
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidating the roles of acid site nature and strength in the direct conversion of levulinic acid into ethyl valerate: the case of Zr-modified beta zeolite-supported Pd catalysts†
M.
Muñoz-Olasagasti
a,
I.
Martínez-Salazar
 a,
M. López
Granados
a,
C.
López-Aguado
b,
J.
Iglesias
b,
G.
Morales
b and
R.
Mariscal
*a
a,
M. López
Granados
a,
C.
López-Aguado
b,
J.
Iglesias
b,
G.
Morales
b and
R.
Mariscal
*a
aGroup of Sustainable Energy and Chemistry (EQS), Institute of Catalysis and Petrochemistry (IPC-CSIC), C/Marie Curie 2, Cantoblanco, 28049 Madrid, Spain. E-mail: r.mariscal@icp.csic.es
bChemical and Environmental Engineering Group, Universidad Rey Juan Carlos, C/Tulipán s/n, 28933 Móstoles, Madrid, Spain
First published on 18th January 2022
Abstract
The effects of the nature and strength of surface acid sites on the properties of the catalyst used for the one-pot conversion of levulinic acid to valeric biofuels have been investigated. The acid supports were prepared from a commercial beta zeolite (HBEA) modified by partial substitution of Al by Zr atoms. This procedure enables changes in the nature and strength of the acidic sites of the starting zeolite. A constant amount of Pd (2 wt%) was incorporated on these acid supports by the incipient wetness impregnation method. These catalysts were tested in liquid phase reactions and the Pd/HBEA catalyst showed the best catalytic performance, achieving an 80% yield of valeric biofuel after 2 h of the reaction. The fresh catalysts were characterized by DRIFT spectroscopy using pyridine, CD3CN, and CO as probe molecules and by XRD to explain the differences in catalytic performance. The presence of strong Brønsted acid sites (BAS) was shown to be critical for reaching high ethyl valerate yields since the differences in catalytic behavior and the concentration of these acid sites parallel each other. Up to three types of Lewis acid sites (LAS) of different strengths were also identified. These LAS have also been shown to be active, although their intrinsic activities decrease as they are weaker. Reutilization tests of the Pd/HBEA catalyst were also carried out and mild deactivation was observed. The spent catalyst underwent a simple calcination treatment at 773 K and this was adequate for the recovery of the surface acidity, but the reducibility of the Pd species was affected. This prevented complete recovery of catalytic activity.
1. Introduction
The high rate of consumption of petrochemical resources constitutes a current global concern. These resources are exploited for their use as fuels and as feedstocks for the synthesis of many chemicals. Biomass is capable of fulfilling both of these functions. First-generation biomass-derived biofuels, such as ethanol from corn or fatty acid methyl esters from plant oils, have the disadvantage of competing with human food needs.1 Lignocellulosic biomass avoids this competition since it is nonedible, renewable, abundant and has a limited economic value.2,3 Utilizing this feedstock requires prior processing to obtain platform molecules such as levulinic acid (LA) or furfural.4–6 LA has great potential due to its functionality, since its keto and carboxylic acid groups are useful for multiple reactions used to upgrade it. Among the products derived from the hydrogenation of LA are 2-methyl tetrahydrofuran, 1,4-pentanediol, gamma-valerolactone (GVL) and valeric biofuels.7–10By focusing on upgrading LA to valeric biofuels, valeric acid and valerate esters, many developments have been realized since Lange et al.11 proposed the procurement of valeric biofuels from lignocellulose via LA in 2010. This process consists of a cascade reaction, in which every step requires specific catalytically active sites, as has been recently reported by Yu et al.12Fig. 1 shows a simplified scheme for the reaction pathway and detailed catalytic requirements. The first step is the conversion of LA to GVL, which proceeds via two steps and can follow two different routes: (i) when the reaction is conducted at 473 K the transformation proceeds via hydrogenation of LA (catalyzed by metal sites) to provide 4-hydroxyvaleric acid/ester, which is then dehydrated (catalyzed by acid sites) to produce GVL; and (ii) at temperatures above 473 K, the reaction proceeds via dehydration of LA (acid sites) to give angelica lactone and this is then hydrogenated (metal sites) to obtain GVL. In the third step, acid sites are required for the GVL ring-opening reaction to give pentenoic acid/ester, which is hydrogenated (at metal sites) to valeric acid (VA) in the fourth and final step. If water is used as the solvent, an additional step converts VA into the corresponding valerate ester by acid-catalyzed esterification with alcohol, a reaction usually promoted by strong Lewis acidity.
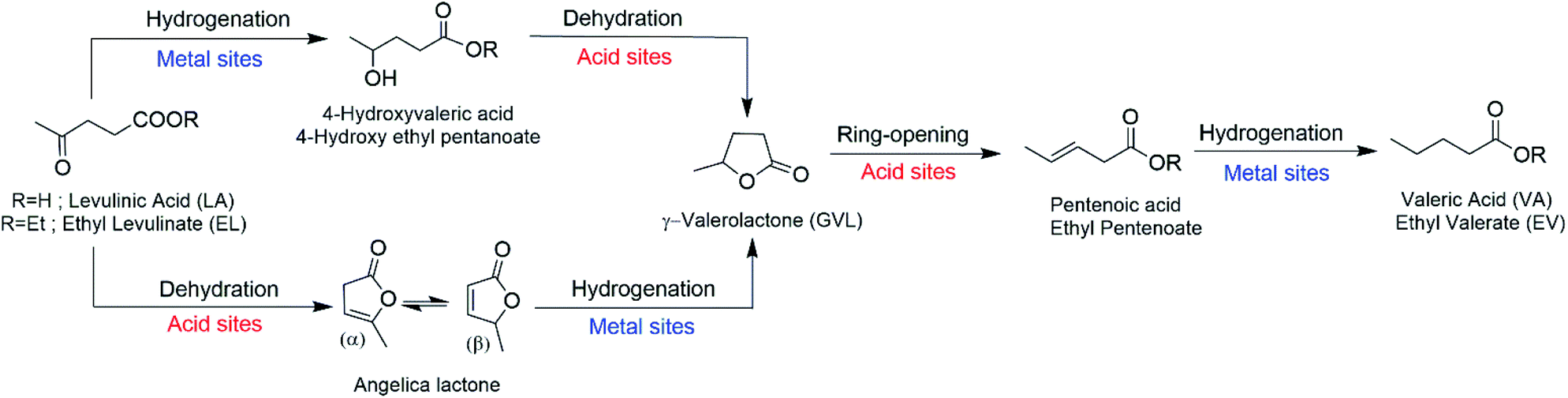 | ||
| Fig. 1 Scheme of the reaction pathway for conversion of LA/EL into VA/EV in aqueous or ethanol solution (adapted from ref. 12). | ||
Therefore, it is evident that a bifunctional catalyst with hydrogenating/metallic and acid functions is required to accomplish this reaction under one-pot conditions. Most of the work regarding the metallic function has studied the type of metal used, such as noble (Pt, Ru and Pd) and non-noble (Co, Ni and Cu) metals. At this point, some other investigations on the selective hydrogenation of levulinic acid to γ-valerolactone should also be emphasized like those based on Cu-ZrO2,13 supported Ru14 and bimetallic Ru–Ni/MMT15 catalysts.
The acid function has also been worthy of numerous studies because acidic sites have been identified as the active sites in the rate determining step, specifically the ring-opening reaction of GVL.16 Recently, Yi et al.17 reported that a metal can regulate the selectivity for conversion of LA into GVL or valeric biofuels using 1,4 dioxane as the solvent. The Ru/HZSM-5 catalyst produces a high yield (85.7%) of valeric biofuels, while the Ni/HZSM-5 catalyst achieves a GVL yield of 93.1% with a negligible formation of valeric acid or esters at 473 K and 3 MPa H2. The introduction of Ru into HZSM-5 increased the number of strong acidic sites and enhanced the ring-opening reaction of the GVL intermediate.
The acid function is generally supplied by the oxide support and does not usually depend on the metal used. Pan et al.18 reported the effects of different supports and acidity levels on the hydrogenation of LA to valerate esters with Ru-based catalysts. A 94% combined yield of ethyl valerate and valeric acid (EV + VA) was achieved after 10 h, using a Ru/SBA-SO3H catalyst at 523 K and 4 MPa H2 and ethanol as a solvent. The catalyst was primarily deactivated by leaching of sulfonic acid sites. The acid support most frequently used for the one-pot reaction has been the HZSM5 zeolite with different supported metals such as Pt,19,20 Ru,21 Pd22 and Co23 under distinct reaction conditions. Regulation of the acidity on the Ni/HZSM-5 catalyst and increases in the potassium content were also studied. Potassium decreased the acid strength and adjusted the ratio of the Brønsted/Lewis centers in the flow reactor at 513 K and with 3.0 MPa of H2.24 In general, all these studies show that a cooperative mechanism involving metal and strong BAS is required to carry out the GVL ring-opening reaction. While the role of LAS has been associated with their activity in another step of the tandem reaction, no clear role has been assigned in this essential step. Zhou et al.25 reported for the first time that strong LAS are also active in the ring opening of GVL. These authors have described a dual catalyst consisting of Pd/C and Hf(OTf)4 in n-octane solution. At a relatively low temperature of 423 K and with 5.0 MPa H2, 92% VA selectivity and almost complete LA conversion were obtained. The recycling experiment showed that the catalysts continued to exhibit satisfactory activity after five uses. Recently Muñoz-Olasagasti et al.26 reported the relevance of the LAS in the gas phase conversion of LA into EV using a continuous fixed-bed reactor and ethanol as the solvent for CoSBA-xAl catalysts. The best catalyst was that with the highest Al content, CoSBA-2.5Al, which reached an EV yield of up to 70%. This result is associated with the presence of LAS attributed to the presence of Co2+ surface species that, although with low intrinsic activity in the selective GVL ring-opening reaction, are highly concentrated in this sample.
Many of the studies described above have been conducted under H2 pressure and autogenous solvent pressure in the liquid phase in a batch reactor. However, despite all of these studies, it is not clear whether, in the liquid phase, the BAS, LAS or both acid centers are responsible for the direct conversion of LA to EV. Moreover, the effects of the strengths of these acid sites on intrinsic activity also require attention.
This is precisely what we have addressed in this paper, in which we evaluate the effects of the acid site nature (B/L). In other words, it is important to identify the importance of Brønsted and Lewis sites (or both) and whether their different strengths affect their intrinsic activity in the direct conversion of LA to EV under liquid phase conditions. For this, we selected Pd as the reducting metal. Although it is more expensive than other metals such as Co and Ni, its choice is justified because it does not leach into the reaction medium.22 Moreover, Pd has been described as an efficient metal for the catalytic hydrogenation of levulinic acid in γ-valerolactone27 where the alloy of Ru–Pd on titanium dioxide favors the activity and stability of the catalyst. Yan et al.28 have also reported a bifunctional Pd/HY catalyst that achieved a 60% yield of the biofuel pentyl valerate in one pot synthesis. However the acid support was the commercial beta zeolite (with an atomic ratio Si/Al = 22) which contains Brønsted and Lewis acid sites. The nature and strength of these centers can be controlled through the substitution of Al atoms by Zr atoms.29–31 In acidic zeolites, the negative charge induced by Al is neutralized with protons, creating Brønsted acid sites. After replacing Al by Zr, the Brønsted acidity provided by the former gives rise to Lewis acidity due to the latter. This approach allows us to change the acid function without modifying the structural properties and thermal stability of the support.32
This particular research is designed to correlate the nature and strength of acid sites in the catalyst with their catalytic behavior in the conversion of LA to EV. Hydrogenation functionality was achieved by incorporating consistent levels of Pd into the zeolite by impregnation, so the effect of such Pd loading can be ignored in the comparison study. Moreover, structural variations have also been eliminated by using the same beta zeolite structure for all materials, in which the only modification is the progressive substitution of Al by Zr. This showed a strong impact on the acidity of the zeolite but not on other relevant textural and structural properties33 and is shown in Fig. S1 and S2.† This series of zeolites were tested in the reaction, and their acidic properties were characterized by diffuse reflectance infrared Fourier transform (DRIFT) spectroscopy to elucidate the correlation between acidity and activity. In this work, we found that strong BAS are required to perform the GVL ring-opening reaction. However, LAS also show activity in this reaction but to a lesser extent. Regarding the strength of LAS, the weaker the Lewis centers are, the less activity they exhibit for the LA to EV conversion. Interestingly, these weak acids associated with Zr are also less active in coke formation.
2. Experimental
2.1. Preparation of catalysts
Zr–Al-Beta zeolite acid supports were prepared by substitution of Al atoms with Zr atoms in a commercial Al-Beta zeolite (CP814C, Si/Al ratio = 19, Zeolyst International). An efficient and optimized two-step procedure was followed. Partial dealumination was carried out by treating the commercial zeolite in aqueous HNO3 with concentrations in the range of 0.1–10 M (room temperature, 1 h, 20 mL g−1). To achieve total dealumination, harsher conditions were needed (373 K, 20 h, 20 mL g−1). After filtration and washing with deionized water until reaching the neutral pH, the material was dried overnight at 383 K. Dealumination was followed by post-synthetic grafting of Zr species into the generated vacancies. The zirconium precursor (Zr(NO3)4, Chemical Point) and water concentration for impregnation were optimized to achieve the highest Zr dispersion. Thus, the Zr precursor was suspended in deionized water (10 mL g−1), and the corresponding amount of the zirconium salt was added. The resultant solid was heated under vacuum to remove the remaining water, and then it was dried overnight at 473 K. Finally, calcination in air at 823 K (for 6 h, with a heating ramp of 3 °C min−1) yielded the zeolites in the active form. More details of these preparations can be found in previous work.30,31,34 The starting commercial beta zeolite was labeled HBEA and three Zr–Al-Beta supports were prepared and labeled ZAB-1.6, ZAB-0.1 and ZAB-0.0, where the number denotes the Al/Zr molar ratio.Afterward, the incipient wetness impregnation method was used to incorporate 2 wt% Pd into the synthesized supports. An aqueous solution of (NH4)2PdCl4 (Sigma-Aldrich, ≥99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 995%) was used as the palladium precursor. Once impregnation with the palladium salt was completed, the solid was dried for 10 h at 393 K and then calcined at 823 K for 2 h. No further treatment was applied, as it is assumed that reduction of the metal occurred under the reaction conditions due to the temperature and hydrogen pressure used (513 K, 4 MPa of H2). The resultant catalysts were labeled Pd/HBEA, Pd/ZAB-1.6, Pd/ZAB-0.1 and Pd/ZAB-0.0.
995%) was used as the palladium precursor. Once impregnation with the palladium salt was completed, the solid was dried for 10 h at 393 K and then calcined at 823 K for 2 h. No further treatment was applied, as it is assumed that reduction of the metal occurred under the reaction conditions due to the temperature and hydrogen pressure used (513 K, 4 MPa of H2). The resultant catalysts were labeled Pd/HBEA, Pd/ZAB-1.6, Pd/ZAB-0.1 and Pd/ZAB-0.0.
2.2. Activity measurements
For catalytic measurements, a stainless-steel autoclave (100 mL, Parr-series 4590 reactor) was loaded with 15 g of ethanol, 0.25 g of levulinic acid and 0.1 g of the catalyst. The autoclave was purged several times, first with nitrogen and then with hydrogen, to remove air and nitrogen, respectively. The system was pressurized with H2 up to 4.0 MPa and then heated to the reaction temperature (513 K). Once the set temperature was reached, agitation was performed at 700 rpm. This time was considered as the time zero of the reaction.The liquid reaction medium was analyzed by gas chromatography (Agilent 6890 N) using a capillary column (ZB-WAX: 30 m × 0.32 mm × 0.50 μm) equipped with a flame ionization detector (FID). The quantified products were ethyl levulinate (EL), gamma-valerolactone (GVL), ethyl valerate (EV) and valeric acid (VA). Signals attributed to ethanol (solvent) and other minor products, such as ethyl pentenoate isomers, were observed in the chromatogram, but they were not quantified due to their low abundances. An aliquot of the reaction medium was mixed with a known amount of cyclohexanol, which was employed as a standard for analysis during sample preparation. Eqn (1) shows that the conversion of LA is defined as the molar ratio of LA consumed in the reaction relative to the initial amount of LA. Yields are defined for every product as the molar ratio of the respective product relative to the initial amount of LA (eqn (2)).
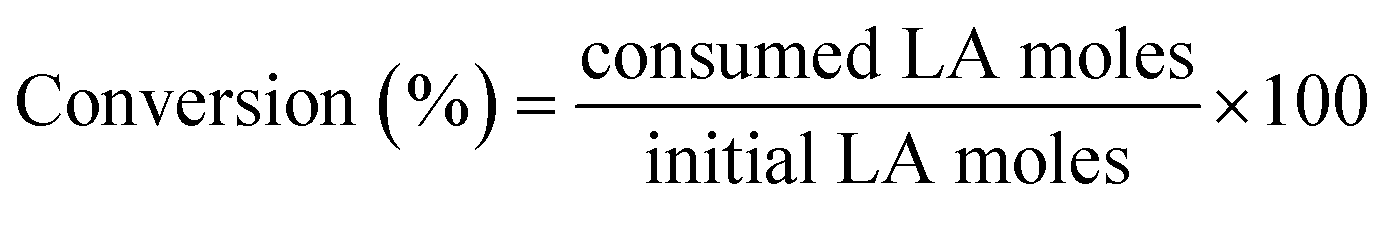 | (1) |
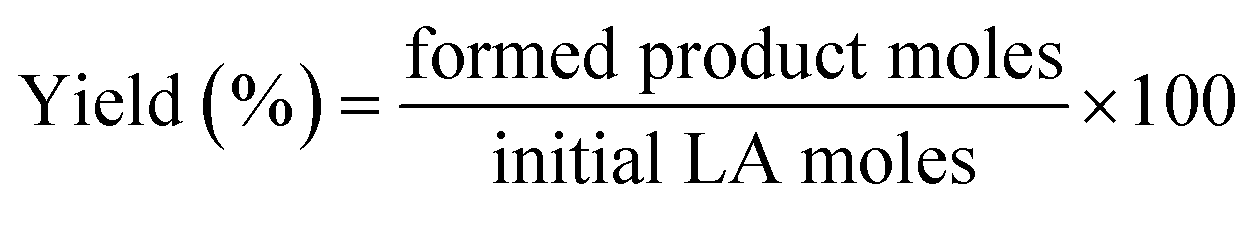 | (2) |
In reutilization tests, the catalyst was recovered after the reaction. The mother liquor was filtered, and the solid was rinsed several times with ethanol. The solid was dried overnight at 333 K, and when necessary, the fresh catalyst was added to compensate for losses in the recovery process. Reutilization reactions were carried out as previously described but with the recovered catalyst. After several reaction cycles, catalysts were reactivated by calcination employing a heating ramp of 1° min−1 up to 773 K, and this temperature was maintained for 1 h. After cooling, the catalyst was retested under identical reaction conditions.
2.3. Catalyst characterization
Elemental analysis of the zirconium and aluminum contents of the modified supports was performed by inductively coupled plasma optical emission spectroscopy (ICP-OES) with a Varian Vista AX apparatus. Powder X-ray diffraction (XRD) patterns were recorded using an X'Pert Pro PANalytical diffractometer in the Bragg–Brentano reflection geometry with the CuKα radiation line (λ = 1.54194 Å) in the 2θ angle range from 4° to 90° (step size 0.0335°)DRIFT spectra were collected with a Nicolet 5700 spectrometer equipped with a high sensitivity Hg–Cd–Te cryo-detector operating in the spectral range of 4000–650 cm−1. A diffuse reflectance accessory (Praying Mantis-Harrick Co.) was used as an optical mirror accessory. Approximately 30 mg of the previously ground sample was placed in a high-temperature catalytic reaction chamber (HVC-DRP Harrick Scientific Products, NY) that allows treatment in situ at high temperatures. To evaluate the surface acidity, pyridine (Py) and deuterated acetonitrile (CD3CN) were used as complementary probe molecules. The sample was first dried at 393 K for 1 h under an Ar flow (50 mL min−1) to clean the surface and then cooled to 298 K. Then, the probe molecule was added in sufficient quantity to saturate the sample. The physisorbed fraction was removed by flushing with an Ar flow (50 mL min−1) at 393 and 298 K for 1 h and 15 min for Py and CD3CN, respectively. To identify the state of oxidation and aggregation of the palladium surface species, carbon monoxide (CO) was also used as a probe molecule. Before CO adsorption, fresh or calcined catalysts were reduced under 10% H2/Ar flow (50 mL min−1) at 523 K for 30 min. After that, H2 was replaced by Ar (50 mL min−1) and the sample was cooled to room temperature. Then, 5% CO/He flow (30 mL min−1) was fed for adsorption. Physisorbed CO was removed by flushing with Ar at this temperature for 2 min. The net spectrum was obtained by subtracting the reference spectrum of the solid catalyst. All spectra were recorded with 128 scans and a resolution of 4 cm−1.
3. Results and discussion
3.1. Catalytic activity
Fig. 2 shows the catalytic behavior of these catalysts in the conversion of levulinic acid (LA) into valeric biofuels (EV + VA) after one hour of the reaction. Complete conversion of levulinic acid was observed for all catalysts under our reaction conditions. The Pd/HBEA catalyst exhibited the highest yield of valeric biofuels, achieving a remarkable value close to 60%; the rest of the series retained a more significant proportion of gamma-valerolactone (GVL), the main intermediate of the integrated reaction (see Fig. 1). Samples with a lower Al/Zr ratio gave worse yields of valeric biofuels, and the most unsatisfactory results were those obtained for the sample without aluminum (Pd/ZAB-0.0). This catalyst provided the highest yield of GVL instead. Therefore, the distribution products will depend on the Al/Zr atomic ratio of the bimetallic zeolitic supports where Al has been exchanged for Zr. As this exchange increases, the catalysts in our reaction present worse behavior. Accordingly, the Pd/HBEA catalyst is the sample with the best catalytic performance.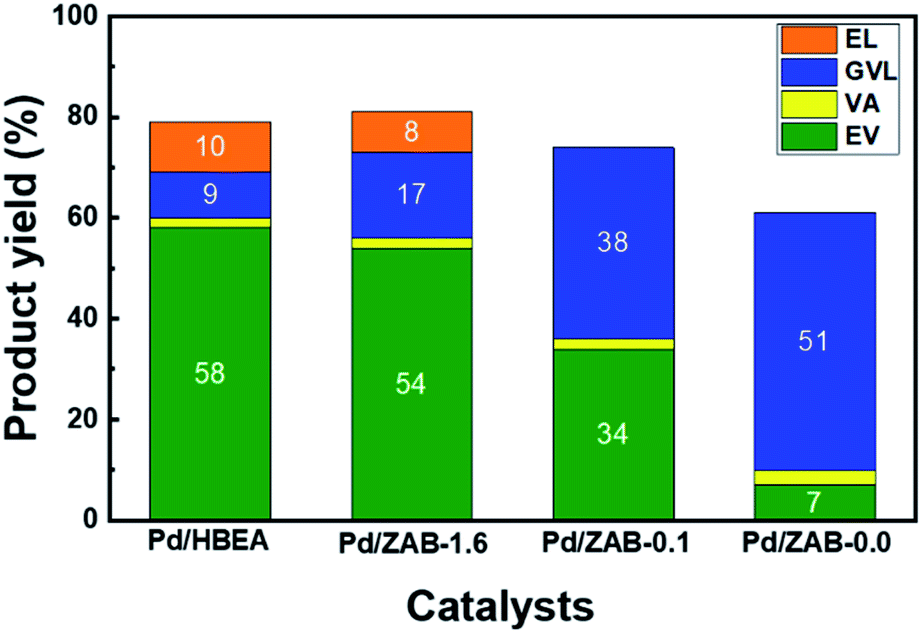 | ||
| Fig. 2 Reaction product yields for the Pd/Zr–Al-Beta catalysts. Reaction conditions: 15 g ethanol, 0.25 g LA, 0.1 g catalyst, T = 513 K, 4.0 MPa H2, 1 h and 700 rpm. | ||
Notably, some additional small peaks were also present in the chromatograms, indicating the appearance of other byproducts. In this way, methyl tetrahydrofuran, 1-pentanol and ethyl pentenoate isomers were identified, representing less than 3–5% of the total products coming from starting LA, which is not enough to achieve the carbon balance. Therefore, carbonaceous coke-like products, which are undetectable by gas chromatography, were most likely formed during the reaction. Some reaction mixtures were analyzed with GC-MS to identify these products, but it was unsuccessful, indicating that they might be high-boiling or coke-like products, hard to detect by GC. Indeed, it is well known that coke can be formed when organic compounds are hydrotreated at temperatures above 500 K and in the presence of acidic sites.35,36 As will be discussed below, these carbonaceous residues are partially responsible for the deactivation of the active centers in the catalyst by fouling.
According to the scheme displayed in Fig. 1, EL and GVL are intermediate products. These intermediates were present after one hour of the reaction (see Fig. 2), and therefore, these catalysts can continue to convert them into the products of interest (EV + VA) if they are allowed sufficient time to carry out the complete reaction. Thus, experiments with longer reaction times (2 h and 4 h) were conducted, and the results are shown in Fig. 3. All the catalysts improved the yields of valeric biofuels with an increased reaction time. Nevertheless, there were apparent differences in the progress of the reactions. For Pd/HBEA, an optimum value was reached at two hours, with a yield of 76% for the combined EV + VA. For the rest of the catalysts, the yield of valeric biofuels continued to increase after 4 h of the reaction. This result is attributed to the continued conversion of intermediates, which increased the yields accordingly.
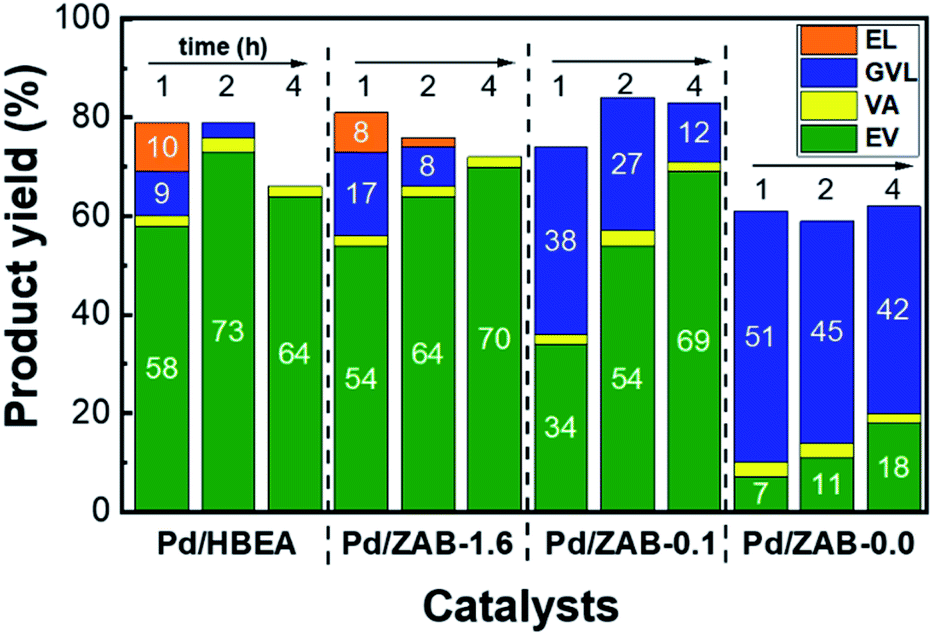 | ||
| Fig. 3 Reaction product yields for the Pd/Zr–Al-Beta catalysts with reaction times of 1, 2 and 4 hours. The reaction conditions are the same as those shown in Fig. 2. | ||
As mentioned, this trend is different for the Pd/HBEA catalyst, which reached a maximum yield of target products after two hours on stream. After that time, not only did the yield of these products decrease, but the carbon balance also worsened. A similar decrease in the carbon balance was also observed for the Pd/ZAB-1.6 sample. The significant difference between these two catalysts is that over Pd/ZAB-1.6, the yield of valeric biofuels did not decrease after four hours, maintaining over 70%. Pd/ZAB-0.1 exhibited much slower reaction rates for the first two hours, but after that, it reached the same yield as Pd/ZAB-1.6 but with a better carbon balance. Finally, for the Pd/ZAB-0.0 catalyst, the carbon balance was much lower from the beginning (61%), but it remained stable. After the first hour, the only process underway was the conversion of intermediate GVL to EV. The maximum attainable yield of EV with this catalyst would be just 60%; after 4 hours of the reaction, it only reached 18%.
To evaluate the stability of the Pd/HBEA catalyst, reutilization tests were conducted. For these assays, a two hour reaction time was selected and the yield of the desired products (EV + VA) reached a maximum of 76%. Fig. 4 displays the results and shows a slight deactivation of the catalyst in each recycling step. Thus, the yield of combined VA and VE decreases from 76% with the fresh catalyst to 56% after three cycles. After the third cycle, the catalyst was calcined at 773 K for one hour, and then a further reutilization test was conducted. This calcination procedure changed the downward trend by significantly improving the yield up to 63%, but the initial activity was not recovered.
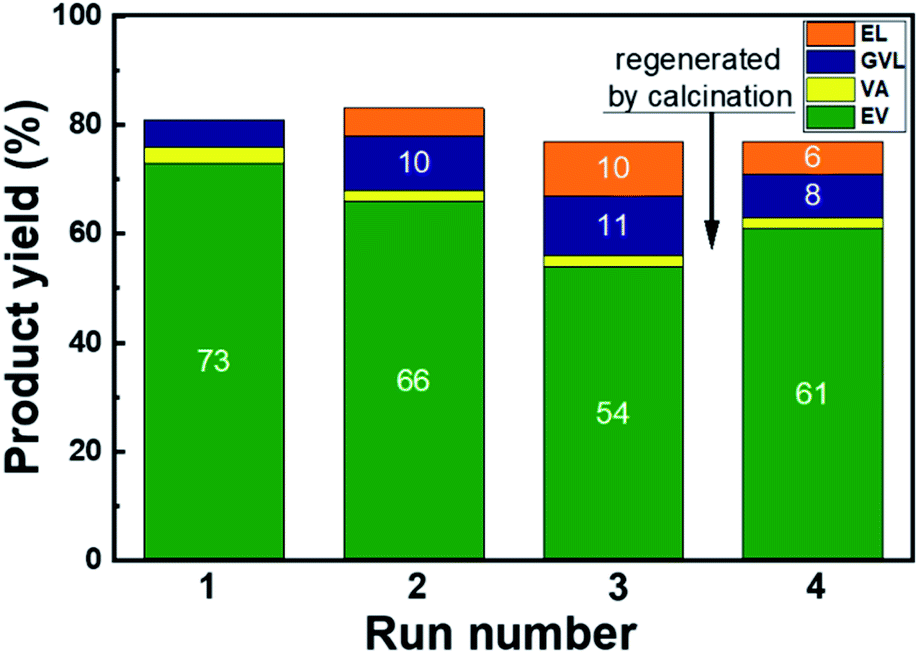 | ||
| Fig. 4 Product yields during the reutilization tests of the Pd/HBEA catalyst. The time of the reaction was 2 h, and other conditions are the same as those shown in Fig. 2. | ||
These results need further explanation and characterization experiments are designed to obtain additional information regarding the reason for the deactivation observed, potential approaches to avoid it and the impact of regeneration by calcination on the active sites.
3.2. Characterization of the catalysts
| Support | Al (wt%) | Zr (wt%) | Al/Zr (mol mol−1) |
|---|---|---|---|
| HBEA | 2.0 | 0.0 | ∞ |
| ZAB-1.6 | 1.4 | 2.9 | 1.6 |
| ZAB-0.1 | 0.2 | 6.9 | 0.1 |
| ZAB-0.0 | 0.0 | 6.3 | 0.0 |
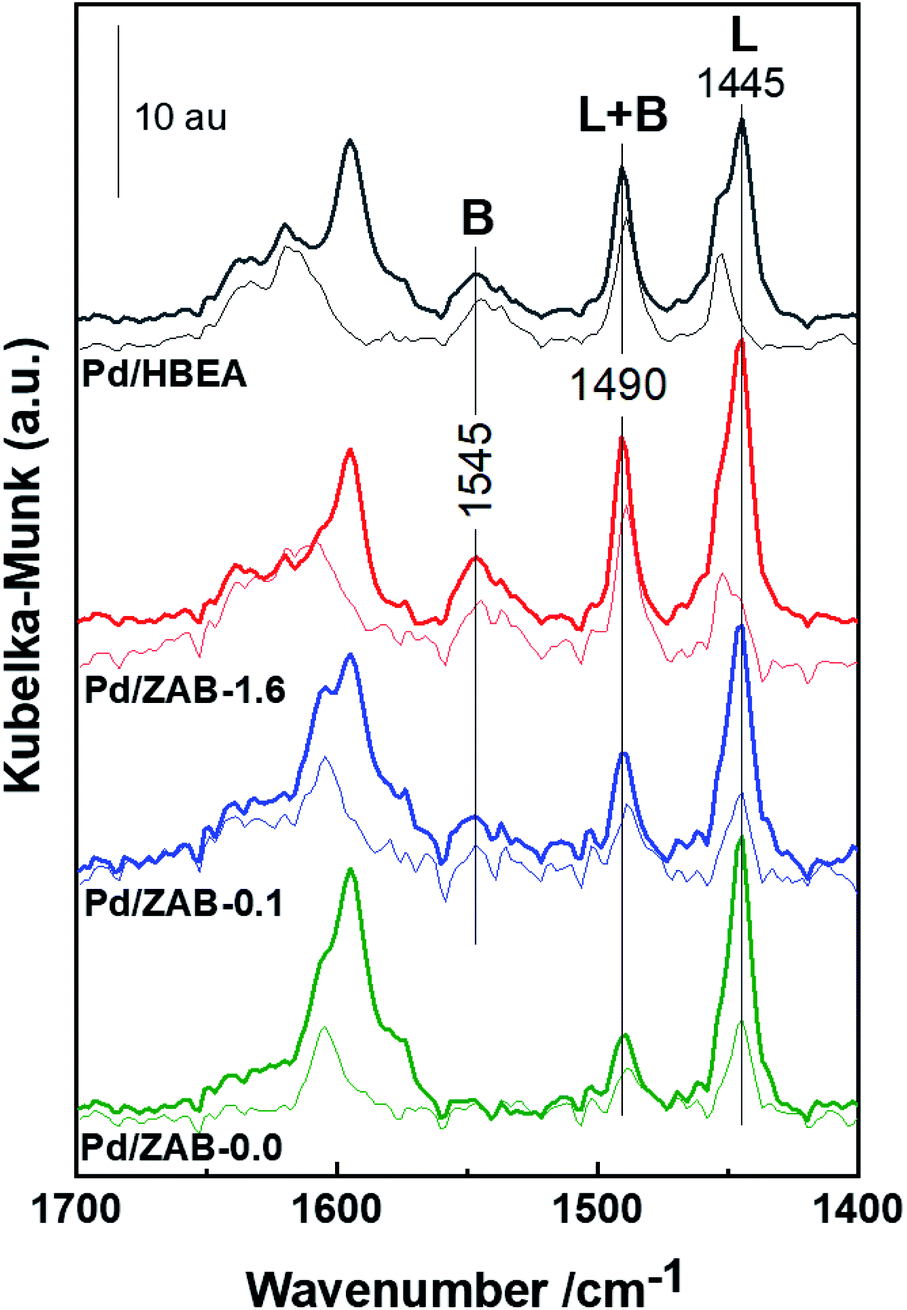 | ||
| Fig. 5 DRIFT spectra of adsorbed Py on Pd/ZrAl-Beta catalysts. The thick and thin lines represent flushing with an Ar flow at 393 and 573 K, respectively. | ||
After desorption at 393 K for one hour (thick lines shown in Fig. 5), some physisorbed/pseudo liquid Py remained, while at above 573 K for 30 min (thin lines), only chemisorbed pyridine was present. Al contributes to both Brønsted and Lewis acidity, while Zr only provides Lewis acid centers.34 From Fig. 5, it can be concluded that the Pd/HBEA and Pd/ZAB-1.6 catalysts showed the most significant numbers of Brønsted acid sites (BAS), whereas this amount was reduced in Pd/ZAB-0.1 and non-existent in the Al-free Pd/ZAB-0.0 catalyst. This confirms the complete removal of all the aluminum from this sample, as indicated by the elemental chemical analysis.
Next, the study of surface acidity was completed by using CD3CN as a probe molecule. The corresponding DRIFT spectra of the different catalysts are shown in Fig. 6, where the deconvolution of the simple infrared bands is also included. Four infrared bands were observed for the Pd/HBEA catalyst, one small peak at 2332 cm−1 and three broad intense peaks at 2318, 2294 and 2272 cm−1. The replacement of Al by Zr atoms had a strong impact on the DRIFT spectra, in which the number and intensities of the absorption bands were modified. At the other end of the series, only two bands are observed for the Pd/ZAB-0.0 catalyst, at 2301 and 2272 cm−1. In the intermediate catalysts Pd/ZAB-1.6 and Pd/ZAB-0.1, which contain both ions (Al and Zr), up to five infrared bands can be observed, representing a combination of the other two catalysts of the series. Two bands were identified in Pd/ZAB-0.0 at 2272 and 2301 cm−1, but correct deconvolution required the inclusion of a band at 2294 cm−1 and two bands at 2318 and 2332 cm−1. These last three infrared bands were also present in the Pd/HBEA catalyst samples where the support only contained Al and the intensities of these three bands decreased with the dealumination of the supports.
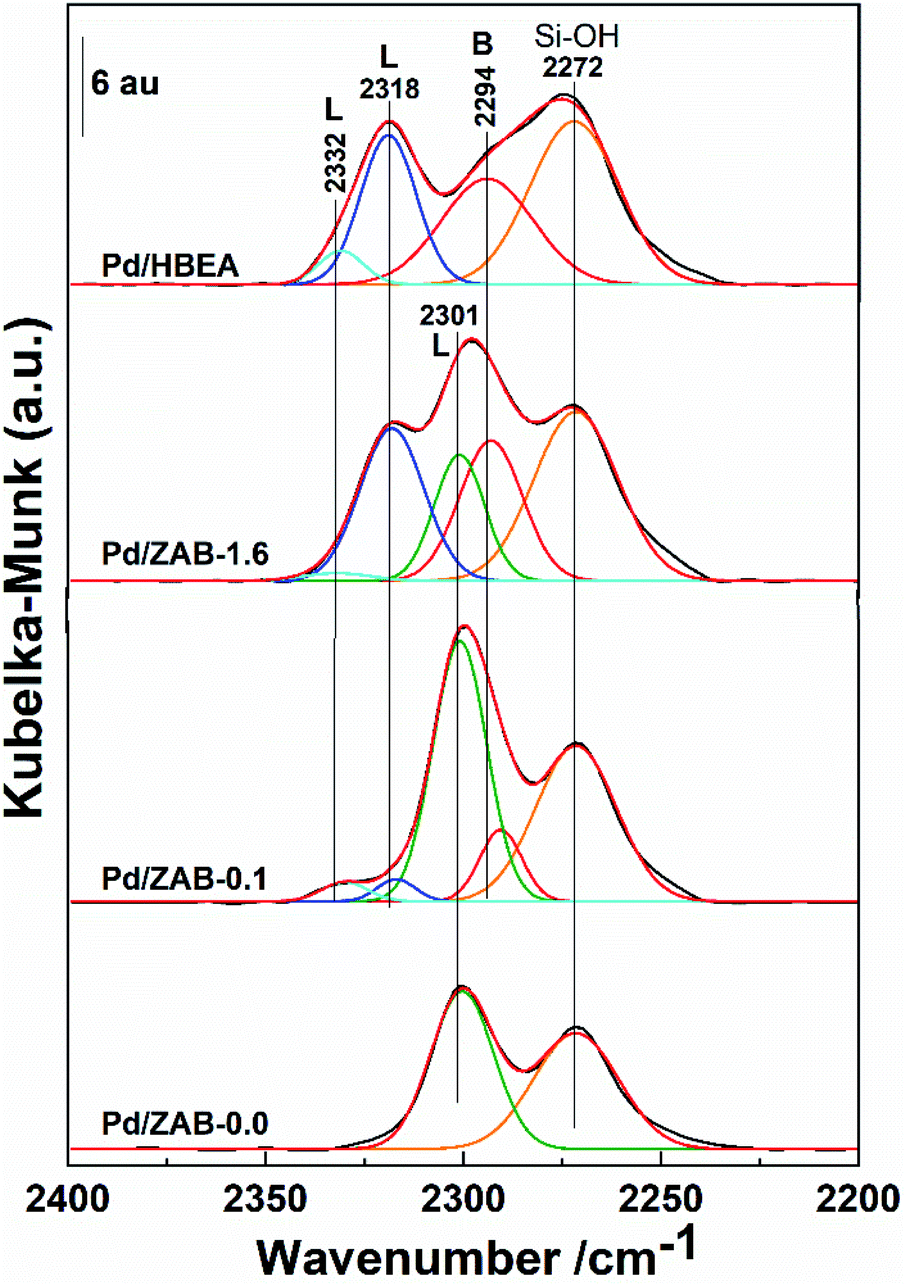 | ||
| Fig. 6 DRIFT spectra of deuterated acetonitrile on Pd/ZrAl-Beta catalysts. Deconvolution in the infrared bands is also displayed. | ||
Regarding the different CD3CN absorption band assignments, it is known that when the band maxima are at frequencies ≥2300 cm−1 they are associated with Lewis acid sites (LAS), while the other bands with maxima at frequencies <2300 cm−1 are associated with Brønsted acid sites (BAS). Moreover, the higher the frequencies of these peaks are, the greater the strength of the indicated acid centers.40–42 Concerning the Brønsted region, very weak BAS associated with silanol groups, with an IR peak at 2272 cm−1, remains in all samples, consistent with the silicic nature of the zeolitic supports29,34 On the other hand, the progressive removal of Al provides a concomitant reduction in the number of strong Brønsted acid sites, so the intensity of the 2294 cm−1 adsorption band decreases throughout the series. Regarding the Lewis acid region, the peaks at 2332 cm−1 and 2318 cm−1 are attributed to strong Lewis acid sites.22,40 Both peak intensities decreased with the replacement of Zr by Al in the catalyst support. Finally, when more Zr was incorporated, an infrared band at 2301 cm−1, associated with weak LAS and the presence of Zr atoms,34,43 prevailed.
The deconvolution shown in Fig. 6 was supported by the results obtained with the adsorption of Py as a probe molecule. Both molecules are complementary in indicating the evolution of surface acidity in terms of the nature and strength of the acid sites. Moreover, it is necessary to mention that the acidity of the catalytic supports was also studied prior to incorporation of Pd (spectra shown in Fig. S3†), and no significant differences were observed for the Pd/ZrAl-Beta catalysts. Therefore, we can confirm that the incorporation of Pd did not affect the surface acidity of the support in this case.
The information regarding surface acidity for this series of catalysts, derived from DRIFT data obtained after absorption of Py and CD3CN, can be summarized as follows: the substitution of Al by Zr in the acidic supports (HBeta zeolite) has consequences for the Lewis and Brønsted surface acid centers. In principle, a strong LAS band (2318 cm−1) appears at a higher frequency (2332 cm−1) with the generation of strong Lewis acid sites. However, when the level of substitution is sufficient, the LAS associated with zirconium atoms are very weak (infrared band centered at 2301 cm−1). The strong BAS band at 2994 cm−1 decreased in intensity throughout the series and was no longer present when Zr had entirely replaced the Al in the parent zeolite. Finally, the weak Brønsted acid sites associated with the presence of silanol groups were essentially maintained throughout the series.
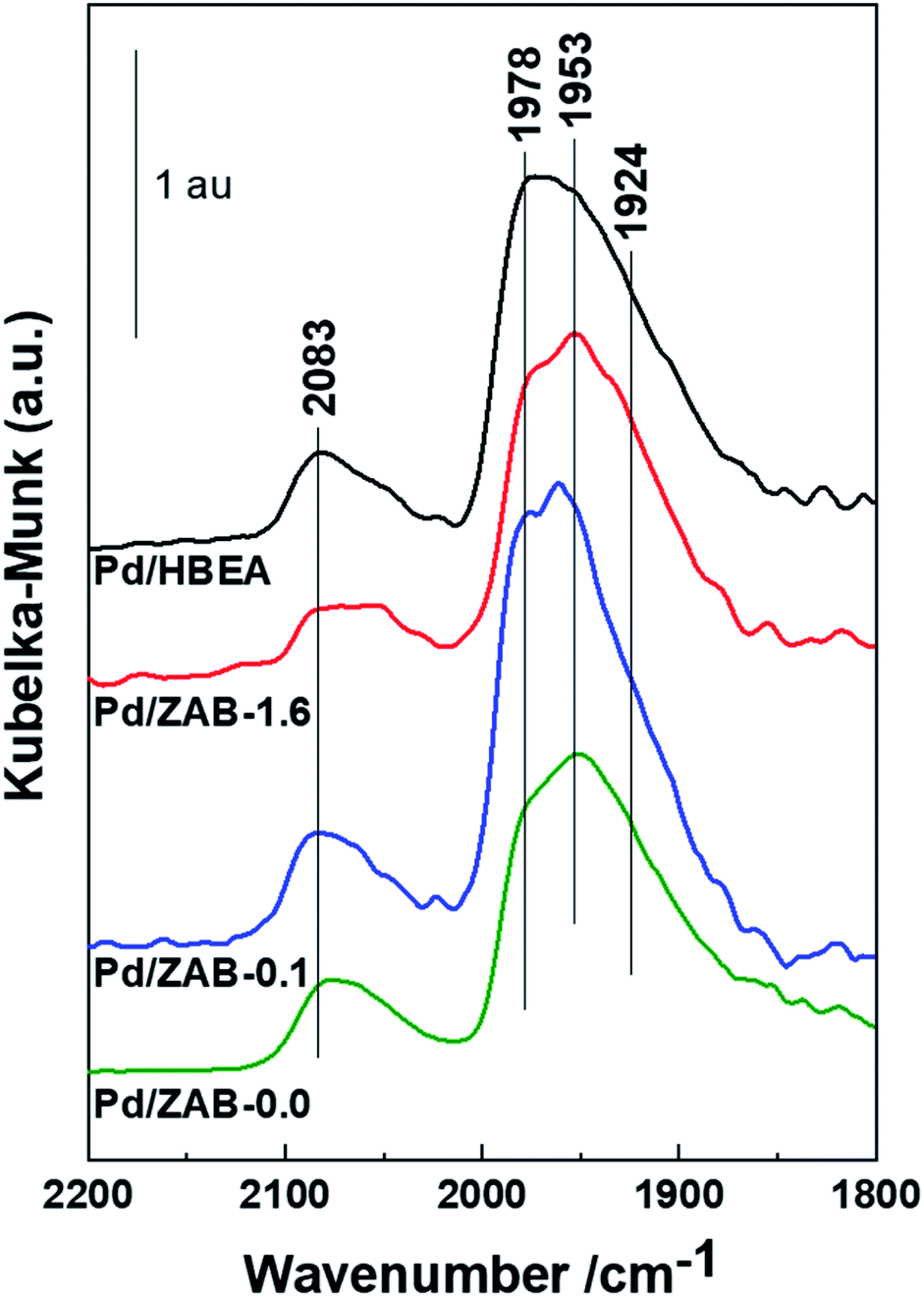 | ||
| Fig. 7 DRIFT spectra of CO adsorbed on reduced Pd/ZrAl-Beta catalysts at room temperature. | ||
The spectra of the different samples are similar in terms of the bands and intensities, and no significant changes were observed. Therefore, we can affirm that the Pd surface species generated after incorporation via impregnation were not affected by the different surface acidities of the supports. This suggests that the previously discussed differences observed in the catalytic comportment of the different materials must be attributed to the nature (Brønsted/Lewis) and strength of the surface acid centers rather than to the exposed metal Pd species.
Therefore, after analyzing the DRIFT spectra of the acid sites and the Pd metallic species, it can be established that neither the incorporation of Pd affects the acidity of the zeolitic support nor the different acidities of the prepared Zr–Al-Beta supports modify the Pd metallic species.
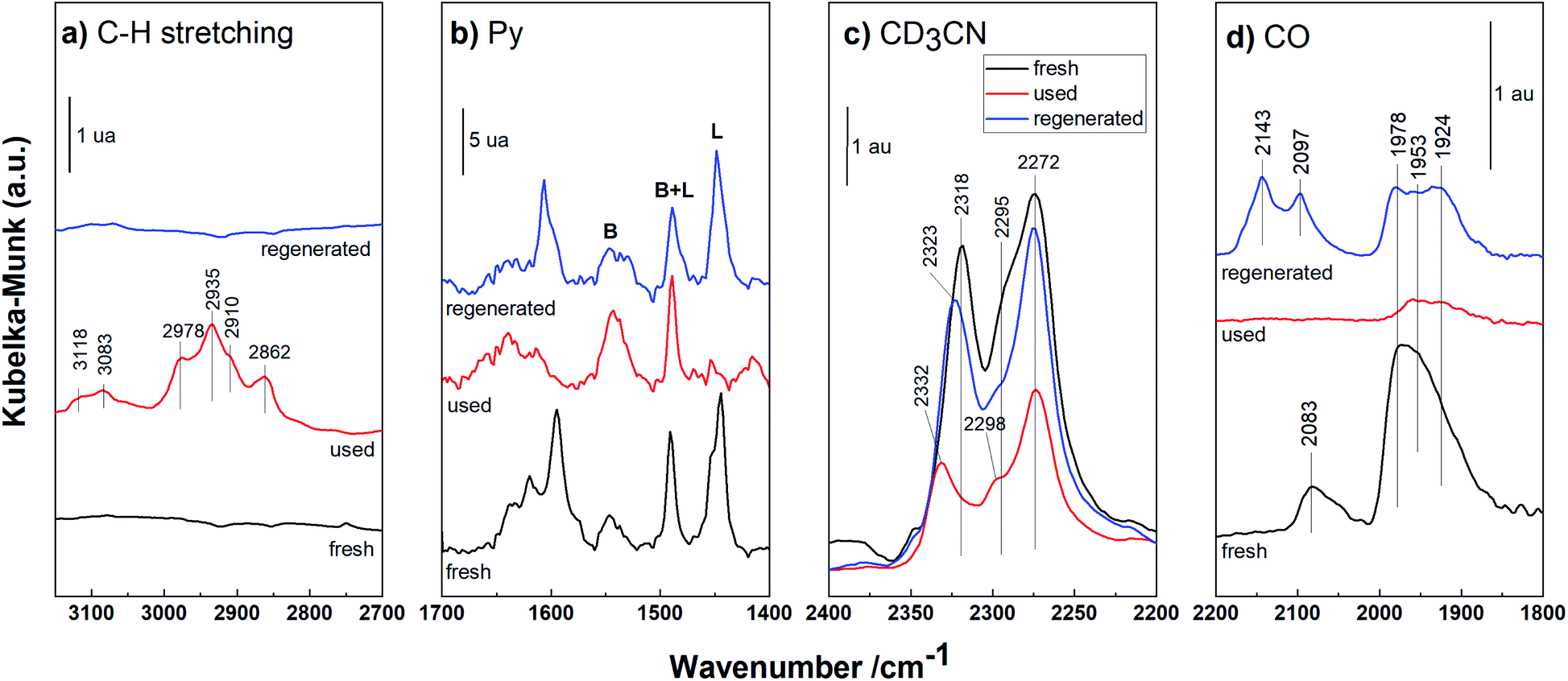 | ||
| Fig. 8 DRIFT spectra of the fresh, used and regenerated Pd/HBEA. (a) C–H stretching region; (b) Py, (c) CD3CN and (d) CO like probe molecules. | ||
DRIFT spectra in the C–H stretching region (3150–2700 cm−1) are presented for the fresh, used and regenerated Pd/HBEA sample shown in Fig. 8a. The fresh catalyst does not present infrared bands in this region, as expected. In contrast, for the used sample, several absorption bands at 3118, 3083, 2978, 2935, 2910 and 2862 cm−1 can be attributed to carbonaceous species with unsaturated bonds (3118 and 3083 cm−1 infrared bands) and with saturated C–H bonds (the rest of the bands).47,48 After exposing the used catalyst to regeneration by calcination, practically all the carbonaceous deposits were removed; see the spectrum of the regenerated catalyst in Fig. 8a.
Fig. 8b shows the spectra of the Pd/HBEA samples (fresh, used and regenerated) obtained by using Py as a probe molecule. In this case, the decreasing acidity for the used catalyst was mainly due to the superficial Lewis acid sites present in the catalyst; once they underwent the regeneration process, they were largely recovered. This suggests that these centers are the main promoters for formation of carbonaceous residues fouling the catalyst surface. This same sequence of DRIFT experiments was also performed for another basic probe molecule, deuterated acetonitrile (CD3CN), and the DRIFT spectra are compiled and shown in Fig. 8c. For the used Pd/HBEA catalyst, a strong drop in the intensity of the bands was observed (relative to the fresh catalyst), which was almost fully reversed after regeneration. Therefore, it can be surmised that the used Pd/HBEA sample underwent a drastic decrease in the number of exposed acid sites, as evidenced by the low intensities of the infrared bands. This is mainly due to the loss of LAS, as these centers can be fouled by carbonaceous species generated during the reaction. Such carbonaceous species can be removed with a high degree of efficiency with regeneration by calcination, as shown with both probe molecules (Py and CD3CN).
On the other hand, the Pd surface species in used and regenerated catalysts have also been studied by DRIFT spectroscopy using CO as the probe molecule, and the spectra are shown in Fig. 8d. It is observed that the intensities of bands due to the Pd surface species diminished drastically because with the acid sites, the carbonaceous deposits also covered the metallic Pd. After the regeneration of this sample, a part of the isolated metallic Pd was recovered during calcination due to coke removal. However, partially oxidized Pd species (band at 2143 cm−1)49 and slightly sintered Pd46 species (the band at 1924 cm−1 is proportionally more intense) were also indicated.
Therefore, it can be concluded that the deactivation of the catalyst occurs mainly due to the fouling of its surface by carbon deposits, which affect both the acidic and metallic exposed active centers. After regeneration employing calcination in air, the surface acidity was practically recovered in its entirety, whereas the metallic Pd species were reoxidized. These reoxidized species were more resistant to reduction than the PdO species present in the precursor catalyst. This may be the main reason that the catalyst does not reach the valeric biofuel yield of the fresh catalyst after regeneration (see Fig. 4).
To investigate the causes of deactivation and the incomplete regeneration of the used catalyst, the samples were characterized by the XRD technique. The diffractograms of the Pd/ZAB catalysts are shown in Fig. 9.
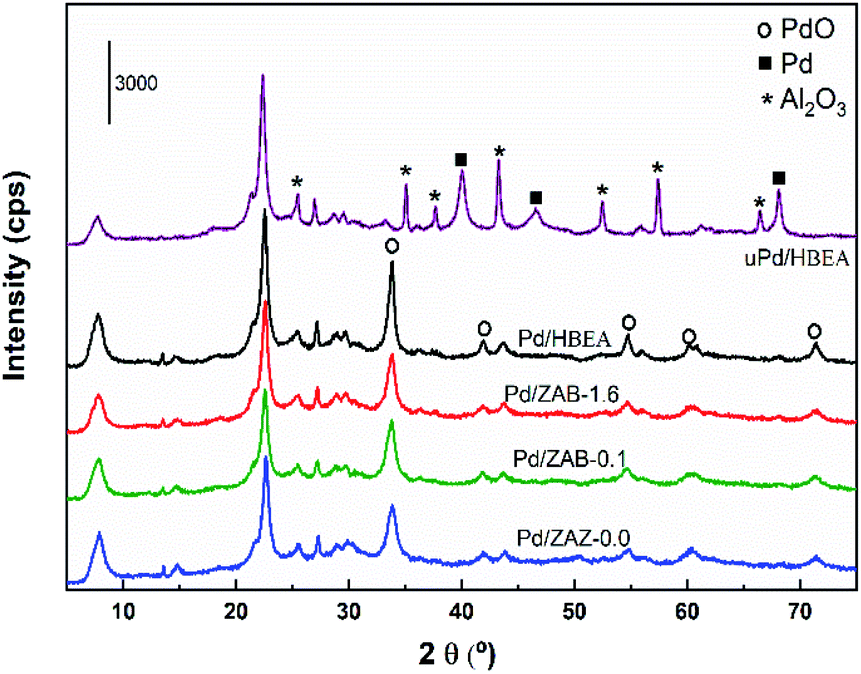 | ||
| Fig. 9 XRD patterns of the fresh Pd/ZAB and used Pd/HBEA catalysts. | ||
The peaks corresponding to the PdO phase (JCPDS 006-0515) are observed for the fresh catalysts. The rest of the peaks are associated with the zeolitic support (see Fig. S2 in the ESI†). However, when we observe the spectrum of the used Pd/HBEA catalyst (uPd/HBEA), peaks corresponding only to metallic Pd (JCPDS 001-1201) are observed, which confirms that under the reaction conditions the palladium oxide is extensively reduced to Pd as expected. This is in accordance with the DRIFT technique after CO adsorption as shown in Fig. 8d. However, a part of the surface Pd may have been reoxidized during the handling of the sample after the reaction is finished. On the other hand, it should also be mentioned that diffraction peaks assigned to the crystalline phase Al2O3 (JCPDS 01-080-0786) are observed in the used catalyst, possibly since a partial extraction of the lattice aluminum occurs during the progress of the reaction. This fact together with the presence of carbonaceous residues is the most important cause of deactivation.
3.3. Impact of acid site nature and strength on catalytic performance and stability
This research is designed to find the direct relationship between the nature and strength of the surface acid sites and their catalytic behavior under liquid phase conditions. For this purpose, an acid support (H-beta zeolite) that was modified by progressively substituting Al for Zr atoms has been used. The correctness of this approach was confirmed by the analysis of DRIFT spectra. This indicated that the incorporation of Pd does not change the acidity of the supports. At the same time, the different acidities of the modified supports do not affect the dispersion and reducibility of the incorporated Pd. Therefore, the differences observed in the catalytic behaviors have to be fully attributed to the changes observed in the surface acidity of these bifunctional catalysts.It is necessary to highlight the complexity of the challenge addressed. On the one hand, we have acid centers with different natures and strengths coexisting in the catalyst, and on the other hand, the acid centers catalyze various steps of the cascade reaction (see the scheme in Fig. 1). These acid sites will be more or less active depending on the step in which they participate (dehydration, GVL ring-opening, or even esterification), their nature and strength, and the reaction conditions.
The yield of valeric biofuels (EV + VA) follows the trend Pd/HBEA ≅ Pd/ZAB-1.6 > Pd/ZAB-0.1 > Pd/ZAB-0.0. This same qualitative tendency is indicated by the intensities of the IR bands for the strong BAS associated with Al sites on the surface of the catalysts (2294 cm−1 with CD3CN and 1545 cm−1 with Py). Therefore, these results suggest that this type of acid site, associated with Al present in the catalysts, is mainly responsible for the best catalytic behavior. Such strong Brønsted centers are required for the GVL ring-opening reaction, which is the rate-determining step of the process.16 This is consistent with the fact (see Fig. 2) that Pd/HBEA, Pd/ZAB-1.6 and Pd/ZAB-0.1 show Brønsted acid sites and gave up to 70% yields for valeric biofuels. However, Pd/ZAB-0.1 required 4 hours to reach this yield, possibly due to slower kinetics attributed to the low concentration of Brønsted acid sites in this sample relative to other samples. The performance is considerably poorer for the Pd/ZAB-0.0 catalyst that does not have strong BAS, even after 4 hours of the reaction. On the other hand, all the catalysts contain other types of weaker BAS associated with silanol groups (Si–OH, at 2272 cm−1 with the CD3CN probe) whose presence and IR intensities do not change significantly in the series of supports. These weak acid sites are irrelevant for the catalytic conversion of LA into EV.50
Moreover, the roles of the LAS as related to their strengths should be analyzed. Three Lewis acid sites were identified from the DRIFT spectra, including the strongest with a band at 2332 cm−1, another strong site shown at 2318 cm−1 and a weak site shown at 2301 cm−1 (see Fig. 6). It was observed that the weak LAS exhibited limited activity in opening the ring of the GVL: no active Brønsted sites were present in sample Pd/ZAB-0.0, which showed some GVL ring opening activity, but much less than the samples with strong BAS and an IR band at 2294 cm−1. However, these centers were more active in the dehydration reaction giving the GVL intermediate. In the case of strong LAS (bands at 2332 and 2318 cm−1), they are clearly more involved in this dehydration reaction and important in producing the intermediates (EL and GVL). Therefore, the capacity of the LAS for the dehydration reaction depends on their strength. On the other hand, the effect of the strong LAS (2332 and 2318 cm−1 bands with CD3CN) on the ring opening reaction of the GVL is clear although the decrease in strong LAS, with the increase in the substitution of Al by Zr, is also accompanied by a reduction of the strong BAS. The presence of these strong LAS centers (together with the presence of strong BAS) is the main difference between the first three samples in the series and the Pd/ZAB-0.0 sample (see Fig. 5 and 6). This difference is critical in the activity of the Pd/ZAB-0.0 catalyst, since the absence of these LAS gives rise to more unknown products and less than 20% conversion to valeric biofuels even after four hours of the reaction. For the rest of the catalysts, even Pd/ZAB-0.1 provides 70% yield of the desired valeric products. In summary, weak and strong LAS are also active in the GVL ring-opening reaction but exhibit less intrinsic activity than BAS. Moreover, the activities of these centers for this reaction depend on their acid strengths.
The surface acid sites play an important role in determining the product distribution. Kumar et al.51,52 investigated the vapor-phase hydrogenation of LA to VA on the Ni/TiO2 catalyst. An improvement in catalytic behavior was observed after incorporating W and increasing the number of BAS due to the W interactions with Ni/TiO2. These authors reported that under their conditions, 543 K and atmospheric pressure, sites other than LAS were mainly responsible for the high selectivity for GVL, whereas BAS were prone to enhance the GVL ring-opening reaction. We observed a similar behavior under liquid phase conditions and applied pressures, but we also observed the ability of LAS to carry out the ring-opening reaction of GVL, although with lower intrinsic activity than strong BAS.
Another aspect that should be mentioned is the role of the surface acid sites in the deactivation and selectivity of the intermediates and products of interest. The strong BAS, mostly present in the Pd/HBEA catalyst, were active in the subsequent transformation of EV into unidentified products; see the drop in the yield of EV after 4 hours of the reaction in Fig. 3. Therefore, these strong acid sites appear to be responsible for the unwanted progression of the reaction beyond the desired products at long reaction times, leading to worse performance at 4 h compared to 2 h. The causes of deactivation of the Pd/HBEA catalyst (Fig. 4) are the formation of carbonaceous residues that foul the acidic and metallic centers (see Fig. 8) on the catalyst surface and the slight formation of the Al2O3 phase (Fig. 9). Regarding regeneration by calcination in air at 773 K for 1 h, practically all the acid sites covered by these carbonaceous residues were refreshed and re-exposed, but during the regeneration process some species of metallic Pd were oxidized to species more difficult to reduce than those in the fresh catalyst. The possible presence of oxidized Pd species in the regenerated catalyst could prevent the recovery of the initial activity. This is why the catalyst presents a partially reversible deactivation due to removal of the carbonaceous residues.
4. Conclusions
The combination of the catalytic activity and the DRIFT spectral data leads to the following conclusions concerning the nature and strength of surface acid sites in fresh catalysts. The importance of strong BAS in catalytic behavior is great because these centers are mainly responsible for the GVL open-ring reaction. However, these acid sites can also promote the undesirable reaction of EV to give other unidentified products at long reaction times. Concerning LAS, three different sites have been identified. Weak LAS associated with Zr species (infrared band at 2301 cm−1 for CD3CN) exhibit low activity in the production of valeric biofuels, but they can be relevant in the deactivation process. When only weak Lewis acidity is present, the catalyst cannot accomplish the full transformation, attaining a limited EV yield. The dramatic difference in the performances of Pd/ZAB-0.1 and Pd/ZAB-0.0 indicates the crucial effects of strong LAS (infrared bands at 2318 and 2332 cm−1 for CD3CN) in driving the reaction, although there are BAS centers also in this sample that can contribute. Consequently, both types of strong centers (BAS and LAS) are active in the cascade conversion of LA into EV. However, the intrinsic activity of the former is more significant because when their concentration is increased, less time is required to achieve expected yields of EV.Regarding the deactivation and regeneration of Pd/HBEA, it was observed that the catalytic activity decreased slightly in each successive use, most likely due to the formation of carbonaceous deposits and the formation of the Al2O3 phase. After regeneration by calcination in air, the catalyst showed a partial recovery of the starting activity due to the full regeneration of acid sites, in agreement with the DRIFT results. However, Pd species were affected by the regeneration procedure and this prevented the catalyst from exhibiting its initial catalytic performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grants RTI2018-094918-B-C41 and RTI2018-094918-B-C42 by MCIN/AEI/10.13039/501100011033 and the Regional Government of Madrid (project S2018/EMT-4344) are kindly acknowledged for funding this research. MMO and IMS, due to the CSIC-UAM framework agreement, thank Ph.D. Program in Applied Chemistry, Doctoral School of Autonomous University of Madrid.References
- G. Dautzenberg, M. Gerhardt and B. Kamm, Holzforschung, 2011, 65, 439–451 CAS.
- C. Antonetti, D. Licursi, S. Fulignati, G. Valentini and A. Raspolli Galletti, Catalysts, 2016, 6, 196 CrossRef.
- J. C. Serrano-Ruiz, R. Luque and A. Sepúlveda-Escribano, Chem. Soc. Rev., 2011, 40, 5266–5281 RSC.
- E. I. Gürbüz, S. G. Wettstein and J. A. Dumesic, ChemSusChem, 2012, 5, 383–387 CrossRef PubMed.
- F. D. Pileidis and M. M. Titirici, ChemSusChem, 2016, 9, 562–582 CrossRef CAS PubMed.
- S. G. Wettstein, D. M. Alonso, Y. Chong and J. A. Dumesic, Energy Environ. Sci., 2012, 5, 8199–8203 RSC.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS.
- D. M. Alonso, S. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- Z. Xue, Q. Liu, J. Wang and T. Mu, Green Chem., 2018, 20, 4391–4408 RSC.
- J. Iglesias, I. Martínez-Salazar, P. Maireles-Torres, D. Martin Alonso, R. Mariscal and M. López Granados, Chem. Soc. Rev., 2020, 49, 5704–5771 RSC.
- J. P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- Z. Yu, X. Lu, J. Xiong and N. Ji, ChemSusChem, 2019, 12, 3915–3930 CrossRef CAS PubMed.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC.
- G. B. Kasar, R. S. Medhekar, P. N. Bhosale and C. V. Rode, Ind. Eng. Chem. Res., 2019, 58, 19803–19817 CrossRef CAS.
- W. Luo, U. Deka, A. M. Beale, E. R. H. Van Eck, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2013, 301, 175–186 CrossRef CAS.
- Z. Yi, D. Hu, H. Xu, Z. Wu, M. Zhang and K. Yan, Fuel, 2020, 259, 3–6 CrossRef.
- T. Pan, J. Deng, Q. Xu, Y. Xu, Q. X. Guo and Y. Fu, Green Chem., 2013, 15, 2967–2974 RSC.
- K. Kon, W. Onodera and K. I. Shimizu, Catal. Sci. Technol., 2014, 4, 3227–3234 RSC.
- X. M. Gu, B. Zhang, H. J. Liang, H. Bin Ge, H. M. Yang and Y. Qin, J. Fuel Chem. Technol., 2017, 45, 714–722 CrossRef CAS.
- W. Luo, P. C. A. Bruijnincx and B. M. Weckhuysen, J. Catal., 2014, 320, 33–41 CrossRef CAS.
- M. Muñoz-Olasagasti, A. Sañudo-Mena, J. A. Cecilia, M. L. Granados, P. Maireles-Torres and R. Mariscal, Top. Catal., 2019, 62, 579–588 CrossRef.
- P. Sun, G. Gao, Z. Zhao, C. Xia and F. Li, ACS Catal., 2014, 4, 4136–4142 CrossRef CAS.
- P. Sun, G. Gao, Z. Zhao, C. Xia and F. Li, Appl. Catal., B, 2016, 189, 19–25 CrossRef CAS.
- J. Zhou, R. Zhu, J. Deng and Y. Fu, Green Chem., 2018, 20, 3974–3980 RSC.
- M. Muñoz-Olasagasti, M. López Granados, C. P. Jiménez-Gómez, J. A. Cecilia, P. Maireles-Torres, J. A. Dumesic and R. Mariscal, Catal. Sci. Technol., 2021, 11, 4280–4293 RSC.
- W. Luo, M. Sankar, A. Beale, Q. He, C. Kiely, P. C. A. Bruijnincx and B. Weckhuysen, Nat. Commun., 2015, 6, 6540 CrossRef CAS PubMed.
- K. Yan, T. Lafleur, X. Wu, J. Chai, G. Wu and X. Xie, Chem. Commun., 2015, 51, 6984–6987 RSC.
- M. M. Antunes, P. Neves, A. Fernandes, S. Lima, A. F. Silva, M. F. Ribeiro, C. M. Silva, M. Pillinger and A. A. Valente, Catal. Sci. Technol., 2016, 6, 7812–7829 RSC.
- B. Hernández, J. Iglesias, G. Morales, M. Paniagua, C. López-Aguado, J. L. García Fierro, P. Wolf, I. Hermans and J. A. Melero, Green Chem., 2016, 18, 5777–5781 RSC.
- J. A. Melero, G. Morales, J. Iglesias, M. Paniagua, C. López-Aguado, K. Wilson and A. Osatiashtiani, Green Chem., 2017, 19, 5114–5121 RSC.
- T. Pang, X. Yang, C. Yuan, A. A. Elzatahry, A. Alghamdi, X. He, X. Cheng and Y. Deng, Chin. Chem. Lett., 2021, 32, 328–338 CrossRef CAS.
- J. A. Melero, G. Morales, J. Iglesias, M. Paniagua, C. López-Aguado, K. Wilson and A. Osatiashtiani, Green Chem., 2017, 19, 5114–5121 RSC.
- M. Paniagua, G. Morales, J. A. Melero, J. Iglesias, C. López-Aguado, N. Vidal, R. Mariscal, M. López-Granados and I. Martínez-Salazar, Catal. Today, 2021, 367, 228–238 CrossRef CAS.
- D. M. Bibby, R. F. Howe and G. D. McLellan, Appl. Catal., A, 1992, 93, 1–34 CrossRef CAS.
- I. Van Zandvoort, Y. Wang, C. B. Rasrendra, E. R. H. Van Eck, P. C. A. Bruijnincx, H. J. Heeres and B. M. Weckhuysen, ChemSusChem, 2013, 6, 1745–1758 CrossRef CAS PubMed.
- G. A. H. Mekhemer, A. K. H. Nohman, N. E. Fouad and H. A. Khalaf, Colloids Surf., A, 2000, 161, 439–446 CrossRef CAS.
- S. Jolly, J. Saussey and J. C. Lavalley, J. Mol. Catal., 1994, 86, 401–421 CrossRef CAS.
- F. Cabello Galisteo, R. Mariscal, M. López Granados, M. D. Zafra Poves, J. L. G. Fierro, V. Kröger and R. L. Keiski, Appl. Catal., B, 2007, 72, 272–281 CrossRef CAS.
- A. G. Pelmenschikov, R. A. Van Santen, J. Jänchen and E. Meijer, J. Phys. Chem., 1993, 97, 11071–11074 CrossRef CAS.
- D. Fuentes-Perujo, J. Santamaría-González, J. Mérida-Robles, E. Rodríguez-Castellón, A. Jiménez-López, P. Maireles-Torres, R. Moreno-Tost and R. Mariscal, J. Solid State Chem., 2006, 179, 2182–2189 CrossRef CAS.
- D. T. Bregante, A. Y. Patel, A. M. Johnson and D. W. Flaherty, J. Catal., 2018, 364, 415–425 CrossRef CAS.
- V. L. Sushkevich, A. Vimont, A. Travert and I. I. Ivanova, J. Phys. Chem. C, 2015, 119, 17633–17639 CrossRef CAS.
- Y. Shen, X. Bo, Z. Tian, Y. Wang, M. Xie, F. Gao, M. Lin, X. Guo, X. Guo and W. Ding, Green Chem., 2017, 19, 2646–2652 RSC.
- T. Lear, R. Marshall, J. A. Lopez-Sanchez, S. D. Jackson, T. M. Klapötke, M. Bäumer, G. Rupprechter, H. J. Freund and D. Lennon, J. Chem. Phys., 2005, 123, 174706 CrossRef PubMed.
- G. Agostini, R. Pellegrini, G. Leofanti, L. Bertinetti, S. Bertarione, E. Groppo, A. Zecchina and C. Lamberti, J. Phys. Chem. C, 2009, 113, 10485–10492 CrossRef CAS.
- B. I. Mosqueda-Jiménez, A. Jentys, K. Seshan and J. A. Lercher, Appl. Catal., B, 2003, 46, 189–202 CrossRef.
- P. Ivanov and H. Papp, Langmuir, 2000, 16, 7769–7772 CrossRef CAS.
- H. Tiznado, S. Fuentes and F. Zaera, Langmuir, 2004, 20, 10490–10497 CrossRef CAS PubMed.
- G. Novodárszki, H. E. Solt, G. Lendvay, R. M. Mihályi, A. Vikár, F. Lónyi, J. Hancsók and J. Valyon, Catal. Today, 2019, 336, 50–62 CrossRef.
- V. V. Kumar, G. Naresh, M. Sudhakar, J. Tardio, S. K. Bhargava and A. Venugopal, Appl. Catal., A, 2015, 505, 217–223 CrossRef CAS.
- V. V. Kumar, G. Naresh, S. Deepa, P. G. Bhavani, M. Nagaraju, M. Sudhakar, K. V. R. Chary, A. Venugopal, J. Tardio and S. K. Bhargava, Appl. Catal., A, 2017, 531, 169–176 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1se01802g |
| This journal is © The Royal Society of Chemistry 2022 |