DOI:
10.1039/D2SD00099G
(Paper)
Sens. Diagn., 2022,
1, 1176-1184
An engineered organic electrochemical transistor (OECT) platform with a highly ammonia-sensitive mesoporous membrane†
Received
2nd June 2022
, Accepted 28th July 2022
First published on 28th July 2022
Abstract
The development of an improved sensor design methodology via an organic electrochemical transistor (OECT)-based organic transistor device is presented here. A polyhydroxyl layer is introduced strategically in the OECT sensor assembly which is composed primarily of poly(3,4-ethylenedioxythiophene) doped with polystyrene-sulfonate (PEDOT:PSS). By introducing this layer, an increased selectivity for ammonia compared to other tested analytes in an aqueous environment has been found. A very low order of magnitude of ammonia concentration was detected by this device at very low operating voltages at room temperature. This superior performance of the OECT device is attributed to the formation of a smart mesoporous bed with electronic properties, which helped to obtain 71.6 ppb as the limit of detection (LOD) for ammonia. This approach incorporates an OECT device platform wherein the sensitivity of the sensor system is enhanced such that improved device performance is achieved while maintaining the cost-effectiveness.
Introduction
Although innumerable sensors for ammonia detection are available for use in various industries and environmental monitoring, the sensitivity of the device for medical applications is very important. Particularly, the detection of ammonia present in a very low concentration generated during the course of diseases or human metabolism is challenging. Among several techniques, the hybrid nanostructures formed by blending of nanoparticles of metal/metal-oxides with a polymer or its derivatives have been utilized in estimating environmental hazards.1–3 These air and environmental monitoring systems can detect analytes within the limit of <1 ppb. Certain optical gas sensors are suitable to detect ammonia but they are large and expensive, and the reagent consumption and maintenance requirements pose additional problems, making them less favorable.4,5 In this aspect, research on the improvement of sensitivity, selectivity, response time and reliability, and miniaturization and reduction of cost and power consumption are factors to be considered. Nanostructured materials are widely used as gas sensors due to their high volume ratio, good morphology and excellent electrical conductivity. These sensors are either metal oxide-based or conducting polymer sensors.6–8 Conducting polymers have been found useful because of their advantages like easily tunable chemical structures, simple processing and promising morphologies compared to other materials. In this context, conjugated polymers and their nanocomposites have been proposed as active sensing materials to gain advantages in optoelectronic properties and better sensitivity towards a number of acidic or basic gases to be utilized in state-of-the-art devices and at room temperature operation. The high demand for analytical devices necessitates the development of smart technologies that enable faster and efficient detection of desired analytes. Currently, various device architecture combinations and improved methods of the existing technology platforms are being developed to enhance the analyte detection sensitivity and selectivity.9–16 The main goal for the development of such devices is to achieve greater device performances in an affordable and less economic cost. Examples of analytical devices include portable and easily accessible glucose sensors used extensively in medical practices as well as for home testing. However, the development of such analytical devices involves a trade-off between the cost of production and device sensitivity. Hence, device improvements without compromising the sensitivity of the analytical device can be further explored. Organic electrochemical transistors (OECTs) are promising in this regard due to their low operating voltages, ability to work in aqueous environments and ease of fabrication. In addition, OECTs can detect desired analytes specifically through active material functionalization or a chemical reaction involving the analytes as a reactant or by-product. Further, OECTs based on poly (3,4-ethylenedioxythiophene) doped with polystyrene sulfonate (PEDOT:PSS) can be integrated into microfluidic systems which are used for logic circuits. Such integration facilitates sensing of biomolecules such as glucose, deoxyribonucleic acid (DNA), neurotransmitters and certain other biomarkers. These devices follow the ion-to-electron converter mechanism, providing an effortless route to interface biology with electronics.17–24
Thus, these devices offer the advantage of detecting large changes in the electronic current even on a relatively small ionic drift. As such, herein, we have introduced a polyhydroxyl layer (PHD) to design a PEDOT:PSS based OECT for exploring its capability towards improved transistor performance. Apart from being selective to ammonia, the PHD layer also provides device stability, protects the transistor in an aqueous environment and provides room-temperature operation under ambient conditions. We have confirmed the presence of strong hydrogen bonds, N–H stretching and cavity condensation. Together, all these notable features along with the mesoporous nature of the PHD film contribute to achieving the remarkable response of the solution processed PHD-OECT towards ammonia at low operating voltages of <1 V. Table 1 represents a comparative study of room temperature ammonia detection using various techniques.
Table 1 A comparative study of room temperature ammonia detection considering varied techniques
| Sl no. |
Ref. |
Active material |
Detection method |
Temperature |
LOD |
| 1. |
Hussain et al.,15aAnal. Chem., 2016, 88, 12453–12460 |
Ionic liquid (RTIL) 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide([C2mim]-[NTf2])/microarray thin film electrode (MATFE) |
Voltammetric |
Room temperature |
0.1 to 2 ppm |
| 2. |
Mackin et al.,14aACS Appl. Mater. Interfaces, 2018, 10, 16169–16176 |
Graphene |
Chemiresistive |
Room temperature |
160 ppm |
| 3. |
Kannan et al.,16J. Mater. Chem. A, 2014, 2, 394 |
α-Fe2O3 |
Impedimetric |
Room temperature |
100–1000 ppm |
| 4. |
Meng et al.,11bJ. Mater. Chem. A, 2015, 3, 1174 |
Cu2O nanorods/rGO |
Surface-controlled |
Room temperature |
200 ppm |
| Adsorption |
| 5. |
Ghosh et al.,36 |
RGO–SnO2 hybrid |
Conducting hetero junctions |
Room temperature |
25–2800 ppm |
| 6. |
Kim et al.,11aJ. Mater. Chem. A, 2017, 5, 19116 |
Chemically fluorinated graphene oxide (CFGO) |
Chemoresistive |
Room temperature |
100–500 ppb |
| 7. |
Wang et al.,37 |
SnO2/bionic porous (BP) carbon composites |
Microwave transduction technology |
Room temperature |
10–200 ppm |
| 8. |
Singh et al.,38 |
MoSe2 crystalline nanosheets |
Resistive |
Room temperature |
1 ppm |
| 9. |
Kanaparthi et al.,39 |
Polyaniline (PANI) |
Chemiresistive |
Room temperature |
200 ppb to 3.15 ppm |
| 10. |
Seekaew et al.,40 |
Sn–TiO2@rGO/CNT |
Resistive |
Room temperature |
250 ppm |
| 11. |
Murugappan et al.,13bElectrochem. Commun., 2011, 13, 1435–1438 |
Room temperature ionic liquids (RTILs) |
Electrochemical |
Room temperature |
50 ppm and 185 ppm |
| 12. |
Liu et al.,41 |
Biomass hydrogel poly-L-glutamic acid and L-glutamic acid (PGA/GA) |
Impedance |
Room temperature |
0.5 ppm |
| 13. |
Yin et al.,42 |
Mesoporous NiO |
Resistive |
Room temperature |
0.001 ppm (1 ppb) |
| 14. |
Chen et al.,43 |
Core–shell Au@SiO2 nanocrystals doped PANI |
Resistive |
Room temperature |
10 ppb |
| 15. |
Tang et al.,44 |
Graphene oxide (GO) |
Love mode surface acoustic wave (SAW) |
Room temperature |
500 ppb |
| 16. |
Song et al.,45 |
Ultrathin silicon nanowires (SiNWs) |
Field-effect transistor |
Room temperature |
100 ppb |
| 17. |
Garg et al.,46 |
Zeolite imidazole framework/reduced graphene oxide (ZIF-67/rGO) composite |
Chemoresistive |
Room temperature |
74 ppb |
| 18. |
Present work |
PEDOT:PSS/polyhydroxyl derivative (PHD) |
Organic electrochemical transistor (OECT) |
Room temperature |
71.6 ppb |
Experimental section
Materials and methods
PEDOT:PSS, acrylic acid, and 30% ammonia solution were purchased from Sigma-Aldrich. Glycerol was purchased from Merck and p-toluene sulfonic acid from LobaChemie. CV measurements were carried out using a CH instruments Model 700D series. Ag/AgCl was used as the reference electrode and 0.1 M NaCl as the aqueous electrolytic solution. The thickness of the deposited films was optimized using a Profilometer (Dektat-150). Field emission scanning electron microscopy (FESEM) images were recorded on a Sigma Carl ZEISS SEM instrument. FT-IR was recorded on a Perkin Elmer spectrometer with samples prepared using KBr pellets.
Synthesis
A polyhydroxyl derivative was synthesized by mixing 0.03 mol of glycerol, 0.09 mol of acrylic acid and 0.05 g of p-toluene sulfonic acid together for 30 min. A viscous solution was formed after mixing which was used for coating the device. The material has been found to be chemically, thermally, and mechanically stable for room temperature experiments which can be attributed to the cross-linked homogeneous texture.25
Sensor fabrication
Microscopic glass slides of dimensions 1 cm × 2 cm were utilized as substrates which were cleaned in piranha solution for 1 hour and washed several times with deionised water followed by sanitation prior to use. The cleaned substrates were then dried, ionized and subsequently 100 nm thickness aluminium contacts were thermally deposited on them inside the glove box. A channel of dimensions 30 mm length (L) and 2 mm width (W) was obtained which was used as the source (S) and drain (D). 30 nm thick PEDOT:PSS films were coated on these aluminium deposited substrates. The coated substrates were again heated at 130 °C for 1 hour. The synthesized polyhydroxyl layer was coated over the PEDOT:PSS region and heated at 50 °C for 30 minutes. An ammonia concentration of 1 μM was used for all the experiments.
Characterization
a. Electrical measurements
All the electrical characterizations of the devices were carried out under ambient conditions at room temperature using a Keithley2614B. A positive gate bias (Vg) was applied with the Ag/AgCl electrode and the two aluminium electrodes served as a source (S) and drain (D). The ammonia sensing experiments were performed at drain voltage Vd = −0.2 V with a gate voltage Vg sweep ranging from 0 to 1 V in 5 ml electrolyte solution at pH = 7. The analyte and ammonia solutions were confined in a 10 mL beaker and mixed with the electrolyte prior to testing.
b. Field emission scanning electron microscopy (FESEM) and X-ray diffraction (XRD)
FESEM images of the PHD film on glass slides were recorded on a Sigma Carl ZEISS scanning electron microscope at an accelerating voltage of 15 kV at different magnifications. XRD was recorded on a Bruker D8 Advance model (provided in ESI†).
c. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH)
BET/BJH experiments of PHD were performed on an Autosorb-IQ MP instrument to understand the porosity of the selective layer.
d. Fourier transform infrared analysis (FT-IR)
FT-IR was recorded on a Perkin Elmer spectrometer with solid samples prepared using KBr pellets. Selected IR peaks with tentative assignments (nmax/cm−1) were recorded.
Results and discussion
a. Electrical measurements
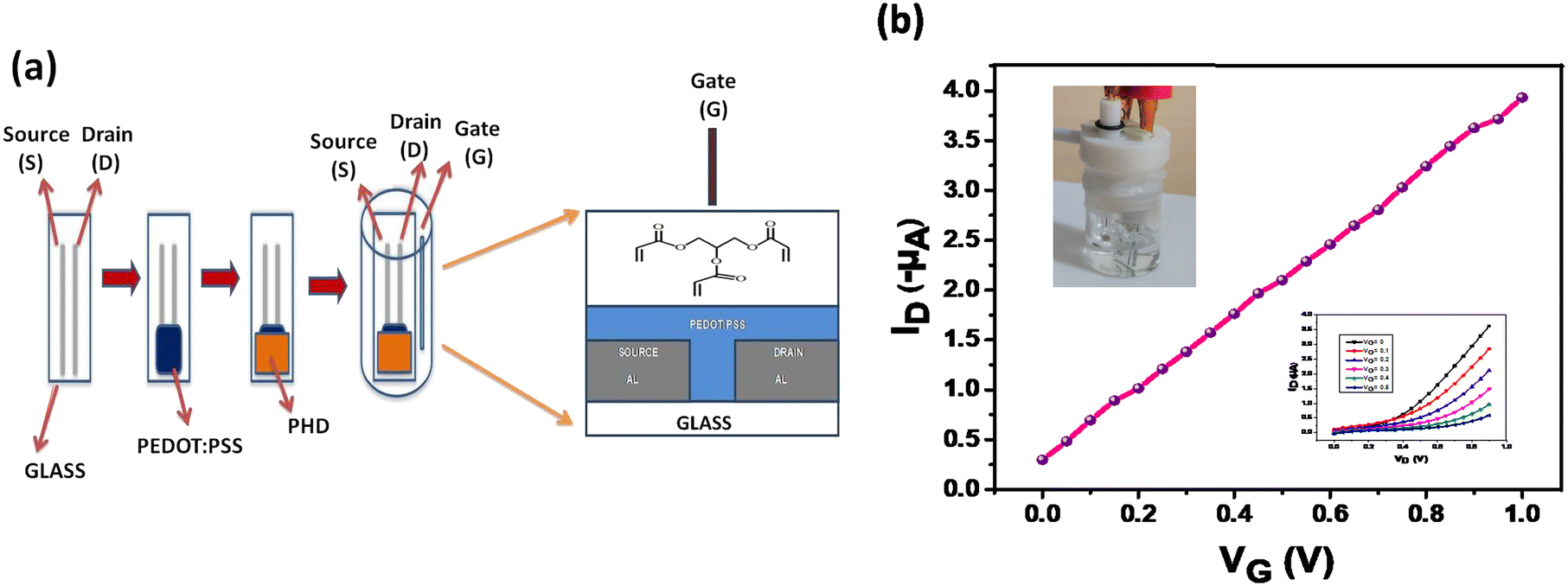
The OECT and polyhydroxyl derivative used for coating are shown in Fig. 1a. The electronic transfer characteristic is indicated at Vd = −0.2 V and drain characteristics at different gate voltages are shown in Fig. 1b. The measurements were done under positive gate bias which subsequently changes the channel conductance through injection of ions from the aqueous medium. This process, in turn, affects the conductivity of the PEDOT:PSS layer which is referred to as doping/de-doping of PEDOT:PSS as depicted in eqn (1).| | | PEDOT+:PSS− + M+ + e− ⇌ PEDOT0 + M+:PSS− | (1) |
where M+ represents a cation and e− an electron.
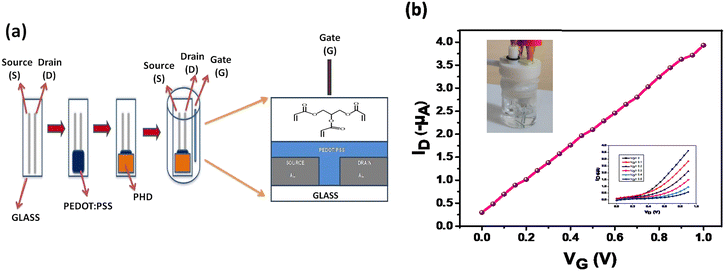 |
| | Fig. 1 Schematic illustration and digital image of the ammonia sensing device. a) Schematic drawing of the device showing various layers and incorporation of a mesoporous layer immersed in an electrolyte solution. b) Transfer characteristics (ID–VG) (inset top left device setup; inset down right output curves). | |
The transistor characteristics of the PHD-OECT are due to ions acting as charged carriers contributing to its high sensitivity. Therefore, these characteristics result in a built-in amplification entity (Fig. 1b). This system is found to be highly sensitive towards ammonia, i.e., it forms an ammonia sensitive layer. Hence, the presence of small concentration of ammonia is selectively detected. It is found that the electron transfer curve shifts significantly upon exposure to different concentrations of ammonia. Various analytes, such as urea, thiourea, ethanolamine, diethylamine, FeCl3, hydrazine, NaCl and KCl, have been tested with this sensor assembly.
b. FESEM and XRD
The surface morphology of the PHD film layer before and after exposure to ammonia can be seen in Fig. S1(a–c) (ESI).† In Fig. S1a and S1b (ESI†), the PHD layer before exposure to ammonia at 100× and 500× magnification respectively shows well-defined grooves. Exposing it to ammonia changes the structural pattern (Fig. S1c).† The XRD data shows a shift of peaks after exposure to ammonia. Both in powder and film form, PHD is mostly amorphous in nature (Fig. S1d and S1e, ESI†). This could be one of the factors contributing to the PHD-OECT device's high performance at low operating voltages. However, the peak at 20.8° for PHD powder (Fig. S1d, ESI†) shows a shift to 28.48° after exposure to ammonia. Similarly, the peak at 23.04° for the PHD film (Fig. S1e, ESI†) shows a shift to 32.08° after exposure to ammonia (provided in the ESI†).
c. Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH)
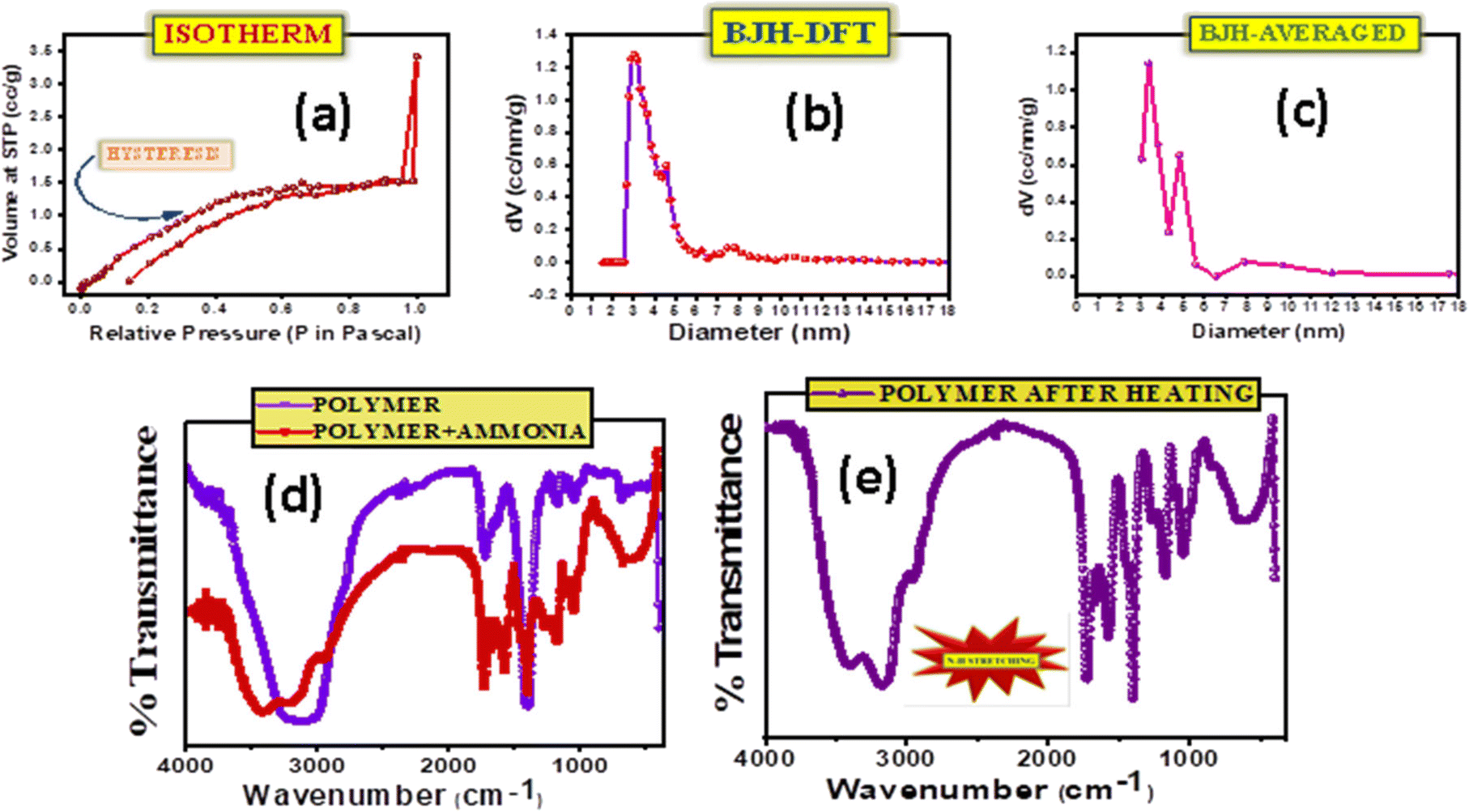
The adsorption and desorption of N2 in an isotherm determines the presence of pores in the material and the likeable cavity condensation. The appearance of hysteresis highlights the presence of mesoporous pores in the material (Fig. 2a). Since pores of a specific size are filled at higher pressures and expelled at lower pressures, cavity condensation takes place during adsorption; and cavity evaporation takes place during desorption. The BET experiment performed on the samples revealed the porosity of the selective layer. In Fig. 2b and c, the pore distributions of the selective layer are observed which is in direct contact with the electrolyte. Fig. 2b shows the pore size distribution calculated through the DFT method, while in Fig. 2c it is an averaged data. In both figures, the presence of different pore sizes is observed; however, a maximum of 3 nm pore size was noticed. Thus, the ions in the electrolyte could easily pass through the porous selective layer to dope or de-dope the PEDOT:PSS layer. Although it is possible for the ions of the analyte to infiltrate the layer, the selectivity factor, along with attraction and repulsion forces, comes into play and allows more ammonium ions to be detected.26–28
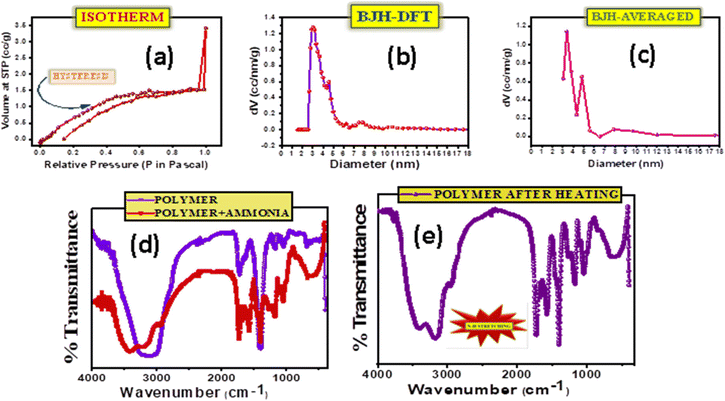 |
| | Fig. 2 Surface area evaluation for determination of pores as well as functional groups and characterizing chemical bond formation. a) Isotherm of a mesoporous PHD layer when exposed to nitrogen. b) Pore size distribution of PHD calculated using the BJH-DFT method. c) Pore size distribution of PHD calculated using the BJH-averaged method. d) FTIR analysis of PHD before and after exposure to ammonia. e) FTIR analysis of PHD after heating and exposure to ammonia resulting in N–H stretching. | |
d. Fourier transform infrared (FT-IR) analysis
FT-IR analysis performed at room temperature on the selective layer (Fig. 2d and e) on pristine and after ammonia exposure confirmed the changes occurring after the addition of ammonia and revealed broad peaks at ∼3406 cm−1 for PHD w.r.t. 3138 cm−1 for PHD without ammonia corresponding to the –OH groups. The peaks at 3172 cm−1 and 2947 cm−1 present in PHD with ammonia are not seen in PHD without ammonia, and the peaks at 1732 cm−1 for PHD without ammonia and at 1724 cm−1 for PHD with ammonia confirm the formation of ester. The idea of detecting a hydrogen bond (HB) solely on one single site can be done by quantum chemical calculation. However, the prediction of H-bonding can be made by observing the easily accessible basic sites of PHD for interacting with NH3. First, we assume that the interaction with these sites results in H-bonded complexes. It could also be demonstrated from the classification of acceptor atom types in order to form hydrogen-bonded complexes and also for forming more effective sites. In this context, three regions may be recommended to associate with NH3 through hydrogen bonds with specific acceptor atoms, say –C–O–C– and –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. Accordingly the utility of these sites as a criterion for sensing of NH3 through H-bonds can be understood. These groups can be connected to a H-atom of NH3 by a HB and the strength of the bond as well as the sensitivity depends on the electron density of these groups to attract hydrogen. HBs are ubiquitous in this structure and have been the important focus in sensing processes. Many studies have shown that sensing mechanisms can be well-explained by HB interactions.29–34 The probe PHD shown in Fig. 2d and e, taken by the IR vibration analysis before and after interaction with NH3, shows that all the relevant vibrational peaks are distinctly shifted. The FTIR spectra of functional groups, –CO, –C–O–C– and H–N related to HBs, show the red-shift in vibration frequencies of up to 268 cm−1 (3406 → 3138 cm−1) (Fig. 2d and e). This red shift indicates the existence of a hydrogen bond (HB), whereas the blue shift indicates its weakening. Analysis of sensing mechanisms can be demonstrated from the six nucleophilic sites of PHD. Out of these sites, the most electronegative sites attract the H of NH3 although these sites cannot be distinguished from experimental results. Thus, the vibrational spectra confirm the mechanism of HB in the ground state. The presence of hydrogen bonding between the –CO group of PHD and ammonia results in an increase of –CO as well as H–N bond length. Fundamentally, the vibrational frequency for these bonds will decrease leading to a change in IR stretching frequencies of –CO (PHD) and H–N (ammonia). The IR spectra of PHD after addition of ammonia have also been examined. On heating for 20 minutes the IR spectra were again taken, showing a slight impression of ammonia in the spectra. In Fig. 2e, the difference after application of heat could be ascertained. The original IR of PHD could be observed with N–H stretching at 3175 cm−1 which was not found before exposure to ammonia. This can be attributed to the formation of strong hydrogen bonds between PHD and ammonia. It proves stable hydrogen bond formation between –CO (PHD) and ammonia at the exposed –CO groups of the material. Moreover, the formation of hydrogen bonds may weakly polarise the ammonia molecule which can facilitate the accumulation of more NH3 molecules inside the pores of the material and thereby induce cavity condensation. Thus, both hydrogen bonding and cavity condensation operate in this system. The possibility of reversibility in this configuration indicates that the device can be reusable with a baseline correction. Aggregation of NH3 at the nucleophilic sites i.e. –CO and –C–O–C– can significantly affect the extent of reactivity or selectivity at these regions. Here, the presence of NH3 in IR spectra on heating reveals the importance of H-bond networks where it forms a strong HB with the analyte.
O. Accordingly the utility of these sites as a criterion for sensing of NH3 through H-bonds can be understood. These groups can be connected to a H-atom of NH3 by a HB and the strength of the bond as well as the sensitivity depends on the electron density of these groups to attract hydrogen. HBs are ubiquitous in this structure and have been the important focus in sensing processes. Many studies have shown that sensing mechanisms can be well-explained by HB interactions.29–34 The probe PHD shown in Fig. 2d and e, taken by the IR vibration analysis before and after interaction with NH3, shows that all the relevant vibrational peaks are distinctly shifted. The FTIR spectra of functional groups, –CO, –C–O–C– and H–N related to HBs, show the red-shift in vibration frequencies of up to 268 cm−1 (3406 → 3138 cm−1) (Fig. 2d and e). This red shift indicates the existence of a hydrogen bond (HB), whereas the blue shift indicates its weakening. Analysis of sensing mechanisms can be demonstrated from the six nucleophilic sites of PHD. Out of these sites, the most electronegative sites attract the H of NH3 although these sites cannot be distinguished from experimental results. Thus, the vibrational spectra confirm the mechanism of HB in the ground state. The presence of hydrogen bonding between the –CO group of PHD and ammonia results in an increase of –CO as well as H–N bond length. Fundamentally, the vibrational frequency for these bonds will decrease leading to a change in IR stretching frequencies of –CO (PHD) and H–N (ammonia). The IR spectra of PHD after addition of ammonia have also been examined. On heating for 20 minutes the IR spectra were again taken, showing a slight impression of ammonia in the spectra. In Fig. 2e, the difference after application of heat could be ascertained. The original IR of PHD could be observed with N–H stretching at 3175 cm−1 which was not found before exposure to ammonia. This can be attributed to the formation of strong hydrogen bonds between PHD and ammonia. It proves stable hydrogen bond formation between –CO (PHD) and ammonia at the exposed –CO groups of the material. Moreover, the formation of hydrogen bonds may weakly polarise the ammonia molecule which can facilitate the accumulation of more NH3 molecules inside the pores of the material and thereby induce cavity condensation. Thus, both hydrogen bonding and cavity condensation operate in this system. The possibility of reversibility in this configuration indicates that the device can be reusable with a baseline correction. Aggregation of NH3 at the nucleophilic sites i.e. –CO and –C–O–C– can significantly affect the extent of reactivity or selectivity at these regions. Here, the presence of NH3 in IR spectra on heating reveals the importance of H-bond networks where it forms a strong HB with the analyte.
Gaussian studies
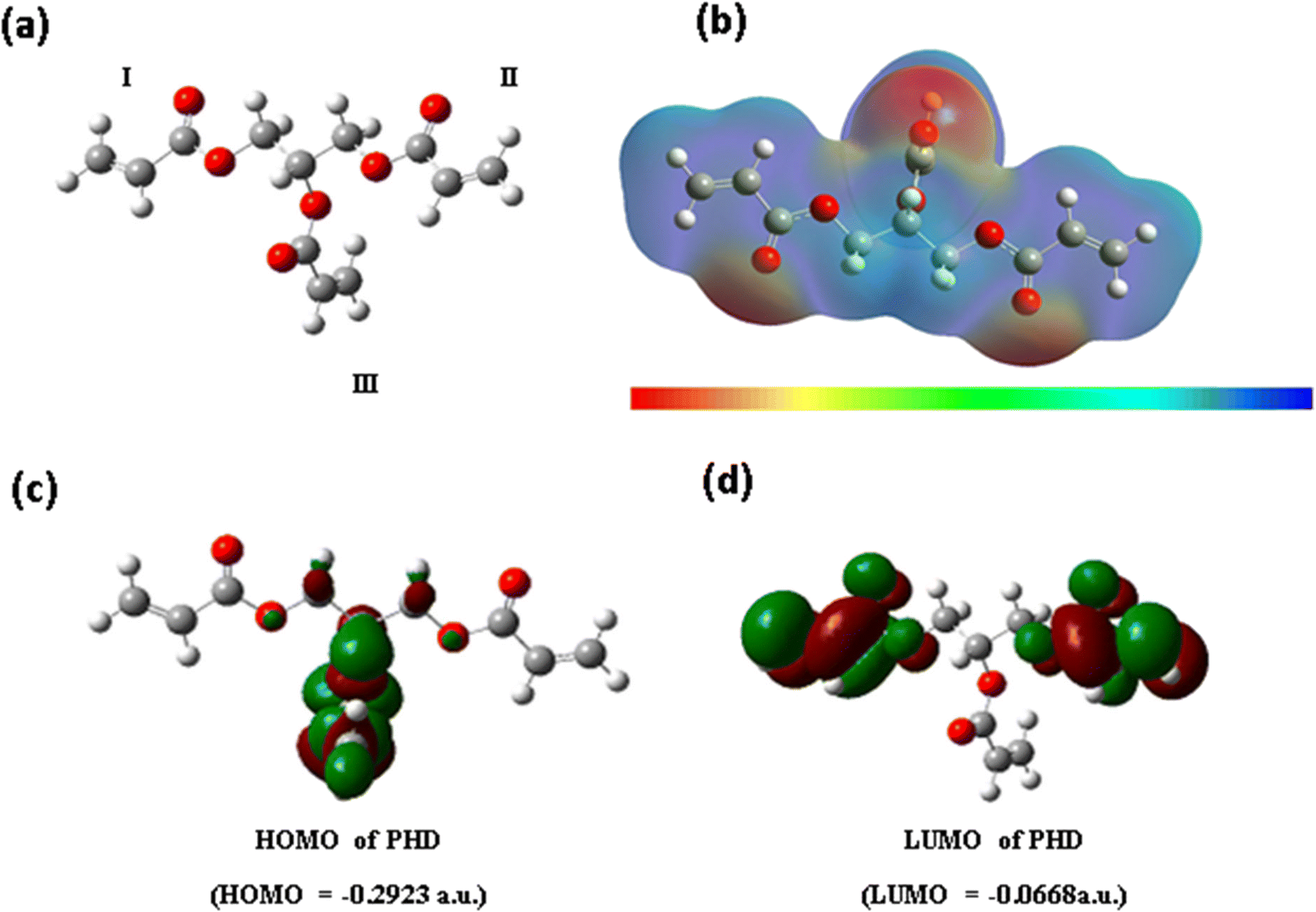
The structure of the PHD derivative may be analysed in three subunits I, II and III (Fig. 3a). The configuration of subunits I and II remains along the same axial plane, whereas unit III lies perpendicular to this plane. The acrylate groups are bonded to primary carbon centres in subunits I and II, whereas in subunit III the acrylate group is attached to the tertiary carbon centre through a carboxylate bond. Hence, the nature of electrostatic potential around this monomer PHD may indicate distinguishable variation in electron density. The completely optimized polyhydroxyl derivative with the B3LYP/6-31G(d,p) method was used for calculating electrostatic potential (Fig. 3b). Thus, we have computed the EPS profile in the 3D structure of monomer PHD.
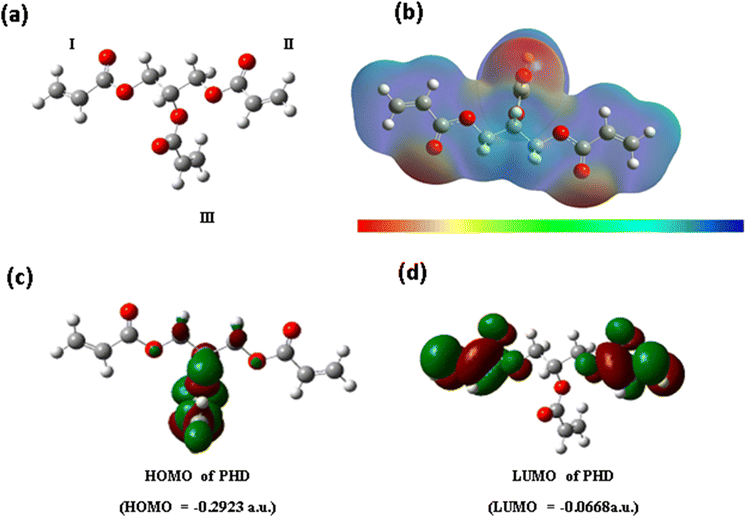 |
| | Fig. 3 Computational study and molecular electronic structure calculations of PHD. a) Gaussian model of the 3D structure of PHD with three acrylate subunits. b) Electronic cloud density distribution of PHD. c) HOMO of PHD. d) LUMO of PHD. | |
More negative values are found around –CO of these three regions and less negative or positive values are found around the rest of the regions. The maximum negative value in regions I and II is approximately −4.31 × 10−2 a.u., whereas the value for region III is −4.58 × 10−2 a.u. Hence, region III may be more perceptive of NH3 or NH4OH (in aqueous solution) than regions I and II. This may be the reason why the PHD material is very sensitive towards NH3 or NH4OH (in aqueous solution).The electron labile property of the polyhydroxyl derivative can be related to the energy gap of frontier orbitals, i.e. HOMO–LUMO (Fig. 3c). The optimized structure of the PHD derivative using B3LYP/6-31G(d,p) was used for calculating the HOMO and LUMO energies and Fig. 3c and d show the electron density distribution. The HOMO–LUMO gap is found to be −6.1 eV, but the value is much larger for an electronically conductive material. According to the electronic property of the PHD derivative, –CC– and –CO groups generally undergo n–π* and π–π* transitions, but such transitions occur only in the excited states. Hence, electronic conduction may not occur in the process of NH3 or NH4OH (in aqueous solution) sensing. It is essential to analyse electrostatic potential in the 3D structure of the polyhydroxyl derivative.
Sensing mechanism
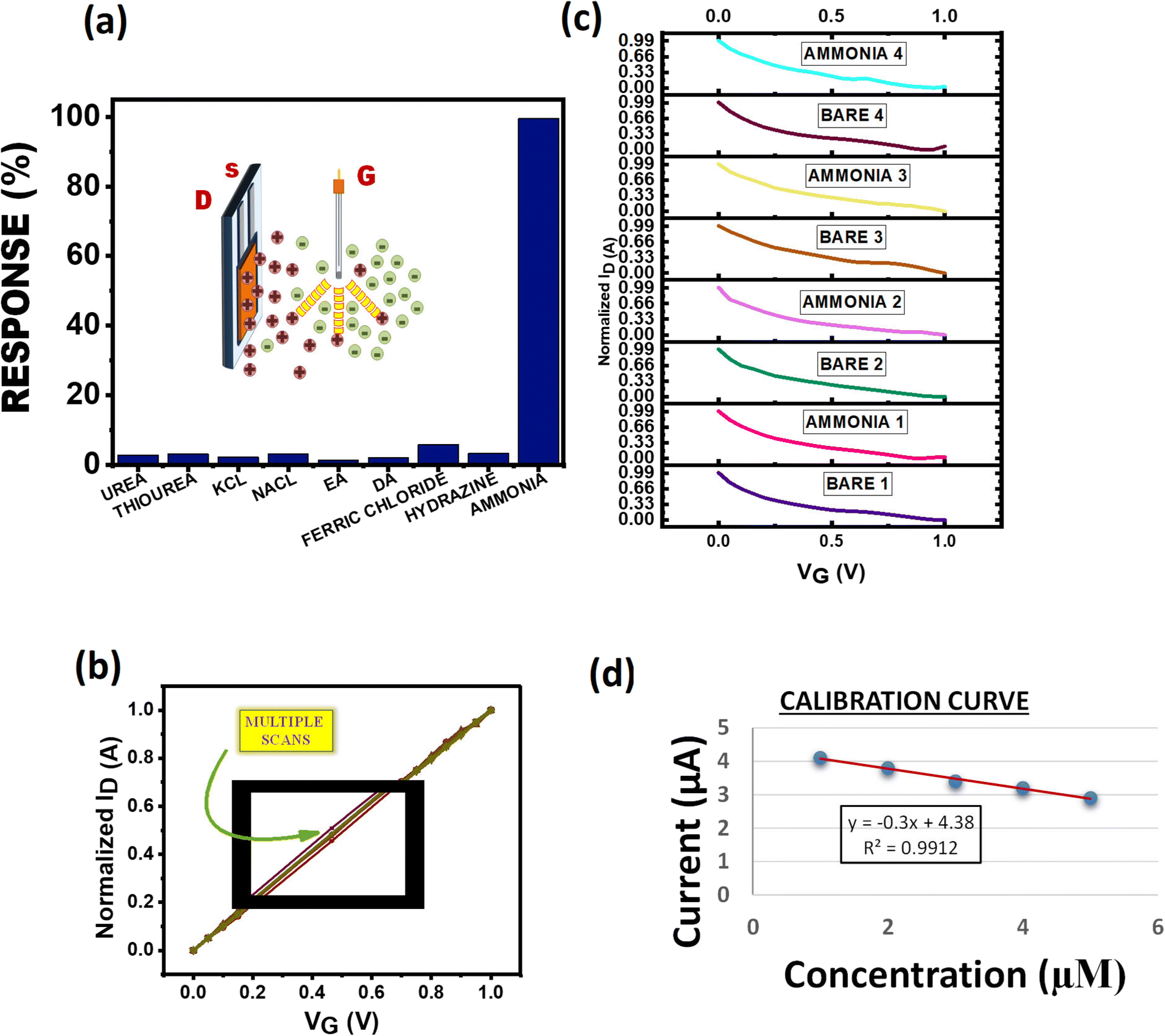
The sensing of a transistor is based on integration of ions in aqueous solution repelled by the positively charged gate and attracted by the negative channel. The species sensed acquires a positive charge in aqueous medium which then associates with the polyhydroxyl layer and gets absorbed by it to further de-dope the PEDOT:PSS polymer film to create changes in the I–V characteristics of the device. The absorption/association with the acrylate polymer film involves different intermolecular interactions like H-bonding, dipolar interaction, London forces, dispersion forces and individual ionic conductivities of ions furnished by various species taken into consideration. In this study, we have analysed a large set of species commonly found in industrial wastes and biological processes, such as ammonia, urea, thiourea, diethylamine (DA), ethanolamine (EA), hydrazine, and ionic salts like NaCl, KCl, and FeCl3 in order to extract the highest sensitivity. The sensitivity profiles of these species depend on attraction forces, repulsion forces, and different intermolecular interactions between the acrylate polymer and ions furnished by the species taken into consideration. As per the results obtained in the process, ammonia shows the maximum sensitivity due to H-bonding between the acrylate and NH3 which after absorption through the polymer film de-dopes the PEDOT:PSS polymer up to the maximum extent (Fig. 4a). In ionic cases like NaCl and KCl the sensitivity is lower due to poor interaction between the polymer and Na+/K+ ions. The sensitivity profile clearly shows approximately similar values for NaCl and KCl which can be explained on the basis of almost similar ionic properties and dissociation of both salts in water. As NaCl is slightly more soluble in water hence it shows a higher value than KCl. In the case of ionic salts like FeCl3, due to higher solubility than NaCl and KCl, FeCl3 shows a slightly higher sensitivity but the overall sensitivity when compared to NH3 is fairly low due to rapid complexation of Fe3+ ions with water molecules to form [Fe(H2O)6]3+ hexaaquairon(III) complex ions. Hence, the interaction of Fe3+ has been reduced with the sensing polymer layer which led to the sensitivity profile seen in Fig. 4a. In the case of other organic samples taken into consideration for this study, namely urea and thiourea molecules, due to the high electronegativity of oxygen (O) in the urea molecule polarisation takes place leading to lower electron density on both –NH2 groups. This results in less association of the urea molecule with the acrylate group as indicated by the reduced sensitivity of urea as compared to thiourea. However, overall they have a very high tendency to form hydrogen bonding with the polymer; hence their sensing profile is high as compared to those of other organic samples. In the case of hydrazine, the molecule exists as NH2–NH3+ in water which will associate with the polymer but repulsions due to a nearby second –NH2 group led to lower sensitivity than ammonia, urea and thiourea but can be comparable to that of ionic salts like KCl and NaCl. The sensitivity of ethanolamine is least as compared to the rest due to the presence of intra-molecular hydrogen bonding between the –OH group and –NH2 group which will result in less association of ethanolamine with the acrylate group of the PHD layer. In the case of diethylamine, the association is somewhat more due to the absence of such an electron rich group. It is less repelled and more associated to the polymer as compared to ethanolamine as seen in the sensitivity profile.
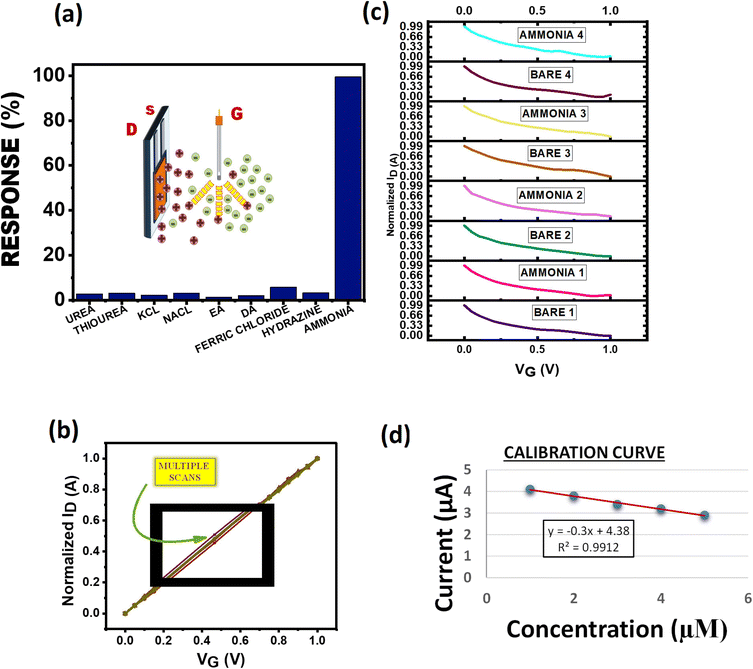 |
| | Fig. 4 Response of PHD-OECT to common industrial and biological waste products. a) Sensitivity of PHD compared to other analytes. b) Repeatability of the device kept in electrolytic solution. c) Analysing different devices in the same electrolytic solution. d) Calibration curve of the PHD modified device after exposure to ammonia. | |
Repeatability and reproducibility are two distinguished features in a sensor. The device characteristics were measured for the same device multiple times in the same electrolyte solution (Fig. 4b). The device does not deter away from its original value and is repeatable even after successive 10 scans. Four different devices fabricated under the same conditions scanned in the same electrolyte solution indicate that our device is reproducible (Fig. 4c). The values for different concentrations of ammonia were plotted to get the calibration curve for the calculation of the limit of detection (LOD) (Fig. 4d). Increasing concentration of ammonia is injected into the electrolyte solution where the device is immersed as seen in Fig. 1a and the LOD was calculated to be 71.6 ppb. Exposure to ammonia of about 25 ppm for 8 hours is hazardous to health.35 As such, this approach to fabricate OECTs could be further explored for improved detection of various analytes in a cost-effective manner.
Conclusion
In conclusion, a mesoporous film platform using a vertical OECT has been developed utilizing poly(3,4-ethylenedioxythiophene) doped with polystyrene-sulfonate (PEDOT:PSS), and ammonia sensing at the 71.6 ppb level has been achieved. We have demonstrated that the sensor assembly has excellent transistor characteristics and can be operated with voltages <1 V. The polyhydroxyl film is optimal in an aqueous environment at room temperature due to its cross-linked homogeneous texture. The developed PHD-OECT is highly sensitive and selective for ammonia with the presence of strong hydrogen bond formation and N–H stretching. It has been observed that there were mesoporous pores on the PHD film that enhance the sensing attributes of PHD-OECT towards ammonia with excellent electrical response. Due to the economic viability of this PHD-OECT sensor, the present approach can be further explored to detect other biological and chemical analytes efficiently.
Conflicts of interest
The authors declare that the research was conducted in the absence of conflict of interest.
Acknowledgements
The authors thank the Department of Electronics & Information Technology, DeitY Project No. 5(2)/2022-NANO, ICMR, Grant no. 5/3/8/20/2019-ITR and DST-Max Planck Society, Germany (No. IGSTC/MPG/PG (PKI)/2011A/48) for financial support. The Centre for Nanotechnology, Department of Chemistry and the Central Instruments Facility, IIT Guwahati are acknowledged for instrument facilities.
References
-
(a) B. Timmer, W. Olthuis and A. van den Berg, Sens. Actuators, B, 2005, 107, 666–677 CrossRef CAS;
(b) J. F. M. Oudenhovena, W. Knobena and R. van Schaijka, Procedia Eng., 2015, 120, 983–986 CrossRef;
(c) R. A. Michaels, Environ. Health Perspect., 1999, 107, 617–627 CrossRef CAS PubMed;
(d) D. Leduc, P. Gris, P. Lheureux, P. A. Gevenois, P. De Vuyst and J. C. Yernault, Thorax, 1992, 47, 755–757 CrossRef CAS PubMed.
-
(a) J.-C. Jin, J. Wu, G.-P. Yang, Y.-L. Wu and Y.-Y. Wang, Chem. Commun., 2016, 52, 8475–8478 RSC;
(b) A. Kalita, S. Hussain, A. H. Malik, U. Barman, N. Goswami and P. K. Iyer, ACS Appl. Mater. Interfaces, 2016, 8, 25326–25336 CrossRef CAS PubMed;
(c) N. Sharma, N. Sharma, P. Srinivasan, S. Kumar, J. B. B. Rayappan and K. Kailasam, J. Mater. Chem. A, 2018, 6, 18389 RSC;
(d) L. R. Adil, P. Gopikrishna and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 27260–27268 CrossRef CAS PubMed.
-
(a) T. Someya, Z. Bao and G. G. Malliaras, Nature, 2016, 540, 379–385 CrossRef CAS PubMed;
(b) M. Berggren and A. Richter-Dahlfors, Adv. Mater., 2007, 19, 3201–3213 CrossRef CAS;
(c) N. Zehra, D. Dutta, A. H. Malik, S. S. Ghosh and P. K. Iyer, ACS Appl. Mater. Interfaces, 2018, 10, 27603–27611 CrossRef CAS PubMed.
-
(a) T. Grady, T. Butler, B. D. MacCraith, D. Diamond and M. A. McKervey, Analyst, 1997, 122, 803–806 RSC;
(b) W. Cao and Y. Duan, Sens. Actuators, B, 2005, 110, 252–259 CrossRef CAS.
- Z. Jin, Y. Su and Y. Duan, Sens. Actuators, B, 2001, 72, 75–79 CrossRef CAS.
-
(a) N. R. Stradiotto, H. Yamanaka and M. V. B. Zanoni, J. Braz. Chem. Soc., 2003, 14(2), 159–173 CrossRef CAS;
(b) U. Yogeswaran and S.-M. Chen, Sensors, 2008, 8, 290–313 CrossRef CAS PubMed;
(c) G. Saikia, A. K. Dwivedi and P. K. Iyer, Anal. Methods, 2012, 4, 3180–3186 RSC.
-
(a) F. Rigoni, S. Tognolini, P. Borghetti, G. Drera, S. Pagliara, A. Goldoni and L. Sangaletti, Analyst, 2013, 138, 7392–7399 RSC;
(b) A. Kalita, S. Hussain, A. H. Malik, N. V. V. Subbarao and P. K. Iyer, J. Mater. Chem. C, 2015, 3, 10767–10774 RSC.
-
(a) P. K. Sekhar and J. S. Kysar, J. Electrochem. Soc., 2017, 164(4), 113–117 CrossRef;
(b) N. Zehra, A. Kalita, A. H. Malik, U. Barman, M. A. Afroz and P. K. Iyer, ACS Sens., 2019, 5, 191–198 CrossRef PubMed.
-
(a) S. Stříteský, A. Marková, J. Víteček, E. Šafaříková, M. Hrabal, L. Kubáč, L. Kubala, M. Weiter and M. Vala, J. Biomed. Mater. Res., Part A, 2018, 106(4), 1121–1128 CrossRef PubMed;
(b) V. Rani and K. S. V. Santhanam, J. Solid State Electrochem., 1998, 2, 99–101 CrossRef CAS.
-
(a) M. Chen, D. Nilsson, Th. Kugler and M. B. T. Remonen, Appl. Phys. Lett., 2002, 81(11), 2011–2013 CrossRef CAS;
(b) C.-S. Lee, S. K. Kim and M. Kim, Sensors, 2009, 9, 7111–7131 CrossRef CAS PubMed;
(c) S. Mondal, N. Zehra, A. Choudhury and P. K. Iyer, ACS Appl. Bio Mater., 2020, 4, 47–70 CrossRef PubMed;
(d) N. Meher, S. R. Chowdhury and P. K. Iyer, J. Mater. Chem. B, 2016, 4, 6023–6031 RSC;
(e) N. Meher and P. K. Iyer, Nanoscale, 2017, 9, 7674–7685 RSC.
-
(a) Y. H. Kim, J. S. Park, Y.-R. Choi, S. Y. Park, S. Y. Lee, W. Sohn, Y.-S. Shim, J.-H. Lee, C. R. Park, Y. S. Choi, B. H. Hong, J. H. Lee, W. H. Lee, D. Lee and H. W. Jang, J. Mater. Chem. A, 2017, 5, 19116–19125 RSC;
(b) H. Meng, W. Yang, K. Ding, L. Feng and Y. Guan, J. Mater. Chem. A, 2015, 3, 1174–1181 RSC.
-
(a) S. Pandey, G. K. Goswami and K. K. Nanda, Sci. Rep., 2013, 3, 2082 CrossRef PubMed;
(b) S. Arya, M. Riyas, A. Sharma, B. Singh, Prerna, P. Bandhoria, S. Khan and V. Bharti, Appl. Phys. A: Mater. Sci. Process., 2018, 124, 538 CrossRef;
(c) A. S. Tanwar, S. Patidar, S. Ahirwar, S. Dehingia and P. K. Iyer, Analyst, 2019, 144, 669–676 RSC;
(d) S. Hussain, A. H. Malik and P. K. Iyer, ACS Appl. Mater. Interfaces, 2015, 7, 3189–3198 CrossRef CAS PubMed.
-
(a) S. C. Hernandez, D. Chaudhuri, W. Chen, N. V. Myung and A. Mulchandani, Electroanalysis, 2007, 19, 2125–2130 CrossRef CAS;
(b) K. Murugappan, J. Lee and D. S. Silvester, Electrochem. Commun., 2011, 13, 1435–1438 CrossRef CAS.
-
(a) C. Mackin, V. Schroeder, A. Zurutuza, C. Su, J. Kong, T. M. Swager and T. Palacios, ACS Appl. Mater. Interfaces, 2018, 10, 16169–16176 CrossRef CAS PubMed;
(b) N. Meher and P. K. Iyer, Angew. Chem., Int. Ed., 2018, 57, 8488–8492 CrossRef CAS PubMed.
-
(a) G. Hussain and D. S. Silvester, Anal. Chem., 2016, 88, 12453–12460 CrossRef CAS PubMed;
(b) A. Dey, A. Singh, D. Das and P. K. Iyer, Phys. Chem. Chem. Phys., 2016, 18, 32602–32609 RSC.
- P. K. Kannan and R. Saraswathi, J. Mater. Chem. A, 2014, 2, 394 RSC.
- H. S. White, G. P. Kittlesen and M. S. Wrighton, J. Am. Chem. Soc., 1984, 106, 5375–5377 CrossRef CAS.
- D. A. Bernards, G. G. Malliaras, G. E. S. Toombes and S. M. Gruner, Appl. Phys. Lett., 2006, 89, 053505–053508 CrossRef.
-
(a) A. Dey, A. Singh, D. Dutta, S. S. Ghosh and P. K. Iyer, J. Mater. Chem. A, 2019, 7, 18330–18337 RSC;
(b) S. Pecqueur, S. Lenfant, D. Guérin, F. Alibart and D. Vuillaume, Sensors, 2017, 17, 57 CrossRef PubMed.
-
(a) M. Ghittorelli, L. Lingstedt, P. Romele, N. I. Crăciun, Z. M. Kovács-Vajna, P. W. M. Blom and F. Torricelli, Nat. Commun., 2018, 9, 1441–1451 CrossRef PubMed;
(b) M. J. Donahue, A. Williamson, X. Strakosas, J. T. Friedlein, R. R. McLeod, H. Gleskova and G. G. Malliaras, Adv. Mater., 2018, 30, 1705031–1705035 CrossRef PubMed.
- N. Stutzmann, R. H. Friend and H. Sirringhaus, Science, 2003, 299, 1881–1884 CrossRef CAS PubMed.
- M. Uno, Y. Tominari and J. Takeya, Appl. Phys. Lett., 2008, 93, 173301–173303 CrossRef.
- J. Rivnay, P. Leleux, M. Ferro, M. Sessolo, A. Williamson, D. A. Koutsouras, D. Khodagholy, M. Ramuz, X. Strakosas, R. M. Owens, C. Benar, J.-M. Badier, C. Bernard and G. G. Malliaras, Sci. Adv., 2015, 1, 1400251–1400255 CrossRef PubMed.
- M. Demelas, E. Scavetta, L. Basiricò, R. Rogani and A. Bonfiglio, Appl. Phys. Lett., 2013, 102, 193301–193304 CrossRef.
- P. Dutta, B. Kalita, B. Gogoi and N. Sen Sarma, J. Phys. Chem. C, 2015, 119, 17260–17270 CrossRef CAS.
- R. Bardestani, G. S. Patience and S. Kaliaguine, J. Chem. Eng., 2019, 97, 2781–2791 CAS.
- P. Kuhn, A. Forget, D. Su, A. Thomas and M. Antonietti, J. Am. Chem. Soc., 2008, 130, 13333–13337 CrossRef CAS PubMed.
- J. C. Groen, L. A. A. Peffer and J. Pérez-Ramírez, Microporous Mesoporous Mater., 2003, 60, 1–17 CrossRef CAS.
- R. J. C. Brown, J. Mol. Struct., 1995, 345, 77–81 CrossRef CAS.
- B. Kojić-Prodić and K. Molčanov, Acta Chim. Slov., 2008, 55, 692–708 Search PubMed.
- A. D. Buckingham, J. E. Del Bene and S. A. C. McDowell, Chem. Phys. Lett., 2008, 463, 1–10 CrossRef CAS.
- P. A. Colman and L. C. Allen, Chem. Rev., 1972, 72, 283–303 CrossRef.
- M. C. Etters, Acc. Chem. Res., 1990, 23, 120–126 CrossRef.
- I. V. Alabugin, M. Manoharan, S. Peabody and F. Weinhold, J. Am. Chem. Soc., 2003, 125, 5973–5987 CrossRef CAS PubMed.
- T. N. I. F. O. S. A. H. (NIOSH), Ammonia, https://www.cdc.gov/niosh/pel88/7664-41.html Search PubMed.
- R. Ghosh, A. K. Nayak, S. Santra, D. Pradhan and P. Kumar Guha, RSC Adv., 2015, 5, 50165 RSC.
- N. Wang, N. Zhang, T. Wang, F. Liu, X. Wang, X. Yan, C. Wang, X. Liu, P. Sun and G. Lu, Sens. Actuators, B, 2022, 350, 130854 CrossRef CAS.
- S. Singh, J. Deb, U. Sarkar and S. Sharma, ACS Appl. Nano Mater., 2020, 3, 9375–9384 CrossRef CAS.
- S. Kanaparthi and S. G. Singh, Org. Electron., 2019, 68, 108–112 CrossRef CAS.
- Y. Seekaew, W. Pon-On and C. Wongchoosuk, ACS Omega, 2019, 4, 16916–16924 CrossRef CAS PubMed.
- L. Liu, T. Fei, X. Guan, X. Lin, H. Zhao and T. Zhang, Sens. Actuators, B, 2020, 320, 128318 CrossRef CAS.
- M. Yin and Z. Zhu, J. Alloys Compd., 2019, 789, 941–947 CrossRef CAS.
- G. Chen, Y. Yuan, M. Lang, Z. Lv, W. Ma, N. Gu, H. Liu, J. Fang, H. Zhang and Y. Cheng, Appl. Surf. Sci., 2022, 598, 153821 CrossRef CAS.
- Q. B. Tang, Y. J. Guo, Y. L. Tang, G. D. Long, J. L. Wang, D. J. Li, X. T. Zu, J. Y. Ma, L. Wang, H. Torun and Y. Q. Fu, J. Mater. Sci., 2019, 54, 11925–11935 CrossRef CAS.
- X. Song, R. Hu, S. Xu, Z. Liu, J. Wang, Y. Shi, J. Xu, K. Chen and L. Yu, ACS Appl. Mater. Interfaces, 2021, 13, 14377–14384 CrossRef CAS.
- N. Garg, M. Kumar, N. Kumari, A. Deep and A. L. Sharma, ACS Omega, 2020, 5, 27492–27501 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab