
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced electrocatalytic activity of low-cost NiO microflowers on graphene paper for the oxygen evolution reaction†
Luca
Bruno
 *ab,
Mario
Scuderi
c,
Francesco
Priolo
a and
Salvo
Mirabella
*ab
*ab,
Mario
Scuderi
c,
Francesco
Priolo
a and
Salvo
Mirabella
*ab
aDipartimento di Fisica e Astronomia “Ettore Majorana”, Università degli Studi di Catania, via S. Sofia 64, 95123, Catania, Italy
bIMM-CNR (Catania Università), via S. Sofia 64, 95123, Catania, Italy
cIMM-CNR, VIII strada 5, 95121, Catania, Italy
First published on 8th September 2022
Abstract
Electrochemical water splitting represents a promising source of renewable energy. Since electrochemistry is a useful method, it should be fast and cost-effective. However, the half-reaction for the oxygen evolution reaction (OER) represents a limiting factor because of its slow kinetics and large energy barrier. Typically, although critical raw materials such as IrO2 and RuO2 can be used as catalysts, sustainable and efficient catalysts are urgently required. Among oxides of transition metals, NiO represents a promising candidate as a high-performance catalyst for the oxygen evolution reaction. Here, a low-cost, high-throughput, environmentally friendly chemical methodology is used to produce NiO microflowers composed of very thin sheets (20 nm thick) intertwined like petals of a desert rose, as shown by electron microscopy and X-ray diffraction analysis. These microflowers, dispersed onto a graphene paper substrate by spin coating or drop casting, were carefully tested as electrocatalysts for the OER. The optimized electrode based on the NiO microflowers exhibited an overpotential of 314 mV at a current density of 10 mA cm−2 under alkaline conditions (1 M KOH). The intrinsic activity of the catalyst was evaluated by measuring the Tafel plot (as low as 40 mV dec−1) and turnover frequencies (0.01 or 6.98 s−1 for bulk or redox determination). The reported results prove that Ni based nanostructures are promising materials for efficient anodes in sustainable water splitting electrocatalysis.
Introduction
Efficient and cheap renewable energy production from water electrolysis is a crucial challenge for a sustainable society.1–4 Electrochemical water splitting represents a promising way to produce renewable energy. Water splitting consists of two half reactions: the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER), taking place at the anode and cathode, respectively.5,6 The OER is considered the limiting process of the overall water electrolysis, because it involves four sequential electron transfers, thereby having slow kinetics and a large energy barrier for water splitting.7–9 In alkaline solution, the OER can be described by the following electrochemical reaction:8| 4OH− → O2 + 2H2O + 4e− | (1) |
Over the past few decades, considerable research effort has been devoted to designing, synthesizing, and characterizing different oxygen evolution anode materials with the aim of achieving acceptable rates of active oxygen production at the lowest possible overpotentials.8 Optimizing the kinetics of the OER is crucial for the generation of hydrogen as a renewable fuel. Accordingly, the development of efficient, abundant, and inexpensive catalysts is essential. Nowadays, one important issue is the availability of raw materials for large-scale production of energy through electrolysis.10–12 Although they are of significant economic importance for key sectors in the global economy, they have a high supply risk and there is a lack of (suitable) substitutes, due to their unique and reliable properties. The development of new non-precious metal catalysts and understanding the origin of their activity for the OER are essential for rationally designing highly active low-cost catalysts as alternatives to the state-of-the-art critical raw materials.13,14
Oxides and hydroxides of non-critical transition metals (e.g., Fe, Co, Ni) have been extensively investigated and are currently the prevailing electrocatalysts used in the OER.6,15–17 Although they possess inferior electrocatalytic activity for the OER when compared with the state-of-the-art materials (RuO2 and IrO2), they have relatively low cost and high corrosion resistance in alkaline solution as anode materials for the OER.18,19 Among these, Ni oxide represents a promising candidate as an anode material with enhanced electrochemical performances.20–25 Both experimental and theoretical studies suggested that Ni2+ has more efficient interactions with OH− for the OER compared with other metal (Fe, Mn, Co) oxides which is ideal for catalysts with superior performance.23,26 Furthermore, NiO nanostructures (such as oriented arrays, multilayers, or interconnected networks) increase electrolyte permeability through the active material, facilitating mass transport at the interface.24 Thanks to the unique size dependent properties, mass diffusion, and high surface area, nanostructured NiO is often used as a high-performance OER catalyst.21–23,25,27–29 Finally, graphene supported NiO nanostructures have been demonstrated in the literature to be a very good option, since graphene enhances electron transport and hence catalytic performance,30 promotes nanostructure dispersion and inhibits their aggregation.
The electrochemical performance of the catalyst should be carefully evaluated in terms of the balance between mass loading and intensive quantities (turnover frequencies and mass activities). The frequently used overpotential value at a fixed current density (as an indicator for the catalytic activity of the electrodes) is largely dependent on the catalyst mass and surface to volume ratio. A careful determination of turnover frequency and mass activity, independent of catalyst mass, is a key step towards a solid evaluation of the electrochemical properties of new OER catalysts.17 They provide a clear and unequivocal indication of the real intrinsic catalytic properties of the materials used, decoupling them from the effects linked to the catalyst mass loading.
In this work, NiO microflowers (μFs), synthesized by a chemical solution method,31 were dispersed onto a graphene paper (GP) substrate with different mass loadings. The effect of catalyst amount is evaluated through electrochemical analyses and the role of active sites at the surface is elucidated.
Experimental
Synthesis of NiO microflowers
NiO microflowers (μFs) were obtained from a chemical solution method through a bain-marie configuration at 50 °C by mixing 0.42 M NiSO4·6H2O (Merck, Darmstadt, Germany, 98%), 0.07 M K2S2O8 (Alfa Aesar, Kandel, Germany 97%) and 3.5 wt% ammonia (Merck, Darmstadt, Germany, 30–33 wt% NH3 in H2O) in 50 mL deionized water (Milli-Q, 18 MΩ cm). After 20 minutes the solution was centrifuged and washed several times with a mixture of deionized water and ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) until a dark blue precipitate (made of Ni(OH)2 μFs) had totally separated from solution. The μFs were then dried in air in an oven for 24 hours and then annealed at 350 °C for 60 min in Ar leading to the conversion of Ni(OH)2 into NiO.31,32 A μF solution was then prepared by mixing the obtained powder with deionized water and ethanol (15 mg of μFs in 8 mL 1:1 ratio of water and ethanol) followed by sonication for 15 minutes at room temperature in order to avoid nanostructure agglomeration.
:1) until a dark blue precipitate (made of Ni(OH)2 μFs) had totally separated from solution. The μFs were then dried in air in an oven for 24 hours and then annealed at 350 °C for 60 min in Ar leading to the conversion of Ni(OH)2 into NiO.31,32 A μF solution was then prepared by mixing the obtained powder with deionized water and ethanol (15 mg of μFs in 8 mL 1:1 ratio of water and ethanol) followed by sonication for 15 minutes at room temperature in order to avoid nanostructure agglomeration.
Preparation of the electrodes
Graphene paper (GP) substrates (240 nm thick, Sigma Aldrich, St. Louis, MO, USA) were rinsed with deionized water and dried in N2 to clean the surface from any impurity.The electrode is realized by spin coating (200 rpm, 4 minutes) the μF solution onto GP in an area of 1 cm2. Electrodes were dried on a hot plate at 80 °C for 10 minutes. The amount of catalyst was varied by adding another drop of solution and repeating the spin coating and drying steps. NiO μFs loaded on GP by spin coating were labelled NiOS1 and NiOS2. To further increase the μF mass on GP we deposited 10 μL of μF solution via drop casting in an area of 1 cm2, followed by drying on a hot plate at 80 °C for 10 minutes. Also in this case, the amount of catalyst was varied through the addition of different numbers of drops of solution. The electrodes prepared by the drop casting method were labelled NiOD1, NiOD2 and NiOD3. The mass of μFs on GP was measured using a Mettler Toledo MX5 microbalance (sensitivity: 0.01 mg). The values obtained for the catalyst mass were 0.06 mg, 0.18 mg, 0.30 mg, 0.50 mg, and 0.75 mg for NiOS1, NiOS1, NiOD1, NiOD2, and NiOD3, respectively. Table 1 summarizes the deposition method and the mass loading of the prepared samples.
| Sample | Deposition method | Catalyst loading [±0.01 mg] |
|---|---|---|
| NiOS1 | Spin coating | 0.06 |
| NiOS2 | Spin coating | 0.18 |
| NiOD1 | Drop casting | 0.30 |
| NiOD2 | Drop casting | 0.50 |
| NiOD3 | Drop casting | 0.75 |
Characterization of NiO microflowers on graphene paper substrates
Surface morphology was analyzed using a scanning electron microscope (Gemini field emission SEM Carl Zeiss SUPRA 25, FEG-SEM, Carl Zeiss Microscopy GmbH, Jena, Germany) combined with energy dispersive X-ray spectroscopy (EDX). SEM images were analyzed using ImageJ software.33The amount of NiO μFs on GP was evaluated by Rutherford backscattering spectrometry (RBS, 2.0 MeV He+ beam at normal incidence) with a 165° backscattering angle using a 3.5 MV HVEE Singletron accelerator system (High Voltage Engineering Europa, Netherlands). RBS spectra were analyzed using XRump software.34
XRD patterns were recorded using a Rigaku Smartlab diffractometer, equipped with a rotating anode producing Cu Kα radiation operating at 45 kV and 200 mA, in grazing incidence mode (0.5°).
Transmission electron microscopy (TEM) analyses of NiO μFs dispersed on a TEM grid were performed with a Cs-probe-corrected JEOL JEM ARM200F microscope at a primary beam energy of 200 keV operated in scanning TEM (STEM) mode. STEM images were analyzed using DigitalMicrograph® software.35
Electrochemical measurements were carried out at room temperature using a VersaSTAT 4 potentiostat (Princeton Applied Research, USA) and a three-electrode setup with a platinum counter electrode, a saturated calomel electrode (SCE, HI-5412 glass body refill calomel reference electrode, Hanna Instruments) as a reference electrode, and our samples as the working electrode, without purging with any inert gas. 1 M KOH (pH 14, Sigma Aldrich, St. Louis, MO, USA) was used as a supporting electrolyte. Current density was normalized to the geometrical surface area and measured potentials vs. SCE were converted to the reversible hydrogen electrode (RHE) according to the equation:36
| ERHE = ESCE + 0.059pH + 0.244 | (2) |
Cyclic voltammetry (CV) curves were recorded at a scan rate of 5 mV s−1 in the potential range −0.3–1.0 V vs. SCE in order to stabilize electrodes. OER activities of NiO catalysts were investigated using linear sweep voltammetry (LSV) at a scan rate of 5 mV s−1. Electrochemical impedance spectroscopy (EIS) was performed with a superimposed 5 mV sinusoidal voltage in the frequency range 104 to 10−1 Hz. Nyquist plots were acquired in the so-called turnover region, just after the onset potential of each sample where all the studied electrocatalysts possess appreciable OER activity.38 All measured potentials (η′) were manually corrected by iRu-compensation as follows:
| η = η′ − iRu | (3) |
Results and discussion
Fig. 1a and b show the SEM and STEM images of the NiO μFs. Each μF appears as a porous sphere (with a diameter spanning in the 0.8–1.2 μm range), made of 2D interconnected nanosheets (with a thickness of 20 nm, Fig. 1b) pointing toward a common center. The morphology of the catalysts remains that of a desert rose (inset of Fig. 1a), an intricate rose-like formation of crystal clusters of gypsum or baryte, which include abundant sand grains. After the OER test, NiO μFs do not appreciably change their morphology (Fig. S1,† NiOS1 electrode). | ||
| Fig. 1 (a) SEM image of NiO μFs dispersed on GP (desert rose in the inset); (b) STEM image of a single NiO nanosheet; (c) XRD pattern of NiO μFs; (d) high resolution STEM image of the edge of a nanosheet with an enlarged view of the atomic lattice in the inset; (e) 3-D atomic model of the NiO fcc atomic cell; (f) EDX spectrum of μFs dispersed on GP. | ||
NiO μFs are further characterized using X-ray diffraction (XRD) spectroscopy (XRD pattern in Fig. 1c). All the reflections in the XRD pattern can be indexed to face-centered cubic (fcc) phase NiO (JCPDS card no. #47-1049). The two characteristic peaks at 37.25° and 43.28° correspond to the (111) and (200) diffraction planes, respectively. No peaks from other phases are detected, indicating that the product is of high purity.
An in-plane view STEM micrograph of a single NiO nanosheet is shown in Fig. 1d. The inset is an enlarged view of the NiO nanosheet at atomic resolution that shows the crystal lattice and confirms that it is NiO Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m in the bunsenite form (Fig. 1e shows a 3-D atomic model of the NiO fcc atomic cell). Furthermore, it was found that the nanosheets composing the μFs are (111) planes (Fig. 1b and d).
m in the bunsenite form (Fig. 1e shows a 3-D atomic model of the NiO fcc atomic cell). Furthermore, it was found that the nanosheets composing the μFs are (111) planes (Fig. 1b and d).
The EDX spectrum of NiO μFs on GP is reported in Fig. 1f, indicating the effective deposition of the catalyst on the carbon-based substrate. EDX analysis unravels the presence of a peak at 0.85 keV easily attributed to the Lα of Ni, whereas the two peaks centered at 7.47 and 8.26 keV arise from the Kα and Kβ lines of Ni, respectively. The C peak (Kα, 0.277 keV) is ascribed to the underlying GP substrate.
Mass variation of NiO μFs on GP was confirmed by Rutherford backscattering spectrometry (RBS) analysis (Fig. S2†) since the Ni amount is proportional to the Ni peak (∼1.5 MeV) in the spectrum.37 As expected, Ni amounts on GP increased with the number of drops, both during spin coating and drop casting. The low tail of the Ni peak at low energies is attributable to a 3D cluster configuration of Ni μFs agglomerated on the substrate. Peaks at 0.45 MeV and 0.7 MeV refer to the presence of carbon and oxygen from the substrate and μFs, respectively. Finally, SEM analysis allowed us to assess the presence or absence of NiO μF agglomerates depending on the deposition method. Fig. S3† confirms the presence of large agglomerates of μFs in the case of the drop casting method while almost no agglomeration was detected in the spin coating case.
To measure the performances of the NiO μF electrocatalysts toward the OER, we performed a series of electrochemical measurements in alkaline media (1 M KOH, pH 14). Fig. 2a reports the polarization curves of the samples (obtained from the linear sweep voltammetry technique, LSV) after their stabilization through CV (∼20 cycles).
 | ||
| Fig. 2 (a) Polarization curves, (b) Nyquist plots with the fitting curves plotted, (c) schematic representation of equivalent circuit elements on the NiO catalyst on GP and (d) Tafel plots of NiO μFs deposited on GP substrates. | ||
Two kinds of behavior can be recognized in all the samples:
(i) At low overpotentials all the electrodes show a similar response, with a rapid increase of current density due to oxygen formation.
(ii) At high overpotentials the slope for spin coated samples is much lower than that of other samples.
The OER is somewhat quenched for low mass samples at higher current density. NiOD2 and NiOD3 samples (with a mass of 0.50 and 0.75 mg, respectively) exhibit improved OER performance, as evidenced by the overpotentials of (314 ± 1) mV and (312 ± 1) mV required to reach a current density of 10 mA cm−2, which is lower compared to the ones for samples with lower mass (comparison in Table 2). The similar values for both catalysts suggested to us that not only does the material dispersion play a crucial role in the catalytic performance, but also the amount of catalyst is a fundamental parameter to investigate the electrochemical behavior of the samples. There seems to be a sort of a limiting mass (0.50 mg in our case) beyond which no further increase of overpotential can be detected.
| Sample | Catalyst loading [±0.01 mg] | Overpotential at 10 mA cm−2 [±1 mV] | Tafel slope [mV dec−1] |
|---|---|---|---|
| NiOS1 | 0.06 | 338 | 54–108 |
| NiOS2 | 0.18 | 326 | 48–97 |
| NiOD1 | 0.30 | 324 | 40–50 |
| NiOD2 | 0.50 | 314 | 47–60 |
| NiOD3 | 0.75 | 312 | 56–58 |
To assess the activity trend of our set of electrocatalysts and to further elucidate the electrode kinetics, electrochemical impedance spectroscopy (EIS) was performed (Fig. 2b). This kind of circuit is extensively used in all these cases in which products are formed after the adsorption/desorption of one or more intermediate species at the electrode surface.41–46 Table S1† reports all the parameters extracted from the fit. In this circuit (Fig. 2c), Ru is the uncompensated resistance (linked to the voltage drop in the solution), Rct is the charge transfer resistance for the electrode reaction, CPEdl is the double layer capacitance (in our case all the capacitances are replaced by Constant Phase Elements, CPE, to take into account the non-ideal behavior of the electrodes), Rp is related to the mass transfer resistance (pseudoresistance) of the adsorbed intermediate OH−, and CPEp is the pseudocapacitance (associated with the surface coverage of the adsorbed intermediates).18,20,38–45 The experimental data, supported by the Armstrong and Henderson model, show a reduction in both Rct and Rp as the mass of the catalyst increases, while Ru remains almost constant (Fig. S4a†), as expected. The reduced Rct and Rp prove that the increase in catalyst mass accelerates the electron transfer kinetics at the electrode–solution interface, facilitating the adsorption of OH− on the surface of μFs (and the subsequent O2 formation). The increase in mass leads to an increase in CPEdl indicating an improved capability to adsorb OH− and higher electrocatalytic activity (Fig. S4b†). In contrast, CPEp decreases as the mass of the catalyst increases since the higher the number of active sites, the lower their occupancy.
The electrocatalytic activities of the NiO μFs were further examined by corresponding Tafel plots, extrapolated from polarization curves (Fig. 2d), and the Tafel slopes extrapolated from the linear fit of data points are reported in Table 2. Generally, a smaller Tafel slope implies faster electrochemical kinetics.4,7,8
Moreover, it is possible to have an indication of the rate determining step (RDS) of the process. By considering the different proposed paths for the OER8,19 and the works of Doyle, Brandon and Lyons18,20,47 on the OER on Ni electrodes, our Tafel slopes in the low current density region indicate a RDS represented by the adsorption of OH− ions on the surface of the electrocatalyst (see the ESI† for details of the OER mechanism at Ni-based electrodes). Any change of Tafel slope in this potential region can easily reflect the concentration of active sites and their contribution in the overall process.7 The lower Tafel slope of the drop casted catalysts indicates that the oxygen production rate is reduced for high mass samples. At high current density, the Tafel slopes drastically change for the samples prepared by spin coating (reflecting the trend of current density in LSVs of Fig. 2a). For the drop cast catalyst the Tafel slope values remain nearly constant for the entire investigated potential range. The mentioned increase can be attributed to different mechanisms:
(i) increased mass transport resistances,
(ii) change in the RDS,
(iii) adsorption of the reaction intermediates,20 and
(iv) change of active sites and their contribution.7
Given the above, the small mass deposited on the GP for NiOS1 and NiOS2 does not provide an adequate number of active sites to produce oxygen causing a change in the slope of the polarization curves and an increase of Tafel slopes.
Our results prove that there is a limiting mass value (and a degree of dispersion) below which the catalytic performance of NiO electrodes drastically worsens at high overpotentials. Nevertheless, to more deeply investigate the effect of NiO μFs on the OER mechanism it is essential to take into account the number and especially availability of active sites on the surface of the catalysts.
Relying only on overpotential to evaluate the OER performance can be misleading since it is an extensive quantity and it is not quantitatively linked to the number of active materials involved in the OER. Not only does the number of active sites on the surface of our catalysts influence the oxygen production, but also the rate of adsorption/desorption of OH− ions has an influence. To better investigate the metrics related to the intrinsic activity of the electrodes, the turnover frequency (TOF) must be considered.17,48 The TOF is defined as the rate of production of oxygen molecules per active site:
 | (4) |
The greatest difficulties arise when determining the number of active sites. There is not a general consensus in considering the bulk of the catalyst or only the outer surface as an active element involved in the OER.16 At least two possibilities can be identified: (i) the activity is confined to a near surface region in which there is ionic conductivity and electrolyte intercalated between the μFs and in the multiple planes composing NiO nanosheets, and (ii) bulk activity in which the entire material is active.16,17,47 We determined a minimum (TOFbulk) and a maximum (TOFredox) TOF based on the different assumptions on the number of active sites described above. In particular, TOFbulk assumes that all the deposited moles are active, while TOFredox assumes that only one active site per electron is transferred in the Ni reduction peak of the cyclic voltammogram (CV, see Fig. S5†).16 For the calculation of TOFbulk, the number of moles is given by:
 | (5) |
Concerning the determination of the redox peak, integration is the easiest approach and an efficient way of finding out the number of active catalytic sites. It is possible to use the first backward sweep of a CV scan and integrate the charge under the reduction peak.48 The n for TOFredox can be calculated by the following expression:
 | (6) |
485 C mol−1).
Fig. 3a shows the values for TOFbulk and TOFredox calculated for all the samples at a fixed overpotential. In all cases TOFredox is approximately two orders of magnitude higher than TOFbulk. By considering only TOFbulk, it is easy to understand how the lower mass sample (NiOS1) has a higher TOF.
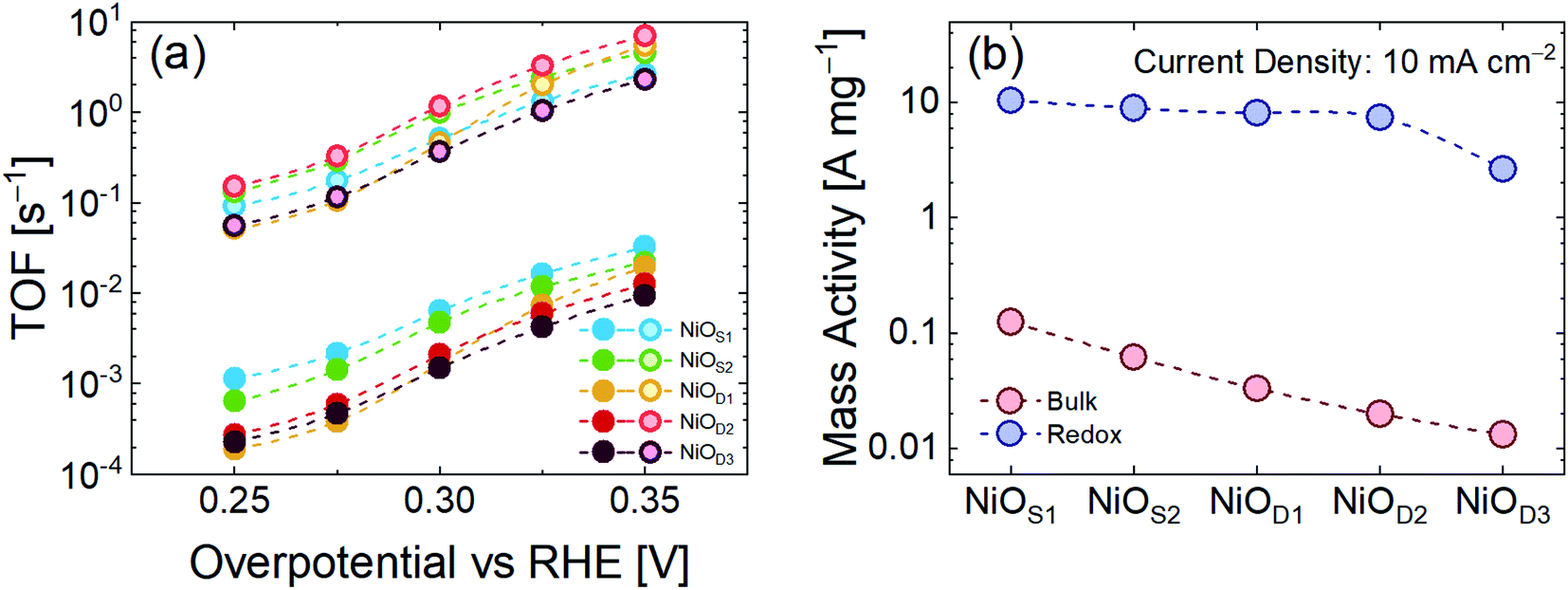 | ||
| Fig. 3 (a) TOFbulk and TOFredox of NiO catalysts on GP; (b) bulk and redox mass activity at a current density of 10 mA cm−2. | ||
On the other hand, TOFredox indicates that the NiOD2 sample is the best electrocatalyst. This discrepancy arises from the consideration that not all the catalyst mass deposited on GP is electrochemically active during the OER, but only a small fraction of the total mass contributes active sites to the reaction.47,48 To evaluate and compare the intrinsic catalytic activity, it is important to look at the activity per unit mass (A mg−1, see the ESI† for details). Fig. 3b compares the mass activity of the NiO catalysts at a current density of 10 mA cm−2 by considering both the total loading of μFs and only the redox contribution to the OER. The mass activity calculated from the redox fraction of the total mass is almost 2 orders of magnitude higher than in the bulk case. Such behavior reveals that increasing the mass of the μFs does not lead to a corresponding increase of the intrinsic OER performance, because of the decrease of the number of electrocatalyst active sites. In the high mass samples, the electrochemical properties are weakened, leading to less effective material utilization.25
The obtained results should be compared with the state of the art. As remarked by Kibsgaard, the state of the art for the OER is not represented by ruthenium, iridium, or ruthenium- and iridium-based materials, and comparison with these precious materials is irrelevant.17Table 3 compares the state of the art for the TOF of NiO-based non-noble metal catalysts for the OER in alkaline media at an overpotential of 350 mV. The reaction rate (TOFredox) of 6.98 s−1 for NiOD2 is, to the best of our knowledge, among the highest reported for non-noble catalysts under alkaline conditions in the literature.16,24,25,48–52 Moreover, we compared the mass activity of our electrocatalysts at a constant current density of 10 mA cm−2 as a function of overpotential with the current state of the art (Fig. S6†). In our samples, an increase of the mass loading leads to a reduction of the overpotential for the OER, but at the same time, a significant decrease of intrinsic activity. This comparison, together with the TOF values, makes our NiO μFs valuable candidates as anode electrodes for the OER.
| Electrocatalyst material | Electrolyte | Overpotential [mV] | Tafel slope [mV dec−1] | TOFbulk [s−1] at η = 350 mV | TOFredox [s−1] at η = 350 mV | Ref. |
|---|---|---|---|---|---|---|
| NiO hollow nanofibers | 1 M KOH | 322 | 78 | 0.07 | NA | 24 |
| NiTi oxide nanosheets | 1 M KOH | 320 | 52 | 0.005 | NA | 52 |
| NiO thin film | 0.5 M KOH | 360 | 54 | 0.07 | NA | 49 |
| Ni(OH)2 nanoplates | 0.1 M KOH | 474 | 87 | 0.015 (490 mV) | NA | 50 |
| Ni/NiO nanoparticles | 1 M KOH | 320 | 61 | 0.11 | NA | 51 |
| NiO nanowalls | 1 M KOH | 345 | 48 | 0.18 | NA | 25 |
| Ni–P | 1 M KOH | 335 | 71 | 0.05 | 0.62 | 48 |
| NiOS1 | 1 M KOH | 338 | 54–108 | 0.03 | 2.64 | This work |
| NiOS2 | 1 M KOH | 326 | 48–97 | 0.02 | 4.57 | This work |
| NiOD1 | 1 M KOH | 324 | 40–50 | 0.02 | 5.44 | This work |
| NiOD2 | 1 M KOH | 314 | 47–60 | 0.01 | 6.98 | This work |
| NiOD3 | 1 M KOH | 312 | 56–58 | 0.009 | 2.31 | This work |
Finally, the durability of the NiOD2 electrode was tested using chronopotentiometry. As displayed in Fig. S7,† the voltage at a current density of 10 mA cm−2 slightly increased up to 15 hours, remaining lower than 0.4 V, demonstrating a good stability of the electrode.
Conclusions
In conclusion, we developed a high-efficiency OER catalyst by dispersing low-cost synthesized NiO microflowers (μFs) onto a GP substrate. By varying the loading of μFs the role of active sites was elucidated with respect to the total mass of the electrocatalyst. The NiO catalyst with optimized mass loading and material dispersion on GP shows an overpotential of only 314 mV at a current density of 10 mA cm−2, and a promisingly low Tafel slope of 47 mV dec−1. The performance of the NiO μFs was also supported by high intrinsic activity, in terms of bulk TOF of 0.01 s−1 at an overpotential of 350 mV and of a record value for redox TOF of 6.98 s−1. The high activity and low cost of the present NiO μFs open the route towards large-scale and long-term applications of NiO OER catalysts.Author contributions
L. B.: investigation, data curation, visualization, writing, original draft preparation, reviewing and editing. M. S.: investigation and reviewing. F. P.: supervision. S. M.: supervision, conceptualization, methodology, investigation, reviewing and editing. All authors contributed to the final version of this paper.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article.Acknowledgements
The authors would like to thank Mr Giuseppe Pantè (CNR-IMM) and Mr Salvo Tatì (CNR-IMM) for their technical support, Dr Sergio Battiato (University of Catania and CNR-IMM), and the Bio-nanotech Research and Innovation Tower (BRIT) laboratory of the University of Catania (Grant no. PONa3_00136 financed by the MIUR) for the Smartlab diffractometer facility. This work was supported by programma di ricerca di Ateneo UNICT 2020-22 linea 2 PIA.CE.RI “NaTI4Smart Sviluppo di Nanomateriali e Tecnologie Innovative per Smart detection”, by PRIN 2017 “CLEAN-Valorizing Sustainable Plastics through a CLEver use of NANoparticles” 20174FSRZS_003 and by “PON MIUR ADAS +” (ARS01_00459).Notes and references
- P. C. K. Vesborg, B. Seger and I. Chorkendorff, J. Phys. Chem. Lett., 2015, 6, 951–957, DOI:10.1021/acs.jpclett.5b00306.
- J. M. Gonçalves, T. A. Matias, K. C. F. Toledo and K. Araki, Adv. Inorg. Chem., 2019, 74 DOI:10.1016/bs.adioch.2019.03.002.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120(2), 851–918, DOI:10.1021/acs.chemrev.9b00248.
- A. R. Zeradjanin, J. Masa, I. Spanos and R. Schlögl, Front. Energy Res., 2021, 8, 613092, DOI:10.3389/fenrg.2020.613092.
- W. Li, D. Xiong, X. Gao and L. Liu, Chem. Commun., 2019, 55, 8744, 10.1039/c9cc02845e.
- S. Anantharaj, S. R. Ede, K. Sakthikumar, K. Karthick, S. Mishra and S. Kundu, ACS Catal., 2016, 6, 8069–8097, DOI:10.1021/acscatal.6b02479.
- G. Li, L. Anderson, Y. Chen, M. Pan and P. Y. Abel Chuang, Sustain. Energy Fuels, 2018, 2, 237, 10.1039/c7se00337d.
- R. L. Doyle and M. E. G. Lyons, in Photoelectrochemical Solar Fuel Production: from Basic Principles to Advanced Devices, 2016 Search PubMed.
- X. Tian, P. Zhao and W. Sheng, Adv. Mater., 2019, 31, 1808066, DOI:10.1002/adma.201808066.
- S. Kiemel, T. Smolinka, F. Lehner, J. Full, A. Sauer and R. Miehe, Int. J. Energy Res., 2021, 45, 9914–9935, DOI:10.1002/er.6487.
- N. T. Suen, S. F. Hung, Q. Quan, N. Zhang, Y. J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337, 10.1039/c6cs00328a.
- A. H. Tkaczyk, A. Bartl, A. Amato, V. Lapkovskis and M. Petranikova, J. Phys. D: Appl. Phys., 2018, 51, 203001, DOI:10.1088/1361-6463/aaba99.
- G. Carollo, A. Garbujo, F. Mauvy and A. Glisenti, Energy Fuels, 2020, 34, 11438–11448, DOI:10.1021/acs.energyfuels.0c01678.
- A. I. Inamdar, H. S. Chavan, S. M. Pawar, H. Kim and H. Im, Int. J. Energy Res., 2019, 44, 1789–1797, DOI:10.1002/er.5026.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426, DOI:10.1016/0254-0584(86)90045-3.
- C. Roy, B. Sebok, S. B. Scott, E. M. Fiordaliso, J. E. Sørensen, A. Bodin, D. B. Trimarco, C. D. Damsgaard, P. C. K. Vesborg, O. Hansen, I. E. L. Stephens, J. Kibsgaard and I. Chorkendorff, Nat. Catal., 2018, 1, 820–829, DOI:10.1038/s41929-018-0162-x.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433, DOI:10.1038/s41560-019-0407-1.
- R. L. Doyle, I. J. Godwin, M. P. Brandon and M. E. G. Lyons, Phys. Chem. Chem. Phys., 2013, 15, 13737, 10.1039/c3cp51213d.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800, 10.1039/c4cy00669k.
- M. E. G. Lyons and M. P. Brandon, Int. J. Electrochem. Sci., 2008, 3, 1425–1462 CAS.
- P. Manivasakan, P. Ramasamy and J. Kim, RSC Adv., 2015, 5, 33269, 10.1039/c5ra01739d.
- D. Takimoto, S. Hideshima and W. Sugimoto, ACS Appl. Nano Mater., 2021, 4, 8059–8065, DOI:10.1021/acsanm.1c01328.
- W. Yuan, C. Li, M. Zhao, J. Zhang, C. M. Li and S. P. Jiang, Electrochim. Acta, 2020, 342, 136118, DOI:10.1016/j.electacta.2020.136118.
- V. D. Silva, T. A. Simões, J. P. F. Grilo, E. S. Medeiros and D. A. Macedo, J. Mater. Sci., 2020, 55, 6648–6659, DOI:10.1007/s10853-020-04481-1.
- S. Cosentino, M. Urso, G. Torrisi, S. Battiato, F. Priolo, A. Terrasi and S. Mirabella, Mater. Adv., 2020, 1, 1971, 10.1039/d0ma00467g.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557, DOI:10.1038/nmat3313.
- B. P. Reddy, K. Mallikarjuna, M. Kumar, M. C. Sekhar, Y. Suh and S. H. Park, Ceram. Int., 2021, 47, 3312–3321, DOI:10.1016/j.ceramint.2020.09.172.
- A. Mondal, A. Paul, D. N. Srivastava and A. B. Panda, Int. J. Hydrogen Energy, 2018, 43, 21665–21674, DOI:10.1016/j.ijhydene.2018.06.139.
- P. T. Babar, A. C. Lokhande, M. G. Gang, B. S. Pawar, S. M. Pawar and J. H. Kim, J. Ind. Eng. Chem., 2018, 60, 493–497, DOI:10.1016/j.jiec.2017.11.037.
- Z. Pu, Q. Liu, A. M. Asiri and X. Sun, J. Appl. Electrochem., 2014, 44, 1165–1170, DOI:10.1007/s10800-014-0743-6.
- K. O. Iwu, A. Lombardo, R. Sanz, S. Scirè and S. Mirabella, Sens. Actuators, B, 2016, 224, 764–771, DOI:10.1016/j.snb.2015.10.109.
- M. Urso, G. Pellegrino, V. Strano, E. Bruno, F. Priolo and S. Mirabella, Nanotechnology, 2018, 29, 165601, DOI:10.1088/1361-6528/aaacb6.
- ImageJ. ImageJ. Available online: https://www.imagej.nih.gov Search PubMed.
- M. Thompson, Xrump. Available online: https://www.genplot.com Search PubMed.
- DigitalMicrograph®. Available online: https://www.gatan.com/products/tem-analysis/gatan-microscopy-suite-software Search PubMed.
- M. B. Stevens, L. J. Enman, A. S. Batchellor, M. R. Cosby, A. E. Vise, C. D. M. Trang and S. W. Boettcher, Chem. Mater., 2017, 29, 120–140, DOI:10.1021/acs.chemmater.6b02796.
- T. L. Alford, L. C. Feldman and J. W. Mayer, in Fundamentals of nanoscale film analysis, 2007, DOI:10.1007/978-0-387-29261-8.
- S. Anantharaj and S. Noda, ChemElectroChem, 2020, 7, 2297–2308, DOI:10.1002/celc.202000515.
- J. E. B. Randles, Faraday Discuss., 1947, 1, 11–19, 10.1039/DF9470100011.
- R. D. Armstrong and M. Henderson, J. Electroanal. Chem., 1972, 1, 81–90, DOI:10.1016/S0022-0728(72)80477-7.
- E. B. Castro, C. A. Gervasi and J. R. Vilche, J. Appl. Electrochem., 1998, 28, 835–841, DOI:10.1023/A:1003488409147.
- B. E. Conway and T. C. Liu, Langmuir, 1990, 6, 268–276, DOI:10.1021/la00091a044.
- E. A. Franceschini, G. I. Lacconi and H. R. Corti, Electrochim. Acta, 2015, 159, 210–218, DOI:10.1016/j.electacta.2015.01.110.
- N. Krstajić, M. Popović, B. Grgur, M. Vojnović and D. Šepa, J. Electroanal. Chem., 2001, 512, 16–26, DOI:10.1016/S0022-0728(01)00590-3.
- G. Wu, N. Li, D. R. Zhou, K. Mitsuo and B. Q. Xu, J. Solid State Chem., 2004, 177, 3682–3692, DOI:10.1016/j.jssc.2004.06.027.
- M. E. G. Lyons and M. P. Brandon, J. Electroanal. Chem., 2010, 641, 119–130, DOI:10.1016/j.jelechem.2009.11.024.
- S. Anantharaj, P. E. Karthik and S. Noda, Angew. Chem., Int. Ed., 2021, 60, 2–19, DOI:10.1002/anie.202110352.
- S. Battiato, M. Urso, S. Cosentino, A. L. Pellegrino, S. Mirabella and A. Terrasi, Nanomaterials, 2021, 11, 3010, DOI:10.3390/nano11113010.
- A. C. Pebley, E. Decolvenaere, T. M. Pollock and M. J. Gordon, Nanoscale, 2017, 9, 15070, 10.1039/c7nr04302c.
- X. Zhou, Z. Xia, Z. Zhang, Y. Ma and Y. Qu, J. Mater. Chem. A, 2014, 2, 11799, 10.1039/c4ta01952k.
- F. E. Sarac Oztuna, T. Beyazay and U. Unal, J. Phys. Chem. C, 2019, 123, 28131–28141, DOI:10.1021/acs.jpcc.9b07460.
- Y. Zhao, X. Jia, G. Chen, L. Shang, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung, D. Ohare and T. Zhang, J. Am. Chem. Soc., 2016, 138(20), 6517–6524, DOI:10.1021/jacs.6b01606.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, methods, and supplementary figures and tables. See https://doi.org/10.1039/d2se00829g |
| This journal is © The Royal Society of Chemistry 2022 |