
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The potential scarcity, or not, of polymeric overall water splitting photocatalysts†
Benedict
Saunders
ab,
Liam
Wilbraham
ac,
Andrew W.
Prentice
 a,
Reiner Sebastian
Sprick
d and
Martijn A.
Zwijnenburg
*a
a,
Reiner Sebastian
Sprick
d and
Martijn A.
Zwijnenburg
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: m.zwijnenburg@ucl.ac.uk
bDepartment of Chemistry, University of Warwick, Coventry CV4 7SH, UK
cExscientia, Oxford Science Park, Oxford, OX4 4GE, UK
dDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK
First published on 21st March 2022
Abstract
We perform a high-throughput virtual screening of a set of 3240 conjugated alternating binary co-polymers and homo-polymers, in which we predict their ability to drive sacrificial hydrogen evolution and overall water splitting when illuminated with visible light. We use the outcome of this screening to analyse how common the ability to drive either reaction is for conjugated polymers loaded with suitable co-catalysts, and to suggest promising (co-)monomers for polymeric overall water splitting catalysts.
Introduction
In recent years organic materials, particularly conjugated polymers, have come to the foreground as potential photocatalysts for splitting water into molecular hydrogen and oxygen.1–5 The hydrogen produced in this way would be renewable and green, emitting nothing but water upon combustion, making it a viable candidate for a sustainable replacement of current carbonaceous fuels. While more than a hundred polymers and other organic materials are known to be experimentally active for photocatalytic hydrogen evolution when studied in the presence of a metal co-catalyst and a sacrificial electron donor (SED),2,6,7 a much smaller number has been observed to oxidise water in the presence of a co-catalyst and a sacrificial electron acceptor (SEA),8–11 and a smaller number still to drive overall water splitting when illuminated by solar light.2,12–17 Examples of the latter include carbon nitride,12,13,16 covalent triazine-based framework materials14,15 and linear poly(dibenzo[b,d]thiophene sulfone).17 Here, we computationally screen a very large dataset of organic (co-)polymers to understand if this apparent experimental scarcity of organic materials that, under illumination with visible light, can oxidise water and drive the overall splitting of water is due to thermodynamic constraints inherent to the material class or is kinetic in nature. In the latter case materials that appear inactive could potentially become active in the presence of the right co-catalyst(s) or when part of a heterojunction, accelerating otherwise sluggish water oxidation kinetics and/or minimising undesired electron–hole recombination.From a thermodynamic perspective, for a material to reduce protons and oxidise water when illuminated, the material's ionisation potential (IP, valence band maximum) and electron affinity (EA, conduction band minimum) should at least straddle the solution proton reduction and water oxidation potentials, the latter defined – as is convention – in terms of the potential of the equivalent reduction reaction (see Fig. 1a). Similarly, to reduce protons and oxidise a SED or oxidise water and reduce a SEA the material's IP and EA should straddle the proton reduction and SED oxidation or the SEA reduction and water oxidation potentials, respectively, once again defining the oxidation potentials in terms of that of the equivalent reduction half-reactions. It is highly desirable not only for the material to be active, but to be active under visible light, as most of the sunlight is concentrated in this part of the spectrum. Therefore, the material's optical gap, the energy below and wavelength above which the material is transparent to light, needs to be as small as possible. However, at the same time the materials' optical gap needs to be larger than the difference between the potentials of the relevant solution reduction and oxidation half-reactions, 1.23 eV in the case of overall water splitting. Otherwise, the photons would not deliver enough energy for the relevant overall reaction to be thermodynamically favourable. The fact that the exciton binding energy in organic materials, even after immersion in water, is not negligible and that the excitons, excited electron–hole pairs, formed initially by the absorption of light do not spontaneously dissociate into free electrons and holes away from the polymer particle–solution interface complicates the above picture slightly.18 Conceptually, in the case of materials with a non-negligible exciton binding energy we need to consider besides IP and EA also IP* and EA*, the ionisation potential and electron affinity of the excitons, respectively.19,20 This would allow us to describe exciton dissociation on the polymer particle–solution interface, where the exciton donates an electron or hole to drive one of the solution half-reactions, after which the other component of the exciton remains on the particle to take part in another solution half-reaction at some later point. However, in practice when screening materials the effect of the exciton binding energy, typically 0.1–0.2 eV18 can be absorbed in a requirement for a similar overpotential on top of the thermodynamic solution potentials, meaning that the computationally expensive calculation of IP*/EA* can be avoided. Other properties than a material's IP, EA and optical gap, such as its wettability and dispersibility in water or its particle size distribution, will clearly influence its photocatalytic activity. However, if a material is thermodynamically unable to oxidise water or reduce protons, or doesn't absorb sun light, it simply won't act as a water splitting photocatalyst, irrespective of how well it's wetted by or disperses in water or how big or small the particles are. This binary quality of IP, EA and the optical gap, makes them key parameters to screen when trying to understand how common the ability to photocatalytically split water is in a class of materials such as conjugated polymers.
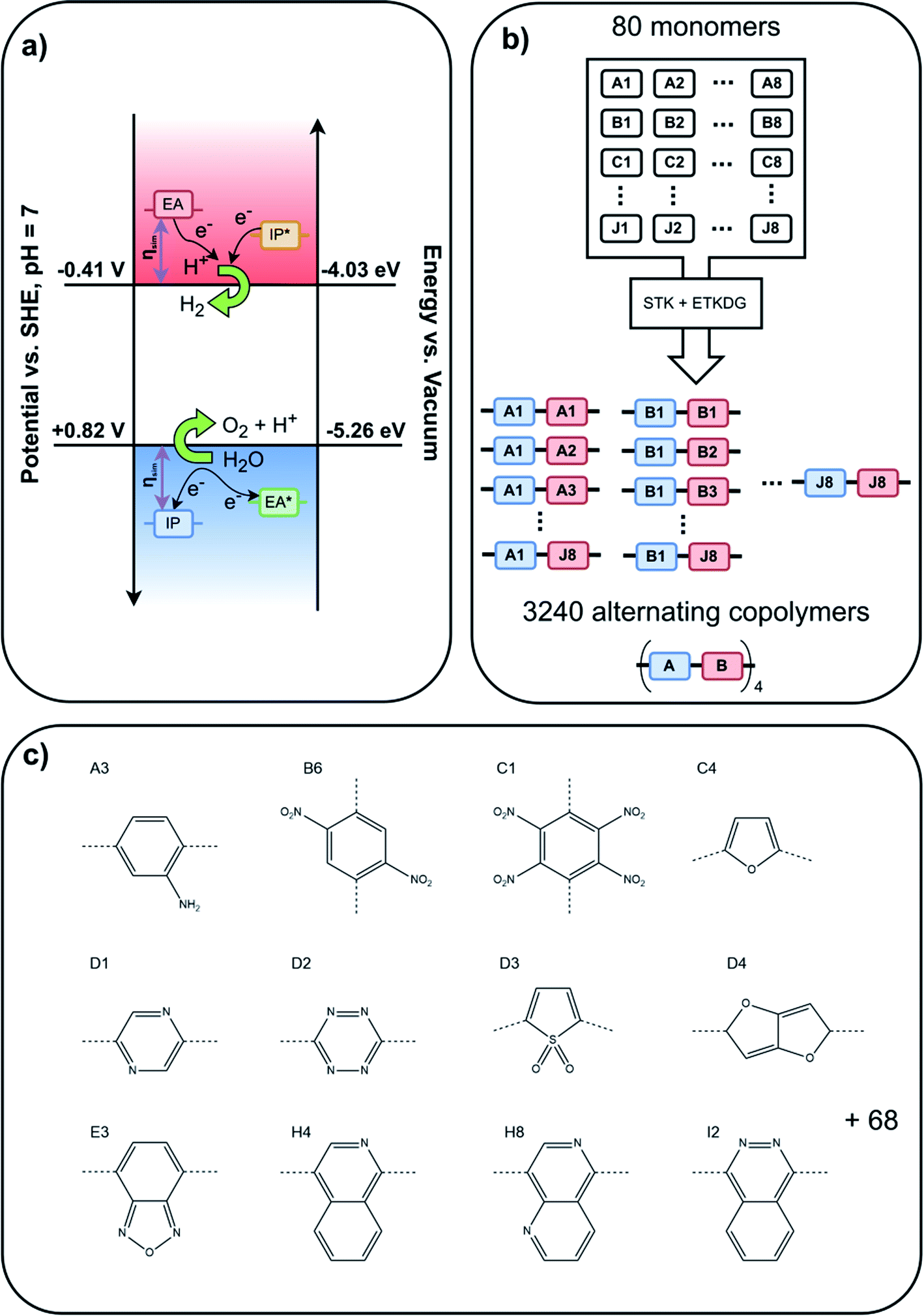 | ||
| Fig. 1 (a) Schematic illustration of the alignment of IP and EA, and their excited state counterparts IP* and EA*, relative to the potentials for proton reduction and water oxidation required for overall water splitting, (b) oligomer assembly methodology, (c) subset of the 80 monomers studied herein, where the dashed lines indicate how the monomers are linked to other monomers in the (co)polymers (all 80 monomers are shown in Fig. S1 in the ESI†). | ||
Previous work by our group demonstrated that density functional theory (DFT) calculations on a single polymer chain embedded in a dielectric continuum can accurately predict the IP and EA values of dry conjugated polymer solids measured by experimental photoelectron spectroscopy when using a relative dielectric permittivity of 2 (organic solid) for the continuum.21,22 We also showed that these DFT predicted IP and EA values, when using a relative dielectric permittivity of 80.1 (water), can be successfully used to explain the trends in the activity of such polymeric solids for sacrificial hydrogen evolution from water/SED mixtures7,23–26 and sacrificial oxygen evolution from water/SEA mixtures.11 However, calculating the IP and EA, as well as optical gap of thousands of polymers, with DFT and its time-dependent extension (TD-DFT) would be prohibitively computationally expensive. Conveniently, we demonstrated previously that calculations using the xTB family of tight-binding DFT methods,27 developed by Grimme and co-workers, after calibration to (TD-)DFT results give very similar results for a fraction of the computational cost28 and allow for the efficient calculation of properties such as IP, EA and optical gap, of tens of thousands of molecules and/or polymers.6,29–31 The xTB calculations, GFN1-xTB32 for ground-state geometries, IPEA-xTB33 for IP and EA, and sTDA-xTB34 for the optical gap, are, just like the DFT calculations discussed above, performed on a single polymer chain, embedded in the case of GFN1-xTB and IPEA-xTB in a dielectric continuum with the relative dielectric permittivity of water. As after calibration the IPEA-xTB predicted IP and EA values closely reproduce the equivalent DFT predicted values, these calibrated IPEA-xTB IP and EA values inherit the ability to rationalise and thus predict trends in the hydrogen and oxygen evolution activity of polymeric solids. Using calibrated xTB we predict the IP, EA and optical gap values of a dataset consisting of 3240 conjugated alternating binary co-polymers and homo-polymers built from a library of 80 monomers, see Fig. 1b and c, and analyse what fraction of the dataset can evolve hydrogen in the presence of a commonly used SED, triethylamine (TEA), and what fraction of polymers can drive the overall splitting of water. We discuss how these numbers change when we require the polymer to work using visible light and, finally, present the monomers which are overrepresented in the polymers that are active in the visible. Such monomers would make good candidates to explore experimentally when synthesising new polymer photocatalysts.
Methodology
We assembled 3240 oligomers from all possible binary AB combinations of the library of 80 monomers, including 80 homo-oligomers (see Fig. 1b). A selection of the monomers is shown in Fig. 1c; the full library is displayed in Fig. S1 in the ESI.† For each oligomer, a SMILES representation was generated using the stk35,36 python package, such that each oligomer contained 16 individual monomers (8-AB- repeat units for the alternating co-polymers, 16-A- repeat units for the homo-polymers). During the computational assembly of the oligomers in stk bromine atoms are used to control where monomers are coupled together and therefore Fig. S1† shows the monomers in their dibromo form. The same dibromo versions of the monomers can be used experimentally to synthesize the corresponding polymers via Suzuki coupling. For asymmetric monomers only one of the possible two orientations was considered during the polymer assembly. As SMILES strings contain no explicit geometry information, a subsequent conformer search was required which we performed using the ETKDG37 algorithm as implemented in RDKit,38 a cheminformatics library, generating 500 conformers for each oligomer and optimising each with the MMFF94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 39 forcefield. The conformer with the lowest forcefield energy was selected and optimised further with GFN1-xTB,32 as implemented in xtb 5.6.4SE,40 in preparation for the calculation of IP, EA and optical gap. The total workflow was automated in a locally modified version41 of the polyHTS code.42
39 forcefield. The conformer with the lowest forcefield energy was selected and optimised further with GFN1-xTB,32 as implemented in xtb 5.6.4SE,40 in preparation for the calculation of IP, EA and optical gap. The total workflow was automated in a locally modified version41 of the polyHTS code.42
The IPEA-xTB32 variant of xTB was used to predict the vertical IP and EA values of the oligomer, with solvation in water represented by the generalised Born surface area (GBSA) model.43 The singlet excitation spectra of the oligomers were calculated with the sTDA-xTB34 method, the lowest of which was taken to be the optical gap. The sTDA-xTB calculations do not include solvation as that is not implemented for sTDA-xTB. The GFN1/IPEA/sTDA-xTB predicted IP, EA and optical gap values for each oligomer were then transformed to values that can be directly compared to previous (TD-)DFT predictions through a linear scaling model, calibrated in prior work by us28 to (TD-)DFT results for oligomers immersed in water. An independent linear model was used for each of the three properties, see Table S1† for the model parameters used. The mean average deviation (MAD) of these models based on the initial training set were 0.08, 0.06 and 0.13 (e)V for IP/EA/optical gap respectively, increasing to 0.16/0.14/0.16 (e)V for a validation set of polymers not included in the training set. These MAD values are reasonable such that the calibration should be transferrable to the wide range of polymers considered herein.
Some of the monomers considered contain groups that can be protonated at sufficiently low pH values, such as the aromatic nitrogen atoms in aniline, diazine, benzopyridine and naphthyridine based monomers (e.g. monomers A3, D1, H4 and H8 in Fig. 1, respectively). We are unable to consider the effect of protonation of these groups in a high-throughput fashion and hence neglect the effect of protonation in the screening. Considering the typical pKa values of the conjugate acids of such groups, this is only an approximation at pH values less than ∼ pH 6, above which such groups will generally not be protonated.
A locally adapted version44 of the MolZ python package45 allows us to query our dataset and assign a z-score to each of the 80 monomers, which provides an insight into the contribution of each monomer to the queried properties. To calculate the z-score we describe the statistics of the scenario where we have a dataset of N polymers, k of which contain a particular monomer i, and n polymers in a sub-set of the dataset with desirable properties, p of which contain i, in terms of a hypergeometric distribution with a mean (![[x with combining macron]](https://www.rsc.org/images/entities/i_char_0078_0304.gif) ) and variance (σ2) of:
) and variance (σ2) of:
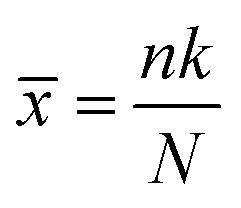

From the mean and variance of the distribution we can then calculate the z-score for a particular value of p via:
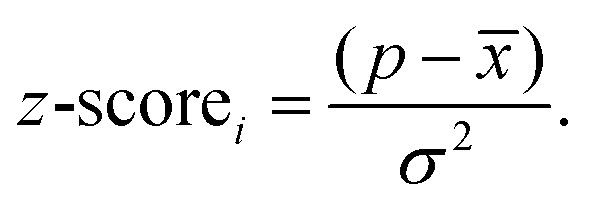
Large positive z-scores indicate that monomer i is overrepresented in the sub-set of polymers relative to other monomers and hence likely a good monomer choice for achieving the properties representative of that subset.
Results and discussion
First, we analyse what fraction of the 3240 polymers can evolve hydrogen in the presence of TEA as the SED. As can be seen from Fig. 2a more than 95% of the polymers in the dataset, even if limiting ourselves to polymers with an absorption onset in the visible region (Fig. 2b), are predicted to be thermodynamically able to drive the reduction of protons and the 2-electron oxidation of TEA to diethylamine and acetaldehyde at pH 12.3 (−0.76 V), which is the experimentally determined pH of the mixture of TEA, water and methanol used in photocatalysis experiments.46 Next, we repeated the analysis, considering the effect of requiring an additional overpotential on top of the equilibrium potentials of each of the half-reactions. This (i) allows us to probe the sensitivity of our predictions and, as discussed in the introduction, compensate for focussing only on IP and EA, and (ii), take into account that an additional potential on top of the equilibrium potentials might be required for meaningful hydrogen evolution and TEA oxidation rates. As can be seen from Fig. 2, the percentage of polymers able to drive both half-reactions decreases steadily when we require such an additional overpotential. Still for an overpotential value of, for example, 0.3 V, proton reduction and 2-electron oxidation of TEA is predicted to be favourable for more than 80% of the dataset. | ||
| Fig. 2 Percentage of (co-)polymers in the dataset that are predicted to be able to drive the reduction of protons and the two-electron (orange line) and one-electron oxidation of TEA (blue line) at pH 12.3 for different optical gap ranges. Panel (a) does not include a limitation on the optical gap. Panel (b) introduces a limitation such that the predicted optical gap must be ≤ 3.5 eV, which is reduced to 2.5 eV in plot (c) and finally 2.0 eV in plot (d). The grey dashed lines in panels (b–d) indicate the number of polymers in the entire dataset that fall within each optical gap constraint. See Fig. S2† for a zoomed in version of (a–d). | ||
Proton reduction and hydrogen–hydrogen bond formation is typically catalysed by added platinum nanoparticles or palladium nanoparticles remaining from the catalyst used in polymer synthesis.47,48 Polymers can also potentially act as a catalyst for proton reduction themselves but in the presence of palladium or platinum nanoparticles the activity of those will very likely outcompete any inherent catalytic activity of the polymer.49 In contrast, TEA oxidation most likely is not catalytic, and probably takes the form of two sequential out-of-sphere electron-transfer steps between the polymer and TEA. First TEA gets oxidised to TEA+, which after deprotonation, gets oxidised in the second electron-transfer step to diethylamine and acetaldehyde, the expected 2-electron oxidation products. To efficiently oxidise TEA a polymer's IP should thus probably not only be more positive than the potential of the half-reaction associated with the 2-electron oxidation of TEA but also be more positive than that for the 1-electron oxidation of TEA (0.64 V at pH 12.3), as otherwise the 1-electron oxidation step might act as a thermodynamic barrier.25 Indeed, we observed in previous combined computational and experimental studies that polymers which are experimentally highly active for sacrificial hydrogen evolution in the presence of TEA have such predicted positive IP values.7,24–26,28
Going back to Fig. 2a we can see that a smaller percentage of the dataset can drive both proton reduction and the 1-electron oxidation of TEA at pH 12.3, between 60 and 70% in the absence of an overpotential requirement and approximately 40% for an overpotential of 0.3 V. Fig. 2c and d finally demonstrate that if we require a polymer not only to be able to reduce protons and oxidise TEA but also to have an optical gap that is small enough to absorb a large part of the visible spectrum the percentages get truly small. This reduction, especially in the case when considering the 2-electron oxidation of TEA and proton reduction, is mainly the direct results of the optical gap restriction. Of the 3240 polymers in the dataset 3197 (98.7%) are predicted to have an optical gap below 3.5 eV, 1184 (36.5%) an optical gap below 2.5 eV and only 267 (8.2%) to have an optical gap below 2.0 eV (see dashed lines in Fig. 2b–d). However, when considering the 1-electron oxidation of TEA and proton reduction there is a clear further reduction, which probably stems from the fact that polymers with a small optical gap also will have a small fundamental gap, the difference between IP and EA, which might straddle the potentials for the 2-electron oxidation of TEA and proton reduction but not those for the 1-electron oxidation of TEA and proton reduction.
Fig. 3 shows the case of overall water splitting but now as a function of pH. For neutral water, pH 7, more than 50% of polymers are predicted to straddle the proton reduction and water oxidation potentials, which when requiring, for example, an additional 0.3 V overpotential for both half-reactions reduces to a still respectable more than 30%. This 30% includes poly(dibenzo[b,d]thiophene sulfone) (P10, monomer J8, polymer 3240), the linear conjugated homopolymer known experimentally to be active for overall water splitting in the presence of an iridium oxide co-catalyst,17 as well as those linear conjugated polymers that have been previously reported in the literature11 to be experimentally active for sacrificial water oxidation using a cobalt co-catalyst and a silver salt as the SEA. In addition to the P10 homopolymer, mentioned above in the context of overall water splitting, Suzuki coupled co-polymers of 1,4-dibromophenylene and 2,5-dibrompyridine (P24, monomers C6 and C7, polymer 1472) and 1,4-dibromophenylene and 2,5-dibromopyrazine (P28, monomers C6 and D1, polymer 1474) are present within this subset. Conversely, the poly(p-phenylene) (P1, monomer C6, polymer 1471) and poly(thiophene) (P17, monomer C5, polymer 1411) homopolymers, which experimentally show no sign of sacrificial water oxidation activity under the same conditions, are not included in the 30 or 50% because their IP values are predicted to be not sufficiently positive to oxidise water. The fact that our approach predicts polymers known to be active for sacrificial water oxidation to have IP values that are sufficiently positive to oxidise water, even when requiring an additional overpotential, and polymers known to be inactive to have IP values that are not sufficiently positive, gives us added confidence in our approach.
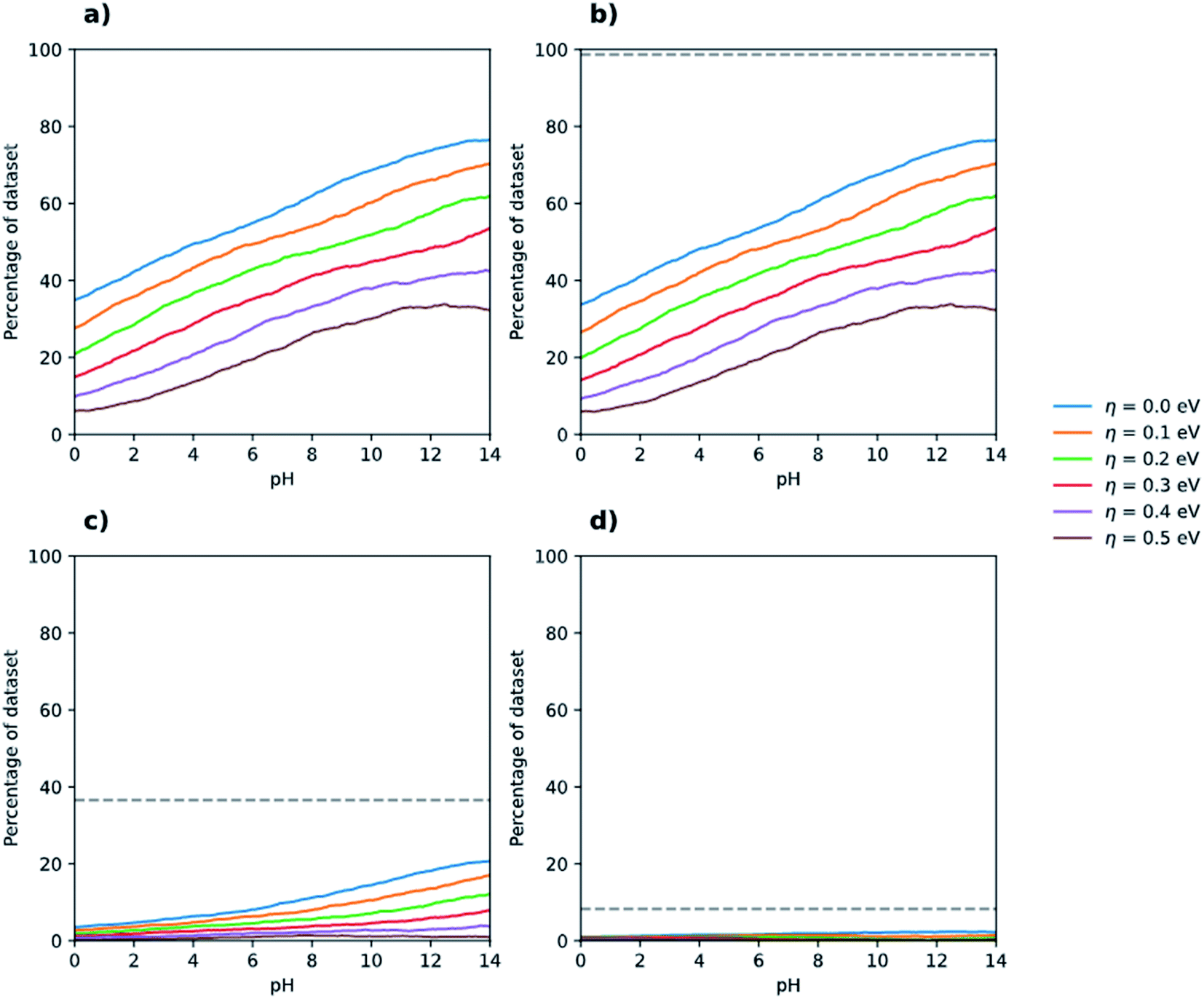 | ||
| Fig. 3 Percentage of (co-)polymers in the dataset that are predicted to be able to drive the overall splitting of water as a function of pH for different overpotential values and optical gap ranges. Panel (a) includes all polymers predicted to have an optical gap between 0.0 and 3.5 eV, panel (b) those with an optical gap between 1.23 eV and 3.5 eV f, panel (c) those with an optical gap between 1.23 eV and 2.5 eV, and 1.23 eV and 2.0 eV for panel (d). The grey dashed lines in panels (b–d) indicate the number of polymers in the entire dataset that fall within each optical gap constraint. See Fig. S3† for zoomed in version of (a–d). | ||
The fraction of polymers able to both reduce protons and oxidise water, for each overpotential value, increase steadily with pH. At pH 12.3, the same pH as for when using TEA as SED, we predict that approximately 70% of polymers straddle the proton reduction and water oxidation potentials and when requiring an additional 0.3 V overpotential still more than 40% do so. These numbers are surprisingly similar to those discussed above for the case of where TEA instead of water is used as electron donor. Also, similar to when TEA is used as the SED, Fig. 3c and d show that if we require additionally for the polymer to have an optical gap small enough to absorb a large part of the visible spectrum relatively few polymers remain. Of the 3240 polymers in the dataset 3141 (96.9%) are predicted to have an optical gap between 1.23 and 3.5 eV, 1128 (34.8%) an optical gap between 1.23 and 2.5 eV and only 211 (6.5%) to have an optical gap below 2 but above 1.23 eV (see dashed lines in Fig. 3b–d). Moreover, just as for the case of the 1-electron oxidation of TEA and proton reduction, discussed above, only a fraction of those polymers with an optical gap between 1.23 and 2.5 eV and 1.23 and 2.0 eV will straddle the water splitting potentials. Even if all polymers with an optical gap between 1.23 and 2.5 eV or 1.23 and 2.0 eV would have an optical gap larger than 1.23 V, their IP and EA still might not straddle the water oxidation and proton reduction potentials because their IP is too deep or their EA to shallow. In contrast, polymers with a large optical gap will likely also have large fundamental gaps and thus will span the water splitting potentials for most realistic IP and EA values.
The origin of the experimental scarcity of polymers that split water appears thus not to find its origin in any thermodynamic limitations of the material class. Many polymers are known to evolve hydrogen in the presence of TEA and very few to perform overall water splitting, even if in terms of thermodynamics similar numbers of polymers can drive both half-reaction pairs. We thus propose that the origin of the scarcity of polymer photocatalysts for overall water splitting is instead kinetic in nature. We previously19 speculated this to be the case for specific polymers such as poly(pyridine) based on calculations on poly(p-phenylene) and it. However, at that time it was computationally intractable to accurately calculate the properties for the large number of polymers required to perform adequate statistics, and thus demonstrate how common a property the thermodynamic ability to overall split water is for polymers, making the case for kinetics even stronger.
Overall TEA oxidation involves two electrons (holes) and water oxidation four, and while after the first electron transfer step TEA+ likely diffuses away from the polymer, reducing the chance of the back electron transfer, the water oxidation intermediates probably stay adsorbed on the co-catalyst surface until oxygen is formed, making back electron transfer more likely. Non-catalytic water oxidation by means of out-of-sphere electron transfer in analogy to the case of TEA is very unlikely because of the extreme potential of the half-reaction associated with the initial one-hole oxidation of water (2.15 V at pH 7). In the case of a sluggish water-oxidation co-catalyst, electron–hole recombination hence likely outcompetes water oxidation. Similarly, sacrificial electron acceptors used in the case of sacrificial water oxidation, e.g., Ag+ and Ce4+, generally are thermodynamically easier to reduce than protons, i.e., have a more positive reduction potential, and only involve one rather than two electrons, meaning that in the presence of a sluggish water-oxidation co-catalyst, sacrificial water oxidation might be possible but overall water splitting not. The good news associated with this scenario is that this would mean that by choosing the right water oxidation co-catalyst a significant number of polymers should be active for overall water splitting. Indeed, experimentally the small number of polymers active for overall water splitting, were generally first reported to be only active for sacrificial water oxidation8,10 and only after optimisation of the water oxidation co-catalyst became active for overall water splitting.13,15 It would also suggest that combining polymers or polymers and small molecule acceptors together to make heterojunctions, splitting the excitons, and separating electrons and holes on separate sub-systems, reducing electron–hole recombination, should be a worthwhile strategy. As a matter of fact, bulk heterojunctions have been shown to yield promising results in the case of sacrificial hydrogen evolution.50 Even in the presence of a heterojunction, however, the presence of efficient hydrogen and oxygen evolution co-catalysts is probably beneficial in terms of the long-term stability of the polymer. Stability is not generally an issue in sacrificial hydrogen evolution with continuous hydrogen evolution for multiple days demonstrated for selected polymer materials without obvious signs of material degradation during this time.22–24 However, the build-up of charges in the polymer,51 especially holes, during overall water splitting in the presence of a sluggish water oxidation co-catalyst could result in material degradation and loss of activity. Reversely, a very active water oxidation co-catalyst, which very efficiently extracts charges from the polymer, might result in very stable polymer photocatalysts under overall water splitting conditions. For this reason, we also rule out that stability or lack thereof is the/a reason for the scarcity of overall water splitting photocatalysts.
One possible hurdle to efficient overall water splitting with polymer photocatalysts, even in the presence of optimal co-catalysts, is that relatively few (co-)polymers are, as discussed above, predicted to have the required driving force for proton reduction and water oxidation and absorb green to red light. However, it is important to stress here that such (co-)polymers are predicted to exist, discussed in more detail below, and this is not an issue unique to polymeric photocatalysts. As a matter of fact, many state-of-the-art inorganic solid-state photocatalysts, such as doped SrTiO3, only absorb blue, violet and ultraviolet light52,53 and developing systems that are active on the red-side of the visible spectrum is also a challenge for inorganic materials.52,53
Next, we analysed, by means of z-scores, which monomers are overrepresented amongst those binary AB co-polymers that can drive overall water splitting and absorb a large part of the visible spectrum. Fig. 4a and b show the monomers with the highest z-score as a function of pH for polymers that can drive overall water splitting with an overpotential of 0.3 V and a predicted optical gap between 1.23 eV and 2.5 eV or 1.23 eV and 2.0 eV, respectively. Below we discuss the z-scores for the case of fresh water, pH 6–8, in detail. Analysis for water splitting under alkaline and acidic conditions can be found in the ESI.† For pH 6–8, 4,7-coupled dibromobenzo[c][1,2,5]oxadiazole (E3, see Fig. 1c) and 2,5-coupled dibromothiophene-1,1-dioxide (D3, see Fig. 1c) have relatively high z-scores for polymers with an optical gap below 2.5 eV. E3 occurs in 35 out of 107 co-polymers that can drive overall water splitting at pH 7 and have an optical gap of less than 2.5 eV, while D3 occurs in 19 out of the 107. For polymers with an optical gap below 2.0 eV, D3, 3,6-coupled 1,2,4,5-tetrazine (D2, see Fig. 1c), 1,4-coupled 2,5-dinitrobenzene (B6, see Fig. 1c), and 1,4-coupled isoquinoline (H4, see Fig. 1c) have corresponding high z-scores, although that is skewed by the fact that only 8 out of the 3240 polymers are predicted to have an optical gap of less than 2 eV and can drive overall water splitting with an overpotential of 0.3 V at pH 7.
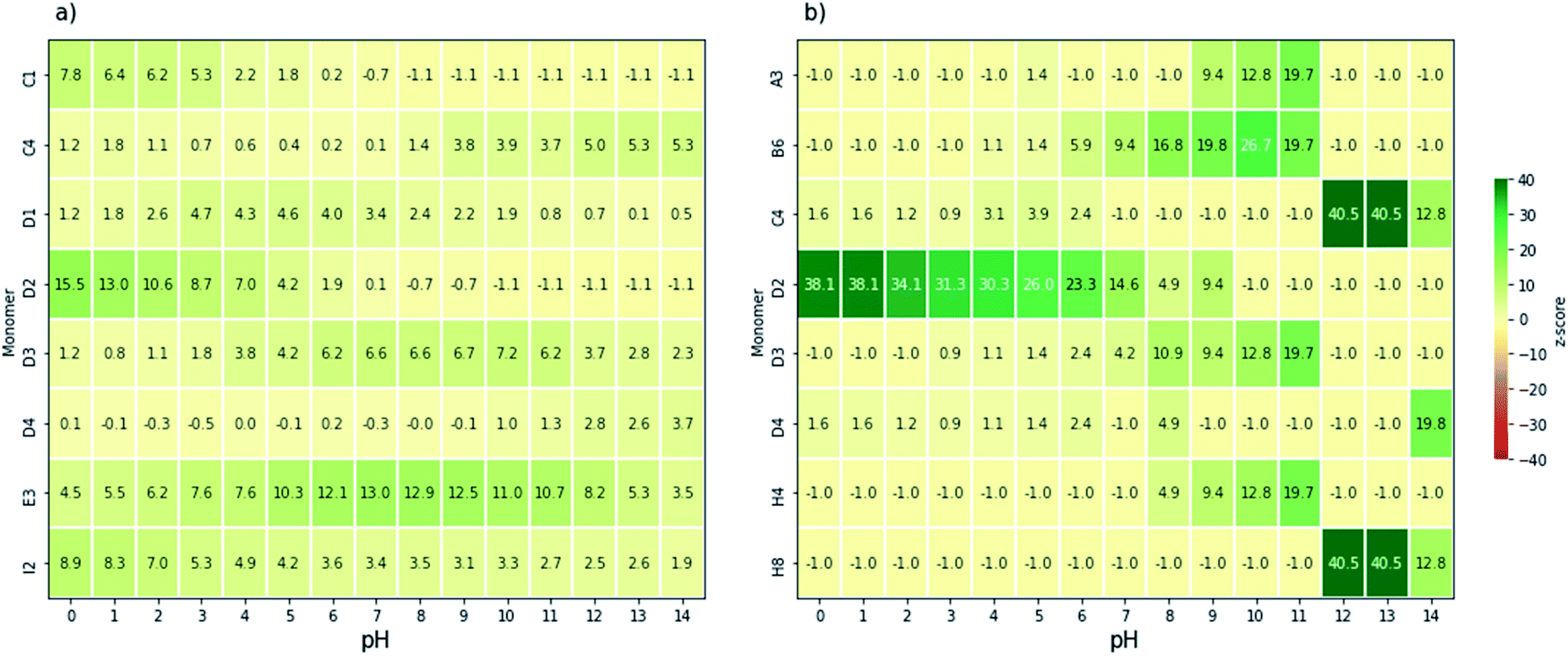 | ||
| Fig. 4 Heatmaps of the monomers with the highest z-score for watersplitting with overpotentials of 0.3 V, constrained by optical gap ranges (a) 1.23–2.5 eV and (b) 1.23–2.0 eV. The monomers which feature here correspond to those illustrated in Fig. 1c. | ||
Polymers based on E3 and D3, for which a large number of (co)-polymers are predicted to be able to drive overall water splitting at pH 7 and have an optical gap < 2.5 eV: 44% of the 80 (co-)polymers containing E3 and 18% of the (co-)polymers containing D3 in the dataset, should make especially good choices for future synthetic studies towards overall water splitting polymer photocatalysts. Such co-polymers can be obtained, for example, by Suzuki coupling the dibromo versions of E3 and D3 with suitable dibromo co-monomers. Promising leads in term of monomers to co-polymerise E3 with via Suzuki coupling to yield polymers predicted to be able to split water at neutral pH include 1,4-dibromo-2-fluorobenzene (A2), 2,5-dibromobenzonitrile (A4), 1,4-dibromo-2,5-difluorobenzene (A6), 1,4-dibromo-2-nitrobenzene (A8), 1,4-dibromo-2-(trifluoromethyl)benzene (B1), 2,5-dibromoterephthalonitrile (B2), 1,4-dibromo-2,5-bis(trifluoromethyl)benzene (B7), 2,5-dibromopyridine (C7), 2,5-dibromopyrimidine (C8), 2,5-dibromopyrazine (D1), and most nitrogen substituted naphtalenes (e.g. E6 and F2). Similarly, promising co-monomers to obtain active polymers via Suzuki coupling of D3 include A8, B1, B2, B7, C8, D1, 1,4-dibromo-2,5-dinitrobenzene (B6) and a variety of naphtalenes substituted with two nitrogens (e.g. F3 and F8). 1,4-dibromo-2,3,5,6-tetranitrobenzene (C1) is also predicted to be a promising co-monomer for D3. However, it is likely to be explosive and best avoided for that reason.
Fig. 5, finally, shows the 14 polymers with the lowest > 1.23 eV optical gap of the subset of polymers that can drive overall water splitting at pH 7 with an overpotential of 0.3 V. Just like the co-polymers of E3 and D3 discussed above, some of which are also included in this 14, these, with exception of the D3-C1 co-polymer for the reasons outlined above, are promising synthesis targets for future work on overall water splitting polymer photocatalysts.
 | ||
| Fig. 5 The 14 polymers with the lowest > 1.23 eV optical gap of the subset of polymers that can drive overall water splitting at pH 7 with an overpotential of 0.3 V. | ||
Conclusions
Thermodynamically a significant number of conjugated polymers are predicted to be able to drive overall water splitting, a similar number as polymers able to drive hydrogen evolution when oxidising triethylamine as a sacrificial electron donor. As many polymers are known experimentally to evolve hydrogen in the presence of triethylamine, we argue that the scarcity of polymers performing overall water splitting is not due to any thermodynamic constraints but rather probably has a kinetic origin, with overall water splitting requiring an efficient co-catalyst for the water oxidation. Moreover, in the presence of such co-catalysts, it is likely that a significant number of polymers that are very active for sacrificial hydrogen evolution, will also be active for overall water splitting. Considering the rapid development of polymer photocatalysts and increase in sacrificial hydrogen production rates we believe that now more effort must be made to study water oxidation with polymer photocatalysts, focusing on developing suitable co-catalysts, given that this is currently a bottleneck in developing systems for overall water splitting and our work suggests that this is a realistic target. A large hurdle for efficient overall water splitting with visible light, finally, is found to be the fact that only a relatively small number of the co-polymers studied are predicted to absorb yellow to red light. However, this is similarly an issue for inorganic photocatalysts, with many state-of-the-art inorganic photocatalysts, e.g. SrTiO3, only absorbing blue, violet and ultraviolet light.Author contributions
BS performed the calculations and analysed the results. BS, LW and MAZ conceived the study. LW assisted in the z-score calculations. AWP assisted in the thermodynamic analysis and RSS provided a synthetic perspective. MAZ supervised the work. All authors contributed to the writing of the manuscript and have approved the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Yang Bai, Dr Isabelle Heath-Apostolopoulos and Prof. Andrew Cooper for discussion and acknowledge the UK Engineering and Physical Sciences Research Council (EPSRC) and the Leverhulme Trust for funding this work through grants EP/N004884/1 and RPG-2019-209, respectively. R. S. S thanks the University of Strathclyde for financial support through The Strathclyde Chancellor's Fellowship Scheme.References
- V. S. Vyas, V. W.-h. Lau and B. V. Lotsch, Soft Photocatalysis: Organic Polymers for Solar Fuel Production, Chem. Mater., 2016, 28, 5191–5204 CrossRef CAS.
- Y. Wang, A. Vogel, M. Sachs, R. S. Sprick, L. Wilbraham, S. J. A. Moniz, R. Godin, M. A. Zwijnenburg, J. R. Durrant, A. I. Cooper and J. Tang, Current Understanding and Challenges of Solar-Driven Hydrogen Generation Using Polymeric Photocatalysts, Nat. Energy, 2019, 4, 746–760 CrossRef CAS.
- J. Kosco, F. Moruzzi, B. Willner and I. McCulloch, Photocatalysts Based on Organic Semiconductors with Tunable Energy Levels for Solar Fuel Applications, Adv. Energy Mater., 2020, 10, 2001935 CrossRef CAS.
- T. Banerjee, F. Podjaski, J. Kröger, B. P. Biswal and B. V. Lotsch, Polymer Photocatalysts for Solar-to-Chemical Energy Conversion, Nat. Rev. Mater., 2021, 6, 168–190 CrossRef CAS.
- Y. Bai, K. Hippalgaonkar and R. S. Sprick, Organic Materials as Photocatalysts for Water Splitting, J. Mater. Chem. A, 2021, 9, 16222–16232 RSC.
- Y. Bai, L. Wilbraham, B. J. Slater, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Accelerated Discovery of Organic Polymer Photocatalysts for Hydrogen Evolution from Water through the Integration of Experiment and Theory, J. Am. Chem. Soc., 2019, 141, 9063–9071 CrossRef CAS PubMed.
- C. B. Meier, R. Clowes, E. Berardo, K. E. Jelfs, M. A. Zwijnenburg, R. S. Sprick and A. I. Cooper, Structurally Diverse Covalent Triazine-Based Framework Materials for Photocatalytic Hydrogen Evolution from Water, Chem. Mater., 2019, 31, 8830–8838 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, A Metal-Free Polymeric Photocatalyst for Hydrogen Production from Water under Visible light, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. Bi, W. Fang, L. Li, J. Wang, S. Liang, Y. He, M. Liu and L. Wu, Covalent Triazine-Based Frameworks as Visible Light Photocatalysts for the Splitting of Water, Macromol. Rapid Commun., 2015, 36, 1799–1805 CrossRef CAS PubMed.
- Z.-A. Lan, Y. Fang, Y. Zhang and X. Wang, Photocatalytic Oxygen Evolution from Functional Triazine-Based Polymers with Tunable Band Structures, Angew. Chem., Int. Ed., 2018, 57, 470–474 CrossRef CAS PubMed.
- R. S. Sprick, Z. Chen, A. J. Cowan, Y. Bai, C. M. Aitchison, Y. Fang, M. A. Zwijnenburg, A. I. Cooper and X. Wang, Water Oxidation with Cobalt-Loaded Linear Conjugated Polymer Photocatalysts, Angew. Chem., Int. Ed., 2020, 59, 18695–18700 CrossRef CAS PubMed.
- J. Liu, Y. Liu, N. Liu, Y. Han, X. Zhang, H. Huang, Y. Lifshitz, S.-T. Lee, J. Zhong and Z. Kang, Metal-Free Efficient Photocatalyst for Stable Visible Water Splitting Via a Two-Electron Pathway, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Overall Water Splitting by Pt/G-C3N4 Photocatalysts without Using Sacrificial Agents, Chem. Sci., 2016, 7, 3062–3066 RSC.
- D. Kong, X. Han, J. Xie, Q. Ruan, C. D. Windle, S. Gadipelli, K. Shen, Z. Bai, Z. Guo and J. Tang, Tunable Covalent Triazine-Based Frameworks (CTF-0) for Visible-Light-Driven Hydrogen and Oxygen Generation from Water Splitting, ACS Catal., 2019, 9, 7697–7707 CrossRef CAS PubMed.
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. Jin, Strong-Base-Assisted Synthesis of a Crystalline Covalent Triazine Framework with High Hydrophilicity Via Benzylamine Monomer for Photocatalytic Water Splitting, Angew. Chem., Int. Ed., 2020, 59, 6007–6014 CrossRef CAS PubMed.
- Z. Pan, M. Liu, G. Zhang, H. Zhuzhang and X. Wang, Molecular Triazine–Heptazine Junctions Promoting Exciton Dissociation for Overall Water Splitting with Visible Light, J. Phys. Chem. C, 2021, 125, 9818–9826 CrossRef CAS.
- Y. Bai, C. Li, L. Liu, Y. Yamaguchi, B. Mounib, H. Yang, A. Gardner, M. Zwijnenburg, N. Browning and A. Cowan, Photocatalytic Overall Water Splitting under Visible Light Enabled by a Particulate Conjugated Polymer Loaded with Iridium, ChemRxiv, 2022 DOI:10.26434/chemrxiv-2022-8vr18.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, Polymer Photocatalysts for Water Splitting: Insights from Computational Modeling, Macromol. Chem. Phys., 2016, 217, 344–353 CrossRef CAS.
- P. Guiglion, C. Butchosa and M. A. Zwijnenburg, Polymeric Watersplitting Photocatalysts; a Computational Perspective on the Water Oxidation Conundrum, J. Mater. Chem. A, 2014, 2, 11996–12004 RSC.
- A. W. Prentice and M. A. Zwijnenburg, The Role of Computational Chemistry in Discovering and Understanding Organic Photocatalysts for Renewable Fuel Synthesis, Adv. Energy Mater., 2021, 11, 2100709 CrossRef CAS.
- P. Guiglion, A. Monti and M. A. Zwijnenburg, Validating a Density Functional Theory Approach for Predicting the Redox Potentials Associated with Charge Carriers and Excitons in Polymeric Photocatalysts, J. Phys. Chem. C, 2017, 121, 1498–1506 CrossRef CAS.
- D. J. Woods, S. A. J. Hillman, D. Pearce, L. Wilbraham, L. Q. Flagg, W. Duffy, I. McCulloch, J. R. Durrant, A. A. Y. Guilbert, M. A. Zwijnenburg, R. S. Sprick, J. Nelson and A. I. Cooper, Side-Chain Tuning in Conjugated Polymer Photocatalysts for Improved Hydrogen Production from Water, Energy Environ. Sci., 2020, 13, 1843–1855 RSC.
- R. S. Sprick, B. Bonillo, R. Clowes, P. Guiglion, N. J. Brownbill, B. J. Slater, F. Blanc, M. A. Zwijnenburg, D. J. Adams and A. I. Cooper, Visible-Light-Driven Hydrogen Evolution Using Planarized Conjugated Polymer Photocatalysts, Angew. Chem., Int. Ed., 2016, 55, 1792–1796 CrossRef CAS PubMed.
- M. Sachs, R. S. Sprick, D. Pearce, S. A. J. Hillman, A. Monti, A. A. Y. Guilbert, N. J. Brownbill, S. Dimitrov, X. Shi, F. Blanc, M. A. Zwijnenburg, J. Nelson, J. R. Durrant and A. I. Cooper, Understanding Structure-Activity Relationships in Linear Polymer Photocatalysts for Hydrogen Evolution, Nat. Commun., 2018, 9, 4968 CrossRef PubMed.
- R. S. Sprick, C. M. Aitchison, E. Berardo, L. Turcani, L. Wilbraham, B. M. Alston, K. E. Jelfs, M. A. Zwijnenburg and A. I. Cooper, Maximising the Hydrogen Evolution Activity in Organic Photocatalysts by Co-Polymerisation, J. Mater. Chem. A, 2018, 6, 11994–12003 RSC.
- R. S. Sprick, L. Wilbraham, Y. Bai, P. Guiglion, A. Monti, R. Clowes, A. I. Cooper and M. A. Zwijnenburg, Nitrogen Containing Linear Poly(Phenylene) Derivatives for Photo-Catalytic Hydrogen Evolution from Water, Chem. Mater., 2018, 30, 5733–5742 CrossRef CAS.
- C. Bannwarth, E. Caldeweyher, S. Ehlert, A. Hansen, P. Pracht, J. Seibert, S. Spicher and S. Grimme, Extended Tight-Binding Quantum Chemistry Methods, WIRES Comput. Mol. Sci., 2021, 11, e1493 CAS.
- L. Wilbraham, E. Berardo, L. Turcani, K. E. Jelfs and M. A. Zwijnenburg, High-Throughput Screening Approach for the Optoelectronic Properties of Conjugated Polymers, J. Chem. Inf. Model., 2018, 58, 2450–2459 CrossRef CAS PubMed.
- I. Heath-Apostolopoulos, L. Wilbraham and M. A. Zwijnenburg, Computational High-Throughput Screening of Polymeric Photocatalysts: Exploring the Effect of Composition, Sequence Isomerism and Conformational Degrees of Freedom, Faraday Discuss., 2019, 215, 98–110 RSC.
- L. Wilbraham, D. Smajli, I. Heath-Apostolopoulos and M. A. Zwijnenburg, Mapping the Optoelectronic Property Space of Small Aromatic Molecules, Commun. Chem., 2020, 3, 14 CrossRef CAS.
- I. Heath-Apostolopoulos, D. Vargas-Ortiz, L. Wilbraham, K. E. Jelfs and M. A. Zwijnenburg, Using High-Throughput Virtual Screening to Explore the Optoelectronic Property Space of Organic Dyes; Finding Diketopyrrolopyrrole Dyes for Dye-Sensitized Water Splitting and Solar Cells, Sustainable Energy Fuels, 2021, 5, 704–719 RSC.
- S. Grimme, C. Bannwarth and P. Shushkov, A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All SPD-Block Elements (Z = 1–86), J. Chem. Theory Comput., 2017, 13, 1989–2009 CrossRef CAS PubMed.
- V. Ásgeirsson, C. A. Bauer and S. Grimme, Quantum Chemical Calculation of Electron Ionization Mass Spectra for General Organic and Inorganic Molecules, Chem. Sci., 2017, 8, 4879–4895 RSC.
- S. Grimme and C. Bannwarth, Ultra-Fast Computation of Electronic Spectra for Large Systems by Tight-Binding Based Simplified Tamm-Dancoff Approximation (sTDA-xTB), J. Chem. Phys., 2016, 145, 054103 CrossRef PubMed.
- L. Turcani, E. Berardo and K. E. Jelfs, Stk: A Python Toolkit for Supramolecular Assembly, J. Comput. Chem., 2018, 39, 1931–1942 CrossRef CAS PubMed.
- https://github.com/lukasturcani/stk .
- S. Riniker and G. A. Landrum, Better Informed Distance Geometry: Using What We Know to Improve Conformation Generation, J. Chem. Inf. Model., 2015, 55, 2562–2574 CrossRef CAS PubMed.
- https://www.rdkit.org/ .
- T. A. Halgren, Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- https://github.com/grimme-lab/xtb .
- https://github.com/benedictsaunders/polyHTS2 .
- https://github.com/LiamWilbraham/polyhts .
- A. V. Onufriev and D. A. Case, Generalized Born Implicit Solvent Models for Biomolecules, Annu. Rev. Biophys., 2019, 48, 275–296 CrossRef CAS PubMed.
- https://github.com/benedictsaunders/PolyMolZ .
- https://github.com/LiamWilbraham/molz .
- Y. Bai, L. Wilbraham, H. Gao, R. Clowes, H. Yang, M. A. Zwijnenburg, A. I. Cooper and R. S. Sprick, Photocatalytic Polymers of Intrinsic Microporosity for Hydrogen Production from Water, J. Mater. Chem. A, 2021, 9, 19958–19964 RSC.
- J. Kosco, M. Sachs, R. Godin, M. Kirkus, L. Francas, M. Bidwell, M. Qureshi, D. Anjum, J. R. Durrant and I. McCulloch, The Effect of Residual Palladium Catalyst Contamination on the Photocatalytic Hydrogen Evolution Activity of Conjugated Polymers, Adv. Energy Mater., 2018, 8, 1802181 CrossRef.
- M. Sachs, H. Cha, J. Kosco, C. M. Aitchison, L. Francàs, S. Corby, C.-L. Chiang, A. A. Wilson, R. Godin, A. Fahey-Williams, A. I. Cooper, R. S. Sprick, I. McCulloch and J. R. Durrant, Tracking Charge Transfer to Residual Metal Clusters in Conjugated Polymers for Photocatalytic Hydrogen Evolution, J. Am. Chem. Soc., 2020, 142, 14574–14587 CrossRef CAS PubMed.
- A. W. Prentice and M. A. Zwijnenburg, Hydrogen Evolution by Polymer Photocatalysts; a Possible Photocatalytic Cycle, Sustainable Energy Fuels, 2021, 5, 2622–2632 RSC.
- J. Kosco, M. Bidwell, H. Cha, T. Martin, C. T. Howells, M. Sachs, D. H. Anjum, S. Gonzalez Lopez, L. Zou, A. Wadsworth, W. Zhang, L. Zhang, J. Tellam, R. Sougrat, F. Laquai, D. M. DeLongchamp, J. R. Durrant and I. McCulloch, Enhanced Photocatalytic Hydrogen Evolution from Organic Semiconductor Heterojunction Nanoparticles, Nat. Mater., 2020, 19, 559–565 CrossRef CAS PubMed.
- A. Sekar and K. Sivula, Organic Semiconductors as Photoanodes for Solar-Driven Photoelectrochemical Fuel Production, Chimia Int. J. Chem., 2021, 75, 169–179 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Particulate Photocatalysts for Light-Driven Water Splitting: Mechanisms, Challenges, and Design Strategies, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- S. Chen, T. Takata and K. Domen, Particulate Photocatalysts for Overall Water Splitting, Nat. Rev. Mater., 2017, 2, 17050 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SMILES representation and pictures of all monomers, calibration parameters used and predicted IP, EA and optical gap for all (co-)polymers. See DOI: 10.1039/d2se00027j |
| This journal is © The Royal Society of Chemistry 2022 |