
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An open-source platform for 3D-printed redox flow battery test cells†
Hugh
O'Connor
 a,
Josh J.
Bailey
abc,
Oana M.
Istrate
b,
Peter A. A.
Klusener
d,
Rob
Watson
b,
Stephen
Glover
b,
Francesco
Iacoviello
c,
Dan J. L.
Brett
c,
Paul R.
Shearing
c and
Peter
Nockemann
*a
a,
Josh J.
Bailey
abc,
Oana M.
Istrate
b,
Peter A. A.
Klusener
d,
Rob
Watson
b,
Stephen
Glover
b,
Francesco
Iacoviello
c,
Dan J. L.
Brett
c,
Paul R.
Shearing
c and
Peter
Nockemann
*a
aThe QUILL Research Centre, School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, 39-123 Stranmillis Rd, Belfast BT9 5AG, UK. E-mail: p.nockemann@qub.ac.uk
bSchool of Mechanical and Aerospace Engineering, Queen's University Belfast, Ashby Building, Stranmillis Rd, Belfast BT9 5AG, UK
cElectrochemical Innovation Laboratory, Department of Chemical Engineering, University College London, London, WC1E 7JE, UK
dShell Global Solutions International B.V., Grasweg 31, 1031 HW Amsterdam, The Netherlands
First published on 21st February 2022
Abstract
The development of new, large-scale stationary energy storage technologies, such as redox flow batteries, is vital to fully utilise renewable energy resources. However, test cells capable of assisting in this development can be prohibitively expensive and unreliable. Here, an open-source, low-cost, customisable 3D-printed test cell is presented as an alternative. These newly developed cells are designed to be printable using affordable desktop 3D-printers and readily available polymers. A simulation-led design optimisation yielded an improved internal manifold geometry that demonstrated improved real-world performance. The polymers used have been tested for chemical compatibility and through the use of advanced X-ray micro-CT, optimised parameters for 3D-printing have been identified. This framework provides a straightforward process enabling researchers to produce robust cells at an extremely low cost, helping to democratise research and widen accessibility to flow electrochemistry.
Introduction
In the midst of a worsening climate crisis, it is widely accepted that the pathway to a sustainable future involves a transition to greater utilisation of renewable energy resources.1 These resources, such as wind, hydro, and solar – whilst being non-polluting and abundantly available – are intermittent and difficult to predict. Coupled with a lack of adequate technology for large-scale energy storage, this makes the use of renewables in a stable and reliable power grid problematic.2 Effective grid-scale energy storage is one of the world's foremost challenges, requiring collaboration between institutions and researchers and the democratisation of new technologies. Recently, significant research has been carried out in an attempt to develop adequate large-scale electrochemical energy storage systems, with the redox flow battery (RFB) among the most promising for longer-term energy storage.3–5A typical redox flow battery consists of redox couples, dissolved in electrolyte solutions, and stored in separate reservoir tanks. During charge and discharge these electrolytes are pumped into half-cells, separated by an ion-exchange membrane where electrons are either consumed or generated within a porous electrode.6Fig. 1 shows a schematic of a basic redox flow battery cell and system. As a result of this unique working principle, power and energy are decoupled, i.e., power is determined by the cell, and energy by the tank size and electrolyte energy density, allowing these devices to be customised to meet a wide range of requirements.7 Furthermore, in conventional RFB systems, such as Fe–Cr and all-vanadium, the electrodes are not chemically converted during charge and discharge, and these systems generally have a longer lifespan than other energy storage technologies such as Li-ion batteries. However, RFBs have several drawbacks that have limited their wider use, primarily their low energy densities,7 high cost of some components required, (e.g., membrane materials),2 and the lack of a “winning” or “best” RFB chemistry.6
 | ||
| Fig. 1 Typical redox flow battery system. (a) Schematic showing electrolyte tanks and two half cells connected to pumps and an external power source or load. (b) Diagram of a typical single-cell assembly: 1. endplates; 2. current collectors; 3. flow frames; 4. porous electrodes; 5. gaskets and 6. ion exchange membrane. | ||
These issues have been addressed by researchers in several ways, most commonly through the development of new electrolyte and membrane technologies.4,8–10 Flow battery test cells used in the development of these new electrolytes tend to be expensive and provide limited scope for re-design, presenting a potential barrier-to-entry into the field of flow battery research. Commercially available test cells with active areas of ∼25 cm2 typically range in price from ∼$2300 to $400011–14 and, in some cases, their sealing can be problematic. Here, we report the development of custom-made test cells for laboratory research utilising fused deposition modelling (FDM) 3D-printing as a compelling alternative to these commercially available cells.
As the accessibility of 3D-printing has increased,15 more researchers are recognising its potential to be used in electrochemical flow systems.16 Recently, 3D-printing has been used to develop electrochemical flow systems which can be used for water splitting, electrosynthesis cells, electroanalysis reactors, fuel cells and electrolysers.17,18 Some of these electrosynthesis and electroanalysis reactors were FDM 3D-printed from polypropylene (PP), incorporating small channels and reaction chambers that are analogous to the features found in RFB test cells.19,20 Another general-purpose reactor was developed for electrosynthesis, electrodeposition, and electrochemical water splitting, with manifolds and turbulence promoters manufactured via FDM 3D-printing.21 As part of its design process, the chemical compatibility of polyethylene-terephthalate and acrylonitrile–butadiene–styrene (ABS) were evaluated in several common chemical compounds and electrolytes.
Elsewhere, the chemical compatibility of a wider range of FDM 3D-printed materials with common solvents22 and chemical solutions23 were also investigated. The chemical resistance of the components was shown to vary between printed and unprinted filaments due to microstructural defects which arise during printing.
3D-printing has also been utilised in the field of electrochemical energy storage, with a range of 3D-printing technologies being utilised to fabricate electrode materials24 for various energy storage devices such as Li-ion batteries and supercapacitors.25 So far, few reports have been published on the potential of 3D-printing technologies applied to RFBs.26 Test cells for use in a Zn–Ce system have been partially 3D-printed via FDM, incorporating ABS flow plates held together by a stainless-steel clamping structure.27 A similar test cell has also been designed for a vanadium28 and a non-aqueous cobalt29 system, with flow and end plates manufactured via stereolithography 3D-printing. This 3D-printing technology has also been used to manufacture turbulence promoters to enhance reactant distribution30 and static mixers in a slurry electrode to improve charge transfer.31 3D-printing has also been employed in a heat-managing miniaturized RFB cell, integrating fluidic networks manufactured via multi-jet fusion which were then coated with nickel.32
These pioneering works have begun to explore the potential of 3D-printing in electrochemical flow reactors.33 However, there is still scope for wider utilisation of the technology, particularly as applied to RFBs.
The freedom of design offered by FDM 3D-printing allows for the investigation of many different complex manifold and cell topologies that are difficult to achieve via traditional manufacturing methods. This advantage has been widely utilised in the full cell fabrication of other rechargeable batteries.24 Additionally, parts can be produced at a low-cost and high speed34 compared to the methods used to manufacture the majority of test cells. Some of these cells have been collated and can be seen in Table S1 in the ESI.† Furthermore, the majority of the existing devices mentioned can be difficult to reproduce without expertise in manufacturing or 3D-design. This study aims to provide researchers with a straightforward framework that can be used to easily 3D-print custom-made, low-cost test cells. These cells and methods are intended to assist in the development of improved flow battery electrolytes, materials, and cell designs.
This 3D-printing approach has been combined with a coupled electrochemical-computational-fluid-dynamics (CFD) model and X-ray micro-CT to generate and print an optimised cell design. To the knowledge of the authors, this is the first time that rapid prototyping and simulation have been employed to converge on a more optimised RFB cell design and demonstrate this improvement with real-world results. This framework provides a platform for altering cell design for parametric studies enabling fast-track development and optimisation of performance for a given electrolyte system. The resultant cell design of this study has been subjected to a set of electrochemical tests to fully assess its performance against commercially available test cells. Remarkably, the total cell cost was estimated to be 5% or less than the selling price of commercially available test cells. The polymers used (ABS and PP) are also tested in common flow battery supporting electrolytes to ensure chemical compatibility. Finally, by providing all source files, this work enables researchers with no expertise in 3D-design to download and print cells without the use of computer-aided design (CAD) software.
Results and discussion
Polymer selection and compatibility testing
Two potentially suitable FDM polymers, ABS and PP, were identified16,17,20,27,35,36 for use in flow batteries. However, it has also been suggested that ABS may be less chemically resistant, and is only suitable for short-term testing with low acid concentrations.27 To address this, and to identify suitable material–electrolyte combinations, a chemical compatibility study was carried out. Inspired by the previous works,21–23 a number of commonly used aqueous and non-aqueous flow battery solvents and supporting electrolytes used in flow battery systems were selected and tested with these polymers. A total of six aqueous solutions (3 M sulfuric acid (H2SO4), 1 M potassium hydroxide (KOH), 2 M sodium chloride (NaCl), deionized water (diH2O), 3 M hydrochloric acid (HCl) and 3 M methanesulfonic acid (MSA)) and four non-aqueous solvents (i.e., acetonitrile (ACN), propylene carbonate (PC), dichloromethane (DCM) and an ionic liquid, trihexyl tetradecyl phosphonium bistriflimide ([P66614][NTf2])) were investigated. A concentration of 3 M sulfuric acid (H2SO4) was chosen as it is the concentration used in the electrochemical modelling in the “test cell design” section and the real-world tests in the “charge–discharge-testing” section. Chemical incompatibility between a chosen polymer and electrolyte could result in leakage or absorption of electrolyte and failure of experiments. As such, three different printed samples of ABS and PP, each from a different manufacturer (i.e.,Verbatim, FormFutura and Ultimaker) were tested. Classification of the levels of degradation was based on a previous study21 which stated that degradation in mass of <1% after 1 week was acceptable, 1–5% was moderate, and >5% was severe. The samples were left in solution and the mass differences between the beginning and end of the test were recorded. A duration of one week was chosen to give a good representation of the degradation that may occur during typical usage identified from literature (see Table S1†). The results are shown in Fig. 2.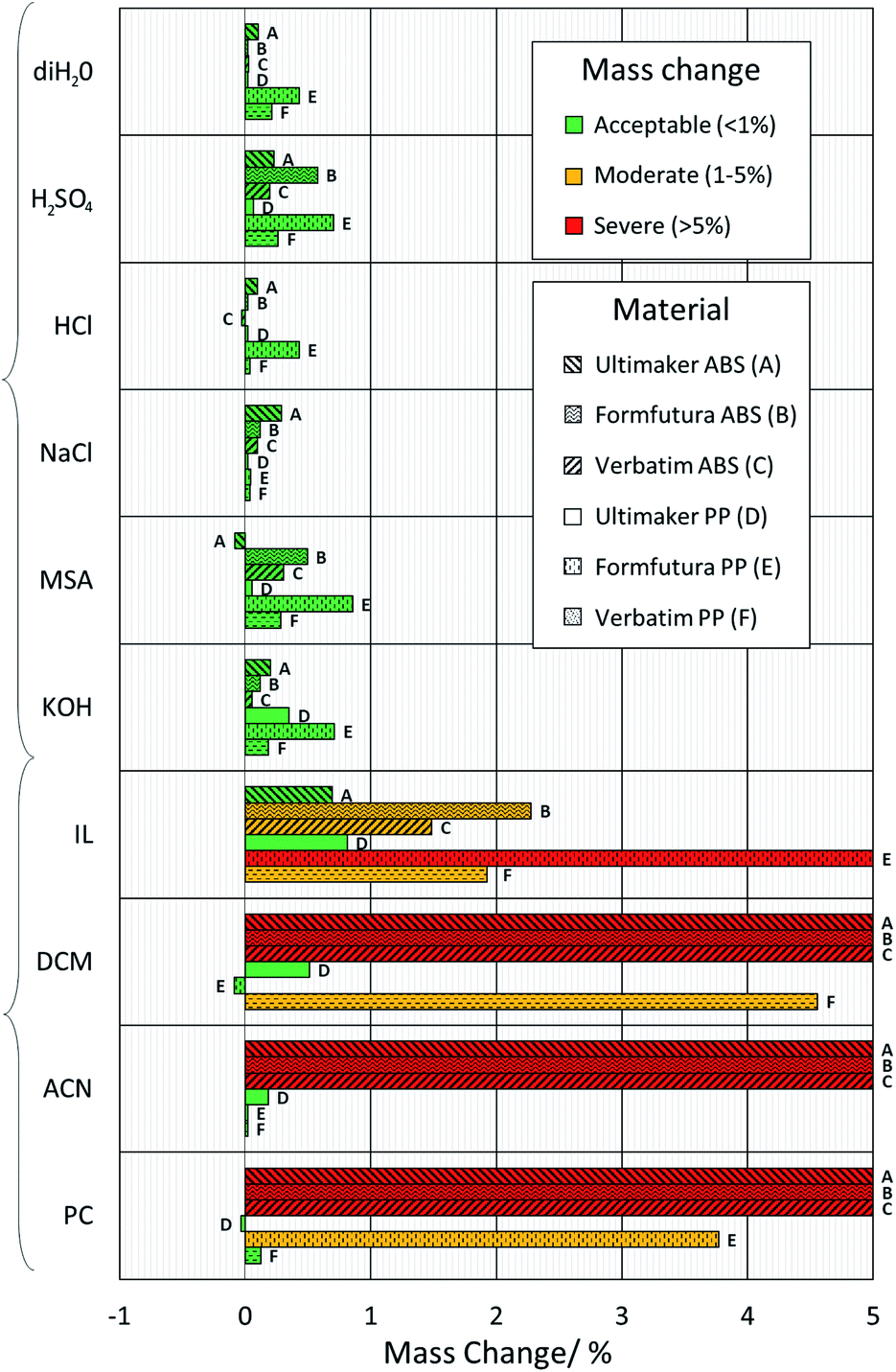 | ||
| Fig. 2 Chemical compatibility tests. Bar graph showing the percentage mass difference of 3D-printed ABS and PP samples after one week in solution at 25 °C, and subsequent washing and drying. | ||
All ABS and PP filaments showed no measurable volume change or swelling in aqueous solutions, and after cleaning and drying, they exhibited less than 1% mass change. As such, they all appeared suitable candidates for use in the 3D-printing of flow battery cells using these electrolyte systems. However, as expected from previous studies,21,22 the 3D-printed ABS rapidly softened and experienced severe mass gain, with dissolution and swelling, when submerged in the organic solvents DCM and ACN. ABS samples submerged in PC also exhibited the same behaviour. Thus, it can be concluded that ABS should not be used with any of these three non-aqueous compounds. Moreover, Formfutura ABS was found to discolour in MSA and H2SO4, alongside a small mass loss of Ultimaker ABS in MSA; however, the mass change in all instances was below 1% and thus deemed acceptable. The discolouration and mass loss are predicted to be due to the different plasticisers and antioxidants used by different manufacturers. These findings indicate the potential for ABS flow frames to be used for long-term lab-scale studies with electrolytes containing higher acid concentrations.
PP showed greater compatibility with these solutions, exhibiting acceptable to moderate mass changes. However, some samples, such as Verbatim PP in DCM and Formfutura PP in PC, showed a larger mass change than the other two PP samples. This trend is repeated across a number of different PP samples, particularly those printed using Formfutura PP. In all aqueous and non-aqueous solutions (with the exception of NaCl) these samples increased in mass more than the other PP filaments tested. This finding, along with a number of other fluctuations between the same types of polymers from different manufacturers, would indicate that the different filament additives can result in a noticeable difference in chemical compatibility and warrants further study.
Test cell design process
An active area of 50 × 50 mm was chosen for the design detailed in this work. These dimensions were chosen based on the dimensions of test cells described in literature (see Table S1†). Another key design choice was the overall structure of the test cell. Although other RFB cell structures exist,37,38 the most common are the conventional “flow-through” and “zero-gap” cell structures (Fig. 3). In flow-through cells, the electrolyte flows through the electrode, which typically consists of a carbon felt material. In zero-gap cells, however, the electrolyte flows over the surface of thinner carbon paper electrodes through flow channels which are typically machined into graphite flow plates.39 Whilst it is widely accepted that designs that include flow fields can achieve higher power densities and reduce overpotentials,40 most commercially available cells41 and larger industrial-size cell stacks42 still use flow-through electrode configurations. Furthermore, zero-gap cells tend to be more expensive than flow-through cells, due to the complex machining and extra material cost of the graphite flow plates.43 As one of the primary objectives of this work was to provide researchers with low-cost, easy-to-manufacture flow cells, a flow-through design was developed, however zero-gap designs will be explored in future work.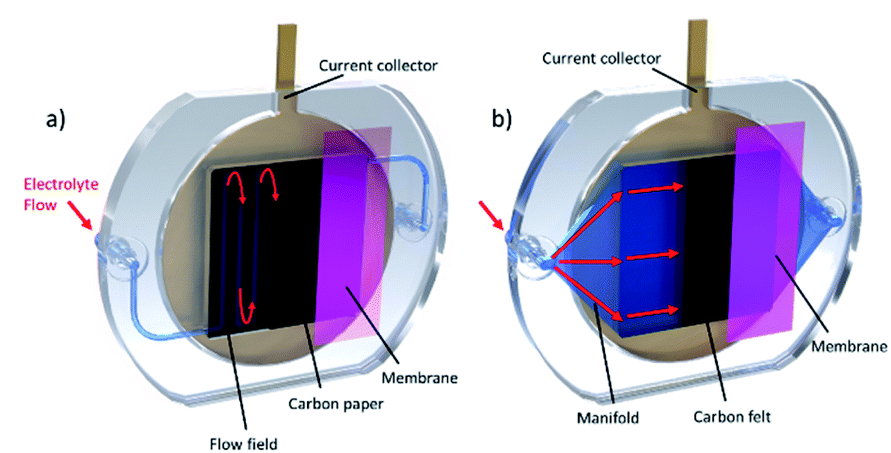 | ||
| Fig. 3 Schematic of different flow battery cell configurations. (a) “Zero-gap” cell structure. Electrolyte flows through channels in a graphite bipolar plate, over the surface of thinner carbon paper electrodes. (b) “Flow-through” cell structure. All of the electrolyte is directed through a thicker carbon felt electrode. | ||
To improve reactant distribution and reduce concentration overpotential in the 3D-printed cell, the manifold design was optimised using a coupled electrochemical-CFD model developed in literature.44–46 This model examined the flow of electrolyte through the porous carbon felt electrode in one half-cell (the negative half-cell) in 3D at a given state-of-charge (SOC), allowing cell designs that have the potential to improve reactant distribution to be investigated. This approach has been successfully employed by other research groups, with novel cell topologies resulting in greatly enhanced reactant distribution.30 The parameters used were selected from literature44,46 and an inlet flow rate and applied current density of 25 mL min−1 and 50 mA cm−2 were chosen, respectively. For the simulations, cell geometry has been simplified in the z-direction to a uniform thickness of 2 mm. The effect of the carbon felt electrode, with micron-sized fibres, is modelled with homogenised macro-parameters where permeability is described by the Kozeny–Carman equation, using the carbon fibre diameter. Key model parameters are detailed in Table S2.†
The cell was designed to be as compact as possible given the 25 cm2 active area. A detailed cell design drawing can be seen in Fig. S3.† A more compact footprint results in a shorter print time, less material used and lower costs. Within these constraints, a simple manifold was designed. This design ensured the avoidance of large overhangs, which can prove difficult to 3D-print, and consisted of four equally spaced, 2 mm × 2 mm flow channels between the electrode compartment and the in- and outlets. A velocity streamline plot of the electrolyte flow through this manifold design is shown in Fig. 4a. Due to the wide range of velocities seen, the logarithm of the velocity magnitude has been contoured in Fig. 4c.
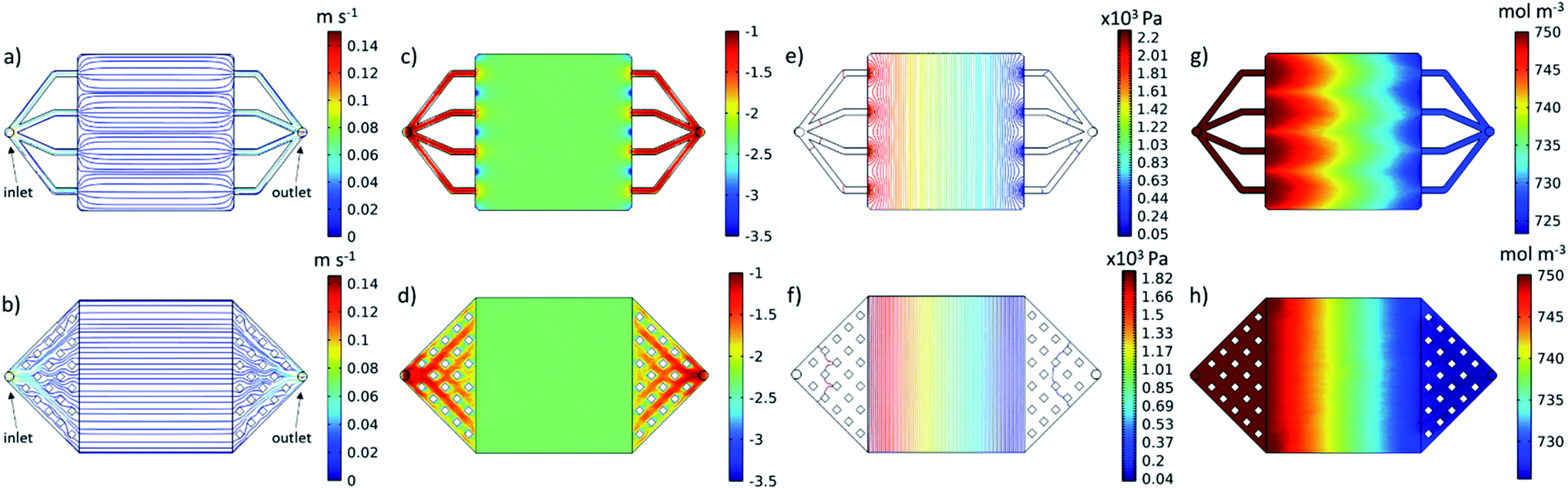 | ||
| Fig. 4 CFD-electrochemical modelling results at the midplane of cell for the unoptimized (top) and optimized (bottom) cell designs, showing electrolyte flow through the manifold channels and within the electrode cavity. It should be noted that, although the electrode is hidden in these results, the microscopic parameters attributable to the carbon felt in the cavity have been modelled as homogenised macro-parameters. (a and b) Velocity streamlines, (c and d) logarithm of velocity magnitude, (e and f) pressure contours and (g and h) vanadium(III) concentration. | ||
Across most of the electrode area, this original manifold design achieved an even velocity profile, which can be seen from both the streamlines in Fig. 4a and from the contour plot of the log of velocity in Fig. 4c. However, regions of both higher and lower velocity are found to exist near the inlet and outlet channels which can be seen in Fig. 4c. The velocity profile close to the inlet into the electrode area appears to be critical, as it can have a dramatic effect on the distribution of the concentration of vanadium(III), V3+, across the entire working section, as can be seen in Fig. 4g. As V3+ enters the cell during charging, it is reduced to vanadium(II), V2+, which results in a drop in V3+ ions moving from inlet to outlet. Due to the regions of low velocity close to the inlet in between the channels, V3+ is locally depleted, and the distribution doesn't recover uniformity. A more even distribution results in a cell with reduced concentration overpotential, allowing for higher power output.47
Based on these results, the manifold design was refined, within the same footprint. The optimised manifold (Fig. 4b) consisted of a “diffuser” type design from the inlet and outlet nozzle to the electrode area. This diffuser was divided using 20 evenly spaced 2 mm × 2 mm pillars in each manifold. These pillars were incorporated to reduce the size of overhangs in the manifold, whilst limiting design complexity to enhance printability. This design results in an increase in cross-sectional area, which decelerates the flow from the inlet, achieving a more even velocity profile at entry to the electrode area, as seen both in the straight and parallel streamlines of Fig. 4b and in the plot of the log of the velocity magnitude in Fig. 4d, in which the disturbances at the inlet from the original manifold design have been completely eliminated. As a result, a more uniform distribution of reactant concentration can be seen in Fig. 4h with the nonuniformity from Fig. 4g having been greatly reduced. This also resulted in an increase in the minimum reactant concentration seen within the domain to 726 mol m−3, from 720 mol m−3.
The optimised design also exhibited a reduction in pressure drop across the cell from 2.2 kPa (Fig. 4e) to 1.82 kPa (Fig. 4f). This pressure difference of 0.38 kPa equates to a reduction in pressure drop of ∼17%. As cell area and flow rates are scaled up to larger, commercial-scale cells and stacks, this pressure drop difference would become increasingly significant, equating to large differentials in parasitic pumping losses when comparing systems. These improvements highlight the potential for this approach of iterative refinement using rapid prototyping and CFD-electrochemical modelling to improve flow cell design, which will be re-visited in future work.
3D-printing parameters
FDM 3D-printing has many desirable traits when it comes to the manufacture of flow battery cells. FDM systems are low cost,34 safe, and easy to use in a desktop or laboratory environment,48 allow for many thermoplastic polymers to be printed, and have the ability to print complex internal geometries. However, an area of concern was the potential porosity of the parts produced. Microporosity of parts produced by FDM can occur as a result of the pores and voids which form as the thermoplastic polymers cool during the printing process,49 potentially allowing electrolyte to permeate into the 3D-printed components. This could be further exacerbated by the back pressures exerted on the walls of the part by the fluid flow,50 and the potentially “aggressive” media used as electrolyte.22 This effect can be mitigated by changing various printing parameters. Previously, increasing the material flow rate by applying an “extrusion multiplier” or “k-value” has been demonstrated to have perhaps the most significant effect on the sealing of 3D-printed parts.50 Increasing this property results in more polymer being forced through the nozzle per unit time, which can fill defects and reduce the permeability of the part.22 In addition, other recommendations have been made, including allowing for diagonal filling in walls and distributing the seams for each layer randomly.50 All of these recommendations have been implemented in the design of the half-cell shown during and after printing in Fig. 5. | ||
| Fig. 5 3D-printed cell structure. (a) The 3D-printed flow frame during the printing process. The optimised manifold design can be seen along with a large, 20 mm-wide brim to ensure build plate adhesion. (b) A 3D rendering of the finished flow frame. | ||
All printed components (both ABS and PP) for flow cells in this work were printed using a 0.4 mm nozzle, 0.2 mm layer height, 100% infill, and 105% flow rate (k = 1.05). The print time for each flow frame is approximately 7.5 hours for ABS and 9.5 hours for PP. Detailed print settings can be found in the Methods section. These settings are easily achievable on any desktop 3D-printer and provide reasonably fast printing times, whilst minimising electrolyte permeation into the components.
X-ray micro-computed tomography
To probe the impact of changing the k-value on the porosity of the 3D-printed parts, six samples (PP and ABS for k = 0.90, 1.00, 1.05) were printed, exposed to vanadium electrolyte, and scanned using X-ray micro-computed tomography (micro-CT). Acquisition and processing parameters are in the Methods section. 3D renderings of the solid plastic and the internal void space are shown in Fig. 6a and b, respectively. For both materials, increasing the k-value had a significant effect on the internal structure of the 3D-printed parts, leading to a decreasing volume of void space, as shown in Fig. 6c. At each k-value, ABS contained more voids than PP and the effect of increasing the k-value from 0.90 to 1.05 led to a 75% decrease in internal porosity from 6.41% to 1.59% and a 96% decrease in internal porosity from 1.9% to 0.07% in ABS and PP, respectively. Although increasing the k-value above 1.00 is associated with a reduction in dimensional accuracy, X-ray micro-CT clearly demonstrates 3D-printed parts with lower porosity were accessible by a small increment in this parameter. Moreover, negligible vanadium (not reliably segmentable from high grayscale noise) was detected in PP samples, whereas ABS showed 0.42%, 0.11% and 0.03% vanadium deposits for k-values of 0.90, 1.00, and 1.05, respectively (see the Fig. S2†). It should be noted that vanadium deposits below the resolution limit may be present but undetected, and vanadium deposits in the ABS k = 1.05 sample were minimal and may not represent the true level of ingress in the component.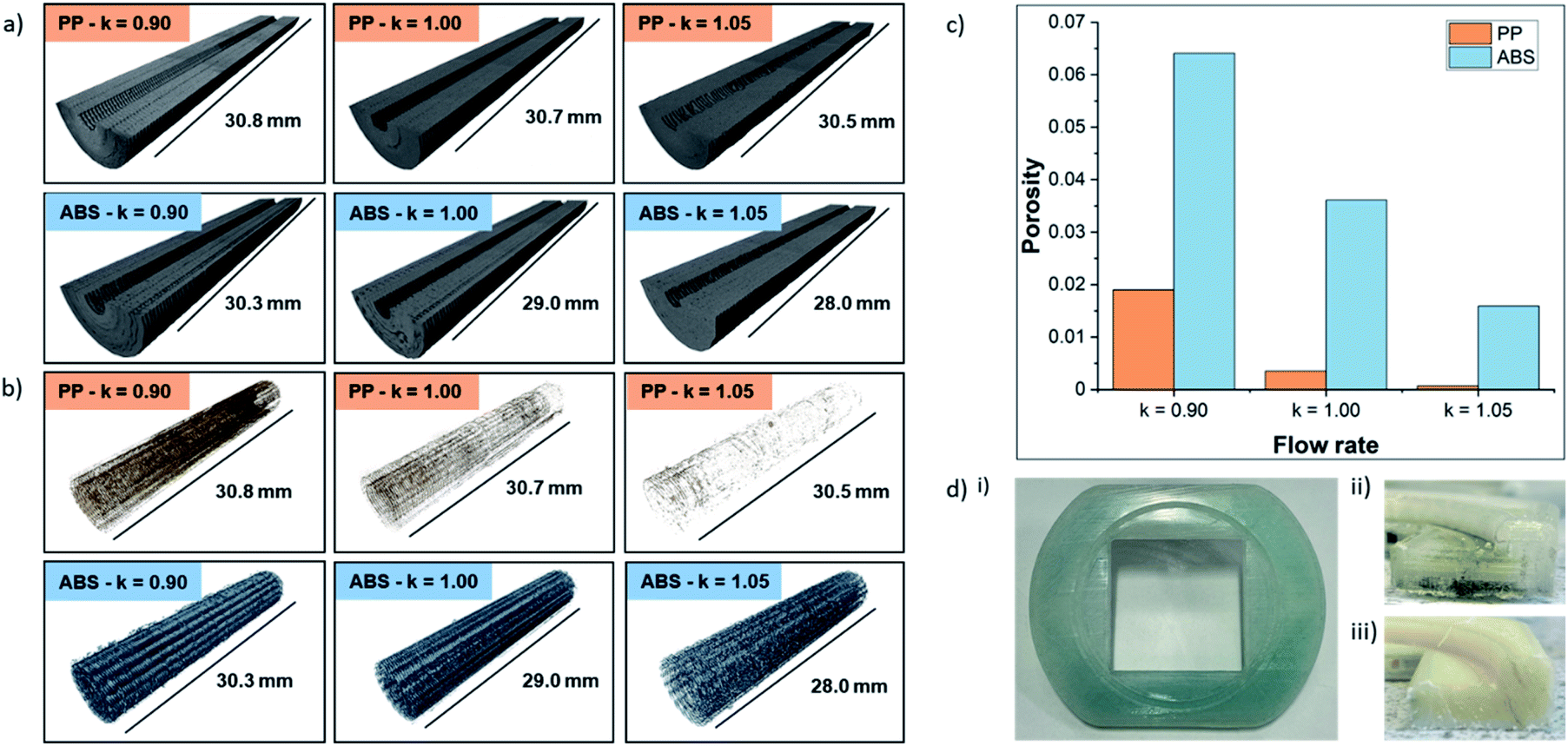 | ||
| Fig. 6 X-ray CT tomography. (a) 3D renderings of solid phase from X-ray micro-CT tomograms of 3D-printed parts. (b) 3D renderings of corresponding porous phase. (c) Bar chart showing effect of k-value on porosity of 3D-printed parts. (d) Permeation of electrolyte into 3D-printed parts. (i) View from membrane side of PP half-cell after operation with V4+. (ii) Cross-section showing permeation of V3+ along the bottom of the PP half-cell. (iii) Cross-section of ABS half-cell, showing no permeation into the 3D-printed flow frame. | ||
Polypropylene electrolyte permeation
Having been previously used in other electrochemistry applications19,20 and comparable RFB test cells using vanadium electrolyte,51 as well as favourable performance in the chemical compatibility tests and low void space during the X-ray CT scans, it was expected that PP would be the ideal material for 3D-printing flow cells. However, when these cells were tested with an electrode compression of 26% and electrolyte flow rate of 25 mL min−1, a high level of permeation was observed within 1 hour. This effect is shown in Fig. 6d(i), where the previously clear polypropylene half-cell was discoloured after absorbing electrolyte. Upon further investigation, the location of electrolyte permeation was identified by examining a cross-section of the half-cell, shown in Fig. 6d(ii). It appears that the permeation occurs at the bottom surface of the half-cell, which is in contact with the heated build-plate (65–85 °C) during the printing process. This permeation was observed with half-cells printed using PP filaments from all three of the manufacturers in this study. The cause of this permeation is unknown at this time, and, to the authors' knowledge, this behaviour has not been reported elsewhere in literature. An in-depth investigation is underway to identify the cause of this permeation along with strategies to prevent it, allowing PP to be used in a wider range of flow electrochemistry applications in the future. ABS cells were used in all subsequent tests in this work as these cells were not susceptible to electrolyte permeation. A cross-section of an ABS cell after 12 hours under the same conditions used in PP testing is shown in Fig. 6d(iii) with no observable electrolyte permeation.Cell assembly
The 3D-printed flow frames were designed to be easily assembled, with grooves for O-rings, gaskets and current collectors. A schematic view of all the components in the cell can be seen in Fig. 7a. Hand-cut expanded polytetrafluoroethylene (ePTFE) with a thickness of 1 mm or ethylene propylene diene monomer (EPDM) sponge gaskets with a thickness of 1.5 mm were used between the flow frame and both the membrane and current collectors. The current collectors consisted of 4 mm thick brass disks, with a 20 mm protrusion to connect the cell to the battery test equipment. To make the cell as compact as possible, the diameter of the current collectors was 75 mm, and the O-ring inner diameter was 71 mm. To protect the current collectors from corrosion, a 0.6 mm thick graphite composite disk (PV15, SGL Carbon) was used. The components were clamped together using a G-clamp which was hand-tightened before adjustment to 10 Nm using a torque wrench. The clamping ensured adequate sealing of the cell and reduced contact resistance of the components, whilst the electrode depth was dictated by the thickness of the 3D-printed flow frames and gaskets (∼8 mm total). More information about these materials and their manufacture is in the Methods section. | ||
| Fig. 7 Exploded view of cell components and bespoke flow battery test rig (a) schematic view of the cell showing how the components stack together: 1. brass current collector; 2. graphite current collector; 3. current collector gasket; 4. 75 mm O-ring; 5. graphite felt electrode; 6. 3D-printed flow frame; 7. membrane gasket; and 8. ion exchange membrane. (b) Schematic view of the components making up the flow battery test rig. (c) Photograph of system during initial charging of the cell, with two parts of the yellow, V5+ in the positive tank and one part of the violet, V2+ in the negative tank. | ||
The circular shape of the current collectors ensured an even clamping force was applied around the perimeter of the electrode compartment. The sealing structure allows for low tolerances in both 3D-printed and non-3D-printed parts, reducing cost and complexity by avoiding the need for expensive manufacturing processes. Coupled with the optimised 3D-printing parameters, confirmed by X-ray micro-CT, these design choices resulted in a robust, leakproof cell that can be reliably used for testing. Furthermore, the assembled cell is compact and lightweight, weighing only 900 g when fully assembled. The cells were re-used multiple times during testing with no signs of mechanical damage, withstanding the clamping force applied. One area of concern regarding durability was the 3D-printed nozzles. The 3D-printed nozzles designed for 4.8 mm bore tubing used in this work were relatively robust, however, certain users may want to ensure a longer lifespan, or require compatibility with different diameters of tubing. To facilitate this, versions of all the cell designs discussed in this work that allow for removable nozzles have been included. File details are in the ESI.†
Charge–discharge testing
To characterise the efficacy of the 3D-printed cells, appropriate operating conditions were identified from literature.41,52–55 A commercially available cell with the same 50 × 50 mm active area and similar electrode cavity thickness (C-Tech Innovation) was tested under the same conditions as the 3D-printed cells. Tests were carried out using a typical vanadium electrolyte, 1.5 M V2+ and V5+ dissolved in 3 M H2SO4, and a commonly used, commercially available Nafion® 212 membrane. Two different electrode compressions, 26% and 42%, were examined using flow rates of 25 and 50 mL min−1. For the charge–discharge tests, the cell was cycled at a constant current density of 50 mA cm−2 between 1.10 V and 1.75 V. These cut-off voltages equate to roughly 10% and 90% SOC of the cell with 26% electrode compression. These tests were all carried out using a bespoke flow battery test rig (shown in Fig. 7b and c) at ∼20 °C. Component information and testing parameters are found in the Methods section.A typical charge–discharge test using the 3D-printed flow cell is presented in Fig. 8a. Representative cycles from various tests can be seen in Fig. 8b–e. Fig. 8b and c show an increased cycle time and capacity in cells that have an optimised flow field design. This effect can be explained by enhanced mass transport and therefore a more effective electrolyte distribution through the electrode (Fig. 8b and c), resulting in reduced concentration overpotential. This can also be observed in the modelling results from Fig. 4g and h, where larger concentrations of spent electrolyte are seen in the unoptimised design.
 | ||
| Fig. 8 Results of the charge–discharge tests carried out using 3D-printed and commercial flow cells. (a) Typical charge–discharge cycles at a flow rate of 25 mL min−1. (b) Voltage vs. time plot for charge–discharge experiments using 42% electrode compression and a flow rate of 50 mL min−1 with three different 50 mm × 50 mm cell designs; optimised 3D-printed cell, unoptimised 3D printed cell, and a commercially available cell (C-Tech Innovation). (c) Voltage vs. capacity for the same three flow cells. (d) Voltage vs. time plot for the same charge–discharge experiments using 26% electrode compression in the optimised 3D-printed cell and the C-Tech cell. (e) Voltage vs. capacity for the same two cells. | ||
A similar pattern is shown when comparing Fig. 8b and c (42% electrode compression) to Fig. 8d and e (26% electrode compression). This performance improvement as electrode compression is increased, has been extensively investigated elsewhere in literature.41,53,56
The commercially available cell had higher overpotentials than the 3D-printed cell. At 42% compression, charging began at a higher voltage in the commercial cell, ∼1.45 V vs. ∼1.40 V, and discharging began at a lower voltage, ∼1.44 V vs. ∼1.50 V. Furthermore, the cut-off voltages were reached more quickly in the commercial cell, ∼20 min sooner during charging and ∼23 min sooner during discharging, with a smaller capacity, 485 mA h less upon charge and 438 mA h less upon discharge. There are various potential explanations for higher overpotentials in the commercially available cell. Once such explanation is higher ohmic resistance of the cell. To investigate this, the cells were assembled without a membrane or electrolyte, and the resistances measured (see Methods section for procedure). The average resistance of the 3D-printed cells was 0.015 Ω, compared to 0.035 Ω for the commercial cell. This discrepancy in electrical resistance may be due to the materials used to manufacture the components (endplates and current collectors), or higher contact resistance between components in the commercial cell due to the different clamping methods. Another contributing factor to the lower overpotentials could derive from better reactant distribution in the 3D-printed cell designs.
Both 3D-printed cell designs consequently achieve similar energy efficiencies (see the ESI† for equations) to the commercially available cell, with energy efficiencies (neglecting the power consumption of the peristaltic pumps) of 81.4% for the optimised design and 79.0% for the unoptimised design, compared to 78.0% for the commercially available cell. These energy efficiencies are plotted in Fig. 9a, along with several other similar cells from literature at 40–50 mA cm−2. Each cell number corresponds to a row in Table S1,† along with more detail about the cell construction and performance. The 3D-printed cells achieved satisfactory energy efficiencies especially considering the lack of optimisation of the membrane and electrodes. This is illustrated in the favourable comparison to cell 7 in (Fig. 9a),60 which also used a Nafion 212 membrane and a current density of 50 mA cm−2. In future work, the performance of the 3D-printed cells will be improved by optimising the selection of membrane and electrode materials. Furthermore, no leaks from the cells occurred during the charge–discharge tests, depicted here or during leak tests which were carried out with electrolyte at a flow rate of 50 mL min−1 and electrode compression of 42% for up to 31 days (744 hours), demonstrating the efficacy of the 3D-printed cells for longer duration lab testing. These tests are compared with the highest reported electrolyte flow rate and longest reported test durations found for a number of similar cells from literature in Fig. 9b.
 | ||
| Fig. 9 Comparison between 3D-printed test cells and existing lab-scale test cells used in literature. (a) Comparison of energy efficiencies. (b) Comparison of maximum reported test duration and electrolyte flow rates. | ||
Cell cost analysis
Detailed cost analyses have been carried out to approximate the cost for researchers to manufacture the 3D-printed cells (details in the ESI†). Key costs are shown in Table 1. The total cell cost is estimated to be $107. Neglecting overheads, this results in a cost-saving of >95% compared to commercially available lab-scale test cells.11–14 This significant cost reduction makes the 3D-printed cell developed and tested here a compelling alternative to researchers working on flow batteries.| Parameter | Value | Unit |
|---|---|---|
| Print time per half-cell | 7.55 | hours |
| 3D-printed half-cell mass | 67 | g |
| 3D-printed material cost (Cmaterial) | 11.19 | $ |
| Associated 3D-printer cost (Cmachine) | 4.70 | $ |
| Labour cost (Clabour) | 10.44 | $ |
| Energy cost (Cenergy) | 0.43 | $ |
| Endplate cost (Cendplates) | 59.32 | $ |
| Sealing cost (Csealing) | 21.35 | $ |
| Total cell cost (Ctotal) | 107.43 | $ |
Methods
Chemical compatibility testing
A similar procedure to that described by Marques-Montes et al.21 was followed to determine the chemical compatibility of the materials used for 3D-printing. 2.85 mm diameter ABS and PP filaments from Ultimaker, Verbatim and ABS were used for the tests and the manufacturers recommended print settings for speed and temperature were used when printing the samples, along with 100% infill, a k-value = 1 and a 10 mm brim. The specimens were put into sample bottles with a height of 40 mm and a diameter of 20 mm which were filled with 20 mL of solution. 4 mm glass rods, with a length of 40 mm were used to submerge the specimens in solution. Each specimen was weighed to an accuracy of ±0.0001 g and inserted into a sample bottle. The samples bottles were stored in an INC/50 incubator (Genlab) at 25 °C for 1 week. The sample bottles were shaken once every 24 hours during the test. The specimens were then rinsed in deionised water and dried in a vacuum oven at 40 °C for 12 h. Any remaining moisture was removed under vacuum for 12 h. The mass of the part was then recorded, and the mass change calculated. More details about the samples used and the preparation of the solutions can be found in the ESI† section.Electrolyte preparation
The electrolyte solution was prepared using a similar method to that described in literature.57 For the supporting electrolyte, a 3 M H2SO4 solution was prepared using concentrated sulfuric acid (99.99 wt%, Alfa Aesar) and deionized water (diH2O). Vanadyl sulfate oxide hydrate (99.9% metals basis VOSO4·4.25H2O, Alfa Aesar) was added to obtain a 1.5 M solution of VOSO4 which was dissolved using a hotplate and magnetic stirrer at 50 °C until the solution turned a clear blue colour. The vanadium(IV), VO2+ solution was then split into three-parts; two of which were put into the positive tank (100 mL) and one-part into the negative tank (50 mL). These solutions were then charged at 50 mA cm−2 (galvanostatic mode) until the voltage reached 1.75 V. The cell was then charged in potentiostatic mode until the electrolyte was fully charged and the current dropped below 4 mA cm−2. This resulted in one-part vanadium(II), V2+ in the negative half-cell and two parts vanadium(IV), VO2+ in the positive half-cell. Half of the VO2+ was removed and equal amounts of V2+ and VO2+, 50 mL in each tank, were used for the subsequent experiments. Both tanks were kept under nitrogen at all times to prevent oxidation of the electrolyte.3D-printing procedure
The cell was designed using Solidworks 2020 CAD software (Dassault Systèmes). Ultimaker Cura slicing software was used to generate G-code which can be read by 3D-printers. All cells printed in this work were manufactured using an Ultimaker S5 (Ultimaker) FDM 3D-printer using single extrusion. To ensure strong build plate adhesion, a 20 mm brim was used along with 3D-printing adhesives (Magigoo Original and PP). All prints used a nozzle diameter of 0.4 mm, a layer height of 0.2 mm and were printed with 100% infill and a flow rate of 105% (k = 1.05). For all ABS filaments, the nozzle temperature was 250 °C and the print speed was 60 mm s−1. A build plate temperature of 85 °C was used for Ultimaker ABS and 100 °C for the Verbatim and Formfutura ABS. For the Verbatim PP material, the nozzle temperature was 250 °C, the print speed was 30 mm s−1, the fan speed was 30%, and the build plate temperature was 85 °C. Similar settings were used for the Formfutura “Centaur” PP; however, a lower build plate temperature of 65 °C was used. The default print speed and temperature settings from the Ultimaker Cura were used for the Ultimaker PP. Retraction settings for the PP prints were also refined to 5 mm and an acceleration of 5 mm s−2. To prevent vertical seams in the 3D-printed components, the “Z-seam” alignment in Ultimaker Cura was set to “random” and “randomize infill” was also selected.Non-3D-printed components
For the current collectors, brass disks with a thickness of 4 mm, diameter of 75 mm and a protrusion of 20 mm × 5 mm were used along with 0.6 mm thick PV15 graphite composite (SGL Carbon). The half-cells were separated by a commercially available Nafion® 212 membrane (Sigma-Aldrich) which was held in place using gaskets hand-cut from either 1 mm thick expanded PTFE Fluolion 200 (James Walker) or 1.5 mm thick EPDM sponge (PAR group). Sealing was ensured by use of 75 mm-diameter nitrile O-rings with a 1.78 mm cross-section (Polymax). All components were held together using a simple clamp. For the tests carried out here, an Irwin G-clamp (RS Components), was used and tightened to 10 Nm using a torque wrench. An insulating material was used between the clamp and the current collectors to prevent a short circuit.Membrane preparation
The Nafion® 212 membrane was prepared based on standard procedures from literature.58 The membrane was first cut to size (ca. 60 mm × 60 mm), allowing for a ∼5 mm overlap around all sides of the electrode compartment. The membrane was first washed in 100 mL diH2O for 1 h, then treated in 100 mL 3 wt% aqueous H2O2 for 1 h. The membrane was then washed in 100 mL diH2O for 1 h and treated in 100 mL 1 M H2SO4 for 1 h. Lastly, the membrane was washed with 100 mL diH2O water for 1 h. These steps all took place at 80 °C. The membrane was stored at room temperature in diH2O water until used in flow cells.Cell testing
Charge–discharge cycling was performed using a Biologic VMP3 potentiostat with a 20 A booster (Biologic) and EC-lab Software. This was carried out at 50 mA cm−2 between 1.1 V and 1.75 V which equates to approximately 10–90% SOC of the cells tested. The flow cells were run at 25 and 50 mL min−1 with the same pumping speed for both the anolyte and catholyte half-cells. Each tank contained 50 mL of electrolyte for the charge–discharge tests. Two different electrode configurations were examined. In the first setup, three pieces of graphite felt (GFD 4.65 EA, SGL Carbon) were placed in each half-cell; this setup was used to evaluate different manifold designs and for long term leak testing of cells. In the second setup, one piece of untreated, 1.12 cm thick graphite felt (Alfa Aesar) was used in each half-cell. The total half-cell thickness with gaskets and O-rings is 8 mm thick resulting in electrode compressions of ∼42% for setup one and ∼26% for setup two. Electrolyte temperature was logged using a TC-08 thermocouple data logger (PicoTech) and a pair of Digi-Sense compact PFA-coated thermocouple probes (Cole Palmer), one in each electrolyte tank. Ambient temperature was also recorded using a K-type thermocouple (RS Components). The electrolyte tanks were kept under nitrogen throughout all tests and each tank was stirred using HI 180 magnetic stirrers (Hanna Instruments). 120S/DV manual control variable speed peristaltic pumps and 4.8 mm bore × 1.6 mm wall thickness silicone tubing (RS components) were used to control the flow of electrolyte through the cell.Ohmic resistance investigation
The commercial test cell (C-tech innovation) and a 3D-printed test cell was assembled in the same configuration as described in the “cell testing” section using three pieces of graphite felt (GFD 4.65 EA, SGL Carbon) in each half-cell, however no membrane or electrolyte was used. The resistances were measured by a 4-point probe method using an Agilent 34401A Digital Multimeter (Agilent technologies). Measurements were taken three times per assembly. Each cell was then taken apart and reassembled three times to account for any variations in assembly. The average resistance of each cell was then calculated.X-ray micro-CT acquisition, reconstruction, processing, and analysis
Six samples in total, with optimised geometries for X-ray micro-CT scanning, were designed and 3D-printed using ABS and PP, each with flow rates of 90%, 100%, and 105% (k = 0.90, 1.00, 1.05). The geometry chosen was a hollow tube with an outer diameter of 6 mm and wall thickness of 1.5 mm, such that each sample could be brought close to the X-ray source of the Nikon XT H 225 instrument used for all scans, giving rise to a voxel dimension of ca. 4.50 μm. The hole provided a pathway to flow vanadium electrolyte through at a flow rate of 10 mL min−1 for approximately 10 min, prior to all scanning, to potentially highlight areas of electrolyte permeation through residual vanadium in the resultant tomograms. All scans were conducted at an X-ray voltage of 60 kV and beam power of 7 W. 1401 projections, each with an exposure time of 1 s, were collected, giving an overall scan time of <25 min per sample. Given the small lateral dimensions of the samples, four scans were taken along the z-axis, a vertical distance of ca. 7.4 mm between them, constituting an overlap of 15% and total scan time for all six samples of <12 h. Reconstruction of the radiographic projections into 3D tomograms was performed in CTPro3D (Nikon) by finding the optimised centre shift but without beam hardening correction. Image processing and analysis was carried out in Avizo 9.4.0 (Thermo Fisher Scientific). Raw images were Gaussian-filtered (3D, kernel size of 1.5) and stitched together manually before segmentation by a standard watershed approach.59 The stitched volumes were cropped at the top and bottom to remove edge effects. Phase fraction calculations were performed by simple voxel counting and all fractions are relative to the entire volume encapsulated by the plastic matrix (excluding the pore space in the hole and outside the cylinder).Conclusions
A process for designing and 3D-printing a low-cost, open-source redox flow battery test cell has been detailed. The cell has been evaluated under demanding test conditions identified from literature (electrolyte flow rates of 50 mL min−1 and electrode compression ratios >40%) and has been shown to be leakproof during long-duration tests (up to 1 month). A coupled electrochemical-CFD model was used to investigate electrolyte flow through different manifold designs to converge on a more effective flow cell design which achieved improved reactant distribution. The 3D-printing platform was then used to validate the simulation results which were reflected in real-world performance, the first time such results have been reported in literature. This highlights the flexibility of this method for flow cell production, allowing different designs to be easily evaluated experimentally. Furthermore, the results of these tests compared favourably to a commercially available redox flow battery test cell. This framework presents a compelling avenue for flow cell development and will be utilised extensively in future work to investigate larger-scale cells and new cell topologies.Many of the techniques investigated in this work are also applicable to the wider field of flow electrochemistry and can help facilitate the wider use of FDM 3D-printing. Methods of refining print parameters to reduce porosity have been validated by using X-ray micro-CT to show the effect of different k-values on part quality. ABS has been identified as a well-suited polymer for 3D-printing flow cells and has also been shown to be chemically compatible with a number of commonly-used aqueous flow battery supporting electrolytes. Although PP showed promise during the evaluation of materials for 3D-printing of flow cells, these parts experienced electrolyte permeation during leak testing. A more thorough examination of the causes and mitigation of this effect is the focus of current investigations.
FDM 3D-printing has been shown to present an extremely cost-effective method of flow battery test cell production. Work is on-going to scale up the 3D-printed test cells detailed here towards larger industrial-scale cells and stacks to investigate their economic viability. This pathway is illustrated in Fig. 10.
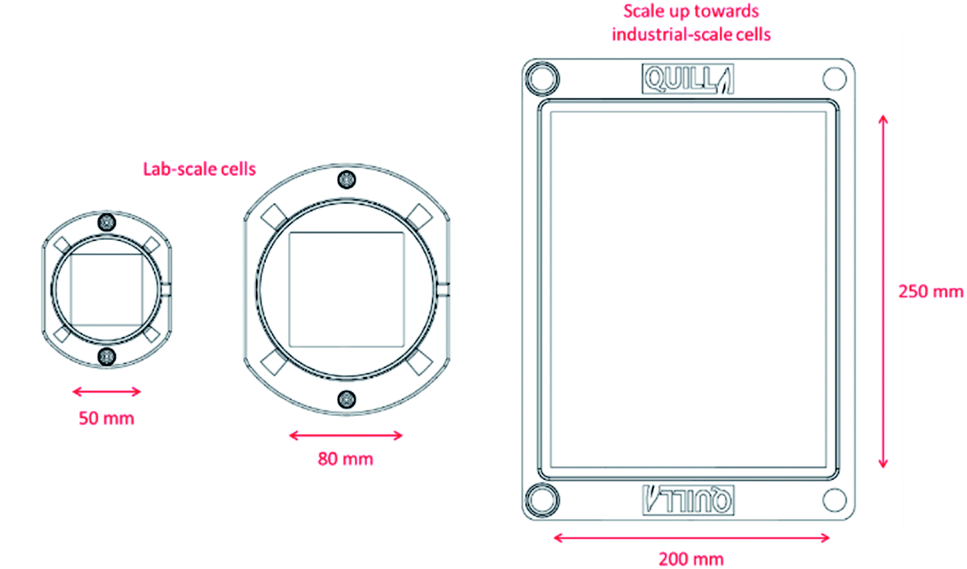 | ||
| Fig. 10 Scale up of 3D-printed flow cells towards industrial-scale. | ||
Author contributions
Hugh O'Connor: conceptualisation, methodology, validation, visualization, investigation, writing – review & editing; Josh J. Bailey: methodology, validation, visualization, investigation, writing – review & editing; Oana M. Istrate: conceptualisation, methodology, writing – review & editing; Peter A. A. Klusener: writing – review & editing; Rob Watson: conceptualisation, writing – review & editing, supervision; Stephen Glover: conceptualization, methodology, writing – review & editing, supervision; Francesco Iacoviello: investigation, writing – review & editing; Dan J. L. Brett: validation, writing – review & editing; Paul R. Shearing: validation, writing – review & editing; Peter Nockemann: conceptualisation, methodology, visualization, writing – review & editing, supervision.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding and support from the Department for Employment and Learning, Northern Ireland for a PhD studentship of H. O'C. Shell Global Solutions International B. V. is acknowledged for financial support of the studentship and for the mentoring of H. O'C. Furthermore, the authors acknowledge the QUILL Research Centre for providing facilities and lab-space. The International Energy Research Centre (IERC) is acknowledged for funding of the “Impress” grant that supported preliminary research leading to this paper. This work was supported by the National Research Facility for Lab X-ray CT (NXCT) through EPSRC grant EP/T02593X/1. JJB's work is supported by the Centre for Advanced Sustainable Energy (CASE). CASE is funded through Invest NI's Competence Centre Programme and aims to transform the sustainable energy sector through business research. PRS acknowledges funding from the Royal Academy of Engineering (CiET1718/59). DB and PRS acknowledge support from the EPSRC (EP/P009050/1, EP/N032888/1, EP/R023581/1). OMI acknowledges funding from the Royal Society (RGS\R1\211407).References
- N. Panwar, S. Kaushik and S. Kothari, Renewable Sustainable Energy Rev., 2011, 15, 1513–1524 CrossRef.
- T. Gur, Energy Environ. Sci., 2018, 11, 2696–2767 RSC.
- R. Ye, D. Henkensmeier, S. J. Yoon, Z. Huang, D. K. Ki, Z. Chang, S. Kim and R. Chen, J. Electrochem. Energy Convers. Storage, 2018, 15, 010801 CrossRef.
- M. Skyllas-Kazacos, M. Chakrabarti, S. Hajimolana, F. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–R79 CrossRef CAS.
- E. Sanchez-Diez, E. Ventosa, M. Guarnieri, A. Trovo, C. Flox, R. Marcilla, F. Soavi, P. Mazur, E. Aranzabe and R. Ferret, J. Power Sources, 2021, 481, 228804 CrossRef CAS.
- X. Ke, J. Prahl, J. Alexander, J. Wainright, T. Zawodzinski and R. Savinell, Chem. Soc. Rev., 2018, 47, 8721–8743 RSC.
- A. Weber, M. Mench, J. Meyers, P. Ross, J. Gostick and Q. Liu, J. Appl. Electrochem., 2011, 41, 1137–1164 CrossRef CAS.
- W. Wang, Q. Luo, B. Li, X. Wei, L. Li and Z. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef CAS.
- P. Leung, X. Li, C. de Leon, L. Berlouis, C. Low and F. Walsh, RSC Adv., 2012, 2, 10125–10156 RSC.
- H. Chen, G. Cong and Y. Lu, J. Energy Chem., 2018, 27, 1304–1325 CrossRef.
- C-Tech Innovation, C-Flow 5 × 5 Electrochemical Cell, https://www.ctechinnovation.com/product/c-flow-5x5/, accessed 08/05/20, 2020.
- Pinflow energy storage and Biologic, Redox Flow Lab Cell, https://www.biologic.net/accessory/redox-flow-battery/.
- Redox-Flow.com, Redox Flow Battery Test Cell, https://redox-flow.com/products/redox-flow-battery-test-cell.
- Mti Corporation, Vanadium Redox Flow Cell (Single Split Unit) for Battery R&D – EQ-VRB-C-LD, https://www.mtixtl.com/VanadiumRedoxFlowCellSingleSplitUnitforBatteryRandD-EQ-VRB-C-LD.aspx.
- J. Tully and G. Meloni, Anal. Chem., 2020, 92, 14853–14860 CrossRef CAS PubMed.
- V. Sans, Current Opinion in Green and Sustainable Chemistry, 2020, 25, 100367 CrossRef.
- A. Ambrosi, R. Shi and R. Webster, J. Mater. Chem. A, 2020, 8, 21902–21929 RSC.
- M. Browne, J. Dodwell, F. Novotny, S. Jaskaniec, P. Shearing, V. Nicolosi, D. Brett and M. Pumera, J. Mater. Chem. A, 2021, 9, 9113–9123 RSC.
- M. Symes, P. Kitson, J. Yan, C. Richmond, G. Cooper, R. Bowman, T. Vilbrandt and L. Cronin, Nat. Chem., 2012, 4, 349–354 CrossRef CAS PubMed.
- P. J. Kitson, M. H. Rosnes, V. Sans, V. Dragone and L. Cronin, Lab Chip, 2012, 12, 3267–3271 RSC.
- R. Marquez-Montes, V. Collins-Martinez, I. Perez-Reyes, D. Chavez-Flores, O. Graeve and V. Ramos-Sanchez, ACS Sustainable Chem. Eng., 2020, 8, 3896–3905 CrossRef CAS.
- K. Erokhin, E. Gordeev and V. Ananikov, Sci. Rep., 2019, 9, 20177 CrossRef CAS PubMed.
- I. Heikkinen, C. Kauppinen, Z. Liu, S. Asikainen, S. Spoljaric, J. Seppälä, H. Savin and J. Pearce, Addit. Manuf., 2018, 23, 99–107 CAS.
- M. Zhang, H. Mei, P. Chang and L. Cheng, J. Mater. Chem. A, 2020, 8, 10670–10694 RSC.
- K. Fu, Y. Yao, J. Dai and L. Hu, Adv. Mater., 2017, 29, 1603486 CrossRef PubMed.
- A. Ambrosi and R. Webster, Curr. Opin. Electrochem., 2020, 20, 28–35 CrossRef CAS.
- L. Arenas, F. Walsh and C. Ponce de León, ECS J. Solid State Sci. Technol., 2015, 4, P3080–P3085 CrossRef CAS.
- H. Sun, H. Takahashi, N. Nishiumi, Y. Kamada, K. Sato, K. Nedu, Y. Matsushima, A. Khosla, M. Kawakami, H. Furukawa, P. Stadler and T. Yoshida, J. Electrochem. Soc., 2019, 166, B3125–B3130 CrossRef.
- C. Armstrong and K. Toghill, J. Power Sources, 2017, 349, 121–129 CrossRef CAS.
- N. Gurieff, D. Keogh, V. Timchenko and C. Menictas, Molecules, 2019, 24, 3877 CrossRef CAS PubMed.
- K. Percin, A. Rommerskirchen, R. Sengpiel, Y. Gendel and M. Wessling, J. Power Sources, 2018, 379, 228–233 CrossRef CAS.
- J. Marschewski, L. Brenner, N. Ebejer, P. Ruch, B. Michel and D. Poulikakos, Energy Environ. Sci., 2017, 10, 780–787 RSC.
- M. Browne, E. Redondo and M. Pumera, Chem. Rev., 2020, 120, 2783–2810 CrossRef CAS PubMed.
- T. Ngo, A. Kashani, G. Imbalzano, K. Nguyen and D. Hui, Composites, Part B, 2018, 143, 172–196 CrossRef CAS.
- J. Hudkins, D. Wheeler, B. Pena and C. Berlinguette, Energy Environ. Sci., 2016, 9, 3417–3423 RSC.
- G. Chisholm, P. Kitson, N. Kirkaldy, L. Bloor and L. Cronin, Energy Environ. Sci., 2014, 7, 3026–3032 RSC.
- A. Bhattarai, N. Wai, R. Schweiss, A. Whitehead, T. Lim and H. Hng, J. Power Sources, 2017, 341, 83–90 CrossRef CAS.
- C. Stolze, C. Schmerbauch, C. Friebe and U. Schubert, Energy Technol., 2017, 5, 225–227 CrossRef CAS.
- D. Aaron, Q. Liu, Z. Tang, G. Grim, A. Papandrew, A. Turhan, T. Zawodzinski and M. Mench, J. Power Sources, 2012, 206, 450–453 CrossRef CAS.
- M. Ulaganathan, V. Aravindan, Q. Yan, S. Madhavi, M. Skyllas-Kazacos and T. Lim, Adv. Mater. Interfaces, 2016, 3, 1500309 CrossRef.
- T. J. Davies and J. J. Tummino, Journal of Carbon Research, 2018, 4, 0008 CrossRef.
- L. Arenas, C. de Leon and F. Walsh, J. Energy Storage, 2017, 11, 119–153 CrossRef.
- D. Reed, E. Thomsen, B. Li, W. Wang, Z. Nie, B. Koeppel and V. Sprenkle, J. Power Sources, 2016, 306, 24–31 CrossRef CAS.
- X. Ma, H. Zhang and F. Xing, Electrochim. Acta, 2011, 58, 238–246 CrossRef CAS.
- A. Shah, M. Watt-Smith and F. Walsh, Electrochim. Acta, 2008, 53, 8087–8100 CrossRef CAS.
- D. You, H. Zhang and J. Chen, Electrochim. Acta, 2009, 54, 6827–6836 CrossRef CAS.
- N. Gurieff, C. Cheung, V. Timchenko and C. Menictas, J. Energy Storage, 2019, 22, 219–227 CrossRef.
- S. Waheed, J. Cabot, N. Macdonald, T. Lewis, R. Guijt, B. Paull and M. Breadmore, Lab Chip, 2016, 16, 1993–2013 RSC.
- X. Wang, L. Zhao, J. Fuh and H. Lee, Polymers, 2019, 11, 1154 CrossRef PubMed.
- E. Gordeev, A. Galushko and V. Ananikov, PLoS One, 2018, 13, 019837 CrossRef PubMed.
- R. Jervis, L. Brown, T. Neville, J. Millichamp, D. Finegan, T. Heenan, D. Brett and P. Shearing, J. Phys. D: Appl. Phys., 2016, 49, 434002 CrossRef.
- J. Kim and H. Park, Renewable Energy, 2019, 138, 284–291 CrossRef CAS.
- L. Brown, T. Neville, R. Jervis, T. Mason, P. Shearing and D. Brett, J. Energy Storage, 2016, 8, 91–98 CrossRef.
- S. Maurya, P. Nguyen, Y. Kim, Q. Kang and R. Mukundan, J. Power Sources, 2018, 404, 20–27 CrossRef CAS.
- D. Kim, S. Yoon, J. Lee and S. Kim, Appl. Energy, 2018, 228, 891–901 CrossRef CAS.
- P. Ghimire, A. Bhattarai, R. Schweiss, G. Scherer, N. Wai and Q. Yan, Appl. Energy, 2018, 230, 974–982 CrossRef CAS.
- E. Kjeang, B. Proctor, A. Brolo, D. Harrington, N. Djilali and D. Sinton, Electrochim. Acta, 2007, 52, 4942–4946 CrossRef CAS.
- R. Kuwertz, C. Kirstein, T. Turek and U. Kunz, J. Membr. Sci., 2016, 500, 225–235 CrossRef CAS.
- A. Rabbani, S. Jamshidi and S. Salehi, J. Pet. Sci. Eng., 2014, 123, 164–171 CrossRef CAS.
- D. Chen, M. Hickner, E. Agar and E. Kumbur, Electrochem. Commun., 2013, 26, 37–40 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01851e |
| This journal is © The Royal Society of Chemistry 2022 |