
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe effects of oxygen pressure on the discharge performance of potassium–oxygen batteries†
Jannis
Küpper
 * and
Ulrich
Simon
* and
Ulrich
Simon
Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg 1a, 52074 Aachen, Germany. E-mail: jannis.nicolas.kuepper@rwth-aachen.de; Tel: +49-24180-90052
First published on 21st March 2022
Abstract
The potassium–oxygen battery (KOB) has distinguished itself from other metal–oxygen batteries by its facile cell chemistry that enables thousands of cycles with negligible performance loss. Recently, the main discharge product KO2 was reported to undergo a reduction towards K2O2 in the absence of oxygen, which was identified as the cause of diminished rechargeability. This raises the question how oxygen partial pressure p(O2) affects the discharge behavior of KOB. In this study, the influence of p(O2) on the discharge performance is assessed between 0.2 atm and 11 atm. The p(O2)-dependent cell failure mechanisms are investigated by post mortem analysis of the discharge product formed on a carbon cathode. Low p(O2) results in insufficient oxygen transport throughout the cathode leading to low rate capability and capacity. In addition, we find that oxygen depletion promotes parasitic reactions involving the electrolyte solvent dimethyl sulfoxide. High p(O2) is identified as an effective approach towards preventing oxygen depletion and subsequent side reactions. Furthermore, high p(O2) also drastically improves the discharge performance by enabling more efficient cathode utilization. These findings suggest that a high-pressure supply of pure oxygen will be advantageous to further increase cell performance of KOB and thus open avenues for yet unexplored applications.
1 Introduction
The development of cost- and resource-efficient energy storage systems is a requirement for the global energy transition from fossil fuels towards sustainable alternatives. Metal–oxygen batteries, and especially the lithium-oxygen battery (LOB), have received much attention over the last decade due to the promise of up to ten times higher specific energy than lithium-ion batteries. Despite intense research efforts, current LOBs suffer from limited cycle life and low practical capacities. It has become increasingly clear that these challenges are difficult to overcome because they originate in the inherent cell chemistry of LOB. Numerous parasitic side reactions including electrolyte decomposition, cathode degradation and singlet oxygen formation have been linked to the main discharge product lithium peroxide (Li2O2) and its formation mechanism.1Potassium–oxygen batteries (KOB) have been shown in recent years to be a promising alternative to LOB.2–5 Up to 3000 limited-capacity cycles with negligible loss in performance and operation in ambient air have been demonstrated.6,7 Furthermore, practically relevant capacities can be achieved with a commercial carbon paper cathode without need for precious metal catalysts.8 This remarkable performance is enabled by the facile cell chemistry of KOB, where potassium superoxide (KO2) is the sole discharge product.9 The formation of KO2 (eqn (1)) is energetically and kinetically favored and a subsequent disproportionation towards potassium peroxide K2O2 (eqn (2)) is not evident.10 To the best of our knowledge, parasitic reactions between KO2 and common electrolytes and cathode materials have not yet been reported and can explain the outstanding cycle life of KOB.
| K(solv.)+ + O2(solv.) + e− → KO2(s) | (1) |
| 2KO2(s) → K2O2(s) + O2(g) | (2) |
However, while K2O2 formation does not occur in typical discharge conditions, KO2 has been further reduced under polarization in model studies.11,12 Recently, K2O2 formation as a secondary reduction step of previously formed KO2 was reported when O2 is completely removed from the cell during discharge (eqn (3)).13
| K(solv.)+ + KO2(s) + e− → K2O2(s) | (3) |
A further reduction of the discharge product is beneficial from an energy perspective because the specific capacity of K2O2 (486 mA h g−1) is considerably higher than for KO2 (377 mA h g−1). However, Wang et al. found that K2O2 formation and subsequent oxidation entails parasitic side reactions resulting in lower current efficiency (CE) and lower round-trip efficiency.13 These side reactions are yet to be elucidated. Nonetheless, it follows that K2O2 formation must be prevented to achieve long cycle life.9 As K2O2 formation is promoted in the absence of O2, the question arises how the oxygen partial pressure p(O2) affects the discharge behavior of KOB. Thus far, the effects of the p(O2) on the discharge performance are largely unexplored.
In KOB research, cells are typically connected to an inexhaustible O2 supply. Cell failure due to complete O2 depletion cannot occur as O2 consumed by the discharge reaction is constantly replenished. Nonetheless, the attainable discharge capacity (Q) is typically well below the capacity of the anode, particularly for high discharge rates. Thus, cathode processes are considered to be the limiting factor. The main challenge of KOB cathode chemistry is the formation of sparingly soluble KO2 and its deposition within the porous cathode structure. In our recent study, we found that KO2 growth induces several rate-dependent failure mechanisms.8 At low discharge rates, large amounts of deposited KO2 cause pore clogging and cathode surface passivation. At high rates, the available O2 is rapidly consumed and the rate of O2 transport cannot support the applied discharge rate. Large sections of the cathode are rendered electrochemically inactive, which leads to high discharge overpotential and a diminished Q. Based on these findings we demonstrated that modifying the cathode wettability enhances transport of gaseous O2 throughout the entire cathode.8 Homogeneous KO2 growth even at high rates enables a Q improvement up to 300%. Nonetheless, the achieved performance is still below the requirements of practical applications.9
Tuning the operation parameter p(O2) is a promising approach to further improve the rate capability.14–18 Aside from accelerating O2 transport through the gas phase of the cathode compartment, higher p(O2) also increases the O2 solubility in the electrolyte.14 For low p(O2), the concentration of dissolved O2 in the electrolyte c(O2) is directly proportional to p(O2) as described by Henry's law (eqn (4)), where H(O2) is the solubility constant with the dimension L atm mol−1.
| p(O2) = H(O2)c(O2) | (4) |
Higher c(O2) were found to reduce discharge overpotentials of LOB by lowering kinetic and transport barriers of the discharge reaction.19 In addition, higher p(O2) and using electrolytes with high O2 solubility were reported to be beneficial for the discharge performance.14,20 However, Henry's law only describes the O2 solubility well for low c(O2) and low p(O2). At p(O2) above ∼1 atm, the O2 solubility substantially deviates from Henry's law.21 A four-fold increase of p(O2) from 1 atm to 4 atm results in a doubling of the O2 solubility.19 Only negligible discharge performance improvement was observed beyond 11 atm for LOB.14,16 These studies reflect a non-trivial relation between p(O2) and LOB performance and it would therefore be interesting to explore how p(O2) will affect the key performance indicators of a KOB, such as discharge capacity and rate capability. Corresponding fundamental investigations have not yet been performed for KOB, which are usually operated at p(O2) of 1 atm to 2 atm. It is critical to quantify the effects of p(O2) on the discharge performance to assess whether an open, “breathing” cell with ambient air as O2 supply is solely feasible, or whether a closed and pressurized cell with high p(O2) will help to significantly enhance the performance of KOB and to, eventually, open new avenues for applications.
In this article, we report for the first time a systematic study of the discharge behavior of KOB in a wide range of p(O2) from 0.2 atm to 11 atm. Battery testing shows that higher p(O2) decreases discharge overpotentials and improves the attainable Q at any tested rate. Cross-sectional scanning electron microscopy reveals that cell failure is induced by multiple p(O2)-dependent mechanisms. Severe O2 transport limitations at low p(O2) cause high overpotentials and KO2 growth is limited to cathode regions close to the O2 supply. In addition, for low and intermediate p(O2) a multistage discharge profile is observed, which we find to be associated with O2 depletion within the cathode. Raman spectroscopy indicates that in O2-depleted conditions parasitic reactions involving the electrolyte solvent DMSO result in the formation of K2SO4 and K2CO3. By contrast, high p(O2) enables uniform KO2 growth throughout the entire cathode. For p(O2) = 11 atm, the discharge performance does not seem to be limited by O2 transport at the tested discharge rates. Furthermore, sufficiently high p(O2) prevents O2 depletion and in turn the formation of unwanted side products at all stages of discharge.
2 Results and discussion
2.1 Effects of p(O2) on discharge performance
To investigate the effects of p(O2) on the discharge performance, battery testing was performed with p(O2) between 0.2 atm and 11 atm. Three test series were performed with a nominal current density (J) of 0.1 mA cm−2, 0.5 mA cm−2 and 1.0 mA cm−2, respectively. In Fig. 1, the combined plot of Q against p(O2) is displayed. Three cells were tested for each combination of J and p(O2) and the error bars indicate the resulting spread of Q.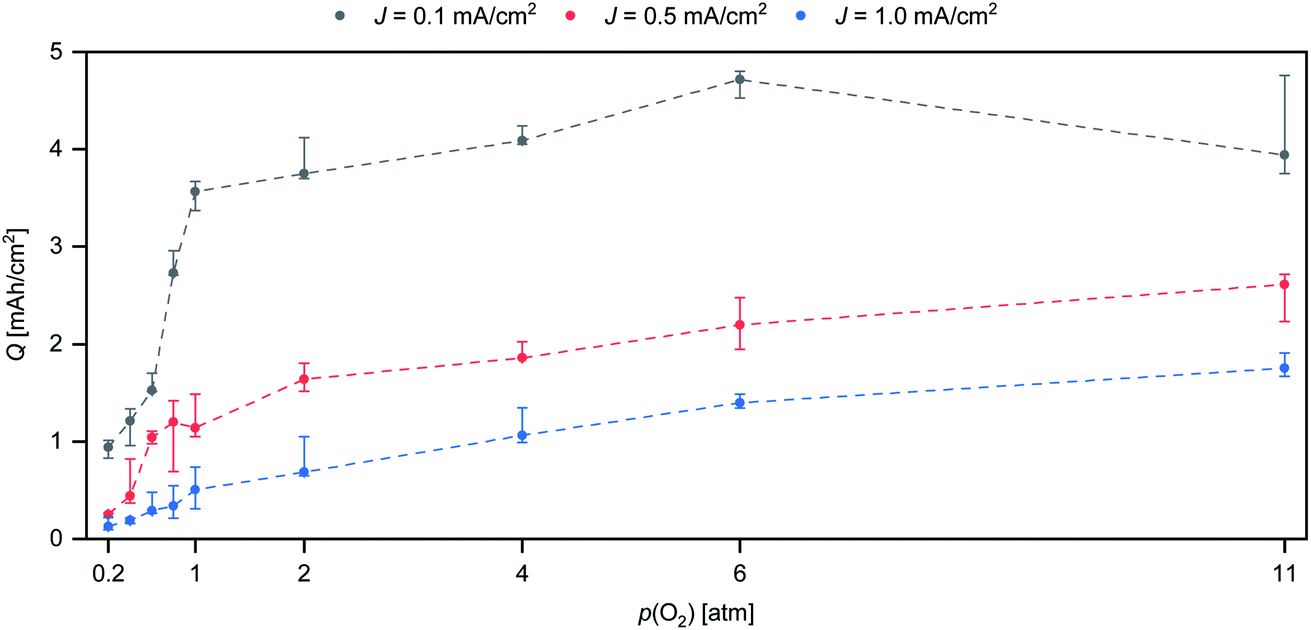 | ||
| Fig. 1 Plot of Q vs. p(O2) at three different discharge rates. Black: J = 0.1 mA cm−2. Red: J = 0.5 mA cm−2. Blue: J = 1.0 mA cm−2. The dashed lines are added as guides for the eye. | ||
The plot in Fig. 1 shows a strong effect of p(O2) on Q for all tested rates. In general, higher p(O2) increases the attainable Q. The test series with the lowest rate (J = 0.1 mA cm−2) shows a near five-fold increase in Q from 0.2 atm to 11 atm. The extent of Q improvement is largest in the range of 0.2 atm to 1 atm with a three-fold Q increase from 1.01 mA h cm−2 to 2.96 mA h cm−2. Pressurizing up to 6 atm provides an additional 62% increase in Q up to 4.80 mA h cm−2. For 11 atm, the highest achieved Q is 4.76 mA h cm−2. The difference between the highest Q for 6 atm and 11 atm is less than 1% of the total Q. The variation between cells tested under the same conditions is on the order of 20%, which is commonly observed for self-made metal–oxygen batteries.16 The 1% difference in maximum Q between p(O2) = 6 atm and p(O2) = 11 atm may be considered to be negligible. Therefore, the maximum attainable discharge capacity is seemingly not affected by raising p(O2) from 6 atm and 11 atm.
At intermediate (J = 0.5 mA cm−2) and high rates (J = 1.0 mA cm−2) the difference between low and high p(O2) is yet more pronounced compared to the slowest rate. At J = 1.0 mA cm−2, Q is raised from 0.13 mA h cm−2 (0.2 atm) up to 1.7 mA h cm−2 (11 atm), which represents a 13-fold increase. While p(O2) beyond 6 atm does not seem to be beneficial for slow rates, p(O2) beyond 11 atm could further improve Q for higher rates, since there are still significant Q gains from 6 atm to 11 atm.
To illustrate the effects of p(O2) on the rate capability, the discharge profiles of cells tested at the same J are displayed in Fig. 2.
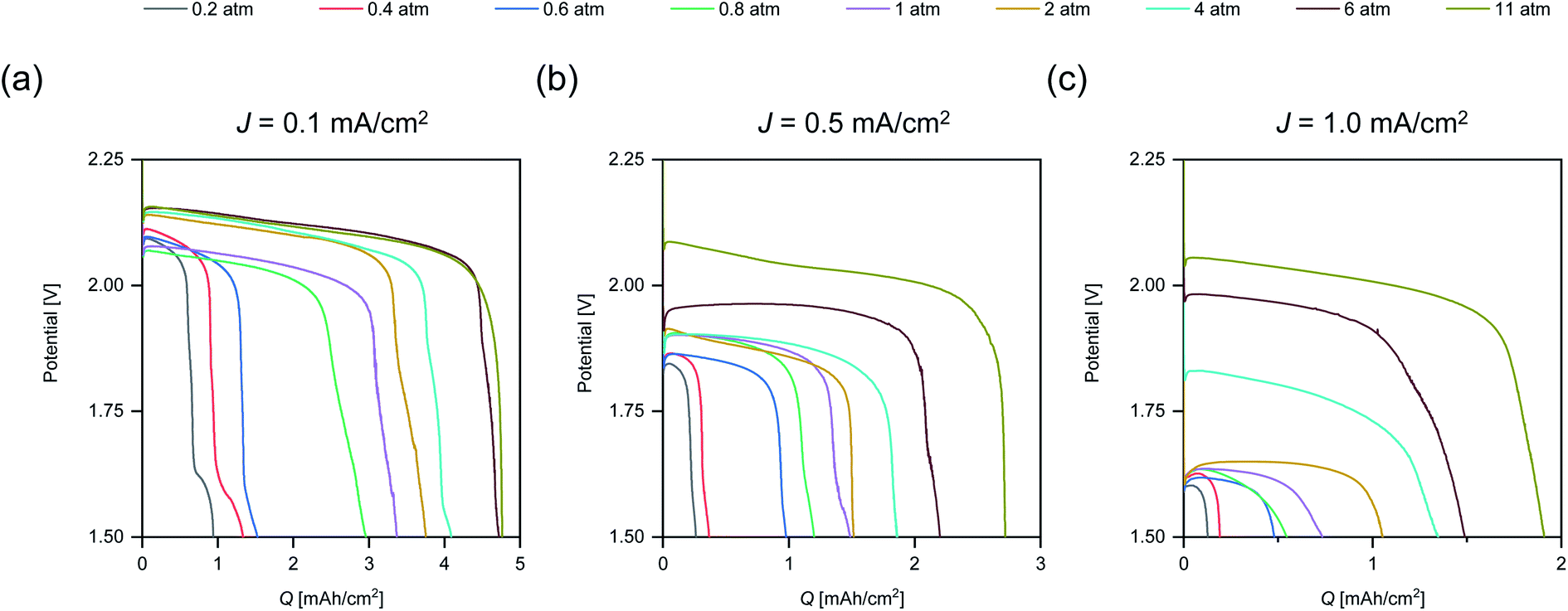 | ||
| Fig. 2 Discharge profiles for cells tested for various p(O2) at three rates: (a) J = 0.1 mA cm−2, (b) J = 0.5 mA cm−2 and (c) J = 1.0 mA cm−2. The x-axis is scaled differently for each plot to improve visibility. | ||
The plots in Fig. 2 demonstrate that increasing p(O2) reduces the discharge overpotential for all tested rates. However, there are some exceptions to this trend, which we ascribe to differences in anode and cathode microstructure leading to slight variations in internal resistances between the cells assembled by hand. Some of the discharge profiles display multiple inflection points towards the end of discharge. This multistage behaviour is seen up to 6 atm and is more pronounced for lower rates. The underlying mechanisms causing multistage behavior are discussed in Section 2.3.
The supportive effect of p(O2) on the discharge overpotential is less pronounced for low rates when compared to high rates. The plateau voltage at J = 0.1 mA cm−2 (Fig. 2a) is increased by ∼30 mV from 2 atm to 11 atm and by ∼400 mV for J = 1.0 mA cm−2 (Fig. 2c). The marginal improvement for the low rate and the strong improvement for high rates matches the observations made for Q vs. p(O2) (Fig. 1). In addition, it is in accordance with recent findings that increasing O2 transport via cathode modification is especially beneficial for high rates.8
Comparing profiles at the same p(O2) and different J shows that the plateau voltage for any given p(O2) is considerably lower for the higher J. The voltage gap between the rates decreases with higher p(O2) and for the highest p(O2) = 11 atm it is <100 mV. Therefore, it might be expected that similar Q is obtained. However, Q is considerably lower at the high rate (1.9 mA h cm−2) compared to the low rate (4.7 mA h cm−2). Furthermore, the obtained Q for 11 atm at J = 1.0 mA cm−2 (1.9 mA h cm−2) is lower than for J = 0.5 mA cm−2 at 6 atm (2.3 mA h cm−2) despite a ∼150 mV higher plateau voltage. Thus, discharge overpotential alone is not indicative of the discharge performance.
To further investigate the relationship between p(O2) and the discharge overpotential, a discharge was performed, where p(O2) was manually increased from 1 atm up to 11 atm in time intervals of 5 min to allow the cell to reach a steady state. Subsequently, the gas outlet of the cell was opened to allow for rapid gas flow throughout the cathode compartment. The plot in Fig. 3 shows the obtained discharge profile, in which the respective p(O2) regions have been highlighted.
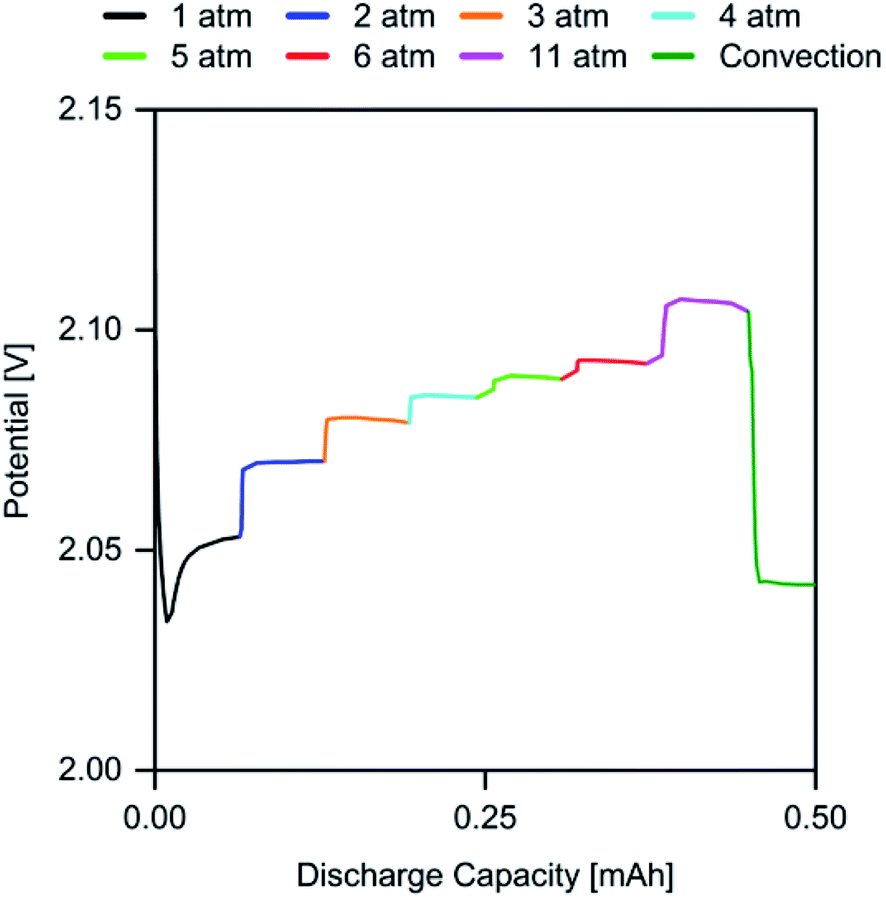 | ||
| Fig. 3 Discharge profile of a cell with J = 0.5 mA cm−2 with dynamic p(O2) in the range of 1 atm to 11 atm. | ||
The stepwise increase of p(O2) leads to an immediate and persistent decrease of the discharge overpotential. The most significant effect is noted for the step from 1 atm to 2 atm, where the cell potential is raised by 17 mV, whereas the 5 atm step from 6 atm to 11 atm increased the cell potential only by 14 mV. This trend matches the prior observations for Q (Fig. 1), where the largest increase was observed for p(O2) between 0.2 atm to 2 atm. However, the significant overpotential reduction from 6 atm to 11 atm indicates that the highest possible p(O2) should be applied to maximize the energy efficiency.
To investigate the extent of the contribution from convection associated with a p(O2) increase towards the cell potential, the cell was opened, leading to rapid gas flow and strong convection within the cathode compartment. While this should promote O2 transport, the cell potential sharply drops after opening the cell. This indicates that the p(O2)-induced increase in O2 concentration within the electrolyte may be responsible for the performance improvement. Based on this finding, an “open” cell design, in which the cell is constantly flushed with (dry) ambient air, may not be suitable to achieve high KOB performance.
This section demonstrates that higher p(O2) is beneficial for the discharge performance by increasing Q and reducing the discharge overpotential, especially for high discharge rates. It follows that the highest possible p(O2) within the safety limits should be applied to achieve the best discharge performance for any KOB system. Nonetheless, despite sufficiently high p(O2), Q was found to be limited for low discharge rates. Since the maximum achievable Q is governed by KO2 growth within the porous cathode,8 the variation of J and p(O2) may affect the deposition of KO2, which could lead to different cell failure mechanisms. Therefore, an investigation of the cathode microstructure is needed to identify strategies to further improve the discharge performance.
2.2 Dependence of cell failure mechanisms on J and p(O2)
To investigate the combined influence of J and p(O2) on the discharge product growth and cell failure mechanisms, scanning electron microscopy (SEM) was performed on the used carbon paper cathodes after discharge. SEM images of the O2 supply-side and cross sections of the cathodes were obtained.In Fig. 4, SEM images of a pristine cathode (a and b) and cathodes after discharge at J = 0.1 mA cm−2 with 0.2 atm (c and d) and 6 atm (e and f) are shown. Spatial histograms of the discharge product distribution were obtained from K element maps via energy-dispersive X-ray spectroscopy and overlaid with the cross sections (Fig. 4e and f). We note that signal intensity cannot be correlated with the absolute amounts of discharge product and only provides information about its spatial distribution.
 | ||
| Fig. 4 SEM images of pristine carbon paper cathode (a and b), after discharge at J = 0.1 mA cm−2 with 0.2 atm (c and d) and 6 atm (e and f). Cross sections after discharge at 11 atm for (g) J = 0.1 mA cm−2, (h) J = 0.5 mA cm−2 and (i) J = 1.0 mA cm−2. Spatial histograms of the discharge product distribution (presented as integrated intensity of K element maps via energy-dispersive X-ray spectroscopy) across the cathode cross sections are overlaid in orange color. | ||
The SEM images for 0.2 atm (Fig. 4c and d) reveal that discharge product deposits are mainly concentrated in the cathode regions close to the O2 supply. This indicates severe O2 transport limitations towards the inner cathode regions. At the start of discharge, O2 in the cathode pores is consumed and not replenished sufficiently. Primarily the accessible regions close to the O2 supply are electrochemically active and the pores between the carbon fibers become filled by discharge product (Fig. 4c). This leads to pore clogging and further reduces the rate of O2 transport. In addition, the deposition of KO2 on the carbon fibers reduces the active cathode surface area due to its electronically insulating properties.22,23 Cell failure occurs when the regions near the O2 supply become electrochemically inactive.
After discharge at 6 atm large deposits of discharge product are visible on the carbon fiber surfaces (Fig. 4e) and within the cathode pores (Fig. 4f). The spatial histogram shows that the discharge product is more evenly distributed across the cathode, which can explain the improved performance at high p(O2): large sections of the cathode remain electrochemically active over the course of discharge. Thus, the active cathode surface area is drastically increased compared to low p(O2) resulting in a lowered discharge overpotential. In addition, high degrees of pore volume filling are visible and can explain the improved Q at high p(O2). These findings indicate that, for high p(O2) and low rates, Q may only be limited by the available pore volume and not by O2 transport.
To illustrate the influence of p(O2) over the entire tested p(O2) range, SEM images for 0.2 atm, 1 atm and 11 atm are displayed in Fig. S1 and S2 in the ESI.† The discharge product distribution at intermediate and high J follows the observed trends for J = 0.1 mA cm−2. Generally, at a given p(O2) the degrees of pore filling are lower for the higher rates.
At J = 0.1 mA cm−2, a similar degree of pore volume filling is observed for 11 atm (Fig. 4g) as for 6 atm (Fig. 4f). No performance improvement was observed for 11 atm compared to 6 atm (see Section 2.1), which supports the notion that cell failure for slow rates is not induced by insufficient O2 transport at 6 atm and further increasing p(O2) to 11 atm is not beneficial.
Comparing the discharge product distribution for the tested three rates at 11 atm (Fig. 4g–i), it is visible that for higher rates the pore volume filling is lower than for slow rates. As noted in Section 2.1, the difference in discharge overpotential between the tested rates is rather small at 11 atm, which indicates that O2 transport is not rate limiting in the early stages of discharge at any rate. However, the proceeding deposition of KO2 within the cathode reduces the active surface area through passivation and is expected to inhibit O2 transport at high depths of discharge. To investigate whether surface passivation or O2 transport limitations lead to cell failure, a second discharge was performed after a 30 min resting step at open-circuit potential (Fig. S3†). This duration is sufficient to equalize c(O2) in the electrolyte and to substantially improve Q when intermittent current discharge is performed.24 However, Q of the discharge after pause was only ∼20 μA h cm−2 for both tested rates, suggesting that O2 transport is not limiting. Therefore, surface passivation is the likely cause of cell failure at high p(O2).
The obtained Q is substantially lower at high rates compared to slow rates, yet surface passivation is the cause of cell failure for both. Thus, a high rate appears to promote KO2 deposition as a surface layer, whereas slow rates enable extensive growths and pore volume filling. Since surface passivation results in cell failure despite sufficient O2 transport at high rates, future cathode design should aim to maximize the cathode surface area to improve the rate capability.
These observations demonstrate the strong influence of p(O2) and J on discharge product formation and deposition. Low p(O2) and high J promote heterogeneous KO2 growth, whereas high p(O2) and low J enable uniform KO2 deposition throughout the entire cathode pore structure resulting in an improved discharge performance.
2.3 Oxygen-depleted discharge
To investigate the underlying mechanisms causing the multistage behavior observed in the discharge profiles in Fig. 2, a pressure monitoring test was performed, which allows to probe the discharge mechanism of metal–oxygen batteries in situ.6,25,26 In Fig. 5a, the discharge profile of a cell with closed O2 supply and the corresponding evolution of p(O2) are displayed. | ||
| Fig. 5 (a) Pressure monitoring test. Black line: discharge profile in a closed cell setup with initial p(O2) of 2 atm at J = 0.5 mA cm−2. Red line: evolution of p(O2) along discharge. The regimes of the first stage and the multistage behavior are highlighted in green and violet. (b) Discharge profiles of cells discharged at J = 1.0 mA cm−2 with the cut-off voltage set to 1.00 V. Blue line: p(O2) = 2 atm. Gray line: p(O2) = 11 atm. The checked line indicates the default cut-off voltage of 1.50 V. | ||
A linear decrease of p(O2) is observed during the first stage of discharge. The ratio of consumed O2 to transferred charge (O2/e−) can be determined from the rate of O2 consumption (see Addendum A1). A O2/e− of 1.0 is obtained, which agrees with sole KO2 formation in the first stage (see eqn (1)).6 In the transition from first stage to second stage O2/e− rapidly approaches zero, signifying a change in discharge reaction mechanism. During second and third stage, discharge proceeds seemingly without O2 consumption. It is evident from the higher discharge voltage of the first stage that O2 reduction and KO2 formation is energetically favorable compared to the reactions occurring during second and third stage. Therefore, the onset of the second stage may be correlated with the depletion of O2 within the cathode.
The discharge profiles in Fig. 2 show that the multistage behavior is observed primarily at low p(O2) and for low J. It is reasonable that multistage behavior is more prevalent for low p(O2) since c(O2) in the electrolyte is proportional to p(O2) and thus the available O2 is depleted faster at low p(O2) than at high p(O2).
For slow rates, high degrees of pore filling were observed at high depths of discharge (see Section 2.2). The pore clogging due to the KO2 deposits leads to O2 depletion despite the low discharge rate, which in turn promotes multistage behavior. However, it is rather counterintuitive that the multistage behavior is not observed for the highest rate: O2 transport was found to be rate-limiting at p(O2) up to 6 atm and therefore severe O2 depletion is expected. An explanation was deduced from the discharge profiles in Fig. 2, in which the onset potential of the second discharge plateau lies in the range of 1.55 V to 1.65 V for the slowest rate. This is only slightly above the set cut-off voltage (1.50 V), which is commonly employed in KOB research. Due to significant discharge overpotential at high rates, the onset potential might be shifted below the cut-off voltage. To verify this assumption, a discharge test was performed at p(O2) = 2 atm with the cut-off voltage set to 1.00 V (Fig. 4b). Indeed, additional inflection points in the cell potential are observed below 1.50 V and show that multistage behavior also occurs at high rates. Yet, it was not apparent in the battery testing due to the set cut-off voltage. When repeating the discharge test with lower cut-off voltage at 11 atm (Fig. 5b), no further discharge plateau is seen despite considerably higher Q. Taking the findings of Section 2.2 into account, sufficiently high p(O2) may prevent O2 depletion and multistage behavior by ensuring O2 availability in the cathode during all stages of discharge.
From the pressure monitoring test (Fig. 5a) it is apparent that faradaic reactions aside from O2 reduction reaction occur in O2-depleted conditions. X-ray powder diffraction (XRD) and Raman spectroscopy were used on the carbon paper cathodes to identify differences in the discharge products after single stage and multistage discharge (Fig. 6).
 | ||
| Fig. 6 Post mortem analysis of cathodes after discharge at J = 0.1 mA cm−2. (a) Powder diffraction patterns. The KO2 reference was obtained from ICSD database (coll. code 38245). (b) Raman spectra of cathodes after discharge. The checked line at 1143 cm−1 indicates the position of the O–O vibration of KO2.27 | ||
The powder diffraction patterns (Fig. 6a) indicate KO2 to be the only discharge product for both single and multistage discharge. Reflexes corresponding to residual electrolyte salt KPF6 are visible, which could not be fully removed by rinsing the samples due to the high degrees of pore filling. The Raman spectrum (Fig. 6b) for the single stage discharge confirms KO2 as the sole discharge product. However, additional signals appeared in a few measuring spots of the cathode after multistage discharge, which correspond to the ν1 symmetric stretching vibrations of potassium carbonate (K2CO3) and potassium sulfate (K2SO4).28,29 Since these side products were not detected via XRD, they might only be present in small amounts compared to KO2.
K2SO4 formation points towards a reaction involving the electrolyte solvent dimethyl sulfoxide (DMSO) because it is the only source of sulfur in the cell. DMSO is susceptible towards oxidation by reactive oxygen species,30–32 yet is stable against the superoxide anion in KOB.12,33 A possible origin of reactive oxygen species might be the further reduction of KO2 towards K2O2.11–13 Recently, the formation of K2O2 has been discovered by in situ ambient-pressure X-ray photoelectron spectroscopy (AP-XPS).13 This study took advantage of the high boiling point of an ionic liquid used as a model electrolyte. However, AP-XPS cannot be performed with DMSO as electrolyte solvent due to its low boiling point, so that we have to find a new approach to elucidate the origins of the side products in DMSO-based electrolytes in our future work.
K2CO3 may be formed in reactions involving carbon-containing components of the cell, i.e., DMSO or the carbon cathode.34 We considered moisture and CO2 contaminations of the electrolyte and O2 supply as potential sources of the side products. The moisture content in the electrolyte was determined to be only 37 ppm, which does not have significant influence on the cell chemistry.7 The introduction of moist ambient air into the cell during battery testing was prevented by always maintaining cell pressure beyond atmospheric pressure. In addition, the presence of considerable amounts of moisture would be expected to cause the formation of KOH, which was neither detected via XRD nor via Raman spectroscopy. Traces of CO2 in the O2 supply does not lead to K2CO3 formation4 and cannot explain K2SO4 formation. Furthermore, the side products are exclusively observed after multistage discharge. Thus, contaminations can be ruled out as the origin of the side products, implying that parasitic reactions occur that are inherent to the cell chemistry. These could also explain the diminished rechargeability that has recently been associated with K2O2 formation:13 K2SO4 likely cannot be oxidized at battery potentials and the formation of K2CO3 may be irreversible, since CO2 is expected to form upon oxidation.35 In addition, substantial charge overpotentials are typically required to oxidize alkali metal carbonates, which could induce further electrolyte decomposition.34,35 It is therefore critical to prevent the formation of these side products. We find that the onset of parasitic side reactions, which is reflected by multistage discharge behavior, does not depend on just a single parameter such as p(O2), J or cathode microstructure. Rather, each of these parameters affects the O2 availability throughout the cathode. Certain conditions, such as low p(O2) and high depths of discharge, facilitate O2 depletion within regions of the cathode, where parasitic reactions towards K2SO4 and K2CO3 become favored. High p(O2) can prevent these side reactions by ensuring O2 availability in all stages of discharge.
An aspect of future research is the study of the influence of p(O2) on the charge behavior of KOB. At present, the impact of a p(O2) variation cannot be distinguished from cathode material specific effects and parasitic side reactions. Therefore, a detailed understanding of these reactions and parameters during discharge is needed before the subsequent processes during charging can be meaningfully investigated. We are currently identifying new approaches to achieve this goal, which includes the study of the temperature dependence and the effects of cathode properties such as surface area and microstructure to discriminate mass transport from material specific effects.
3 Conclusion
By studying the influence of p(O2) on the discharge performance of KOB, we found that increasing p(O2) lowers the discharge overpotential and improves both the attainable capacity and rate capability. These findings suggest that applying high p(O2) will be a requirement for future high-performance KOB. Regarding practical applications, a closed cell filled with pure O2 is likely preferable to an open cell supplied with ambient air, in which five times higher cell pressure is needed to achieve the same p(O2) and discharge performance.Cell failure mechanisms were studied via post mortem analysis. For low p(O2) such as in dry ambient air, severe O2 transport limitations cause O2 depletion within the cathode leading towards cell failure and low discharge performance. By contrast, high p(O2) renders the entire cathode electrochemically active, thereby enabling large degrees of pore volume filling by KO2. For high p(O2) and slow discharge rates, the attainable capacity appears to mainly be limited by the available pore volume. At higher rates, a preference for KO2 deposition as a surface layer is observed. Future cathode design should therefore aim to increase porosity to raise the attainable capacity and increase the cathode surface area to improve the rate capability.
A multistage behavior was observed in discharge profiles at low and intermediate p(O2), which is induced by O2 depletion. A complex interplay of p(O2), the applied discharge rate, and the cathode microstructure can result in low O2 availability within the cathode and facilitate parasitic side reactions. For the first time, we found evidence of non-faradaic reactions occurring in O2-depleted conditions leading to the formation of K2SO4 and K2CO3. Ensuring O2 availability during all stages of discharge is an effective measure to suppress these side reactions and can be achieved by utilizing sufficiently high p(O2). These findings emphasize the critical importance of O2 transport for the discharge performance and cycle life of KOB.
4 Experimental section
4.1 Chemicals and materials
Chemicals were handled in an Ar-filled glovebox (H2O < 0.1 ppm, O2 < 0.1 ppm, MBraun, Germany). Dimethyl sulfoxide (DMSO, 99.9%, anhydrous, Sigma-Aldrich, Germany) and 1,2-dimethoxyethane (DME, 99.5%, anhydrous, Sigma-Aldrich) were stored over 3 Å molecular sieve and H2O contents were determined to be <20 ppm via Karl-Fischer-titration. Biphenyl (Bp, 99%, Sigma-Aldrich) was vacuum-dried at RT and KPF6 (99.5%, Sigma-Aldrich) was vacuum-dried at 110 °C. K metal (99.95%, Sigma-Aldrich) and K-β′′-Al2O3 disks (KBA, diameter Ø = 20 mm, thickness T = 1.0 mm, Ionotec, UK) were used as received. H23 (m = 14.6 mg ± 0.1 mg, Ø = 14 mm, T = 0.211 mm, Freudenberg, Germany), Cu foam (Ø = 16 mm, T = 2.0 mm, 99.5+%, Goodfellow, Germany), Grade GF/B (Ø = 14 mm, Whatman, UK) and Grade GF/A (Ø = 16 mm, Whatman, UK) glass fiber papers were oven-dried at 105 °C overnight and transferred to the glovebox while hot.4.2 Battery cell assembly
The cell assembly procedure was described in detail previously.8 Cells were assembled in the glovebox. The ECC-Std cell housing (EL-CELL, Germany) with a homemade inlay was used. Cu foam was used as anode current collector and GF/B as reservoir for 200 μL 3.0 M KBp in DME, which was the anode active material. KBA served as separator and solid-state electrolyte. GF/A was used as cathode electrolyte reservoir and H23 as cathode. A volume of 60 μL 0.5 M KPF6 in DMSO was added to the cathode, which was previously determined to be the optimum electrolyte volume for a single H23 disk in our setup. Assembled cells were removed from the glovebox.4.3 Electrochemical measurements
Cells were connected to the CTS battery tester (Basytec, Germany) and O2 supply. A mixture of 20.00% O2 in Ar (99.999%, H2O < 3 ppm, Westfalen AG, Germany), corresponding to the O2 content in air, was used for p(O2) between 0.2 atm and 0.8 atm. The maximum p(O2) that could be tested with this O2 supply is 2.2 atm, because the pressure safety limit of the test setup is 11 atm. A pure O2 supply was used (99.999%, H2O < 3 ppm, Westfalen AG, Germany) for p(O2) between 1 atm and 11 atm. Cell tests with p(O2) below 1 atm were not performed with the pure O2 supply, as this might cause unintended solvent losses and gas contamination from the environment. Cells were purged under rapid gas flow for at least 2 min. Battery testing proceeded after a 75 min resting step at open-circuit potential. Currents were normalized to the formal surface area of H23 cathode (A = 1.539 cm2). The cut-off discharge cell voltage was 1.50 V unless noted otherwise. Pressure monitoring was performed by connecting a pressure transducer (CPT2500, WIKA, Germany) to the gas outlet of the cell housing and sealing the O2 supply after purging.4.4 Analysis methods
Cathodes were extracted from cells in the glovebox and thoroughly rinsed with DME. Cross sections of cathodes were produced with a scalpel. Scanning electron microscopy was performed with a LEO Supra35VP device (Carl Zeiss AG, Germany) at an acceleration voltage of 5 kV. Spatial histograms were obtained at 10 kV acceleration voltage from potassium Kα-line elemental mappings via energy-dispersive X-ray spectroscopy. Raman spectroscopy was performed with a LamRAM 300 spectrometer (λ = 633 nm, P = 12 mW, Horiba, Japan) and X-ray powder diffraction with a STOE StadiP powder diffractometer (Stoe & Cie, Germany) with Cu Kα irradiation source (λ = 1.54 Å).Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) – GRK 1856. The authors thank the team of the mechanical workshop of the Institute of Inorganic Chemistry at RWTH Aachen University for the support in design, construction and maintenance of the battery cells and T. Storp for the support in XRD measurements.References
- W.-J. Kwak, Rosy, D. Sharon, C. Xia, H. Kim, L. R. Johnson, P. G. Bruce, L. F. Nazar, Y.-K. Sun, A. A. Frimer, M. Noked, S. A. Freunberger and D. Aurbach, Chem. Rev., 2020, 120, 6626–6683 CrossRef CAS PubMed.
- X. Ren and Y. Wu, J. Am. Chem. Soc., 2013, 135, 2923–2926 CrossRef CAS PubMed.
- W. Yu, K. C. Lau, Y. Lei, R. Liu, L. Qin, W. Yang, B. Li, L. A. Curtiss, D. Zhai and F. Kang, ACS Appl. Mater. Interfaces, 2017, 9, 31871–31878 CrossRef CAS PubMed.
- L. Qin, N. Xiao, S. Zhang, X. Chen and Y. Wu, Angew. Chem., Int. Ed., 2020, 59, 10498–10501 CrossRef CAS PubMed.
- J. Park, J. Y. Hwang and W. J. Kwak, J. Phys. Chem. Lett., 2020, 11, 7849–7856 CrossRef CAS PubMed.
- G. Cong, W. Wang, N.-C. Lai, Z. Liang and Y.-C. Lu, Nat. Mater., 2019, 18, 390–396 CrossRef CAS PubMed.
- W. Wang and Y.-C. Lu, ACS Energy Lett., 2020, 5, 3804–3812 CrossRef CAS.
- J. Küpper, S. Jakobi and U. Simon, Batteries Supercaps, 2021, 4, 1620–1626 CrossRef.
- W. Wang and Y.-C. Lu, Acc. Mater. Res., 2021, 2(7), 515–525 CrossRef CAS.
- P. H. Reinsberg, A. Koellisch and H. Baltruschat, Electrochim. Acta, 2019, 313, 223–234 CrossRef CAS.
- P. H. Reinsberg, A. Koellisch, P. P. Bawol and H. Baltruschat, Phys. Chem. Chem. Phys., 2019, 21, 4286–4294 RSC.
- S. Sankarasubramanian and V. Ramani, J. Phys. Chem. C, 2018, 122, 19319–19327 CrossRef CAS.
- W. Wang, Y. Wang, C.-H. Wang, Y.-W. Yang and Y.-C. Lu, Energy Storage Mater., 2021, 36, 341–346 CrossRef.
- J. Read, K. Mutolo, M. Ervin, W. Behl, J. Wolfenstine, A. Driedger and D. Foster, J. Electrochem. Soc., 2003, 150, A1351 CrossRef CAS.
- X.-h. Yang and Y.-y. Xia, J. Solid State Electrochem., 2010, 14, 109 CrossRef CAS.
- E. J. Nemanick and R. P. Hickey, J. Power Sources, 2014, 252, 248–251 CrossRef CAS.
- H. J. Kwon, H. C. Lee, J. Ko, I. S. Jung, H. C. Lee, H. Lee, M. Kim, D. J. Lee, H. Kim, T. Y. Kim and D. Im, J. Power Sources, 2017, 364, 280–287 CrossRef CAS.
- P. Wunderlich, J. Küpper and U. Simon, Batteries, 2020, 6, 36 CrossRef CAS.
- S. K. Das, S. Xu, A.-H. Emwas, Y. Y. Lu, S. Srivastava and L. A. Archer, Energy Environ. Sci., 2012, 5, 8927 RSC.
- J. Read, J. Electrochem. Soc., 2006, 153, A96 CrossRef CAS.
- F. S. Gittleson, R. E. Jones, D. K. Ward and M. E. Foster, Energy Environ. Sci., 2017, 10, 1167–1179 RSC.
- O. Gerbig, R. Merkle and J. Maier, Adv. Funct. Mater., 2015, 25, 2552–2563 CrossRef CAS.
- Z. Hu, W. Tan, S. Li and F. Pan, Phys. Chem. Chem. Phys., 2020, 22, 24480–24489 RSC.
- F. Wang and X. Li, J. Power Sources, 2018, 394, 50–56 CrossRef CAS.
- B. D. Mccloskey, R. Scheffler, A. Speidel, G. Girishkumar and A. C. Luntz, J. Phys. Chem. C, 2012, 116, 23897–23905 CrossRef CAS.
- P. Hartmann, D. Grübl, H. Sommer, J. Janek, W. G. Bessler and P. Adelhelm, J. Phys. Chem. C, 2014, 118, 1461–1471 CrossRef CAS.
- L. Andrews, J. Chem. Phys., 1971, 54, 4935–4943 CrossRef CAS.
- N. Buzgar and A. I. Apopei, Geologie Tomul L, 2009, 2, 97–112 Search PubMed.
- K. Ben Mabrouk, T. H. Kauffmann, H. Aroui and M. D. Fontana, J. Raman Spectrosc., 2013, 44, 1603–1608 CrossRef CAS.
- D. Sharon, M. Afri, M. Noked, A. Garsuch, A. A. Frimer and D. Aurbach, J. Phys. Chem. Lett., 2013, 4, 3115–3119 CrossRef CAS.
- D. G. Kwabi, T. P. Batcho, C. V. Amanchukwu, N. Ortiz-Vitoriano, P. Hammond, C. V. Thompson and Y. Shao-Horn, J. Phys. Chem. Lett., 2014, 5, 2850–2856 CrossRef CAS PubMed.
- F. Marchini, S. Herrera, W. Torres, A. Y. Tesio, F. J. Williams and E. J. Calvo, Langmuir, 2015, 31, 9236–9245 CrossRef CAS PubMed.
- S. Sankarasubramanian, J. Kahky and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 14899–14904 CrossRef CAS PubMed.
- Y. S. Mekonnen, J. M. Garcia-Lastra, J. S. Hummelshøj, C. Jin and T. Vegge, J. Phys. Chem. C, 2015, 119, 18066–18073 CrossRef CAS.
- B. D. Mccloskey, A. Speidel, R. Scheffler, D. C. Miller, V. Viswanathan, J. S. Hummelshøj, J. K. Nørskov and A. C. Luntz, J. Phys. Chem. Lett., 2012, 3, 997–1001 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2se00182a |
| This journal is © The Royal Society of Chemistry 2022 |