
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA compact non-PGM catalytic hollow fibre converter for on-board hydrogen production
S.
Mazzone
a,
C.
Leishman
a,
G.
Zhang
 b and
F. R.
García-García
*a
b and
F. R.
García-García
*a
aThe University of Edinburgh, School of Engineering, Institute for Materials and Processes, Sanderson Building, The King's Buildings, Mayfield Road, EH9 3JL, Edinburgh, Scotland, UK. E-mail: Francisco.Garcia-Garcia@ed.ac.uk; Tel: +44(0)131 6504860
bState Key Laboratory of Materials-Orientated Engineering, College of Chemical Engineering, Nanjing Tech University, 30 Puzhu Road(S), Nanjing, 211816, PR China
First published on 10th February 2022
Abstract
Hollow fibre-based converters offer an outstanding solution for on-board hydrogen production via ammonia decomposition, representing a more compact, efficient and affordable alternative to traditional packed bed reactors. In this work, Co/Mo-based catalysts supported on three different carbon xerogels were tested during the ammonia decomposition reaction. Co/Mo-NCX, which exhibited the best catalytic performance, was selected to be deposited inside the hollow fibre substrate. At 500 °C and 1 atm the hollow fibre converter was 3.6 times more efficient than the packed bed reactor (i.e. rNH3 = 5.5 × 104 molNH3 m−3 h−1 gcat−1 and rNH3 = 1.5 × 104 molNH3 m−3 h−1 gcat−1, respectively). This can be explained by narrower residence time distribution and minimised mass transfer limitations of the hollow fibre converter. Moreover, the hollow fibre converter showed a noticeably high thermal stability and catalyst-preservability during a 300 h reaction run. Furthermore, it exhibited a significantly lower pressure drop (i.e. >99%), volume (i.e. 70% less) and catalyst loading (i.e. 60% less) compared to the packed bed reactor. On this basis, the hollow fibre converter is especially suitable for on-board hydrogen production in automobiles, where space constraints present a key challenge. The potential of this new technology is enormous, since it will facilitate safe on-board hydrogen production, which is a key step in the decarbonisation of the transport sector in the fuel scenario nowadays.
1. Introduction
Green ammonia has been identified as a promising hydrogen carrier candidate for on-board applications due to its physical and chemical properties, including high hydrogen content (i.e. 17.8 wt%), narrower flammability range in air (i.e. 16–25%) compared to hydrogen (i.e. 4–75%), and its capability to be stored in liquid form under ambient temperature and low pressure conditions.1 However, the commercial use of ammonia as a hydrogen carrier for vehicle applications is hindered by space constraints and the inefficiency in terms of volume, weight, and performance of the most common catalytic reactor employed for the ammonia decomposition reaction, i.e. packed bed reactors (PBRs).2This scenario offers an excellent opportunity for the development of new technologies for on-board hydrogen production via ammonia decomposition. Garcia-Garcia et al. have reported the advances of catalytic hollow fibre reactors (HFRs) which overcome limitations of traditional PBRs.3 Indeed, it has been demonstrated that the use of HFRs narrows the residence time distribution and decreases the catalyst loading and/or the operating temperatures when compared with traditional PBRs.3–5 Moreover, they exhibit an exceptional area/volume ratio without compromising the pressure drop and efficiency. Furthermore, HFRs offer the advantage of improved reaction kinetics due to minimised internal and external diffusion limitations, thereby enhancing the viability of using non-precious metal-based catalysts.6
The most active and extensively studied catalysts reported in the literature for the decomposition of ammonia are ruthenium-based. However, the mass production of ruthenium, is restricted by its high cost and scarcity. This has triggered the catalytic community to search for a potential substitute of ruthenium-based catalysts for the ammonia decomposition reaction.1,7,8 Previous studies investigating the performance of several non-noble bimetallic catalysts, have proposed cobalt/molybdenum (Co/Mo) catalysts, with an optimum Co/Mo atomic ratio of 7/3, as an effective and cheaper alternative to ruthenium counterparts for the ammonia decomposition reaction.8–10
Likewise, it is well known that the ammonia decomposition reaction rate is affected by the chemical and physical properties of the catalyst support. It has been reported that the ideal support for this reaction must exhibit properties such as thermal stability under the reaction conditions, large specific surface area, high thermal and electrical conductivity, low concentration of electron-withdrawing functional groups, and high basicity.1,11–14 Hence, carbonaceous materials including active carbon (AC), high surface area graphite (HSAG), carbon nanotubes (CNTs) and carbon nanofibers (CNFs) have been studied as promising catalyst supports for the ammonia decomposition reaction.8,15,16 Among the several carbon supports investigated so far, CNTs have been proposed as the most efficient catalyst support for this reaction due, in part, to their outstanding electrical properties.8,16–18 However, in addition to their high cost/performance ratio, CNTs are challenging to be uniformly deposited inside the hollow fibre due to their low-density sponge structure.19 In this respect, although some impregnation methods allow the deposition of the powder catalyst inside the hollow fibre support (i.e. dip coating for CNTs), the sol–gel Pechini impregnation method is preferred as it results in the catalyst being homogeneously dispersed.3,20
A promising alternative to CNTs is carbon xerogels, which have already been demonstrated to be efficient catalyst supports.21–23 They also offer a wide range of textural and structural properties that can be tailored either by changing the synthesis conditions or using post-synthesis treatments.24,25 Furthermore, carbon xerogels, which are synthesised by the conventional sol–gel method originally proposed by Pekala,26 can be easily deposited in the hollow fibre substrate via the sol–gel Pechini method and successively impregnated with the metal catalyst solution, offering an exceptional opportunity for tackling the challenges encountered with the deposition of CNTs inside the hollow fibre.
The aim of this work is to design a catalytic hollow fibre converter (HFC), for on-board hydrogen production, which is able to satisfy the demands of ammonia-fuelled vehicles. Our hypothesis is that the HFC will exceed the conversion per unit mass of the catalyst achieved by the PBR, opening the doors to a new line of innovative catalytic converters. With this purpose, this work has been carried out following a bottom-up approach, consisting in three stages. The first stage 1 has focused on the design, synthesis and characterisation of a series of Co/Mo-based catalysts supported on carbon xerogels and on the study of their catalytic activity and stability during the ammonia decomposition reaction in a PBR. To the best of our knowledge, this is the first study in which Co/Mo-based catalysts supported on carbon xerogels are used for the ammonia decomposition reaction. In the second stage, the best performing Co/Mo-based catalyst was deposited in the HFC and tested under the same reaction conditions. Finally, in the stage 3, both the PBR and HFC were designed to target on-board hydrogen production in an ammonia-fuel car and compared in terms of catalyst loading, volume and efficiency.
2. Experimental methods
2.1. Synthesis of Co/Mo catalysts supported on carbon xerogels
Resorcinol–formaldehyde xerogels were synthesised using the conventional sol–gel method.26 A precursor solution containing 30 wt% solids in 60 cm3 of the gel volume was prepared, where the molar ratios of resorcinol to carbonate (R/C) and resorcinol to formaldehyde (R/F) were fixed at 200 and 0.5, respectively. After being stirred for 30 min, the starting solution was gelated and cured for 3 days in a ventilation oven (SciQuip Oven 55S) at 85 °C. The obtained gel was dried for 3 days in the ventilation oven (SciQuip Oven 55S) at 100 °C.The carbon xerogel, labelled as CX, was obtained by carbonisation of the resorcinol–formaldehyde dried xerogels at 800 °C in a nitrogen atmosphere (i.e. 100 cm3 min−1 (STP)), using a tubular furnace (MTI Corporation, OTF-1200X).
In order to study the influence of metal-support interactions on the catalytic performance during the ammonia decomposition reaction, two nitrogen-doped carbon xerogels were also synthesised by employing two different methods, (i) co-precursor method using urea as a nitrogen-containing source, and (ii) post-synthesis activation of CX in a NH3/air atmosphere.
For the first method (co-precursor method), R/F, R/C and Resorcinol/Urea (R/U) ratios were fixed at 200, 0.5, and 2, respectively. The solution was prepared, cured and dried following the same method described above. The nitrogen-doped carbon xerogel, labelled as UCX, was obtained by carbonisation of the nitrogen-containing dried xerogel at 800 °C for 1 h in a nitrogen atmosphere (i.e. 100 cm3 min−1 (STP)) using a tubular furnace (MTI Corporation, OTF-1200X).24
For the second method (post-synthesis activation), the nitrogen-doped carbon xerogel, labelled as NCX, was obtained by activating CX in ammonia and air atmospheres (i.e. 100 cm3 min−1 of NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :air = 1:3 (STP)) at 350 °C for 3 h.
:air = 1:3 (STP)) at 350 °C for 3 h.
The three carbon xerogels were used as supports for Co/Mo catalysts. The catalysts were synthesised by incipient wetness impregnation of the supports with an aqueous solution of (NH4)2MoO4·4H2O and Co(NO3)2·6H2O in order to obtain 1.5 wt% of metal loading, and a Co/Mo ratio equal to 7/3. After impregnation, the catalysts were dried overnight at 120 °C in a ventilation oven (SciQuip Oven 55S).
Catalysts were labelled as Co/Mo-CX, Co/Mo-UCX and Co/Mo-NCX, according to the impregnated supports.
2.2. Carbon xerogel characterisation
2.3. Characterisation of Co/Mo-based catalysts
2.4. Development of the hollow fibre converter
Asymmetric multichannel α-Al2O3 hollow fibre substrates were fabricated by phase inversion, followed by sintering at high temperatures. The detailed procedures for their synthesis can be found elsewhere.27In order to develop the HFC, 10 hollow fibre units, 5 cm long, were glued together using a high temperature ceramic adhesive (Ceramabond 503, AREMCO). The obtained hollow fibre bundle was air dried at room temperature for approximately 1 h and step cured at 100 °C for 1.5 h, 250 °C for 1.5 h, 400 °C for 1.5 h and 600 °C for 1.5 h.
The carbon xerogel deposition onto the hollow fibre bundle substrate followed the same procedure described in Section 2.1: (i) preparation of the carbon xerogel precursor solution, (ii) solution deposition on the hollow fibre substrate, (iii) xerogel gelation, curing and drying in a ventilated oven (SciQuip Oven-55S), and (iv) carbonisation and post-synthesis activation treatment of the xerogel. The amount of carbon deposited on the hollow fibre substrate was calculated by measuring the difference in the weight of the hollow fibre bundle before and after the carbon xerogel deposition.
Based on that, the volume of the aqueous solution of (NH4)2MoO4·4H2O and Co(NO3)2·6H2O required to obtain 1.5 wt% of metal loading was calculated. Finally, prior to wetness impregnation with the Co/Mo-based catalyst solution, the hollow fibre bundle was wrapped with polytetrafluoroethylene (PTFE) tape to avoid catalyst deposition on its outer surface. The impregnated hollow fibre bundle was left drying overnight at 120 °C in a ventilation oven (SciQuip Oven 55S). The metal loading was calculated by measuring the difference in the weight of the hollow fibre bundle before and after the metal impregnation.
2.5. Characterisation of the hollow fibre converter
2.6. Catalytic performance during the ammonia decomposition reaction
Ammonia condensation within the lines was prevented by installing a network of electrical line heating tape and thermal insulating fabric. The temperature of all pipes was maintained above 150 °C, and monitored using several k-type thermocouples distributed throughout the equipment. A computer connected in-line to the mass spectrometer provided real-time readings of the composition of gas circulating through the system.
Prior to each catalytic test, the catalyst was reduced in situ at 600 °C under a flow of 50 cm3 min−1 (STP) of 50 vol% hydrogen in argon. Then, the catalyst was exposed to 10% vol. ammonia balanced in argon, from 100 °C to 600 °C under atmospheric pressure. In all experiments, the feed gas flow rate was set to 100 cm3 min−1 (STP). The whole experiment was designed in such a way that internal and external diffusion limitations were minimised, which allows reliable kinetic data to be obtained. In this respect, the reaction conditions for PBR experiments were optimised to obtain a Thiele modulus ϕ2 ≤0.4 (i.e. catalyst pellets were sieved to 125–250 μm and the Reynolds number was 4.2 × 10−2).
The composition of the exit gases was monitored in-line by using the mass spectrometer. The ammonia conversion xNH3 (%), was estimated using eqn (1):
 | (1) |
In order to study the stability of the catalysts, long-term (i.e. 100 h in the PBR and 300 h in the HFC) reaction experiments were performed at 450 °C. Furthermore, to assess the thermal stability of the HFC during the ammonia decomposition reaction, ten thermal shocks were carried out. During the thermal shock experiments, the HFC was heated using a ramp rate of 20 °C min−1, reaching 450 °C in approximately 20 min. The ammonia reading for the thermal shock experiments at atmospheric pressure was taken at 450 °C.
3. Results and discussion
3.1. Characterization of carbon xerogels
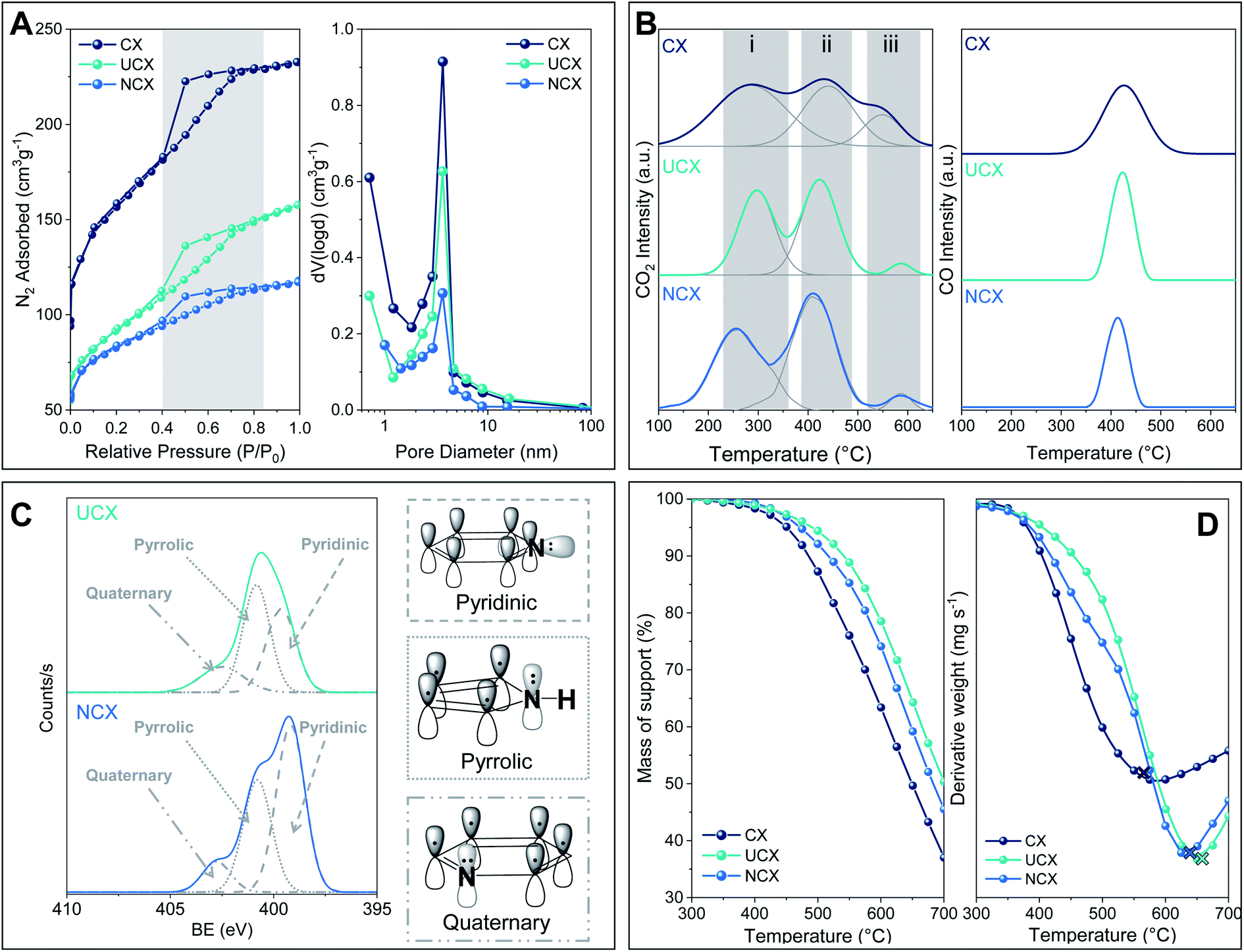 | ||
| Fig. 1 Carbon xerogels studied here: (A) N2 adsorption/desorption isotherms at −196 °C and pore size distributions, (B) CO2 and CO TPD profiles, (C) XPS spectra in the N1s region of UCX and NCX, and (D) air-TGA profiles. | ||
Likewise, it can be observed in Fig. 1A (left) that the nitrogen doping of carbon xerogels induced a reduction of the nitrogen adsorption uptake at a low relative pressure.28,29 The fact that the isotherms of UCX and NCX were shifted downwards with respect to CX, and the hysteresis position and shape did not significantly change, indicates that the nitrogen doping reduced the micropore volume of both carbon xerogels but it did not alter their mesopores.
As is reported in Table 1, both UCX and NCX exhibited a lower specific surface area than CX (i.e. 550, 325 and 1050 m2 g−1, respectively). These results can be explained by the addition of heteroatoms to the carbon lattice during the nitrogen doping, and by the different thermal treatments applied for the synthesis of both UCX and NCX, which are known to affect structural and textural properties of carbon xerogels.14,18
| Support | S BET (m2 g−1) | Pore volume (cm3 g−1) | ||
|---|---|---|---|---|
| Total | Micro | Meso | ||
| CX | 1050 | 0.39 | 0.20 | 0.19 |
| UCX | 550 | 0.27 | 0.11 | 0.16 |
| NCX | 325 | 0.20 | 0.07 | 0.13 |
The CO2–TPD profiles exhibited three peaks, which can be attributed to (i) carboxylic acid groups (i.e. 230–300 °C), (ii) anhydride groups (i.e. 410–420 °C), and (iii) lactone groups (i.e. 530–580 °C). However, the CO–TPD profiles exhibited only one important peak (i.e. 410–420 °C), which can be ascribed to anhydride groups. The CO2 and CO desorption peaks reported here agree with those reported in previous studies wherein the synthesis of carbon xerogels was conducted under similar experimental conditions.21,22,30
It is worth noting that, the total amount of oxygen surface groups decreased after nitrogen doping, regardless of the method used. For UCX, this behaviour can be explained by the longer exposure to high temperatures during the carbonisation stage (i.e. up to 800 °C for 8 h) compared to CX (i.e. up to 800 °C for 1 h).31,32 This behaviour can be further attributed to the formation of various nitrogen functionalities during the pyrolysis of the nitrogen-doped carbon xerogel precursor, which could avoid the formation of certain oxygen surface groups such as lactone surface groups (i.e. 90% less in UCX than that in CX).33 In contrast, the complete absence of lactone groups and the overall decrease of oxygen surface groups in NCX is a consequence of the formation and desorption of amide surface groups during the NH3/air activation treatment (i.e. R1–(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–R2 + NH3 → R1–CONH2 + R2–OH).25,32,34
O)–O–R2 + NH3 → R1–CONH2 + R2–OH).25,32,34
However, although UCX and NCX presented the same types of nitrogen groups, their total and relative amounts are different. For instance, as is reported in Table 2, the total amount of nitrogen groups in UCX is half of that observed in NCX. Likewise, UCX exhibits higher relative contributions of pyrrolic and quaternary nitrogen (i.e. 49% and 28.8%, respectively), whereas NCX showed more pyridinic nitrogen (i.e. 54%). This result can be justified by the different nitrogen-doping methods used to synthesise UCX and NCX. In this respect, Kang et al. found that the relative amount of nitrogen functionalities can be controlled by changing the temperature at which the nitrogen doping occurs.34 Thus, longer exposure to high temperatures during the carbonisation led to a gradual conversion of pyrrolic to pyridinic nitrogen, which is consequently converted into quaternary nitrogen. Similarly, Abidin et al. reported that the carbonisation temperature significantly affects the formation of the nitrogen groups in the carbon xerogel structures.37 Furthermore, Gorgulho et al. showed that relative contributions of nitrogen groups in a sample are not only affected by the temperature at which nitrogen-doped carbon xerogels have been prepared, but also by the nitrogen-containing precursor used.24
As shown by the first derivative of the air-TGA profiles, both UCX and NCX have higher burning temperatures than CX (i.e. 640 °C, 615 °C, and 535 °C, respectively), which suggest that the presence of nitrogen atoms in the lattice of UCX and NCX enhanced their thermal stability. The positive effect of nitrogen doping on the thermal stability of both UCX and NCX can be attributed to the presence of carbon–nitrogen bonds (615 kJ mol−1) which are stronger than carbon–carbon bonds (602 kJ mol−1).27. A similar behaviour was observed by Stohr et al., which reported that nitrogen-doped carbon materials exhibit a higher resistance to oxidation compared to non-doped counterparts.38
It is important to highlight that the lower burning temperature observed for NCX compared to UCX can be explained by its larger amount of pyridine groups (see Table 2), which have been reported to exhibit lower thermal stability than pyrrole and quaternary nitrogen groups.36
3.2. Characterization of Co/Mo-based catalysts
 | ||
| Fig. 2 Co/Mo-based catalysts: (A) TEM images and particle size distributions before and after the reaction experiment, (B) XRD diffractograms, (C) phase diagram of Co–Mo–O2 (generated using FactSage thermochemical software), and (D) TPR profiles. | ||
It is well known that both oxygen surface groups and nitrogen groups can act as anchoring sites for metal atoms, improving their dispersion throughout the support surface.11,12,39–41 However, despite CX presenting the highest amount of oxygen surface groups, Co/Mo-CX exhibited a larger average particle size (i.e. 3.3 nm) compared to both Co/Mo-UCX and Co/Mo-NCX (i.e. 2.7 nm and 2.2 nm, respectively). This result can be explained by a combination of two opposing factors. On one hand, the large amount of acidic electron-withdrawing oxygen surface groups on CX weakens the interaction between the metal atoms and the support, thereby facilitating their mobility and agglomeration into larger particles.12 On the other hand, the presence of basic nitrogen functionalities in both UCX and NCX encourages the anchorage and dispersion of the metal active phase on the support surface due to the higher electronegativity of the support.36,39 All the above supports the fact that catalysts with the lowest average particle size, i.e. Co/Mo-UCX and Co/Mo-NCX, also showed the lowest amount of lactone groups.
It is worth noting that although both Co/Mo-NCX and Co/Mo-UCX catalysts were doped with nitrogen, Co/Mo-NCX showed an average particle size 10% smaller than that of Co/Mo-UCX. According to the XPS results shown in Fig. 1C, this result could be explained by the different nitrogen groups present in both NCX and UCX, which affects the metal-support interaction.36,42,43
Finally, it was found that, after the reaction, the metal particle size distributions did not change significantly, and the average particle size remained the same for all the catalysts. These results suggested that the design approach adopted in this work (i.e. Co/Mo ratio) did not lead to side reactions (i.e. hydrogenation), which might have affected the amount of nitrogen and oxygen groups, thereby the metal particle size.
The main sharp diffraction peak centred at around 26° corresponds to the (002) basal plane diffraction along the graphitic structure.29,44,45 Its sharpness and high intensity suggest that all carbon xerogels show a high crystallisation degree. The apparent crystallite size along the c-axis (Lc) and interlayer spacing along the c-axis (d002) have been calculated by applying Scherrer's equation to the (002) peak. As revealed by the data in Table 3, after nitrogen doping, this diffraction peak shifted to slightly higher angles and its intensity increased, evidencing a shrinkage in the carbon-to-carbon layer distance of both UCX and NCX. However, these results suggested that the incorporation of nitrogen into the carbon xerogel lattice did not induce a significant increase in the structural order, as evidenced by the minimal changes in the crystallite size of CX after nitrogen doping (i.e. 11% bigger for UCX and 10% bigger for NCX). These results agree with the previous work of Kicίnski et al., which studied the effect of the xerogel starting solutions and their carbonisation temperature on the crystallite size and the amount of stacked graphitic layers.29,44 Similarly, Peikolainen et al. reported that the textural and structural properties of carbon xerogels directly affect the degree of order in their carbonaceous structure.45
| Support | 2θ (°) | L C (Å) | d 002 (Å) |
|---|---|---|---|
| CX | 25.9 | 4.3 | 3.44 |
| UCX | 26.0 | 4.8 | 3.41 |
| NCX | 26.1 | 4.7 | 3.41 |
The other major diffraction peaks identified using the ICDD database powder diffraction files (PDF-3) evidenced that the active phase of the three Co/Mo-based catalysts is predominantly composed of a mixture of Co3O4, CoMoO4 and Co2Mo3O8.
Despite the high reduction temperature (i.e. 600 °C), no legible diffraction peaks associated with Co and Mo were observed, suggesting the re-oxidation of the catalysts when exposed to the air atmosphere prior to the XRD analysis. Nevertheless, the absence of Co, Mo and CoO nanoparticle diffraction peaks, and the significantly lower intensities of Co3O4, CoMoO4, Co2Mo3O8 and MoO3 diffraction peaks compared to the distinct diffraction peak associated with the carbon support, can be also attributed to a high metal particle dispersion, which is in agreement with the TEM results.
Overall, the TPR profiles showed five hydrogen consumption peaks: (i) reduction of Co3O4 to CoO (i.e. 280–300 °C), (ii) reduction of CoO to Co (i.e. 430–450 °C), (iii) reduction of CoMoO4 to Co2Mo3O8 (i.e. 520–540 °C), (iv) partial gasification of the carbon support (i.e. 580–600 °C), and (v) reduction MoO3 to MoO2 (i.e. from 700 °C onwards). The decrease in the intensity of peak iv, corresponding to the partial gasification of the carbon support, after the nitrogen doping, can be attributed to the higher thermal stability of both UCX and NCX compared to CX, as evidenced by air-TGA analysis discussed in Section 3.1.5.
3.3. Performance studies of Co/Mo-based catalysts during the ammonia decomposition reaction
The performance during the ammonia decomposition reaction of the three Co/Mo-based catalysts studied in this work is shown in Fig. 3A. All catalysts were found to be highly active during the ammonia decomposition reaction, with a performance comparable to that of previously reported catalysts,9,10,50 as reported in Table 4. It was observed that both Co/Mo-UCX and Co/Mo-NCX exhibited a higher catalytic activity than Co/Mo-CX during the ammonia decomposition reaction. This behaviour can be explained by a combination of two factors, including the lower amount of oxygen surface groups exhibited by both UCX and NCX, and the presence of nitrogen groups incorporated in their carbon lattice. In this respect, it has been widely acknowledged that acidic oxygen surface groups (i.e. carboxyl, anhydride, and lactone) can withdraw electrons from metal atoms, negatively affecting the catalytic activity during the ammonia decomposition reaction.11,51 In contrast, the presence of nitrogen groups benefits the ammonia decomposition reaction by enhancing the local basicity and electron density of the carbon support.16,22,36Fig. 3B shows a schematic diagram of the electron transfer mechanism in the presence of the basic and acidic surface functionalities.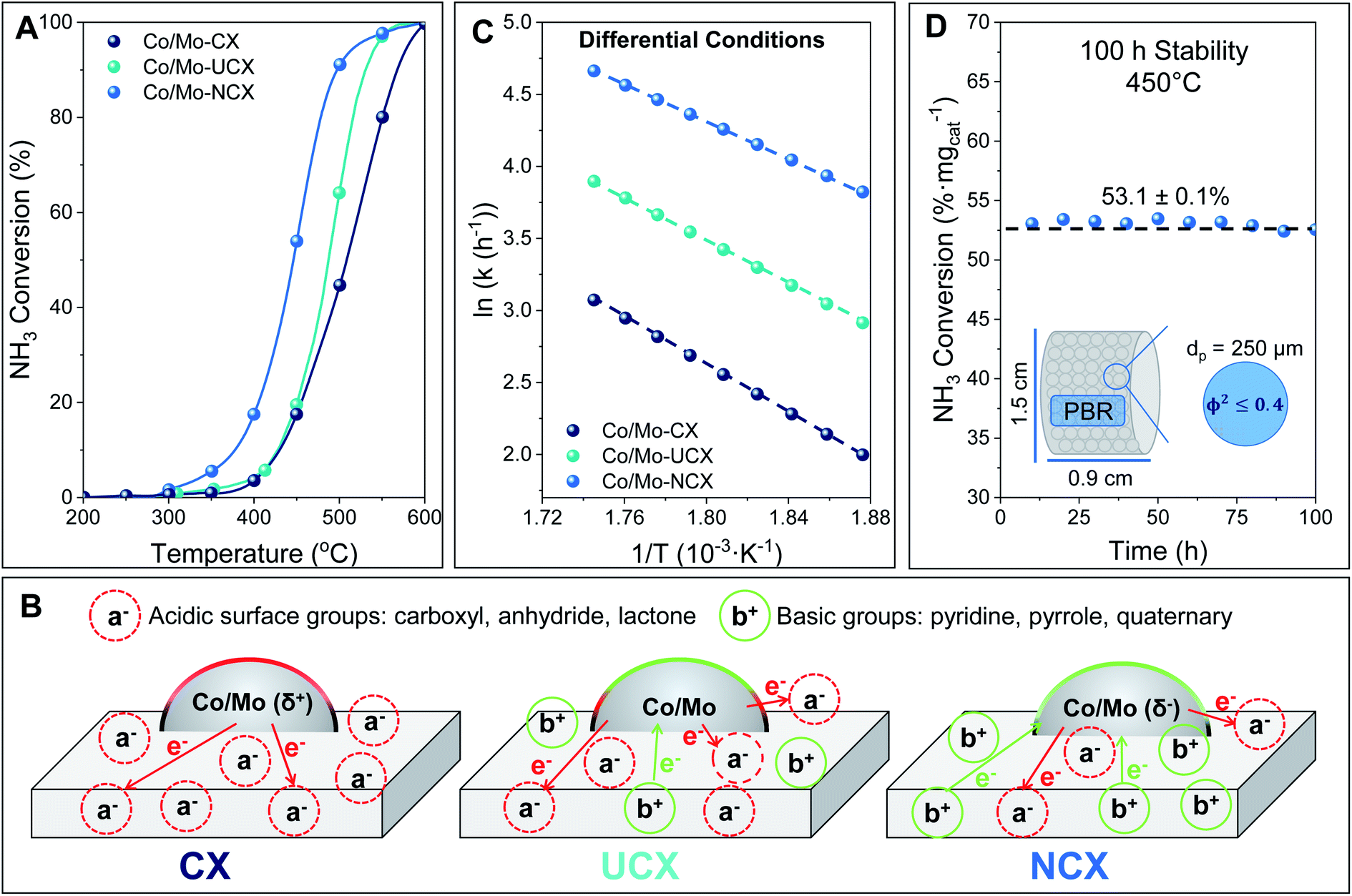 | ||
| Fig. 3 (A) Performance of Co/Mo-based catalysts during PBR experiments, (B) schematic diagram of the electron transfer mechanism in the presence of both oxygen and nitrogen groups decorating NCX, CX and UCX surfaces, (C) Arrhenius plot under differential conditions, and (D) long-term stability study of the PBR at 450 °C. | ||
It is well known that high electron transfer from electropositive elements to the active metal surface is needed to promote the ammonia decomposition reaction rate-limiting step, i.e. the recombinative nitrogen desorption. Therefore, it was expected that the higher charge density of nitrogen-doped carbon xerogels compared to their non-doped counterparts would enhance the catalytic activity of metal particles involved in the ammonia decomposition reaction.
Moreover, this work evidenced an inverse relationship between the performance of the Co/Mo-based catalysts during the ammonia decomposition reaction and their average metal particle size. For instance, among all the catalysts studied, Co/Mo-NCX, which had the smallest metal average particle size (i.e. 2.2 nm), exhibited the highest catalytic activity during the reaction. According to the TEM images in Section 3.1.5., this result can be explained by the different degrees of dispersion of the metal particles over the carbon support. Contrary to the ruthenium-based catalysts, whose activity is maximised when the ruthenium metal particle size is between 2.5 nm and 5 nm,51,52 this result suggests that the optimal metal particle size of a Co/Mo-based catalyst should be around 2.2 nm.
The reaction rate constant (k) for the ammonia decomposition reaction of the three Co/Mo-based catalysts was calculated under differential conditions (i.e. gradient-less ammonia concentration along the reactor), and the regressed parameters shown in the Arrhenius plot presented in Fig. 3C were used to determine the reaction rate constant under the operating conditions and the apparent activation energy. As expected, it was found that both Co/Mo-UCX and Co/Mo-NCX exhibited higher reaction rate constants and lower apparent activation energy than Co/Mo-CX (i.e. 62.3 kJ mol−1, 53.3 kJ mol−1 and 68.2 kJ mol−1, respectively). The higher performance of Co/Mo-NCX compared with Co/Mo-UCX can be explained by both the higher nitrogen content and the smaller average metal particle size of NCX compared to that of UCX (see the XPS and TEM results previously discussed).
Based on these results, Co/Mo-NCX was identified as the most suitable catalyst candidate for the ammonia decomposition reaction. To study the stability of Co/Mo-NCX, a long-term (i.e. 100 h) stability test during the ammonia decomposition reaction was performed at 450 °C. The results presented in Fig. 3D showed that Co/Mo-NCX has an excellent thermal stability, as proven by the constant ammonia conversion achieved during the whole PBR experiment.
3.4. Characterisation of the hollow fibre catalytic unit
 | ||
| Fig. 4 (A) Side pictures of the hollow fibre at different stages of the impregnation process, (B) Hg porosimetry of the hollow fibre before and after Co/Mo-NCX deposition, and (C) SEM images of the hollow fibre substrate and EDX surface mapping (iv) after the deposition of Co/Mo-NCX. | ||
Fig. 4B provides further details of the asymmetric pore structure of the alumina hollow fibre, which exhibited a bimodal nature of the pore size distribution, before and after deposition of Co/Mo-NCX. As can be seen, before the catalyst deposition, two different pore size distributions can be clearly identified, corresponding to the sponge-like region (i.e. Dp = 1.0 μm) and the entrance of the fingers (i.e. Dp = 2.2 μm), respectively.27 After the deposition, the pore size distribution in the sponge-like region and finger-like region decreased from 1.0 μm to 0.6 μm and from 2.2 μm to 1.2 μm, respectively. The reduction in the pore sizes of both regions can be explained by the diffusion of both xerogel and catalyst solutions through the microporous structure of the hollow fibre.3,4,20
EDX surface mapping images presented in Fig. 4C(iv), show that the morphology of the hollow fibre and the use of a precursor liquid solution allowed for uniform dispersion of the catalyst. Based on that, with a catalyst loading of 15 mg in the 5 cm long hollow fibre, it can be assumed that the amount of the catalyst per cm of hollow fibre (WHF) is 3 mg.
3.5. Performance studies of the hollow fibre converter during the ammonia decomposition reaction
Among the three catalysts studied here, Co/Mo-NCX was identified as the most suitable catalyst candidate for deposition in the hollow fibre substrate due to its basicity and high thermal stability, which resulted in enhanced catalytic activity.The beneficial effect of using the HFC in the catalytic activity is shown in Fig. 5A. For instance, at 450 °C, the rate of the reaction in the HFC is 3.6 times higher than that in the PBR (i.e. rNH3 = 3.1 × 104 molNH3 m−3 h−1 gcat−1 and rNH3 = 8.7 × 103 molNH3 m−3 h−1 gcat−1, respectively). This phenomenon can be justified by the presence of the micro-channels in the finger-like region of the hollow fibre substrate which improves the (i) catalyst distribution, (ii) residence time distribution, avoiding typical issues presented by PBRs (i.e. recirculation, generation of preferential paths and stagnant regions), and (iii) mass transfer, resulting in improved reaction kinetics.5,53,54Fig. 5B schematically represents that the use of a HFC avoids the diffusion limitations typically associated with a PBR, which are caused by the tortuosity, porosity and constriction factor of the porous structure of the catalyst pellets. Furthermore, the large area/volume ratio of hollow fibres allows working at lower catalyst metal loading and/or operating temperatures compared to traditional catalytic PBRs. Hence, by using a HFC for the ammonia decomposition, this feature can be exploited to increase the non-precious metal-based catalyst loading to the extent that the catalytic performance of ruthenium-based catalysts is matched, without significant detriment to costs.
 | ||
| Fig. 5 (A) Ammonia decomposition reaction rate vs. temperature of Co/Mo-NCX in both the PBR and HFC, (B) ammonia diffusion mechanism in both the PBR and HFC, (C) long-term stability study of Co/Mo-NCX in the HFC at 450 °C, and (D) thermal shocks at 450 °C after the long-term stability study. | ||
To assess both the stability and adherence in the hollow fibre substrate of the catalyst during the HFC experiment, a 300 h reaction run and 10 thermal shocks at 450 °C were carried out, respectively. In order to provide more realistic data, the experimental conditions adopted (i.e. 300 h and 450 °C) were selected to mimic the operating conditions of a catalytic converter in a car. Likewise, thermal shocks were performed to study the effects of frequent start-up and shutdown cycles on the catalytic activity and assess the catalyst peel off resistance.
From a visual inspection (see Fig. 5C), the HFC maintained a homogeneous colouration after the long-term stability test. As can be seen in Fig. 5C, constant ammonia conversions were achieved at 450 °C in the HFC during the long-term stability test. Furthermore, similar ammonia conversion values were observed after each thermal shock, as shown in Fig. 5D. These results not only suggest that this catalytic system is stable under operating conditions, but also that it has a high catalyst adherence, which prevents the catalyst from peeling off from the hollow fibre substrate. In order to corroborate these data, the HFC unit was weighed before and after the long-term stability test and the thermal shocks, leading to a difference in the weight within the measurement error (i.e. ±0.01%).
4. Reactor design for on-board hydrogen production
The design feasibility study of both the PBR and HFC for on-board applications, targeting a power demand of 100 kW (i.e. 72 m3 h−1 (STP) of hydrogen supply55), relies on the assumptions and constraints listed in Table 5, and had been carried out using the key parameters listed in Table 6. Based on the first two assumptions, the flow rate of ammonia required (VNH3˙) was calculated to be 48.2 m3 h−1 (STP).| Assumptions | i. Isothermal catalytic process |
| ii. The ammonia decomposition reaction is irreversible and follows first order kinetics under operating conditions.56 | |
| iii. Flow rate and fluid properties are uniform over any cross-section normal to the fluid motion | |
| iv. Negligible axial mixing due to either diffusion or convection | |
| v. Ammonia conversion at the equilibrium xNH3 = 99.5% under operating conditions | |
| Constraints | i. Operating conditions: T = 450 °C, P = 1 atm |
| ii. Space constrains in a car: Lmax = 0.9 m, dmax = 0.3 m,57 and Vmax = 63 L |
| PBR | • Density of the bed ρb = 300 kg m−3 |
| • Bed porosity εb = 0.3 | |
| HFC | • Area of one hollow fibre channels Ac = 3 × 10−6 m2 |
| • Area of both the sponge-like and finger-like regions of a single hollow fibre unit A = 6.1 × 10−6 m2 | |
| • Hollow fibre porosity εHF = 0.43.27 | |
| NH3 | • Ammonia density ρf = 0.3 kg m−3 (450 °C, 1 atm) |
| • Dynamic viscosity of ammonia μf = 2.5 × 10−5 Pa s (450 °C, 1 atm) | |
| • Effective diffusion De = 6.1 × 10−6 m2s−1 |
In order to design both the PBR and HFC, the reaction rate constant (k) during the ammonia decomposition reaction for Co/Mo-NCX in both reactors was calculated under the operating conditions, obtaining kNH3 = 4.7 × 103 h−1 gcat−1 for the PBR and kNH3 = 7.5 × 103 h−1 gcat−1 for the HFC.
Likewise, to estimate the catalyst loading required to achieve xNH3 = 99.5% with a pure ammonia feed of 48.2 m3 h−1 (STP) and the volume of both the PBR and HFC, the ammonia mass balance over a differential element for catalyst loading (i.e. dW) was used. Upon integration, it was obtained that
 | (2) |
Hence, the catalyst loading and the reactor volume were calculated to be WPBR = 71.1 kg and VPBR = 237.0 L for the PBR, and WHFC = 23.8 kg and VHFC = 79.3 L for the HFC. However, since the so-designed reactors did not respect on-board space constraints, the effects of both the operating temperature and catalyst metal loading on the volume of the reactors were investigated.
With this scope, starting from the design of the PBR, the operating temperatures were set to 450 °C and 500 °C, and the metal loading ranged between 1.5 wt% and 6.0 wt%, assuming that the performance changes linearly with the metal loading. In addition, the pressure drop and the mass transfer limitations of the different PBR and HFC designs were estimated using the Ergun equation (eqn (3)) and the Hagen–Poiseuille equation (eqn (4)), respectively, and the mass transfer limitations were estimated by using the definition of the Thiele modulus ϕ2 (eqn (5)):
 | (3) |
 | (4) |
 | (5) |
As can be seen in Fig. 6A, only two PBRs have a smaller volume than the maximum volume allowed by the on-board space constraints. More specifically, as shown in Fig. 6B and Table 7, the use of 6.0 wt% metal loading requires a PBR of 59.2 L and 17.8 kg of the catalyst at 450 °C, and a PBR of 33.4 L and 10.0 kg of the catalyst at 500 °C. Nevertheless, working at 450 °C results in lower pressure drop through the reactor (i.e. by 20%) and mass transfer limitations (i.e. by 45%) when compared to working at 500 °C. Hence, the optimal PBR design resulted in: (i) LPBR = 0.88 m, (ii) dPBR = 0.29 m, (iii) WPBR = 17.8 kg, (iv) VPBR = 59.2 L, (v) ΔP = 17.2 bar, and (vi) ϕ2 = 3.1 (see Fig. 6D).
 | ||
| Fig. 6 (A–C) Effect of the temperature and the catalyst metal loading on the volume of both the PBR and HFC, and (D) optimised design of a PBR and HFC for a car with a power demand of 100 kW (i.e. 72 m3 h−1 (STP) of hydrogen supply). | ||
| PBR | HFC | |||
|---|---|---|---|---|
| 450 °C | 500 °C | 450 °C | 500 °C | |
| k (h−1 gcat−1) | 1.4 × 104 | 2.6 × 104 | 3.0 × 104 | 5.2 × 104 |
| V (L) | 59.2 | 33.4 | 19.8 | 11.5 |
| L (m) | 0.88 | 0.73 | 0.90 | 0.90 |
| W cat (kg) | 17.8 | 10.0 | 5.9 | 3.4 |
| ΔP (bar) | 17.2 | 21.6 | 2.8 × 10−3 | 4.8 × 10−3 |
| ϕ 2 | 3.1 | 5.5 | 0.1 | 0.1 |
On the other hand, as shown in Fig. 6C, five HFCs have smaller volume than the one allowed by on-board space constraints. To ease the assembly of single HF units into a larger module, the converter requiring the lowest number of HF units (NHF) has been identified as the most suitable for on-board applications (see Fig. 6B and Table 7). This consideration took into account that the effects of both the pressure drop and Thiele modulus changes with the operating temperature and/or the catalyst loading are negligible. Hence, the optimal HFC design resulted in: (i) LHFC = Lmax = 0.90 m, (ii) WHFC = 3.4 kg, (iii) VHFC = 11.5 L, (iv) NHF = 1040, (v) ΔP= 4.8 × 10−3 bar, and (vi) ϕ2 = 0.1 (see Fig. 6D).
To conclude, this study proved that the HFC represents an economical, compact and efficient technology for on-board hydrogen production, promoting a new line of worthwhile catalytic converters. Note that, as summarised in Table 6, the use of a traditional PBR for vehicular applications turned out to be unfeasible due to its high pressure drop and mass transfer limitations.
5. Conclusion
Among the catalysts studied herein, Co/Mo-NCX was the most suitable catalyst candidate for the ammonia decomposition reaction. This was explained by the improved basicity and electrical conductivity of NCX compared to CX, lowest presence of oxygen groups on the NCX surface, and promoting effects of the nitrogen atoms.The use of the HFC increased the efficiency of Co/Mo-NCX 3.6-fold at 500 °C in comparison to a traditional PBR. Furthermore, the HFC showed high thermal stability and catalyst adherence, as demonstrated by the 300 h reaction experiment.
Likewise, the HFC offered noteworthy advantages compared to the PBR, in terms of cost (i.e. 80% less catalyst), space occupied (i.e. 80% smaller), and efficiency (i.e. >99% less pressure drop and mass transfer limitations).
Finally, the tailorable nature of the HFC technology and the easy power scale-up via integration of units facilitate its adoption for automotive applications, without compromising the design flexibility of vehicles, thereby opening the door for a new line of on-board catalytic converters.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The prolegomena of this work were presented as a communication in the IV UKEM Workshop in October 2020 (https://emissioncontrolukem.wordpress.com/). S. Mazzone would like to thank all who attended that session for their helpful comments and discussion. Moreover, S. Mazzone gratefully acknowledges the funding provided by the School of Engineering at the University of Edinburgh to carry out her PhD. Likewise, C. Leishman gratefully acknowledges the funding provided by the School of Engineering at the University of Edinburgh during her Summer Vacation Internship at the Denbigh Lab in Jun–Aug 2019. Finally, F. R. García-García would like to thank Joseph El-Kadi, and Catalysis and Process Integration Group at the University of Cambridge, for their help and support in the TPR experiments.References
- S. F. Yin, B. Q. Xu, X. P. Zhou and C. T. Au, Appl. Catal., A, 2004, 277, 1–9 CrossRef CAS.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS.
- F. R. García-García, B. F. K. Kingsbury, M. A. Rahman and K. Li, Catal. Today, 2012, 193, 20–30 CrossRef.
- F. R. García-García, M. A. Rahman, B. F. K. Kingsbury and K. Li, Catal. Commun., 2010, 12, 161–164 CrossRef.
- N. Prasetya, Z. Wu, A. G. Gil and K. Li, J. Eur. Ceram. Soc., 2017, 37, 5281–5287 CrossRef CAS.
- N. I. Mahyon, T. Li, R. Martinez-Botas, Z. Wu and K. Li, Catal. Commun., 2019, 120, 86–90 CrossRef CAS.
- T. V Choudhary, C. Sivadinarayana and D. W. Goodman, Catal. Lett., 2001, 72, 197–201 CrossRef.
- T. E. Bell and L. Torrente-Murciano, Top. Catal., 2016, 59, 1438–1457 CrossRef CAS.
- X. Duan, G. Qian, X. Zhou, D. Chen and W. Yuan, Chem. Eng. J., 2012, 207–208, 103–108 CrossRef CAS.
- J. Ji, X. Duan, G. Qian, X. Zhou, G. Tong and W. Yuan, Int. J. Hydrogen Energy, 2014, 39, 12490–12498 CrossRef CAS.
- K. Aika, H. Hori and A. Ozaki, J. Catal., 1972, 27, 424–431 CrossRef CAS.
- H. E. Van Dam and H. Van Bekkum, J. Catal., 1991, 131, 335–349 CrossRef CAS.
- Z. H. Zhong and K. I. Aika, Inorg. Chim. Acta, 1998, 280, 183–188 CrossRef CAS.
- S. F. Yin, Q. H. Zhang, B. Q. Xu, W. X. Zhu, C. F. Ng and C. T. Au, J. Catal., 2004, 224, 384–396 CrossRef CAS.
- X. Duan, J. Zhou, G. Qian, P. Li, X. Zhou and D. Chen, Chin. J. Catal., 2010, 31, 979–986 CrossRef CAS.
- F. R. García-García, J. Álvarez-Rodríguez, I. Rodríguez-Ramos and A. Guerrero-Ruiz, Carbon, 2010, 48, 267–276 CrossRef.
- S. F. Yin, B. Q. Xu, W. X. Zhu, C. F. Ng, X. P. Zhou and C. T. Au, Catal. Today, 2004, 93, 27–38 CrossRef.
- S. F. Yin, B. Q. Xu, C. F. Ng and C. T. Au, Appl. Catal., B, 2004, 48, 237–241 CrossRef CAS.
- M. Melchionna, S. Marchesan, M. Prato and P. Fornasiero, Catal. Sci. Technol., 2015, 5, 3859–3875 RSC.
- F. R. García-García, M. A. Rahman, I. D. González-Jiménez and K. Li, Catal. Today, 2011, 171, 281–289 CrossRef.
- J. P. S. Sousa, M. F. R. Pereira and J. L. Figueiredo, Appl. Catal., B, 2012, 125, 398–408 CrossRef CAS.
- R. P. Rocha, J. Restivo, J. P. S. Sousa, J. J. M. Órfão, M. F. R. Pereira and J. L. Figueiredo, Catal. Today, 2015, 241, 73–79 CrossRef CAS.
- M. Canal-Rodríguez, N. Rey-Raap, J. Á. Menéndez, M. A. Montes-Morán, J. L. Figueiredo, M. F. R. Pereira and A. Arenillas, Microporous Mesoporous Mater., 2019, 109811 Search PubMed.
- H. F. Gorgulho, F. Gonçalves, M. F. R. Pereira and J. L. Figueiredo, Carbon, 2009, 47, 2032–2039 CrossRef CAS.
- M. G. Plaza, F. Rubiera, J. J. Pis and C. Pevida, Appl. Surf. Sci., 2010, 256, 6843–6849 CrossRef CAS.
- R. W. Pekala, J. Mater. Sci., 1989, 24, 3221–3227 CrossRef CAS.
- Z. Shi, Y. Zhang, C. Cai, C. Zhang and X. Gu, Ceram. Int., 2015, 41, 1333–1339 CrossRef CAS.
- J. Rouquerol, F. Rouquerol, P. Llewellyn, G. Maurin and K. S. W. Sing, Adsorption by powders and porous solids: principles, methodology and applications, Elsevier Ltd, 2nd edn, 2014 Search PubMed.
- W. Kiciński, M. Bystrzejewski, M. H. Rümmeli and T. Gemming, Bull. Mater. Sci., 2014, 37, 141–150 CrossRef.
- M. S. Contreras, C. A. Páez, L. Zubizarreta, A. Léonard, S. Blacher, C. G. Olivera-Fuentes, A. Arenillas, J.-P. Pirard and N. Job, Carbon, 2010, 48, 3157–3168 CrossRef CAS.
- W. Shen, Z. Li and Y. Liu, Recent Pat. Chem. Eng., 2008, 1, 27–40 CrossRef CAS.
- M. S. Shafeeyan, W. M. A. W. Daud, A. Houshmand and A. Shamiri, J. Anal. Appl. Pyrolysis, 2010, 89, 143–151 CrossRef CAS.
- J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu and K. M. Thomas, Carbon, 1995, 33, 1641–1653 CrossRef CAS.
- K. Y. Kang, B. I. Lee and J. S. Lee, Carbon, 2009, 47, 1171–1180 CrossRef CAS.
- Q. Zhu, S. L. Money, A. E. Russell and K. M. Thomas, Langmuir, 1997, 13, 2149–2157 CrossRef CAS.
- J. Chen, Z. H. Zhu, S. Wang, Q. Ma, V. Rudolph and G. Q. Lu, Chem. Eng. J., 2010, 156, 404–410 CrossRef CAS.
- A. F. Zainul Abidin, K. S. Loh, W. Y. Wong and A. B. Mohamad, Int. J. Hydrogen Energy, 2019, 44, 28789–28802 CrossRef CAS.
- B. Stöhr, H. P. Boehm and R. Schlögl, Carbon, 1991, 29, 707–720 CrossRef.
- L. F. Mabena, S. S. Ray, S. D. Mhlanga and N. J. Coville, Appl. Nanosci., 2011, 1, 67–77 CrossRef CAS.
- F. R. García-García, E. Gallegos-Suarez, M. Fernández-García, A. Guerrero-Ruiz and I. Rodríguez-Ramos, Appl. Catal., A, 2017, 544, 66–76 CrossRef.
- I. C. Gerber and P. Serp, Chem. Rev., 2020, 120(2), 1250–1349 CrossRef CAS PubMed.
- H. Jin, H. Zhang, H. Zhong and J. Zhang, Energy Environ. Sci., 2011, 4, 3389–3394 RSC.
- L. Perini, C. Durante, M. Favaro, V. Perazzolo, S. Agnoli, O. Schneider, G. Granozzi and A. Gennaro, ACS Appl. Mater. Interfaces, 2015, 7, 1170–1179 CrossRef CAS PubMed.
- W. Kiciński, M. Szala and M. Nita, J. Sol-Gel Sci. Technol., 2011, 58, 102–113 CrossRef.
- A.-L. Peikolainen, M. Uibu, J. Kozlova, H. Mändar, A. Tamm and A. Aabloo, Carbon Trends, 2021, 3, 100037 CrossRef.
- B. Tomić-Tucaković, D. Majstorović, D. Jelić and S. Mentus, Thermochim. Acta, 2012, 541, 15–24 CrossRef.
- P. Arnoldy, J. C. M. De Jonge and J. A. Moulijn, J. Phys. Chem., 1985, 89, 4517–4526 CrossRef CAS.
- K. H. Carpenter, G. E. Whorley and T. W. Lennard, 1985.
- N. Fischer, E. Van Steen and M. Claeys, Catal. Today, 2011, 171, 174–179 CrossRef CAS.
- L. Torrente-Murciano, A. K. Hill and T. E. Bell, Catal. Today, 2017, 286, 131–140 CrossRef CAS.
- Z. H. Zhong and K. I. Aika, J. Catal., 1998, 173, 535–539 CrossRef CAS.
- S. Mazzone, T. Goklany, G. Zhang, J. Tan, E. I. Papaioannou and F. R. García-García, Appl. Catal., A, 2022, 118484 CrossRef CAS.
- M. García-Vázquez, G. Zhang, Z. Hong, X. Gu and F. R. García-García, Chem. Eng. J., 2020, 396, 125379 CrossRef.
- F. R. García-García and K. Li, Appl. Catal., A, 2013, 456, 1–10 CrossRef.
- L. Lelong and R. Rastoin, Fuel Cell, https://energies.airliquide.com/resources-planet-hydrogen/fuel-cell#:%7E:text=PEM%20fuel%20cells%20consume%20about,for%20every%20100%20miles%20covered, accessed 20 November 2021 Search PubMed.
- W. Tsai and W. H. Weinberg, J. Phys. Chem., 1987, 91, 5302–5307 CrossRef CAS.
- M. Burt, New Mirai hydrogen fuel cell electric vehicle – under the skin, https://mag.toyota.co.uk/new-mirai-hydrogen-fuel-cell-electric-vehicle/, accessed 20 November 2021 Search PubMed.
| This journal is © The Royal Society of Chemistry 2022 |