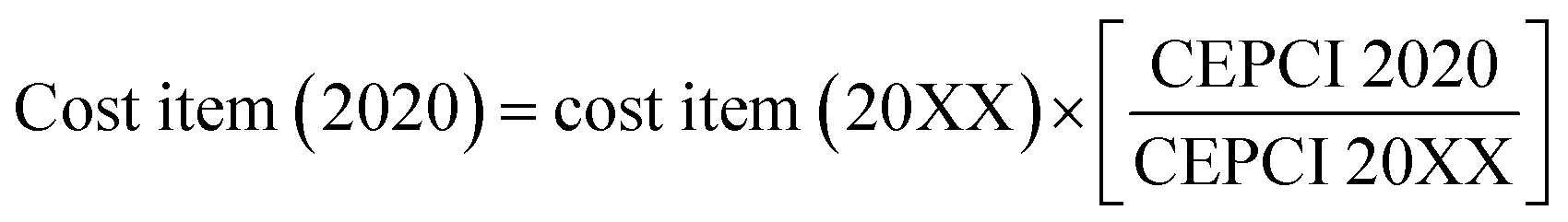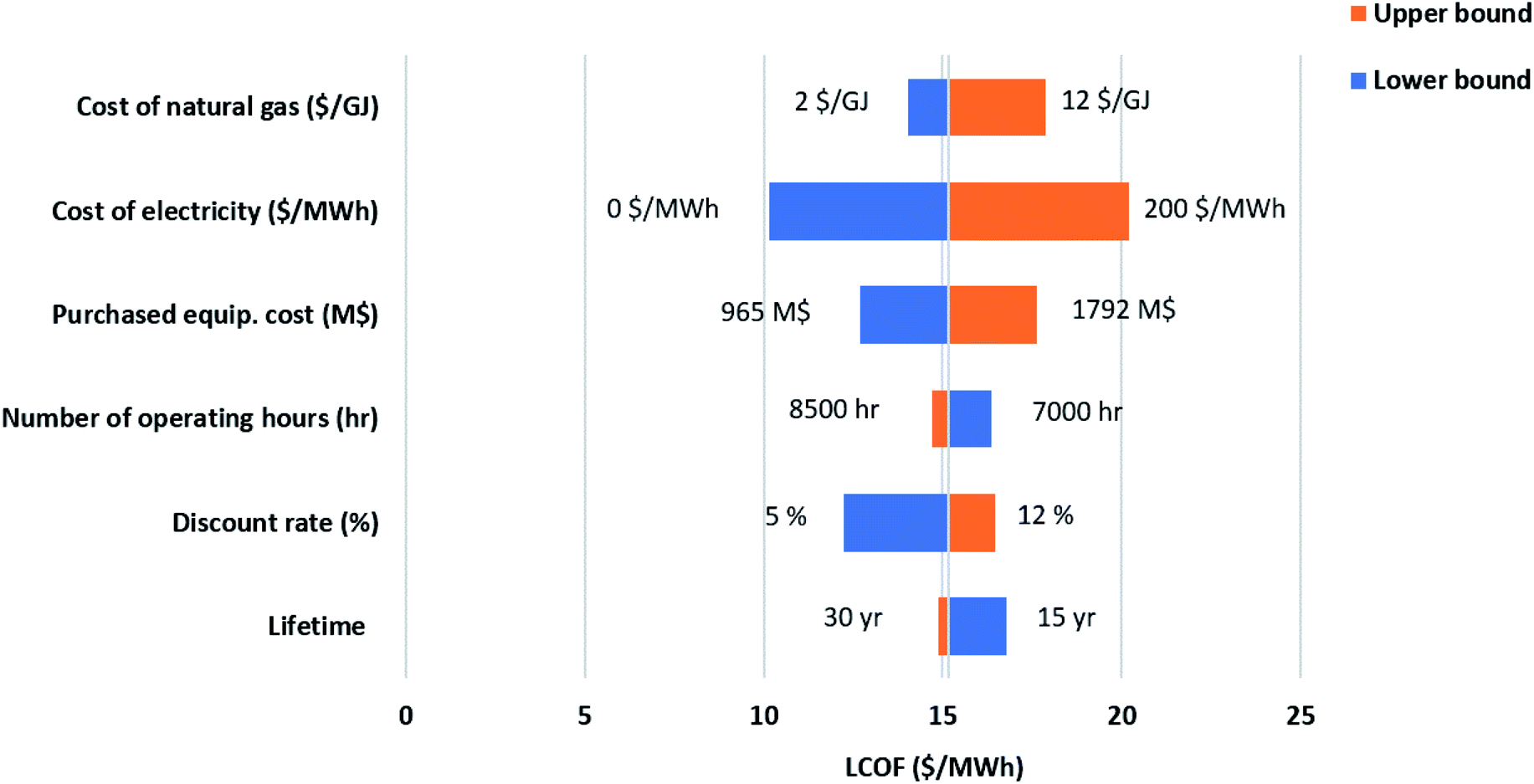Renewable-power-assisted production of hydrogen and liquid hydrocarbons from natural gas: techno-economic analysis†
Received
14th April 2022
, Accepted 5th June 2022
First published on 8th June 2022
Abstract
The declining cost of renewable power has engendered growing interest in leveraging this power for the production of chemicals and synthetic fuels. Here, renewable power is added to the gas-to-liquid (GTL) process through Fischer–Tropsch (FT) synthesis in order to increase process efficiency and reduce CO2 emissions. Accordingly, two realistic configurations are considered which differ primarily in the syngas preparation step. In the first configuration, solid oxide steam electrolysis cells (SOEC) in combination with an autothermal reformer (ATR) are used to produce synthesis gas with the right composition, while in the second configuration, an electrically-heated steam methane reformer (E-SMR) is utilized for syngas production. The results support the idea of adding power to the GTL process, mainly by increased process efficiencies and reduced process emissions. Assuming renewable power is available, the process emissions would be 200 and 400 gCO2 L−1 syncrude for the first and second configurations, respectively. Configuration 1 and 2 show 8 and 4 times less emission per liter syncrude produced, respectively, compared to a GTL plant without H2 addition with a process emission of 1570 gCO2 L−1 syncrude. By studying the two designs based on FT production, carbon efficiency, and FT catalyst volume, a better alternative is to add renewable power to the SOEC (configuration 1) rather than using it in an E-SMR (configuration 2). Given an electricity price of $100/MW h and natural gas price of 5 $ per GJ, FT syncrude and H2 can be produced at a cost between $15/MW h and $16/MW h. These designs are considered to better utilize the available carbon resources and thus expedite the transition to a low-carbon economy.
1 Introduction
To reach the objectives of the Paris agreement1 and the United Nations' Sustainable Development Goals,2 current industrial processes are obliged to move towards reducing their carbon emissions. With the continuous declining cost of renewable power,3 ample opportunities emerge to reduce emissions in different sectors, especially in chemical industries. Among all sectors, the aviation industry will continue to almost entirely depend on high energy density fuels such as kerosene and jet fuel. Aviation fuels are currently mainly petroleum-derived products and the well to wake (WtWa) emissions of fossil jet fuel are reported to be between 80.7 and 109.3 gCO2eq MJ−1.4 However, according to International Energy Agency's Sustainable Development Scenario (SDS), biofuels are anticipated to reach around 10% of aviation fuel demand by 2030, and close to 20% by 2040.5 One practical way to produce jet fuels synthetically is through the Fischer–Tropsch (FT) process. This process produces a wide range of hydrocarbons including kerosene and jet fuel. While this process has many advantages, the main drawback is the low carbon efficiency, meaning that not all carbon in the feed ends up in the product and thus emitted mainly as CO2 to the atmosphere. One way to increase the carbon efficiency of the process is by addition of external carbon-free power.
Integration of renewables into chemical processes results in both increased production of chemicals and concurrent reduction of CO2 emissions. There are two main means of adding renewable power to industrial processes; one is to convert electrical energy to chemical energy of hydrogen by water electrolysis, and the second way is to provide ohmic heat to drive the endothermic chemical processes, such as reforming and pyrolysis reactions.
In the first approach, electricity is used to split water into hydrogen and oxygen. There are plenty of studies in the literature looking at the production of fuels by integration of green hydrogen.6–12 For example, Hillestad et al.13 demonstrated that the carbon efficiency of biomass to liquid (BTL) processes can be greatly improved (i.e. doubled) by adding renewable power in the form of hydrogen and oxygen through electrolysis. Also co-electrolysis of CO2 and H2O to produce syngas and subsequently liquid fuels have also been investigated in the literature.14–17
In the second approach, electricity is used to provide the heat required to drive endothermic chemical processes. Several research reports have been published on this topic and an overview of them is given in Table 1. Abe18 tested an electrically heated reformer that can produce high-purity hydrogen for fuel cell application. Ringler et al.19 disclosed a reformer having electrical heating to produce hydrogen from a hydrocarbon-containing fuel. Alagy et al.20 used an electric furnace for the thermal pyrolysis of hydrocarbons. Lu and Nikrityuk21 proposed a new electrically-heated reactor type for endothermic reactions such as steam-methane reforming (SMR) and dry reforming of methane (DRM). It consists of conducting and non-conducting spherical particles which are electrically heated. Rieks et al.22 used an electrically heated reformer for dry reforming of methane. The catalyst was wash-coated on the heating elements and the experiments were performed at 700–900 °C. More recently, Wismann et al.23,24 used electricity to drive the SMR reaction in an electrically heated catalytic structure. Wash-coated catalyst was integrated into the electrically heated SMR reactor. As a result of proximity between the reaction sites and the heating source, the catalyst effectiveness factor is increased and production of unsought products are limited.23 Electrically heated SMR shows a 100 fold volume reduction compared to a conventional SMR23 and can be deployed in space-constrained environments.
Table 1 Electrically-heated reformers in literature
| Investigation |
Process |
Year |
Ref. |
|
DRM: dry reforming of methane.
|
| Process for the thermal pyrolysis of hydrocarbons using an electric furnace |
Pyrolysis |
1991 |
Alagy et al.20 |
| Reformer and method for operation thereof |
— |
1994 |
Abe18 |
| Reformer system having electrical heating devices |
— |
2005 |
Ringler et al.19 |
| A 2D model for the cylinder methane steam reformer using electrically heated alumite catalyst |
SMR |
2006 |
Zhang et al.25 |
| Investigation of a novel porous anodic alumina plate for methane steam reforming |
SMR |
2009 |
Zhou et al.26 |
| Dry reforming of methane with CO2 on an electron-activated iron catalytic bed |
DRMa |
2011 |
Labrecque and Lavoie27 |
| Axial flow reactor having heating planes and intermediate planes |
— |
2012 |
Mleczko et al.28 |
| Interaction of CO2/CH4 with steel wool in an electrocatalytic dry reforming reactor |
DRM |
2013 |
Banville et al.29 |
| Dry reforming of methane under an electro-catalytic bed: effect of electrical current and catalyst composition |
DRM |
2014 |
Banville et al.30 |
| Experimental study of methane dry reforming in an electrically heated reactor |
DRM |
2015 |
Rieks et al.22 |
| Hydrogen production from steam reforming using an indirect heating method |
Methanol steam reforming |
2018 |
Ji et al.31 |
| A fixed-bed reactor for energy storage in chemicals (E2C): proof of concept |
SMR and DRM |
2018 |
Lu and Nikrityuk21 |
| Electrified methane reforming: understanding the dynamic interplay |
SMR |
2019 |
Wismann et al.24 |
| Electrified methane reforming: A compact approach to greener industrial hydrogen production |
SMR |
2019 |
Wismann et al.23 |
| Integrating CO2 electrolysis into the gas-to-Liquids–Power-to-Liquids process |
Fischer-Trosch |
2020 |
van Bavel et al.32 |
| Improving performance of induction-heated steam methane reforming |
SMR |
2020 |
Almind et al.33 |
| Electrical reverse shift: Sustainable CO2 valorization for industrial scale |
Reverse water gas shift (RWGS) |
2022 |
Wismann et al.34 |
| Scale-up studies on electrically driven steam methane reforming |
SMR |
2022 |
Lu and Nikrityuk35 |
In this study renewable power is added to the gas-to-liquid process, PGTL, to produce heavy hydrocarbons through the Fischer–Tropsch process (Fig. 1). Currently, only Van Bavel et al.32 has explored the possibility of the addition of power to the GTL process. In their hybrid “GTL-PTL” process, CO2 and renewable H2 are co-processed with natural gas to produce liquid fuel. CO2 electrolyser and H2O electrolyser are used for the production of syngas in addition to syngas generated from natural gas reforming. Natural gas was considered as a means of addressing the intermittency of renewables in their hybrid GTL process.
 |
| | Fig. 1 PGTL and GTL concepts with their relative CO2 emissions. | |
Here two realistic designs to reduce carbon emissions of the GTL process through integration of renewable power are investigated. With the rapid decline of cost of renewable power,3 realization of these configurations are becoming extremely viable in the near future.
2 Process modeling and simulation
Aspen HYSYS V10 is used to simulate the process flowsheets and Peng–Robinson equation of state is applied as the thermodynamic model to calculate thermodynamic properties. The specifications of natural gas feeds are shown in Table 2. To examine the effect of presence of CO2 in the feed, a CO2-rich natural gas (10%) is also considered.
Table 2 Specifications of the natural gas feed
|
|
NG1 |
NG2 |
| Temperature [°C] |
50 |
50 |
| Pressure [bar] |
30 |
30 |
| Flow [MMscfd] |
120.2 |
120.2 |
| Molar flow [kmol h−1] |
6000 |
6000 |
| Mole fraction |
|
|
| CH4 |
0.95 |
0.855 |
| C2H6 |
0.02 |
0.018 |
| C3H8 |
0.015 |
0.0135 |
|
n-C4H10 |
0.01 |
0.009 |
|
n-C5H12 |
0.005 |
0.0045 |
| CO2 |
0 |
0.1 |
2.1 Syngas production
Autothermal Reforming (ATR) and Steam Methane Reforming (SMR) constitute two main syngas production methods in the chemical industry to produce high-value chemicals such as methanol, ammonia, etc. Nowadays, more than 50% of the global hydrogen supply is made through SMR.23 In the syngas production step, natural gas is reformed to produce syngas which is a mixture of H2 and CO. To prevent coke formation in the reformer, a pre-reformer is used. Pre-reforming is operated adiabatically at 400–550 °C, and almost all higher hydrocarbons are converted to methane, carbon oxides and hydrogen. The pre-reformer is simulated as a fixed bed adiabatic reactor and chemical equilibrium is assumed to be reached at the outlet.
The H2/CO consumption ratio for the FT synthesis is slightly above 2.36 ATR has the advantage of producing the desired H2/CO ratio by adjusting the operating parameters. In contrast, the syngas generated from E-SMR is hydrogen-rich with a H2/CO ratio greater than 3. To adjust this ratio, H2 needs to be extracted from the syngas. Palladium membrane is chosen here for this separation which operates at high temperatures (>300 °C).
With the ATR configuration, an oxidant is required to derive the oxidation reactions and thus provide heat for the endothermic reactions. The added advantage of using SOEC is the availability of pure oxygen which can be used in the ATR. By using pure oxygen, it is possible to recycle most of the tail gas to the ATR. The SOEC model implemented here is a one-stage adiabatic cell model with a constant conversion of 80%. Thermoneutral operation of SOEC at 850 °C and 40 bar is considered. The unconverted steam is separated by means of cooling and recycled to the SOEC. The energy efficiency of the electrolyser is 83% based on the higher heating value (HHV) of H2, meaning that the electric energy need is around 33 kW h kgH2−1.
With the E-SMR configuration, the heat required to derive the endothermic reforming reactions is provided by electrical power. The E-SMR is simulated by minimizing the Gibbs free energy with an energy flow to supply the required heat for the reforming process and keep its outlet temperature at 850 °C. The generated syngas is passed to a palladium membrane unit to adjust the H2/CO ratio by extracting the excess H2 to have the desired ratio for the FT synthesis.
2.2 Fischer–Tropsch synthesis and hydrogen addition
Nowadays most commercial FT plants are based on cobalt catalyst, due to high selectivity to heavy hydrocarbons, negligible activity towards water-gas shift reaction, and less selectivity to olefins than an iron catalyst and therefore is our preferred catalyst for this process.
The main products of FT reactions are paraffin and olefins. The polymerization reactions taking place are hydrogenation of CO to form n-paraffin, 1-olefins and oxygenates.
| | | nCO + (2n + 1)H2 → CnH2n+2 + nH2O, n = 1, 2,…,∞ | (1) |
| | | nCO + 2nH2 → CnH2n + nH2O, n = 2, 3,…,∞ | (2) |
| | | nCO + 2nH2 → CnH2n+2O + (n−1)H2O, n = 1, 2,…,∞ | (3) |
More than 90% of the products are paraffin and the rest are mainly olefins. The oxygenates are neglected here as they are formed only in limited amounts.
A varying chain growth factor or propagation probability, α is chosen here. It is modeled as a function of temperature and partial pressures of CO, H2 and H2O.37 Anderson–Schulz–Flory distribution predicts the H2/CO consumption ratio to be 3–α. α will change as a function of H2/CO ratio and the temperature, but a typical value of α is 0.94 giving a stoichiometric H2/CO consumption ratio of 2.06.
Outi et al.38 reaction rate model together with a chain growth model published by Ostadi et al.37 are fitted to the Completely Stirred Tank Reactor (CSTR) experimental data of Todic et al.39 and used in this study. The fitted models are reported in Ostadi et al.40
The inlet H2/CO ratio to the FT reactor(s) preferentially is lower than the overall consumption ratio to increase the selectivity to heavier hydrocarbons and suppress the formation of methane and light hydrocarbons. In order to determine the optimal H2/CO ratio of operation, three important factors needs to be considered: the electricity price, with associated investment in electrolysis and E-SMR, and the cost of FT reactors per unit volume as well as the syncrude price.40 However, to limit the scope of the study, an H2/CO ratio of 1.9 is chosen here as the desired ratio. By doing so, this ratio decreases as the reaction proceeds along the reactor. To compensate for the consumption, H2 is added between the stages.
The Fischer–Tropsch synthesis is staged into 3 stages with product withdrawal between the stages. Reactor staging enables higher conversion of syngas and higher selectivity to heavier hydrocarbons.13 The slurry bubble column FT reactors are simulated as CSTR reactors. This type of reactor is characterized by an almost complete mixing of the gases and liquid products, meaning that the catalyst is exposed to the exit gas composition throughout the reactor. The syngas has a pressure of about 26 bar prior to the first FT reactor. In order to control the temperature in FT reactors, boiling water at 210 °C is used as coolant. Once-through conversion in each stage is limited to 60% to have the maximum C5+ selectivity and also preserve catalyst life.41
With 60% conversion in each reactor, the total once-through CO conversion becomes around 94%. Absence of nitrogen in the synthesis loop and a high once-through conversion means a reduction in the need for recycling of unconverted syngas. A small portion of the tail gas is purged to remove trace inerts from the system and also provide part of the heat for the process. This is also where most of the carbon is lost from the system.
3 The considered configurations
Here two realistic designs are considered where renewable power is added to the GTL process: 1 – through SOEC and ATR, 2 – through E-SMR. The main difference is in the syngas and hydrogen generation steps. In configuration 1, an ATR is used to produce syngas and a Solid Oxide Electrolysis Cell (SOEC) is used to generate H2 and O2. In configuration 2, an electrically heated SMR is used to generate syngas. In both designs, there is no need for a costly Air Separation Unit (ASU) or membrane unit to produce oxygen.42 In the suggested processes, absence of N2 in the processing loops allows for high CO conversion in the process.
3.1 Configuration 1: ATR + SOEC
In this design, an Autothermal Reformer (ATR) is used to produce syngas. The O2 required for the reforming process is produced in an SOEC where renewable power is used to split steam to H2 and O2. The Block Flow Diagram (BFD) is shown in Fig. 2 while a more detailed Process Flow Diagram (PFD) is shown in Fig. 3. Steam is added to the pre-reformed gas to have the desired steam-to-carbon ratio (S/C = 0.6) and further heated to 650 °C prior to the ATR. Whether to have pre-heating before ATR or not is investigated and discussed in the results section. The ATR outlet temperature is maintained at 1050 °C by adding enough O2 from the SOEC. The advantage of using O2 is that the produced syngas is nitrogen-free as a result of not using air. The syngas leaving the reformer is then cooled in a multi-step cooling system. The first part of the cooling system is a waste heat boiler followed by a super-heater. The superheated steam is the best candidate to be used as the feed to the SOEC, however it needs to pass through a fired-heater to have the desired temperature before entering the SOEC. The purge stream from the FT synthesis loop is used as fuel for the fired-heater. Syngas leaving the superheater is further cooled to condense water and then reheated to 210 °C and sent to the FT section. With high once-through conversion of syngas in FT section (i.e. 94%) there is no need for direct recycling of FT tail gas to FT section. Since syngas is nitrogen-free, the majority of the tail gas is recycled to ATR (85%) which increases the carbon efficiency of the process. The rest of the tail gas is sent to the fired heater to provide heat for the steam to SOEC. An overview of some important process streams are given in Table S1.† The stream numbers are referred to the process flow diagram in Fig. 3. Temperature, pressure, and mass flows in addition to mole fractions of the important components are chosen to be shown.
 |
| | Fig. 2 Block flow diagram of configuration 1 – SOEC. | |
 |
| | Fig. 3 Process flow diagram of configuration 1 – SOEC. | |
3.2 Configuration 2: E-SMR
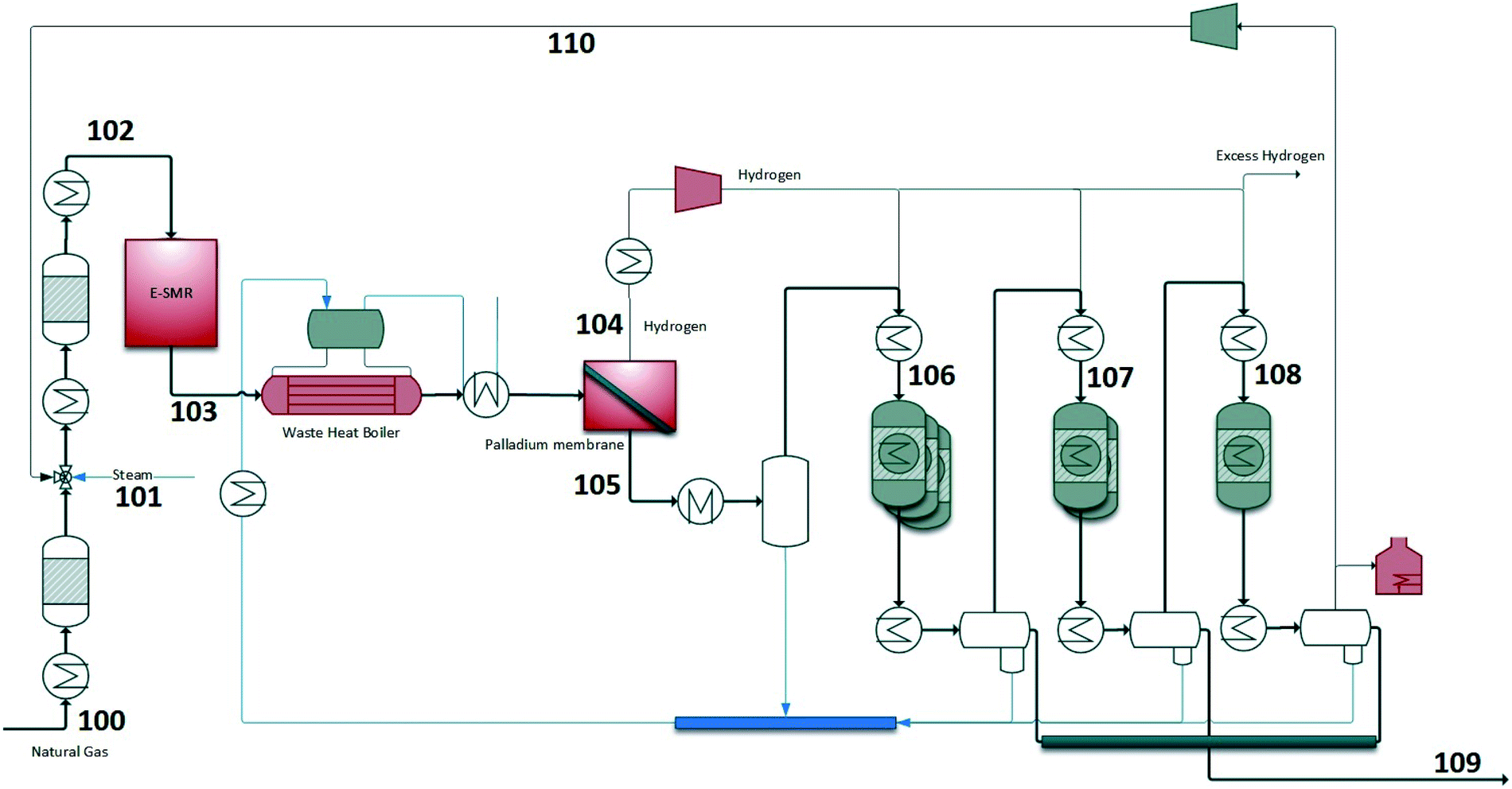
In this configuration, renewable power is used to provide heat in the steam methane reformer and drive the SMR reaction. The BFD and PFD are shown in Fig. 4 and 5. The S/C ratio before the SMR is set to 2.0 which is a reasonable number regarding the cocking potential in the reformer. Natural gas is pre-reformed and heated to 500 °C before the SMR. The electric power input into the SMR (i.e. 632 MW) is high enough to maintain the SMR temperature at 850 °C. The H2/CO ratio at the SMR outlet is high (>3) which is more than the stoichiometric consumption ratio in the FT process. Therefore, part of H2 needs to be separated to have the desired H2/CO ratio before the FT stages (1.9). Palladium membrane is used in this design to separate H2. The temperature upstream of the membrane needs to be at least 300 °C. After H2 separation, syngas is further cooled to condense water and then reheated to 210 °C and sent to the FT section. Since syngas is nitrogen-free (mainly CO2 and methane), the majority of the tail gas is recycled to reformer (90%) which increases the carbon efficiency of the process. The rest of the tail gas is sent to fired heater to supply heat to the plant. An overview of some important process streams are given in Table S2.† The stream numbers are referred to the process flow diagram in Fig. 5. Temperature, pressure, and mass flows in addition to mole fractions of the important components are chosen to be shown.
 |
| | Fig. 4 Block flow diagram of configuration 2 – E-SMR. | |
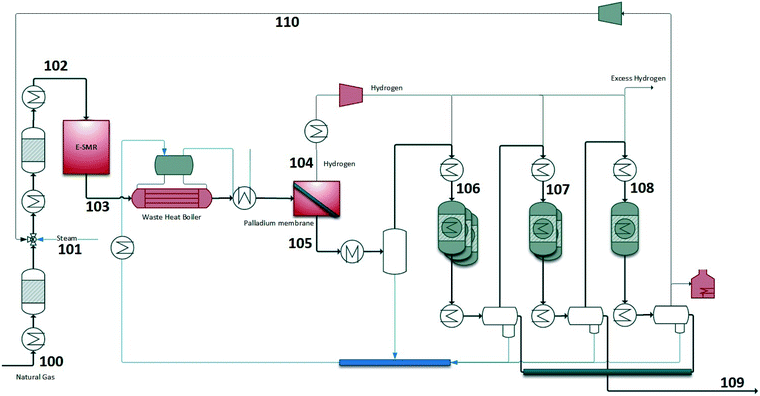 |
| | Fig. 5 Process flow diagram of configuration 2 – E-SMR. | |
4 Results
In E-SMR configuration (configuration 2), excess H2 is produced in the reformer (i.e. syngas has a higher H2/CO ratio than needed in FT reaction). To have a fair comparison between E-SMR and ATR, the same excess H2 is produced in both configurations. The excess H2 is ready to be exported. Moreover, the CO conversion in each FT stage is about 60% which translates to a once-through CO conversion of 94% in all three stages.
The performance of configuration 1 and 2 are shown in Tables 3 and 4, respectively. In each configuration, details of CO conversion, FT production, methane selectivity and H2 addition between stages are shown. In both configurations, the first FT stage has the largest volume and produces more than half of the plant production. The largest H2 addition is at the second stage, mainly as a result of the H2-depleted syngas from the first stage. The CH4 selectivity is around 5% in each stage. Product distribution of C5+ fraction for different FT stages in configuration 1 is shown in Fig. 7.
Table 3 Performance of different FT stages of configuration 1 – SOEC (NG1)
|
|
Stage 1 |
Stage 2 |
Stage 3 |
Total |
| FT volume [m3] |
1250 |
600 |
330 |
2180 |
| C5+ production [t h−1] |
55.1 |
22.7 |
9.1 |
86.8 |
| CO conversion [%] |
60.4 |
60.4 |
60.1 |
93.7 |
| CH4 selectivity [%] |
4.3 |
4.7 |
5.4 |
4.5 |
| H2 addition before each stage [kmol h−1] |
69 |
882 |
364 |
1315 |
| Chain growth factor (α) |
0.94 |
0.93 |
0.92 |
|
Table 4 Performance of different FT stages of configuration 2 – E-SMR (NG1)
|
|
Stage 1 |
Stage 2 |
Stage 3 |
Total |
| FT volume [m3] |
1490 |
890 |
670 |
3050 |
| C5+ production [t h−1] |
51.2 |
20.7 |
8.2 |
80.1 |
| CO conversion [%] |
60.2 |
60.1 |
60.2 |
93.7 |
| CH4 selectivity [%] |
4.8 |
5.7 |
7.2 |
5.2 |
| H2 addition between stage [kmol h−1] |
0 |
870 |
371 |
1241 |
| Chain growth factor (α) |
0.93 |
0.92 |
0.91 |
|
The overall plant results of both configurations are shown in Table 5. The power consumption and the FT production in configuration 1 is slightly higher than configuration 2. With the carbon efficiency being higher in configuration 1, this also indicates that the CO2 emissions are lower. Moreover, the required FT catalyst volume is lower in configuration 1. The main reason for these differences is related to the higher reforming temperature used in ATR (configuration 1) than in the E-SMR (configuration 2) which are 1050 °C and 850 °C, respectively. The reforming temperature affects the CO2 concentration out of the reformer, which in configuration 1 and 2 are 6% and 11%, respectively.
Table 5 Overall plant results in both configurations (NG1). The reason for large difference of catalyst volume is that the syngas in configuration 2 contains more CO2
|
|
Config 1 |
Config 2 |
| Electric power [MW] |
654.2 |
637 |
| Carbon efficiency [%] |
91.8 |
83.7 |
| Excess H2 [kmol h−1] |
7903 |
7914 |
| CO2 emissions [gCO2 Lfuel−1] |
200 |
400 |
| Specific power [kW h Lfuel−1] |
5.6 |
6.3 |
| Methane selectivity [%] |
4.5 |
5.2 |
| CO conversion [%] |
94 |
94 |
| C5+ production [t h−1] |
86.8 |
80.1 |
| FT catalyst volume [m3] |
2180 |
3050 |
| Reactor productivity [t m−3 h−1] |
0.040 |
0.026 |
Configuration 1 is investigated further regarding two important design considerations: 1 – placement of a Reverse Water Gas Shift Reactor (RWGS) reactor after ATR in order to convert CO2 to CO and 2 – the effect of pre-heating level prior to the ATR on the power consumption in the SOEC.
First, placement of a RWGS after ATR is investigated. The CO2 concentration after ATR is about 6%. By assuming that the RWGS reaction reaches equilibrium, the CO2 conversion in RWGS reactor would be about 3% which is quite low. But on the positive side, this converted CO2 increases the FT production by 435 kg h−1 which is equivalent to an increase in production of 5%. However, the low CO2 conversion does not justify the cost of placement of RWGS after ATR.
The second design consideration of configuration 1 is the level of preheating prior to the ATR. A case study is performed in which the temperature of the pre-reformed stream is changed from 365 °C to 650 °C. There is an inverse relationship between the level of pre-heating and the power added to the SOEC, meaning that the higher the preheating level, less power is required in the SOEC (see Fig. 8). The downside of providing the required heat in the ATR through SOEC is that more CO2 is generated in the ATR as a result of combustion with O2 and this reduces the FT production by about 1 t h−1. The results indicate that there is an optimal distribution of natural gas pre-heating and power addition to SOEC. Putta et al. investigated a similar optimization problem for the Biomass and Power to liquid process.43 They found the optimal level of preheating to be about 37–39% of the energy in the biomass feed.
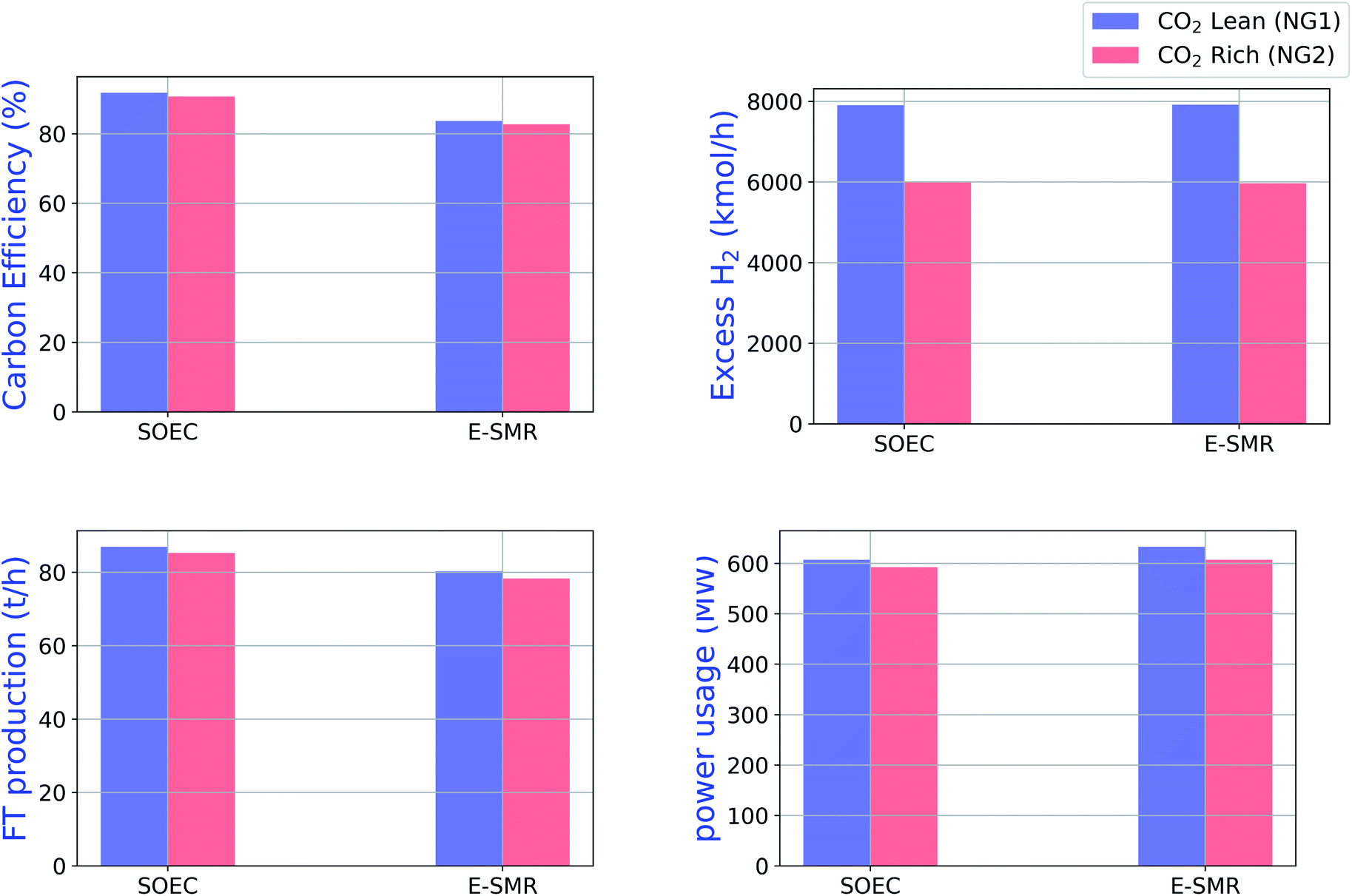
Furthermore, the effect of CO2-rich natural gas is also investigated. The natural gas used so far, NG1 in Table 2, has no CO2. If the natural gas is somewhat richer in CO2, as NG2 in Table 2, what will be the consequences? Comparing the CO2-lean and CO2-rich NG, the power consumption and FT production are lower for CO2 rich cases. The results of simulations with CO2-lean and CO2-rich NG are shown in Fig. 6. The main difference is the amount of excess H2 produced, wherein CO2-rich case, less excess H2 is produced. This is because less hydrogen atoms enter the plant with CO2-rich case due to fixed molar flow of natural gas in both NG1 and NG2.
 |
| | Fig. 6 Carbon efficiency, excess hydrogen, FT production and power usages of the two configurations with CO2 lean and CO2 rich natural gas. | |
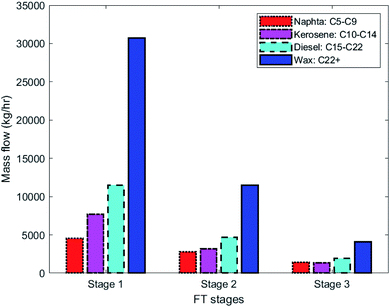 |
| | Fig. 7 Product distribution in FT stages in configuration1-SOEC. The results correspond to stream 107 in Fig. 3. | |
 |
| | Fig. 8 Effect of preheating before ATR on power required in SOEC and FT production. | |
4.1 Carbon and hydrogen flow
The ultimate goal of the proposed processes is to convert hydrogen and carbon atoms in the natural gas and steam into long-chain hydrocarbons. Therefore looking at carbon and hydrogen flows in the process is very informative. Carbon efficiency is considerably improved compared to the previous designs,44,45 from around 60% to above 84% and 91%. The other reason for this improvement aside from the use of clean renewable power is due to the absence of nitrogen (mainly from air) in the synthesis loop which makes it possible to recycle large portion of FT tail gas to reformers. Carbon flows in both configurations are about the same because of having close carbon efficiencies (92% in configuration 1 and 84% in configuration 2). However, the hydrogen balance is more revealing and they are shown in Fig. S1 and S2.† While the Fischer–Tropsch section in both designs show similar numbers, the main difference is in the syngas generation part. The added steam to the E-SMR reformer is five times more than the ATR (configuration 1) and the total added steam in configuration 2 is 80% more. It is interesting to note that the separated water right after the reformer in E-SMR is about 4 times larger than the ATR. It confirms the fact that configuration 2 has poorer hydrogen efficiency compared to configuration 1.
4.2 Water balance
Water is one of the products of FT reaction, and on a molar basis, it is produced equally as CO is consumed (eqn (1) and (2)). Table S3† shows the water balance for the two configurations (in ESI†). There is a net water demand of 35.9 & 26.8 t h−1 in configuration 1 & 2, respectively, mainly consumed in SOEC and reformers. Water retrieved from the FT product may contain some oxygenates and limited amounts of hydrocarbons but the water is perfect to be used as feed to the ATR or E-SMR. These components will be reformed in the reformer. Water retrieved from the syngas is much cleaner, mainly small amounts of CO2 is present, and the water can easily be purified.44
4.3 Effect of grid emission intensity
The two proposed designs are highly dependent on power grid and having access to low-emission-intensity grid is very important when considering advantages with respect to CO2 emissions reductions. By assuming the syncrude density (C5+) to be 800 kg m−3 and grid with zero emission-intensity, the CO2 emissions per liter fuel produced is 200 and 400 gCO2 L−1 syncrude for configuration 1 and 2, respectively. Comparing to the previous designs which had a CO2 emission of 1570 gCO2 L−1 syncrude,44,45 configuration 1 and 2 show 8 and 4 times less emission per liter fuel produced, respectively. By taking into account the H2 lower heating value in addition to FT syncrude, the CO2 emissions per MJ fuel produced is 4 and 8 gCO2 MJ−1 fuel for configuration 1 and 2, respectively, while the previous designs had 38 gCO2 MJ−1 fuel.44,45Fig. 9 shows the emissions of the new designs with respect to grid emission intensity. Configuration 1 has lower emissions than in configuration 2. Having a grid with emission intensity lower than 250 gCO2 kW−1 h−1, both configurations have lower emissions than previous designs.44,45 The reason for insensitivity of the emission profile of previous GTL designs with respect to grid emission intensity is due to the fact that those designs are self-sufficient with respect to power and even export some power to the grid.44,45
 |
| | Fig. 9 Effect of emission intensity on process emissions. | |
4.4 Heat integration
To get a better picture of the amount of heating and cooling required in the proposed processes, the energy composite curves for configuration 1 and 2 are shown in Fig. 10 and 11, respectively. In configuration 1, Fig. 10, there is a requirement for 119 MW of external heating and there is 476 MW of excess heat. In configuration 2, Fig. 11, the external heating and cooling requirements are 74 MW and 620 MW, respectively. In order to have a fair comparison between configuration 1 and 2, it is assumed that the difference in external heating requirement for two configurations is provided by electrical heating. Therefore, 45 MW of electrical heating (119–74 = 45 MW) is required in design 1, which is added to the total power consumption in Table 5. The horizontal line on the hot composite curve at 210 °C represents the steam generated during cooling of the FT reactors. As can be observed, in both processes a large amount of excess heat is available mainly below 200 °C which can be utilized to produce power or hydrogen. Compared to the amount of excess heat required, there is a minor need for external heating in both designs, which can be supplied by electrical heating or changing important operating variables or combusting part of the excess hydrogen.
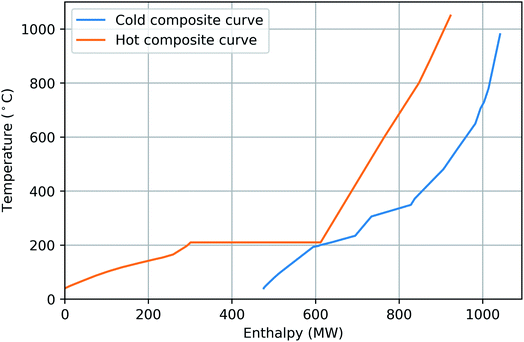 |
| | Fig. 10 Composite curves of configuration 1 - SOEC: the process requires 119 MW of external heating and 476 MW of external cooling. | |
 |
| | Fig. 11 Composite curves of configuration 2- E-SMR: the process requires 74 MW of external heating and 620 MW of external cooling. | |
4.5 Cost estimation
In order to evaluate the economic viability of the two proposed concepts, a levelized cost estimation of the produced fuel (LCOF) (i.e. H2 and FT syncrude) is carried out. The purchase cost of major equipment in the process are calculated based on reported literature values and scaled by using eqn (4) where R is the scaling factor. The costs are brought to $ 2020 by using the Chemical Engineering Plant Cost Index (CEPCI), eqn (5).| |  | (4) |
| |  | (5) |
The results of purchase cost estimates of major equipment are shown in Table 6. The cost estimate for an E-SMR unit is uncertain, as there are no estimates available in the literature. A purchase cost of $400/kW is assumed here with a lifetime of 12.5 years. Due to lack of any literature or industrial cost estimate for E-SMR, there is large uncertainty related to that. For the SOEC, an installed cost of $1000/kW is assumed with a stack lifetime of 5 years.
Table 6 Purchased equipment cost in million US dollars (M$)
|
|
|
ATR + SOEC (M$) |
E-SMR (M$) |
Reference |
| Syngas generation |
Fired heater |
16 |
42 |
Peters et al.46 |
| Reformer |
35 |
570 |
Hamelinck et al.47 |
| Waste heat boiler (WHB) |
70 |
78 |
Hillestad et al.13 |
| H2 generation |
Membrane + H2 compressor |
— |
65 |
Hamelinck et al.47 |
|
|
SOEC (including replacements) |
785 |
— |
Hillestad et al.13 |
| FT synthesis and upgrading |
|
473 |
546 |
Hamelinck et al.47 |
|
Total
|
|
1379
|
1301
|
|
The total capital investment for the two projects are calculated based on the percentage of the delivered equipment method.46 The percentages used to calculate direct and indirect costs are shown in Table S4.† The cost of delivery of each equipment is assumed to be 10% of the purchased cost and the working capital is 10% of the total capital investment.
Having the total investment cost (Cinv), the depreciation cost of capital can be calculated by eqn (6):
| |  | (6) |
where
Cinv is the investment cost,
i is the annual interest rate and n is plant lifetime which is assumed to be 25 years.
The syncrude and hydrogen production costs are calculated by dividing the total annual costs by the produced amount of fuel (i.e. H2 and FT syncrude). The total annual costs consist of annual investment and operating costs. Therefore, the levelized cost of fuel ($ per MW per h) can be defined by eqn (7)
| |  | (7) |
where
PFT and
PH2 are the FT syncrude and H
2 production capacity in MW per h per year. The price of natural gas is assumed to be $5000/MMscf (≈5$ per GJ).
48 The annual operating hours is assumed to be 8000 h which is an availability of 91.3%. The main economic parameters for evaluating
eqn (7) are shown in
Table 7.
Table 7 Main parameters for economic analysis
| Economic parameter |
Value |
| Plant lifetime (years) |
25 |
| Interest rate (%) |
10 |
| Annual operating hours (h per year) |
8000 |
| Capital recovery factor (%) |
11.0 |
| Average electricity price ($ per MW per h) |
100 |
| Natural gas price ($ per GJ) |
5 |
4.6 Sensitivity analysis
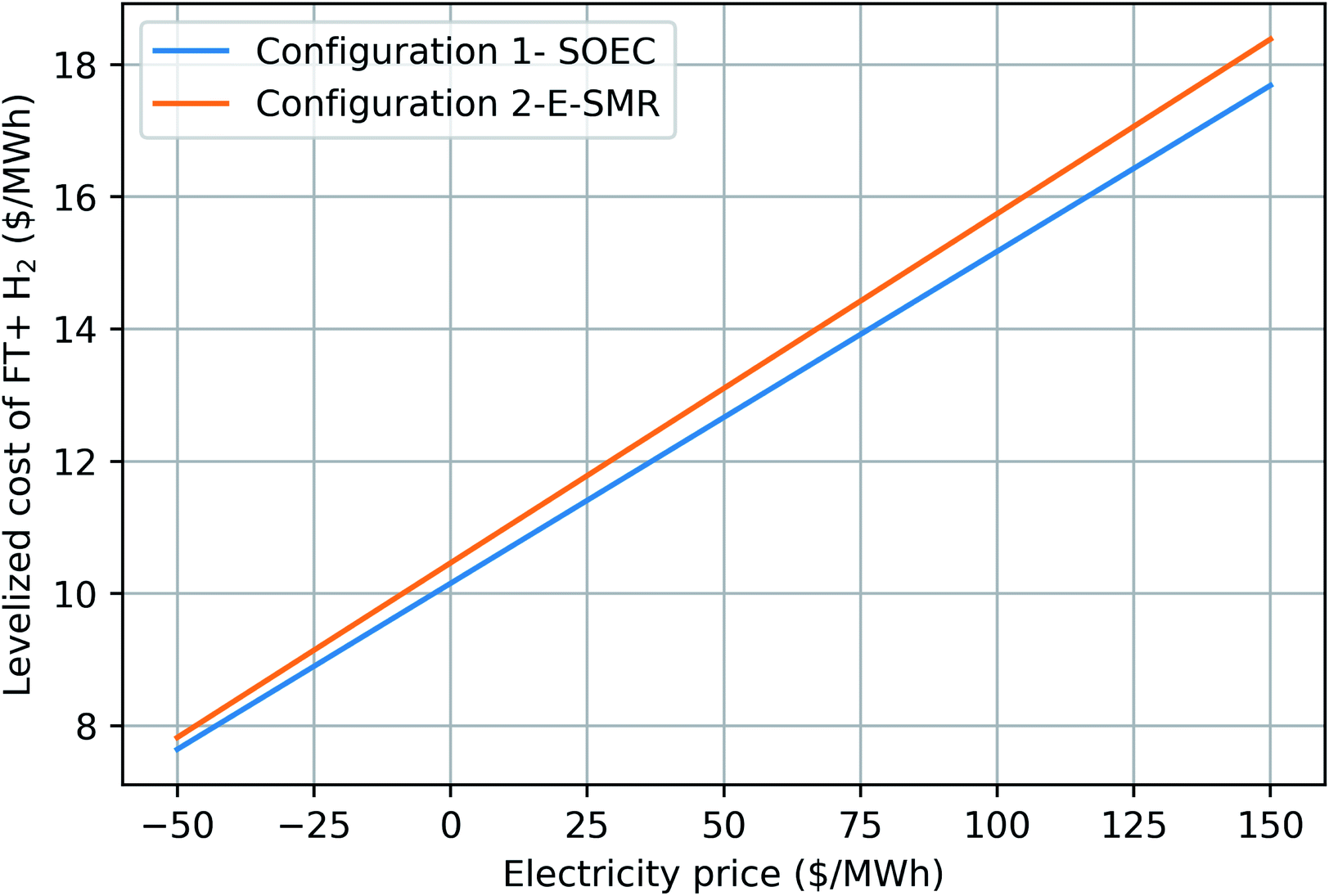
While the base parameters used to calculate the Levelized cost of methanol (LCOMeOH) are given in Table 7, a sensitivity analysis is necessary due to the uncertainty associated with the values used. By changing each variable to its upper and lower limits, as shown in Table 8, Tornado diagrams are generated for both configurations, as shown in Fig. 12 and 13. Electricity price has the largest effect on the levelized cost followed by the natural gas price. Since electricity has a major impact on the cost of produced fuel, it is worthwhile to investigate its variations, as shown in Fig. 14. The negative electricity prices are indicative of periods with renewable power curtailment. The cost of produced fuel through configuration 1 is slightly cheaper. SOEC constitutes about 50% of the purchased cost of equipment in configuration 1, as shown in Table 6, and has a huge impact on the production cost of fuel.
Table 8 Variability ranges in the sensitivity analysis
| Variable |
Base |
Lower limit |
Upper limit |
| Natural gas price ($ per GJ) |
5 |
2 |
12 |
| Interest rate (%) |
10 |
5 |
12 |
| Plant lifetime (year) |
25 |
15 |
30 |
| Electricity price ($ per MW per h) |
100 |
0 |
200 |
| Annual operating hours (h per year) |
8000 |
7000 |
8500 |
| Purchased equipment cost (M$) |
— |
−30% |
+30% |
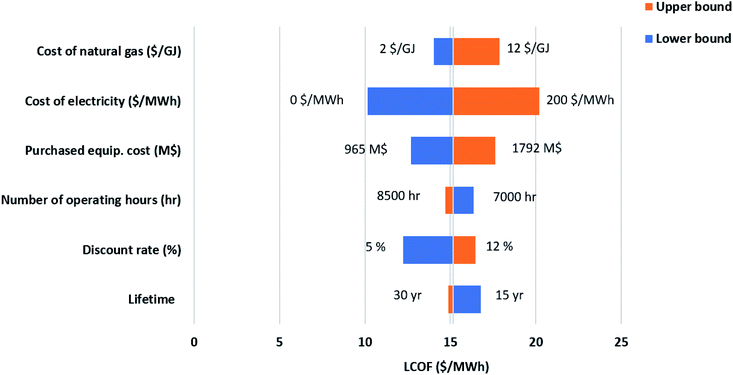 |
| | Fig. 12 Sensitivity analysis-ATR and SOEC. | |
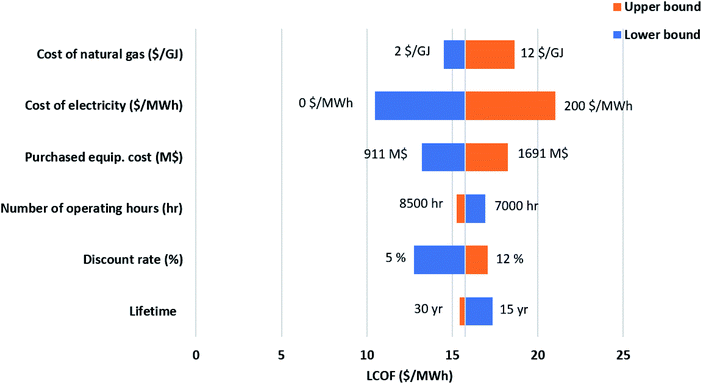 |
| | Fig. 13 Sensitivity analysis-ESMR. | |
 |
| | Fig. 14 Levelized cost of FT and H2 as a function of electricity price. Natural gas price is fixed at 5 $ per GJ. | |
5 Conclusions
In order to improve the carbon efficiency of the GTL process and thus lower process emissions, addition of renewable power to the process is investigated. Two realistic configurations are considered which differ mainly in the syngas generation step. In the first configuration, solid oxide steam electrolysis cells (SOEC) in combination with an autothermal reformer (ATR) is used to produce synthesis gas with the right composition. While in the second configuration, an electrically-heated steam methane reformer (E-SMR) is utilized for syngas production. Addition of renewable power increases the carbon efficiency and thus synthetic hydrocarbon production in both configurations. Carbon efficiency is considerably improved compared to the previous designs,44,45 from around 60% to above 84% and 91%. Moreover, through these processes, simultaneous chemical storage of renewable power is also achieved. In both configurations there is no need for a separate H2 production path to add H2 between the FT stages (for example a parallel SMR process). Additionally, there is no need for an expensive Air Separation Unit (ASU) to produce oxygen. By having the assumption of grid with zero emission-intensity, the CO2 emissions per liter fuel produced is 200 and 400 gCO2 L−1 syncrude for configurations 1 and 2, respectively. Comparing to the previous designs which had a CO2 emission of 1570 gCO2 L−1 syncrude, configurations 1 and 2 show 8 and 4 times less emission for liter syncrude produced, respectively. By taking into account the H2 lower heating value in addition to FT syncrude, the CO2 emissions per MJ fuel produced is 4 and 8 gCO2 MJ−1 fuel for configuration 1 and 2, respectively, while the previous designs had 38 gCO2 MJ−1 fuel.44,45 A relatively large amount of excess heat, mainly below 200 °C, are available in both configurations which can be utilized to produce power or upgraded via heat pumps. Configuration 2 has poorer H2 efficiency than configuration 1 and most of hydrogen in the process leave as water. However, fuel production would be slightly cheaper in configuration 2. Given an electricity price of $100/MW h, FT products and H2 can be produced at a cost between $15/MW h and $16/MW h. Provided that cheap renewable power is available, it is a better choice to add power to the GTL process through SOEC rather than using it in an E-SMR.
These processes are a step forward towards decarbonizing the aviation industry through integration of renewable power in the fuel production step.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Notes and references
-
UNFCCC, Paris Agreement, Decision 1/CP.21, 2015.
-
U. Nations, The sustainable development goals, 2019, https://www.un.org/sustainabledevelopment/sustainable-development-goals/ Search PubMed.
-
D. Ray, Lazard ’s Levelized Cost of Energy Analysis, version 13.0, 2019, https://www.lazard.com/media/451086/lazards-levelized-cost-of-energy-version-130-vf.pdf Search PubMed.
-
R. W. Stratton, H. M. Wong and J. I. Hileman, White Paper, 2010 Search PubMed.
-
IEA, Are aviation biofuels ready for take off?, 2019, https://www.iea.org/commentaries/are-aviation-biofuels-ready-for-take-off Search PubMed.
- D. S. Mallapragada, E. Gençer, P. Insinger, D. W. Keith and F. M. O’Sullivan, Cell Rep. Phys. Sci., 2020, 1(9), 100174 CrossRef.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- P. Schmidt, V. Batteiger, A. Roth, W. Weindorf and T. Raksha, Chem. Ing. Tech., 2018, 90, 127–140 CrossRef CAS.
- M. Ostadi, K. G. Paso, S. Rodriguez-Fabia, L. E. Øi, F. Manenti and M. Hillestad, Energies, 2020, 13, 4859 CrossRef CAS.
- H. Zhang, L. Wang, M. Pérez-Fortes, J. Van herle, F. Maréchal and U. Desideri, Appl. Energy, 2020, 258, 114071 CrossRef CAS.
- M. Martín and I. E. Grossmann, Appl. Energy, 2018, 213, 595–610 CrossRef.
- M. Hillestad, M. Ostadi, G. Alamo Serrano, E. Rytter, B. Austbø, J. Pharoah and O. Burheim, Fuel, 2018, 234, 1431–1451 CrossRef CAS.
- L. J. Comidy, M. D. Staples and S. R. Barrett, Appl. Energy, 2019, 256, 113810 CrossRef CAS.
- G. Cinti, A. Baldinelli, A. Di Michele and U. Desideri, Appl. Energy, 2016, 162, 308–320 CrossRef CAS.
- G. Herz, E. Reichelt and M. Jahn, Appl. Energy, 2018, 215, 309–320 CrossRef CAS.
- C. Graves, S. D. Ebbesen and M. Mogensen, Solid State Ionics, 2011, 192, 398–403 CrossRef CAS.
-
F. Abe, Reformer and method for operation thereof, US Pat., US6641795B2, 2003 Search PubMed.
-
J. Ringler, C. Liebl, M. Preis, J. Huber, J. Kirwan and M. Grieve, Reformer system having electrical heating devices, US Pat., US20070084116A1, 2007 Search PubMed.
-
J. Alagy, P. Broutin, C. Busson and J. Weill, Process for the thermal pyrolysis of hydrocarbons using an electric furnace, US Pat., US5321191A, 1994 Search PubMed.
- Y. R. Lu and P. Nikrityuk, Appl. Energy, 2018, 228, 593–607 CrossRef CAS.
- M. Rieks, R. Bellinghausen, N. Kockmann and L. Mleczko, Int. J. Hydrogen Energy, 2015, 40, 15940–15951 CrossRef CAS.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, F. B. Bendixen, W. L. Eriksen, K. Aasberg-Petersen, C. Frandsen, I. Chorkendorff and P. M. Mortensen, Science, 2019, 364, 756–759 CrossRef CAS PubMed.
- S. T. Wismann, J. S. Engbæk, S. B. Vendelbo, W. L. Eriksen, C. Frandsen, P. M. Mortensen and I. Chorkendorff, Ind. Eng. Chem. Res., 2019, 58, 23380–23388 CrossRef CAS.
-
Q. Zhang, A. B. Iqbal, M. Sakurai, T. Kitajima, H. Takahashi, M. Nakaya, T. Ootani and H. Kameyama, 16th World Hydrogen Energy Conference, Lyon, France, June 13–16, 2006 Search PubMed.
- L. Zhou, Y. Guo, M. Yagi, M. Sakurai and H. Kameyama, Int. J. Hydrogen Energy, 2009, 34, 844–858 CrossRef CAS.
- R. Labrecque and J. M. Lavoie, Bioresour. Technol., 2011, 102, 11244–11248 CrossRef CAS PubMed.
-
L. Mleczko, A. Karpenko, E. Kockrick, A. Tulke, D. Gordon Duff, A. Große-Böwing and W. Vanessa Gepert, Axial flow reactor having heating planes and intermediate planes, WO patent WO2013135660A1, 2013.
-
M. Banville, R. Lee, R. Labrecque and J. M. Lavoie, in Interaction of CO2/CH4 with Steel Wool in an Electrocatalytic Dry Reforming Reactor, WIT Press, Southampton, U.K., 2013 Search PubMed.
-
M. Banville, R. Lee, R. Labrecque and J. M. Lavoie, Dry Reforming of Methane under an Electro-Catalytic Bed: Effect of Electrical Current and Catalyst Composition, WIT Press, Southampton, U.K., 2015 Search PubMed.
- H. Ji, J. Lee, E. Choi and I. Seo, Int. J. Hydrogen Energy, 2018, 43, 3655–3663 CrossRef CAS.
- S. van Bavel, S. Verma, E. Negro and M. Bracht, ACS Energy Lett., 2020, 5, 2597–2601 CrossRef CAS.
- M. R. Almind, S. B. Vendelbo, M. F. Hansen, M. G. Vinum, C. Frandsen, P. M. Mortensen and J. S. Engbæk, Catal. Today, 2020, 342, 13–20 CrossRef CAS.
- S. T. Wismann, K. E. Larsen and P. M. Mortensen, Angew. Chem., Int. Ed., 2022, 61, e202109696 Search PubMed.
- Y. R. Lu and P. A. Nikrityuk, Fuel, 2022, 319 CAS.
- M. Hillestad, Chem. Prod. Process Model., 2015, 10, 147–159 CAS.
- M. Ostadi, E. Rytter and M. Hillestad, Chem. Eng. Res. Des., 2016, 114, 236–246 CrossRef CAS.
- A. Outi, I. Rautavuoma and H. S. van der Baan, Appl. Catal., 1981, 1, 247–272 CrossRef CAS.
- B. Todic, W. Ma, G. Jacobs, B. H. Davis and D. B. Bukur, J. Catal., 2014, 311, 325–338 CrossRef CAS.
- M. Ostadi, E. Rytter and M. Hillestad, Biomass Bioenergy, 2019, 127, 105282 CrossRef CAS.
-
D. Schanke, P. Lian, S. Eri, E. Rytter, B. H. Sannæs and K. J. Kinnari, Natural Gas Conversion VI, Elsevier, 2001, vol. 136, pp. 239 – 244 Search PubMed.
- M. Ostadi and M. Hillestad, Chem. Eng. Technol., 2017, 40, 1946–1951 CrossRef CAS.
- K. R. Putta, U. Pandey, L. Gavrilovic, K. R. Rout, E. Rytter, E. A. Blekkan and M. Hillestad, Front. Energy Res., 2022, 9, 758149 CrossRef.
- M. Ostadi, K. Dalane, E. Rytter and M. Hillestad, Fuel Process. Technol., 2015, 139, 186–195 CrossRef CAS.
- M. Ostadi and M. Hillestad, Chem. Eng. Trans., 2016, 52, 523–528 Search PubMed.
-
M. S. Peters, K. D. Timmerhaus and R. E. West, Plant Design and Economics for Chemical Engineers, McGraw-Hill Professional, New York, 5th edn, 2002 Search PubMed.
- C. N. Hamelinck, A. P. Faaij, H. den Uil and H. Boerrigter, Energy, 2004, 29, 1743–1771 CrossRef CAS.
-
https://www.eia.gov/dnav/ng/hist/n3035us3m.htm
.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a and
Magne
Hillestad
b
*a and
Magne
Hillestad
b