRef CAS.
- K. Hanabusa, Y. Matsumoto, T. Miki, T. Koyama and H. Shirai, Chem. Commun., 1994, 1401–1402 RSC.
- K. Hanabusa, M. Matsumoto, M. Kimura, A. Kakehi and H. Shirai, J. Colloid Interface Sci., 2000, 224, 231–244 CrossRef CAS PubMed.
- Y. Yang, M. Suzuki, M. Kimura, H. Shirai and K. Hanabusa, Chem. Commun., 2004, 1332–1333 RSC.
- Y. Yang, H. Fukui, M. Suzuki, H. Shirai and K. Hanabusa, Bull. Chem. Soc. Jpn., 2005, 78, 2069–2074 CrossRef CAS.
- K. Hanabusa, H. Fukui, M. Suzuki and H. Shirai, Langmuir, 2005, 21, 10383–10390 CrossRef CAS PubMed.
- H. Hoshizawa, Y. Minemura, K. Yoshikawa, M. Suzuki and K. Hanabusa, Langmuir, 2013, 29, 14666–14673 CrossRef CAS PubMed.
- H. Xia, Z. Y. Suo, G. J. Qiang and C. J. Chang, J. Appl. Polym. Sci., 2008, 110, 2168–2178 CrossRef CAS.
- F. Rafiemanzelat, A. F. Zonouz and G. Emtiazi, Polym. Degrad. Stab., 2012, 97, 72–80 CrossRef CAS.
- K. Terada, F. Sanda and T. Masuda, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 789–794 CrossRef CAS.
- K. Terada, E. B. Berda, K. B. Wagener, F. Sanda and T. Masuda, Macromolecules, 2008, 41, 6041–6046 CrossRef CAS.
- K. Terada, T. Masuda and F. Sanda, Macromolecules, 2009, 42, 913–920 CrossRef CAS.
- T. L. Mako, J. M. Racicot and M. Levine, Chem. Rev., 2019, 119, 322–477 CrossRef CAS PubMed.
- A. Buda, C. Sands and M. A. Jepson, Adv. Drug Delivery Rev., 2005, 57, 123–134 CrossRef CAS PubMed.
- K. Nienhaus and G. U. Nienhaus, Chem. Soc. Rev., 2014, 43, 1088–1106 RSC.
- M. S. AlSalhi, J. Alam, L. A. Dass and M. Raja, Int. J. Mol. Sci., 2011, 12, 2036–2054 CrossRef CAS PubMed.
- M. Shimizu, Y. Takeda, M. Higashi and T. Hiyama, Angew. Chem., Int. Ed., 2009, 48, 3653–3656 CrossRef CAS PubMed.
- T. Beppu, S. Kawata, N. Aizawa, Y.-J. Pu, Y. Abe, Y. Ohba and H. Katagiri, ChemPlusChem, 2014, 79, 536–545 CrossRef CAS PubMed.
- T. Beppu, K. Tomiguchi, A. Masuhara, Y.-J. Pu and H. Katagiri, Angew. Chem., Int. Ed., 2015, 54, 7332–7335 CrossRef CAS PubMed.
- X. Chen, Z. He, F. Kausar, G. Chen, Y. Zhang and W. Z. Yuan, Macromolecules, 2018, 51, 9035–9042 CrossRef CAS.
- Q. Zhou, B. Cao, C. Zhu, S. Xu, Y. Gong, W. Z. Yuan and Y. Zhang, Small, 2016, 12, 6586 CrossRef CAS.
- C. Zhu, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, ACS Appl. Bio Mater., 2018, 1, 1768–1786 CrossRef CAS.
- W. R. Dawson and M. W. Windsor, J. Phys. Chem., 1968, 72, 3251–3260 CrossRef CAS.
- R. B. Cundall and D. A. Robinson, J. Chem. Soc., Faraday Trans. 2, 1972, 68, 1133–1144 RSC.
- K. D. Kopple and D. H. Marr, J. Am. Chem. Soc., 1967, 89, 6193–6200 CrossRef CAS PubMed.
- M. E. Jung and J. C. Rohloff, J. Org. Chem., 1985, 50, 4909–4913 CrossRef CAS.
- J. R. Cochrane, J. M. White, U. Wille and C. A. Hutton, Org. Lett., 2012, 110, 2402–2405 CrossRef PubMed.
- S. Manchineella, C. Voshavar and T. Govindaraju, Eur. J. Org. Chem., 2017, 4363–4369 CrossRef CAS.
- N. Hisano, T. Hirao and T. Haino, Chem. Commun., 2020, 56, 7553–7556 RSC.
- O. A. Bell, G. Wu, J. S. Haataja, F. Brömmel, N. Fey, A. M. Seddon, R. L. Harniman, R. M. Richardson, O. Ikkala, X. Zhang and C. F. J. Faul, J. Am. Chem. Soc., 2015, 137, 14288–14294 CrossRef CAS PubMed.
- J.-G. Kang, H.-K. Cho, C. Park, S.-S. Yun, J.-K. Kim, G. A. Broker, D. R. Smyth and E. R. T. Tiekink, Inorg. Chem., 2007, 46, 8228–8237 CrossRef CAS PubMed.
- J. J. P. Stewart, J. Mol. Model., 2013, 19, 1–32 CrossRef CAS PubMed.
- S. Grimme, Chem. – Eur. J., 2012, 18, 9955–9964 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- Compound 1 exhibited negligibly small CD signals in a manner similar to L-tyrosine DKP as shown in Fig. S12. It is assumed that the softness of aggregates is responsible for the small chiroptical activity, as indicated by the MD simulation. Namely, the present diketopiperazine aggregates are weakly assembled, which makes it difficult to show clear CD signals. It is likely that the so-called helix persistence length seems to be not long in the present diketopiperazine aggregates.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan, W. Yang and J. Chem, Theory Comput., 2011, 7, 625–632 CrossRef.
- S. Tsuzuki, K. Honda, T. Uchimaru, M. Mikami and K. Tanabe, J. Am. Chem. Soc., 2002, 124, 104–112 CrossRef CAS PubMed.
- J. R. Grover, E. A. Walters and E. T. Hui, J. Phys. Chem., 1987, 91, 3233–3237 CrossRef CAS.
- The CCSD(T) calculation of 1 was failed due to the limit of computational resource available. It was difficult to estimate the π-stacking-based stabilization energy by the NBO method differently from that of >CO⋯H–N< hydrogen bonding, because many atoms participated in stacking around the benzene rings as shown in Fig. 11.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theor. Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure, additional AFM images, photoluminescence decay, absorption/emission spectra, conformers optimized by the TD-DFT method, representative MO shapes, MD simulation data, and CD/UV-vis absorption spectra. See DOI: 10.1039/d1sm01206a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Kasinan
Suthiwanich
b,
Kasinan
Suthiwanich