
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatch and release strategy of matrix metalloprotease aptamers via thiol–disulfide exchange reaction on a graphene based electrochemical sensor
Vladyslav
Mishyn†
a,
Merve
Aslan
b,
Adrien
Hugo
 a,
Teresa
Rodrigues
c,
Henri
Happy
a,
Rana
Sanyal
bde,
Wolfgang
Knoll
c,
Florence
Baudoux
f,
Vincent
Bouchiat
g,
Rostyslav O.
Bilyy†
h,
Rabah
Boukherroub
a,
Amitav
Sanyal
*bd and
Sabine
Szunerits
*a
a,
Teresa
Rodrigues
c,
Henri
Happy
a,
Rana
Sanyal
bde,
Wolfgang
Knoll
c,
Florence
Baudoux
f,
Vincent
Bouchiat
g,
Rostyslav O.
Bilyy†
h,
Rabah
Boukherroub
a,
Amitav
Sanyal
*bd and
Sabine
Szunerits
*a
aUniv. Lille, CNRS, Centrale Lille, Univ. Polytechnique Hauts-de-France, UMR 8520 – IEMN, F-59000 Lille, France. E-mail: sabine.szunerits@univ-lille.fr
bDepartment of Chemistry, Bogazici University, Bebek, 34342, Istanbul, Turkey. E-mail: amitav.sanyal@boun.edu.tr
cDepartment of Scientific Coordination and Management, Danube Private University, A-3500 Krems, Austria
dCenter for Life Sciences and Technologies, Bogazici University, Istanbul, Turkey
eRS Research, Teknopark Istanbul, Pendik, 34912, Istanbul, Turkey
fCHU Lille, Service Endocrinologie-Diabétologie-Métabolisme-Nutrition, Pôle Médico-Chirurgical, Hôpital Huriez, 4/5ème étage- Aile Ouest, 59500 Lille, France
gGRAPHEAL SAS, 25 avenue des Martyres, 38042 Grenoble, CEDEX, France
hDanylo Halytsky Lviv National Medical University, 79010 Lviv, Ukraine
First published on 11th May 2022
Abstract
Matrix metalloproteases (MMP) are a large family of proteases, which synergistically degrade abundant extracellular matrix and play an essential role in tissue degradation and remodelling. Sensing of MMP levels, notably the pro- and activated matrix metalloproteinases (MMP-2 and MMP-9), would provide an early warning system of chronic inflammation in acute wounds. With this in mind, we developed a graphene-based electrochemical sensor for both of these relevant biomarkers using an innovative surface ligand, 1-pyrene pyridyl disulphide (PDS), engineered to not only capture the analyte with high affinity, but also to allow the regeneration of the whole ligand/analyte construct. The thiol–pyridyl disulphide exchange reaction was used in this work for sequential analysis of MMP-9 and MMP-2 via the engineered 1-pyrene pyridyl disulphide, the PDS surface ligand. The sensor was operational down to 0.1 pM (9.2 pg mL−1) for MMP-9 and allowed to sense MMP-2 on the same surface with a detection limit of 100 pM (950 pg mL−1) once treated with 1-pyrene pyridyl disulphide (PDS) (DTT) and 2-dipyridylsulfide (DPDS), followed by integration of the thiolated MMP-2 aptamer onto the PDS-modified renewable electrode interface. Using swab samples from diabetic patients with open wounds confirmed that the sensors characteristics are adapted for sensing of MMP levels in wound fluids and will open up the future for the development of intelligent bandage.
1. Introduction
The high reactivity of monolayer graphene and graphene edges has been put forward for the chemical functionalization of this large polycyclic material which is witnessing an ever-increasing utilization in a variety of diagnostic devices.1,2 The motivations for chemical modification include modulation of the doping level, opening an electronic band gap, and enabling the fabrication of new surface-modified composite materials for the anchoring of surface receptors for chemical and biological sensing. Pyrene-based hetero-functional surface anchors are widely employed in this respect as the van der Waals interaction between the graphene and the aromatic pyrene moiety ensures their tight binding through π–π stacking. Although traditionally pyrene anchors such as pyrenebutanoic acid succinimidyl ester (PBASE) or 1-pyrenebutyric acid (PBA) have been widely used, other derivatives such as pyrene-maleimide has been lately shown to outperform them.3 While activated esters such as PBASE can undergo facile hydrolysis during their reaction with amine-containing functional ligands, the pyrene-maleimide based linker provides an excellent alternative. The modification of graphene nanosheets with pyrene-maleimide based linkers allows rapid post-functionalization of the graphene interface with thiolated ligands in good yields.3 The thiol-based conjugation reactions are indeed widely employed as thiol groups in form of cysteine functions are either naturally occurring or can be easily engineered into biomolecules using routine molecular biology techniques.4–6Even though the maleimide-based conjugations are widely employed in fabrication of bioconjugated platforms,7–9 including antibody–drug conjugates that are in the clinic, it is known that the highly reactive maleimide group can undergo slow hydrolysis to the non-reactive maleamic acid form in aqueous conditions, which renders it inactive towards the thiol-based conjugation. This potential drawback necessitates seeking other alternative approaches for the covalent anchoring of thiolated biomolecules onto sensing surfaces. The team of Etienne and co-workers immobilized cysteine-tagged dehydrogenases via thiol–ene “click” chemistry on a carbon electrode modified with vinylphenyl groups and used this scaffold for the electroenzymatic enantioselective reduction of D-fructose to D-sorbitol.10 Next to the thiol–ene reaction, thiol-yne chemistry, performed on boron-doped diamond electrodes, allowed the successful “clicking” of thiolated oligonucleotide strands under UV irradiation for 30 min.6 Electrochemical impedance spectroscopy (EIS) was applied to follow the kinetics of hybridization and rehybridization events with a nM sensitivity.6
While these different thiol-based chemistries are efficient, they are non-reversible in nature. Reversibility of ligand attachment can be integrated onto a sensor interface via the application of the thiol–disulphide exchange reaction.4,11 The formed disulphide linkage undergoes effective cleavage to release the conjugated thiol-containing molecules from the attached platform under mild conditions. While this reaction has been largely used to design stimuli-responsive materials that undergo cleavage when exposed to thiol-containing molecules such as glutathione or dithiothreitol (DTT), it has never been investigated for its ability to impart facile renewability of the electrochemical sensing interface. One can also envision that such renewability may play an important role for diagnostic interfaces that are embedded within devices where exchange is not trivial.
In this work, we demonstrate the fabrication of a new reactive handle for functionalization of graphene-based device interface using a synthetic pyrene surface anchor, namely, pyrene-pyridinyl disulphide (PSE) (Fig. 1a). This linker not only provides a robust surface anchoring, but also allows facile modification with thiolated biological receptors. Using ferrocene-thiol as model compound, the concept of catch and release of a surface receptor was exemplified first and allowed optimizing the reaction process. Thereafter, the concept was applied for the integration of thiolated aptamers notably metallopeptidases specific aptamers. In recent years, aptamers have been utilized as detection motifs in biosensing since they can be engineered specifically for target biomolecules and possess high affinities.12 It has been demonstrated that aptamer-functionalized graphene-based devices enable efficient biosensing.3,13–16 Herein, thiolated aptamers for the rapid and sensitive detection of matrix metallopeptidase 9 (MMP-9) and matrix metallopeptidase 2 (MMP-2) will be demonstrated.
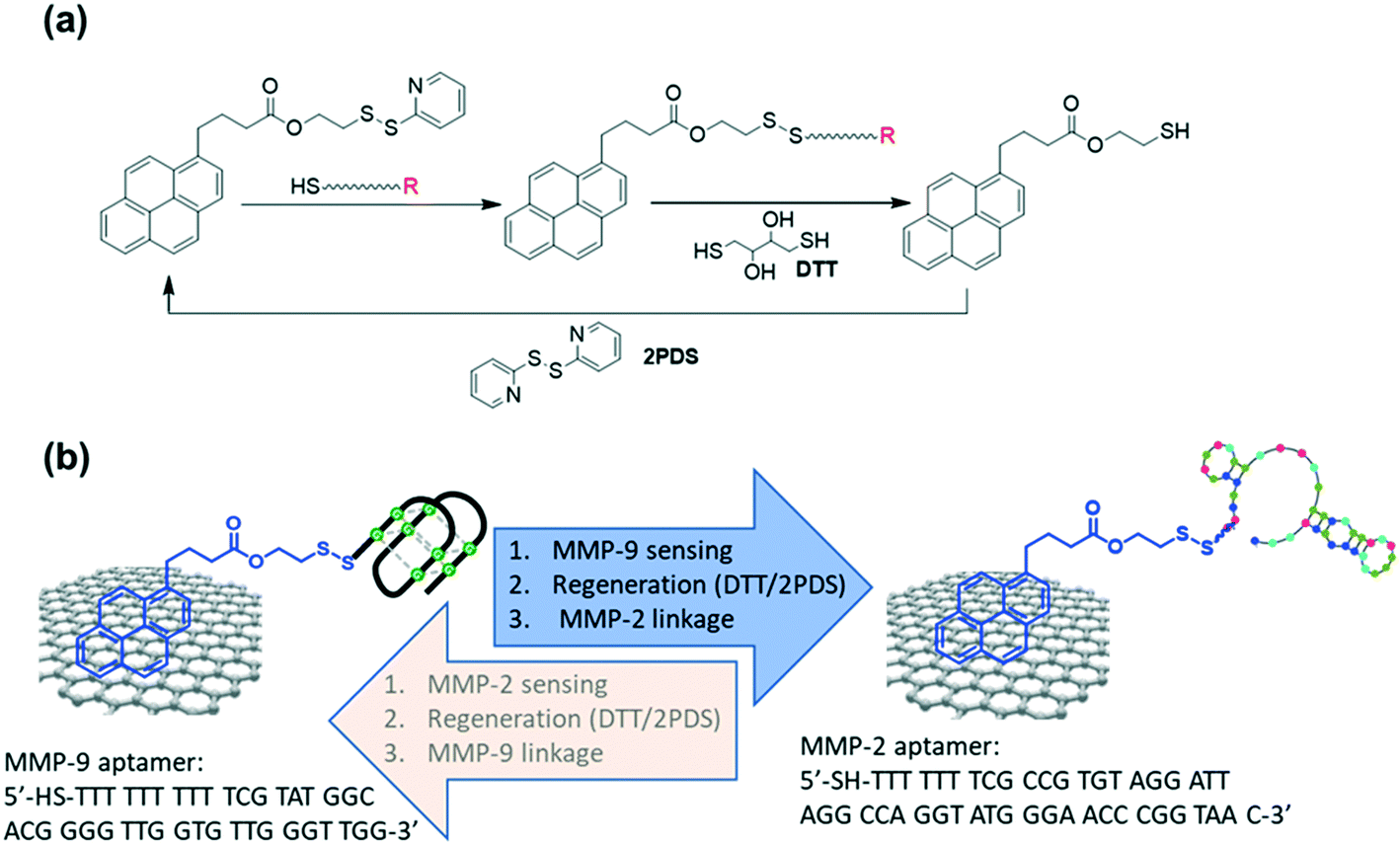 | ||
| Fig. 1 Catch and release strategy using a pyrene-pyridinyl disulphide (PSE) ligand modified graphene-based electrochemical sensor: (a) catch and release approach based on a pyrene-pyridinyl disulphide (PSE) ligand. (b) Schematics of sequential analysis of MMP-9 and MMP-2 using 1-pyrene pyridyl disulphide (PDS) mediated surface reactivation. | ||
Matrix metalloproteases are endopeptidases that not only cleave extracellular matrix (ECM) components, but also process the non-extracellular matrix molecules including growth factors and their binding proteins.17–19 These enzymes play consequently a vital role in regulating angiogenesis during wound healing20,21 through the activation of proangiogenic cytokines (e.g. TNF-α and VEGF) and by generating antiangiogenic peptides (e.g., endostatin from type XVII collagen). Previous analysis of fluids from non-healing wounds indicated that the level of pro- and activated matrix metalloproteinases, notably MMP-2 and MMP-9, are increased in patients with chronic pressure ulcers.22 Indeed, in contrast to the transitional increased level of MMP-9 in an acute surgical wound quickly returning to basal levels,23 the level of MMP-9 remains high in burn wounds and disordered wound healing.24,25 Approaches which would allow for rapid and sensitive detection of MMP-9 and MMP-2 in wound fluids are promising concepts for diagnostics on the wound healing stage. Several studies indeed reported different sensing platforms of these MMPs, with a focus on cancer diagnosis26 or the treatment of renal fibroses.27 Cao et al. used lanthanide-doped up-conversion nanoparticles for the sensing of MMP-7 and MMP-2 in human serum with detection limits of 2.2 and 13.9 ng mL−1, respectively. Detection limits of 9.6 and 25.6 pg mL−1 for respectively MMP-9 and MMP-2 were determined by a fluorescence-based assay using DNase-I-assisted recycling signal amplification.27 More general sensors are those proposed by Kou et al. using platinum nanoparticles (PtNPs) as a scaffold for efficient enzyme cascade amplification, reaching a LOD of 0.03 pg mL−1.28 A hydrogel-framed nanofiber matrix modified with a fluorescent MMP-9 specific peptide, when subjected to MMP-9 mediated cleavage, allowed MMP-9 quantification in a microfluidic device with a detection limit of 0.92 ng mL−1. An electrochemical magneto-immunosensor was applied by Ruiz-Vega et al. for MMP-9 sensing in plasma with a reported limit of quantification of 13 pg mL−1.29
We show here the possibility of sequential analysis of MMP-9 and MMP-2 via the engineered 1-pyrene pyridyl disulphide surface ligand with detection limits of 0.1 pM and 100 pM for MMP-9 and MMP-2, respectively. In addition, in a pre-clinical study on 5 samples obtained from wound fluid from diabetic patients with open non-healing wounds, we validated that the sensitivity of the developed sensor is adapted for sensing of MMP-9 and MMP-2 in non-healing wounds, and one step for an easy sensing concept to understand the occurrence of non-healing wounds.
2. Results and discussion
2.1. Graphene-based sensor modification with PSE
The graphene-based electrodes used in this study are interdigitated gold electrode (IDE) arrays of 90 pairs (each with a width of 10 μm, and separated by 10 μm) coated with CVD grown graphene via chemical transfer.30Fig. 2a depicts an SEM image of the IDE array, indicating the coverage of all the electrodes with graphene. Some folding lines are observed in the image, as expected for chemically transferred graphene sheets. A representative Raman spectrum of the graphene coated electrodes (Fig. 2b) comprises a dominant band at 1580 cm−1 (G band of graphene sp2 carbon lattice), and contributions at 1350 cm−1 (D band), 2706 cm−1 (2D secondary band) and ∼2450 cm−1 (low-intensity D + D′′ contribution) due to defects in the graphene sheet introduced during the transfer. The intensity ratios I2D/IG = 2.04 ± 0.07 and ID/IG = 0.35 indicate high-quality graphene monolayer of low disorder.3 These electrodes were immersed into 5 mM solutions of pyrene-PSE (Fig. 1a) in DMF for different time intervals and the Raman spectra were recorded (Fig. 2b). The band at 1620 cm−1 (D′) increases steadily with immersion time due to the stacking of the pyrene-ligand, as previously observed with other pyrene derivatives such as 1-pyrenebutanoic acid succinimidyl (PBASE) or 1-pyrenbutiruc acid (PBA).3 The I2D/IG ratio (Fig. 2c), related to mono and/or multilayer formation, gradually decreases for the first 4 h and then stabilizes at I2D/IG = 2 after 12 h immersion. Even immersion for 24 h results in I2D/IG > 1. This is different to other investigated pyrene ligands, notably PBASE, and pyrene-maleimide with I2D/IG < 1, indicative of pyrene aggregation on the monolayer graphene sheets, structurally similar to a bilayer graphene.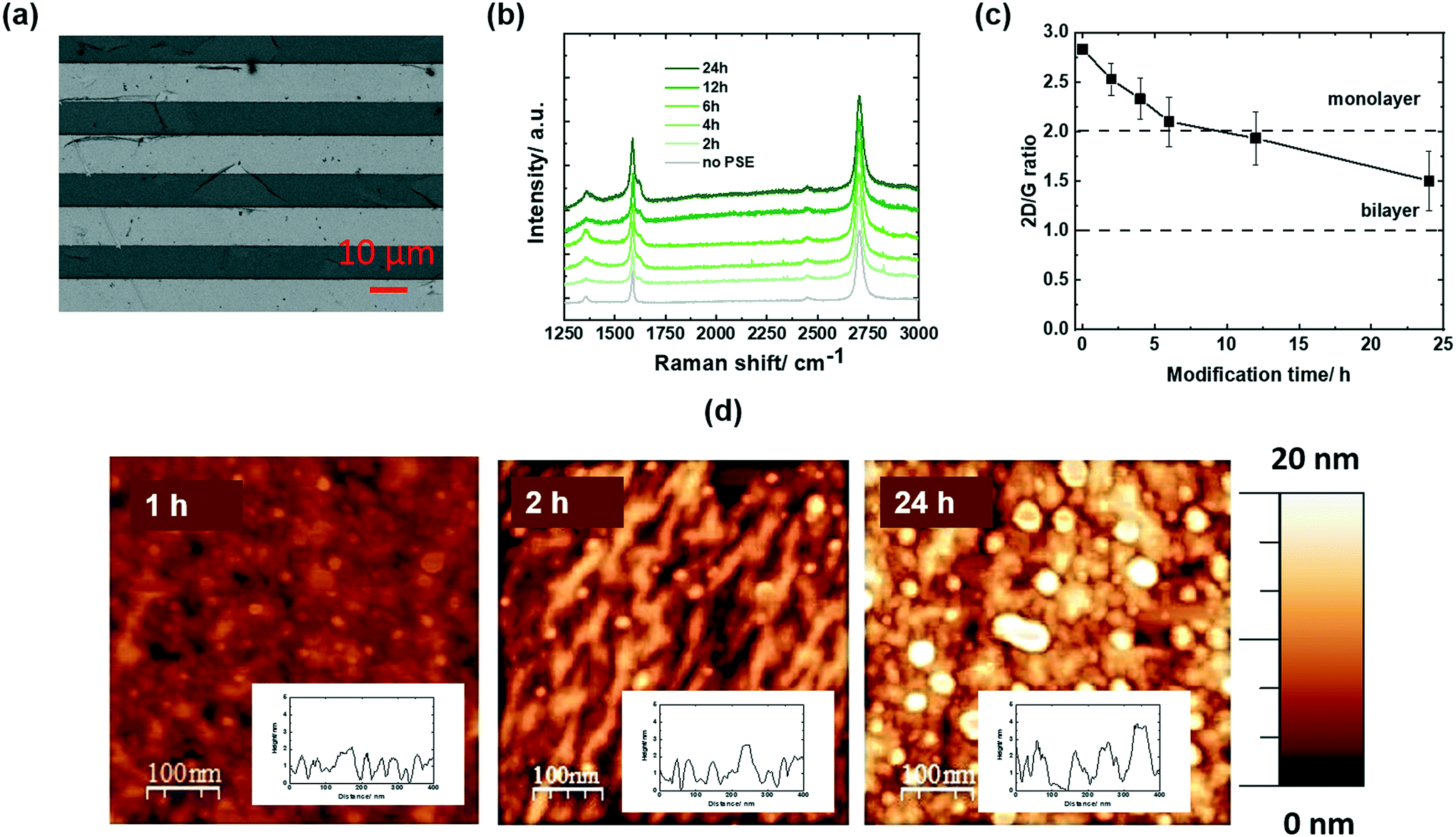 | ||
| Fig. 2 PSE-modified graphene electrodes: (a) SEM image of interdigitated electrodes coated with CVD graphene (dark areas are gold electrodes and bright areas are glass interlayers). (b) Raman spectra of graphene before (grey) and after immersion into PSE (5 mM, DMF) for different time intervals (2, 4, 6, 12 and 24 h). (c) Variation of the 2D/G ratio as a function of modification time extracted from Fig. 2b. (d) AFM images of GFET modified for 1, 2 and 24 h with pyrene-PSE stacks (inset: height step line). | ||
From the AFM images of PSE-modified GFETs for increased time spans (Fig. 2d), the deposition of PSE molecules is clearly visible after 1 h with increasing pyrene molecules deposited after 2 and 24 h. The thickness of the pyrene films increases from 0.6 nm (1 h) to 1.02 nm (2 h) and 1.17 nm (24 h), indicating limited pyrene-stack (multilayer) formation. This is in contrast to pyrene-maleimide where the formation of up to 50 nm thick films was identified by AFM after 24 h.3 PSE assembly seems to be well-controlled and might be due to the presence of the polar pyridinyl group.
2.2. Catch and release of 6-(ferrocenyl)hexanethiol
The PSE-modified graphene electrodes (G-PSE) were investigated for their reactivity with a model thiol compound, in our case 6-(ferrocenyl)hexanethiol (Fig. 3a). The chemical composition of the PSE modified electrode before (G-PSE) and after catching of 6-(ferrocenyl)hexanethiol (G-ferrocene) was validated by XPS (Fig. 3b) and electrochemical (Fig. 4) measurements.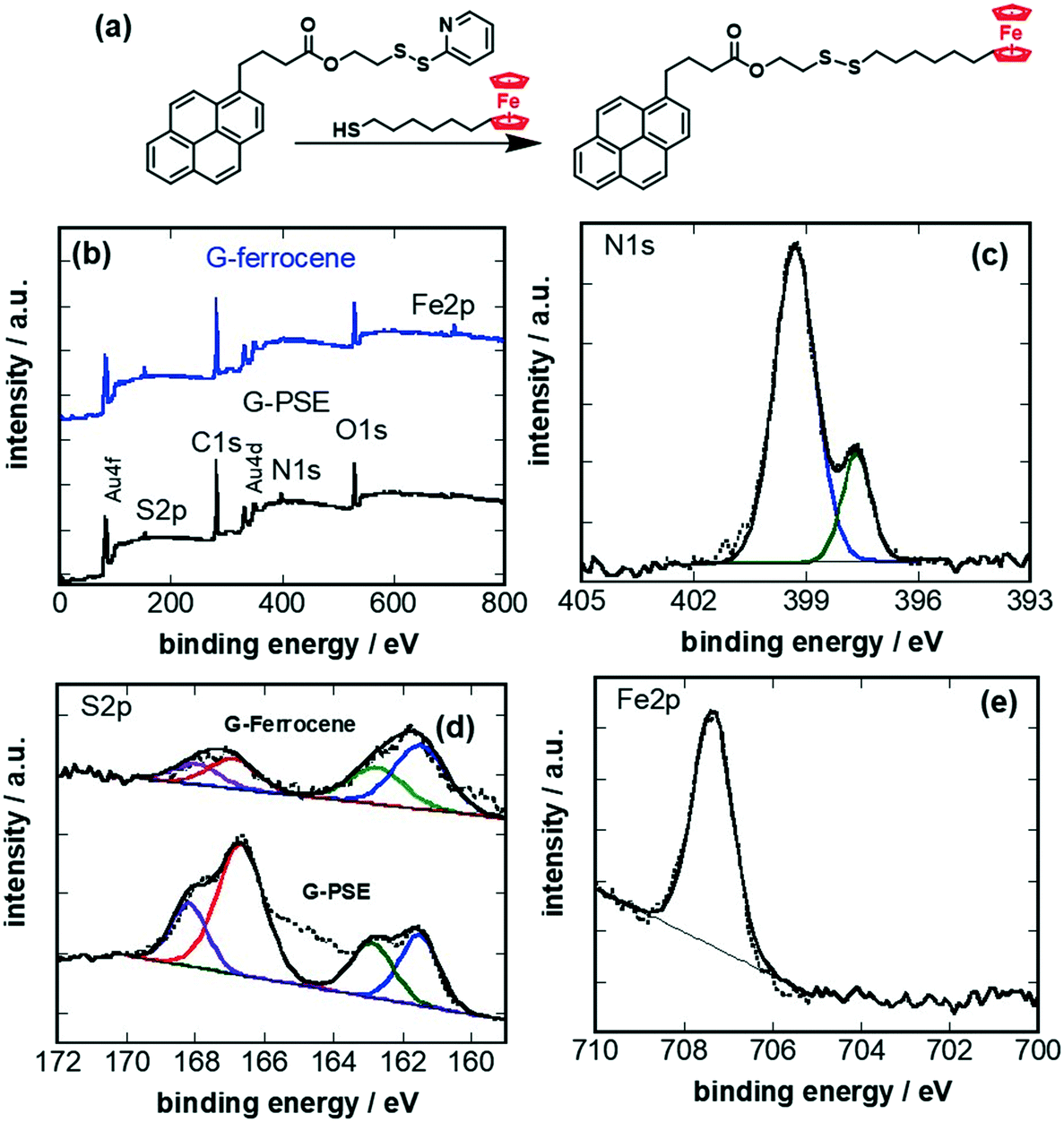 | ||
| Fig. 3 Conjugation of 6-(ferrocenyl)hexanethiol on G-PSE: (a) chemical catch reaction of 6-(ferrocenyl)hexanethiol via replacement of the pyridyl ligand. (b) XPS survey spectra of graphene modified with PSE (G-PSE, black) and after ligand exchange (G-ferrocene, blue). High-resolution XPS spectra of (c) N1s of G-PSE, (d) S2p of G-PSE and G-ferrocene, and (e) Fe2p of G-ferrocene. | ||
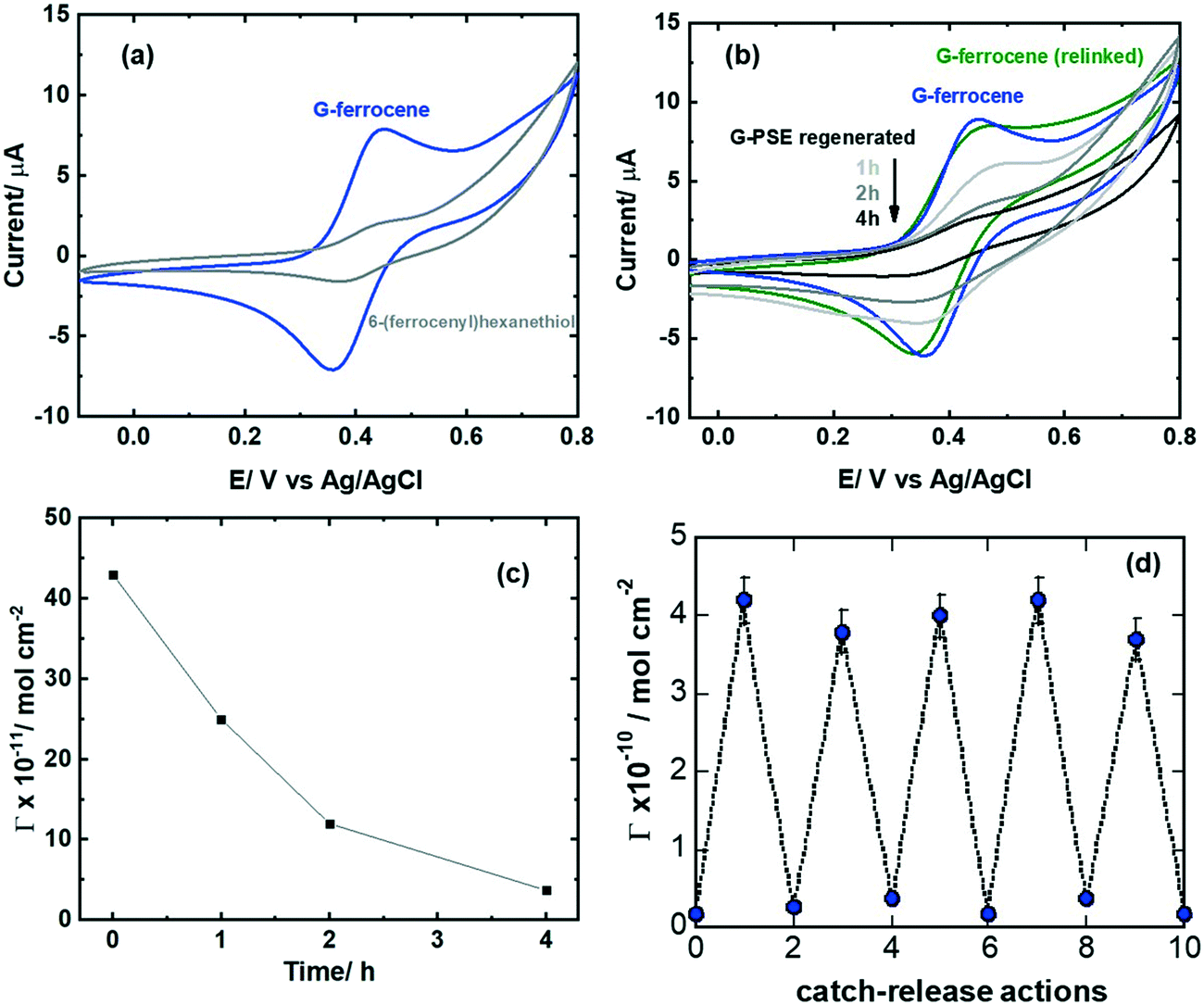 | ||
| Fig. 4 Electrochemical evaluation of the catch/release mechanism via cyclic voltammetry: (a) CVs of ferrocene-modified GFET in acetonitrile/NBu4PF6 (0.1 M) (blue), control: unmodified GFET interface immersed in 6-(ferrocenyl)hexanethiol (grey). (b) CVs of G-ferrocene (blue), after treatment with DTT (1, 2 and 4 h, 10 mM), followed by reintegration of ferrocene-thiol (green). (c) Surface coverage as extracted from Fig. 4b for different DTT (10 mM) treatment times. (d) Catch/release actions: CVs were recorded in all cases in acetonitrile/NBu4PF6 (0.1 M), scan rate = 100 mV s−1. The results are expressed as the mean ± SEM of at least 3 independent samples for each group. | ||
The XPS survey spectrum of G-PSE (Fig. 3b) comprises next to C1s, O1s and Au4f and Au4d bands, contributions due to N1s and S2p, in agreement with the integrated PSE. The bands ascribed to Au4f and Au4d are linked to the gold-base IDE below the graphene, underlining that the thickness of the PSE-graphene film is below 10 nm as expected. The N1s (Fig. 3c) high resolution XPS plot of G-PSE reveals two bands at 398.7 eV (C–N) and 399.5 eV (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). The major features of the S2p spectrum (Fig. 3d) of G-PSE electrodes are located at 162.0 and 163.5 eV and are assigned to the S2p3/2 and the S2p1/2 components of the dithiolane derived groups, respectively. The doublet at higher energy (168.7 eV) is characteristic of oxygenated anions of sulphur. Ligand exchange with 6-(ferrocenyl)hexanethiol resulting in G-ferrocene does not alter the overall S2p signature. Moreover, the presence of iron (ferrocene) on the electrode is evidenced with the additional Fe2p component in the XPS survey spectrum with a broad band at 707.5 eV containing both the Fe2p3/2 and the Fe2p1/2 (Fig. 3e). The loss of N1s indicates, in addition, the ligand exchange with 6-(ferrocenyl)hexanethiol (Fig. 3a).
N). The major features of the S2p spectrum (Fig. 3d) of G-PSE electrodes are located at 162.0 and 163.5 eV and are assigned to the S2p3/2 and the S2p1/2 components of the dithiolane derived groups, respectively. The doublet at higher energy (168.7 eV) is characteristic of oxygenated anions of sulphur. Ligand exchange with 6-(ferrocenyl)hexanethiol resulting in G-ferrocene does not alter the overall S2p signature. Moreover, the presence of iron (ferrocene) on the electrode is evidenced with the additional Fe2p component in the XPS survey spectrum with a broad band at 707.5 eV containing both the Fe2p3/2 and the Fe2p1/2 (Fig. 3e). The loss of N1s indicates, in addition, the ligand exchange with 6-(ferrocenyl)hexanethiol (Fig. 3a).
Cyclic voltammetry (CV) of the G-ferrocene interface clearly shows the presence of the characteristic redox centers (Fig. 4a). The density of ferrocene units is estimated by integrating the oxidation and reduction peaks, considering the one electron oxidation–reduction of ferrocene group using eqn (1):
| Γ = Q/nFA | (1) |
Having validated the successful attachment of 6-(ferrocenyl)hexanethiol on the G-PSE electrodes, amenable release from the electrode surface is validated in the following. Regeneration of the G-PSE electrode occurs in two stages (Fig. 1a):11 first, disulfide exchange via a disulphide reduction agent such as dithiothreitol (DTT), followed by immersion into 2,2-dipyridylsulfide (DPDS), which results in the initial G-PSE electrode and 2-thiopyridone as a side product. Cyclic voltammetry (Fig. 4b) validated the release of the ferrocene molecule by a significant decrease of the redox current related to the surface attached ferrocene ligand after DTT treatment. After 4 h treatment with DTT, from the initial 4.36 ± 0.31 × 10−10 mol cm−2 surface coverage of ferrocene molecules, only 0.36 ± 0.23 × 10−10 mol cm−2 remained (Fig. 4c). Re-integration of 6-(ferrocenyl)hexanethiol into the recovered G-PSE electrode was achieved with almost the same ferrocene loading (green line Fig. 4b) as the first time, noted as G-ferrocene (blue line Fig. 4b). This catch and release strategy can be repeated several times, as indicated in Fig. 4d, with no significant degradation of the surface architecture as evidenced from the surface coverage of ferrocene molecules.
2.3. Application to the sensing of MMP-9 and MMP-2
To validate the interest of G-PSE electrodes for sensing-related applications, two MMP aptamers (Fig. 1b)31,32 were linked sequentially to the electrodes. We opted in this work for a MMP-9 specific DNA aptamer (Fig. 1b) featuring a G-quadruplex structure and first identified by Scarano et al.,32 following a standard SELEX procedure against a fragment of MMP-9 containing the catalytic domain. Using this aptamer in a sandwich-like approach and a piezoelectric transduction, they demonstrated the label-free detection of MMP-9 with a detection limit of 560 pg mL−1 (6.8 pM) in untreated serum. In order to benefit from the high specificity against MMP-9 recorded by this single stranded DNA probe, we used a sequence carrying a cysteine end group for further covalent linkage mediated integration on our G-PSE electrodes.As a first step, the binding kinetics of the derived aptamer to MMP-9 were assessed through surface plasmon resonance (SPR) (Fig. 5a) using a maleimide group modified gold surface to ensure covalent immobilization of the aptamer. A typical binding/unbinding response was observed upon successive exposures of the aptamer-modified surface to the MMP-9 target followed by a washing step with pure buffer, from which a nanomolar affinity constant KD = 2.05 ± 0.68 × 10−11 M, a kon = 2.55 × 104 M−1 s−1 and a koff = 5.23 × 10−7 s−1 were derived. The MMP-2 DNA aptamer used in the work (Fig. 1b) was described by Han et al.31 and reported a KD = 5.59 ± 0.52 × 10−9 M, in line with the SPR results (Fig. 5b).
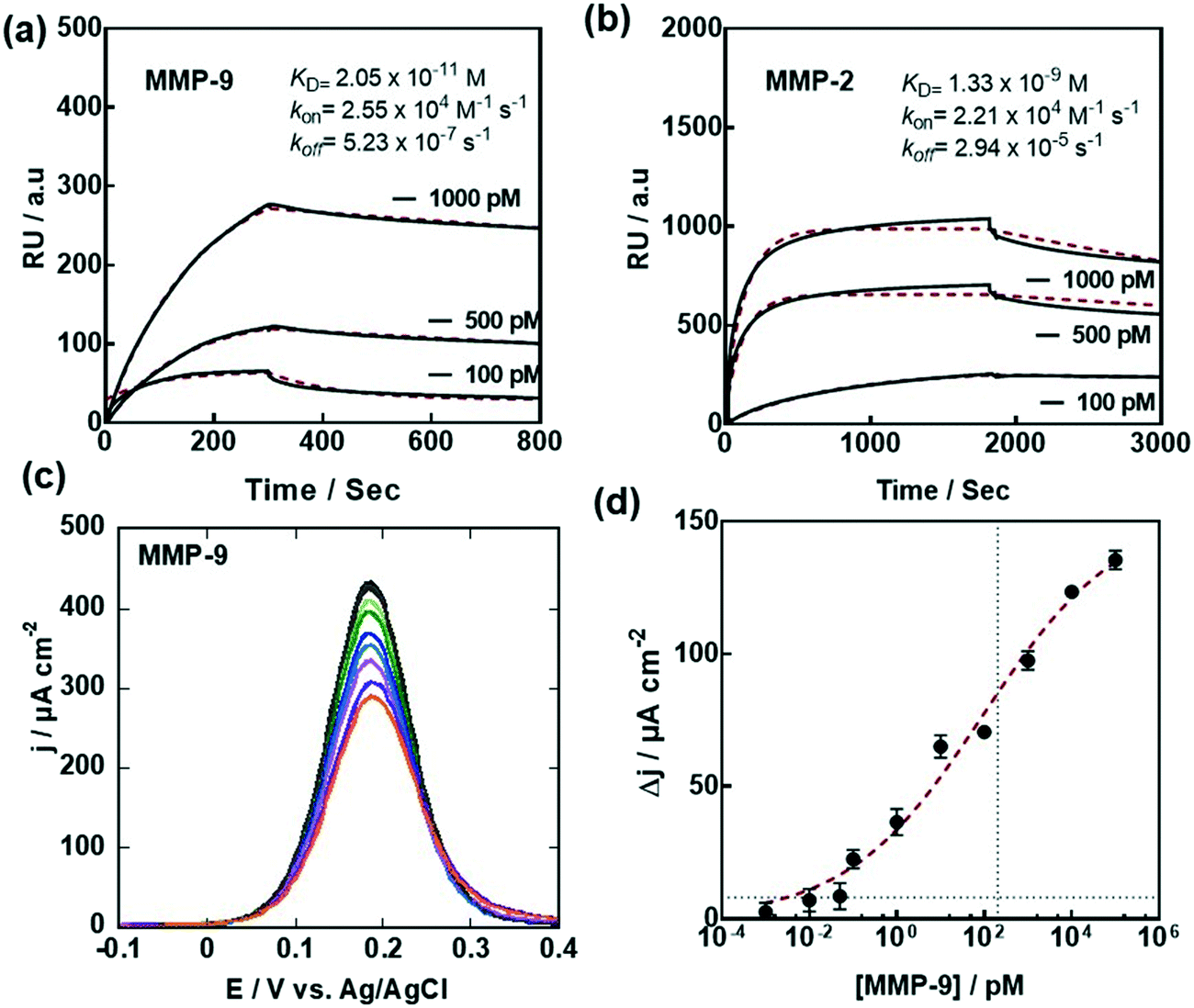 | ||
Fig. 5 Electrochemical sensing of MMP-9: (a) SPR sensogram measuring the apparent binding affinity of MMP-9 to surface immobilized MMP-9 aptamers. (b) SPR sensogram measuring the apparent binding affinity of MMP-2 to surface immobilized MMP-2 aptamers; running buffer for SPR: MOPS (20 mM) + NaCl (150 mM) + CaCl2 (1 mM) + 0.05% Tween20, flow = 30 μL min−1. The binding curve is colored in black and the fit of the data to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 Langmuir based binding curve is colored in red. (c) Differential pulse voltammograms (DPVs) of G-aptamer in ferrocenemethanol (1 mM in 0.1 M PBS, pH 7.4) before (black) and after addition of different MMP-9 concentrations. (d) Dose–response curve of MMP-9. Data were fitted to a Langmuir adsorption isotherm assuming a 1:1 complex. The results are expressed as the mean ± SEM of at least 3 independent samples for each group. :1 Langmuir based binding curve is colored in red. (c) Differential pulse voltammograms (DPVs) of G-aptamer in ferrocenemethanol (1 mM in 0.1 M PBS, pH 7.4) before (black) and after addition of different MMP-9 concentrations. (d) Dose–response curve of MMP-9. Data were fitted to a Langmuir adsorption isotherm assuming a 1:1 complex. The results are expressed as the mean ± SEM of at least 3 independent samples for each group. | ||
Exposure of the MMP-9 aptamer-modified G-PSE to increasing MMP-9 concentrations results in a decrease of the current density (Fig. 5c), as the heterogeneous electron transfer is strongly dependent on the thickness of the functionalization layer and the surface coverage. From the dose–response curve (Fig. 5d), a limit of detection, defined as LoD = (3 × noise)/sensitivity, of 0.1 pM (9.2 pg mL−1) was obtained using an estimated noise level of 2 μA cm−2 and a Δi/log (ΔC) determined as 18.1 μA cm−2. The S-shaped dose–response curve can be fitted to a Langmuir isotherm assuming a 1:1 complex between the aptamer and MMP-9 in solution, with a half saturation point (e.g. analyte concentration to occupy 50% of the receptor) of 55 × 10−12 M, in line with the SPR data. In terms of sensitivity, its performance compares with other MMP-9 sensors such as the FRET-based approach described by Yu et al. with a LOD = 9.6 pg mL−1,27 the piezoelectric biosensor reported by Scarano et al. (100 pg mL−1)32 or the microfluidic-based fluorescence sensor developed by Han and Koh33 with a LOD = 10 pM (92 pg mL−1) (Table 1). One intrinsic advantage of the aptamer sensor is that it allows discrimination between the active and pro-active forms of MMP-9. Furthermore, no amplification is required to ensure the low sensitivity, different to works using fluorescence labels27,33 as well as sandwich assays for read out.28,32 Compared to the sensor proposed by Han and Koh,33 being based on the use of MMP-9 specific peptides labeled with a fluorescent tag, a label-free strategy has been implemented in this work via the use of a MMP-9 surface linked aptamer.
| Sensing concept | LOD [pg mL−1] | Ligand | Ref. |
|---|---|---|---|
| Piezoelectric | 100 | MMP-9 aptamer | 32 |
| Microfluidic based fluorescence | 92 | MMP-9 peptide | 33 |
| Electrochemical magneto-immunosensor | 13 | MMP-9 polyclonal antibody | 29 |
| Electrochemical sensor | 6.0 | MMP-7 peptide decorated Au NPs/carbon nanotubes | 34 |
| Electrochemical | 0.03 | MMP-2 peptide with cucurbit[7]uril modified Pt NPs as amplification | 28 |
| FRET | 9.6 | MMP-9 specific FITC-peptide | 27 |
| Electrochemical graphene-based sensor | 9.2 | MMP-9 aptamer | This work |
The same interface was, in the following, used for the sensing of MMP-2 (Fig. 6). First, the MMP-2 aptamer has to be attached to the surface, and this was achieved by immersing the sensor in dithiothreitol for 4 h followed by immersion into 2,2-dipyridylsulfide to recover the initial G-PSE electrode (Fig. 1a). Thiolated MMP-2 aptamer was immobilized onto this surface after 2 h of incubation and used for the titration of MMP-2 (Fig. 6a). As was the case for MMP-9, a decrease in current was observed upon exposure to increasing MMP-2 concentrations. From the dose–response curve (Fig. 6b), a limit of detection of 100 pM (920 pg mL−1) was obtained. Half-saturation of the current signal was observed at MMP-2 concentration of 10 × 10−9 M, in line with a nM affinity constant. Importantly, titrating the MMP-2 specific aptamer modified electrodes with the MMP-9 protein did not result in significant decrease in the current signal.
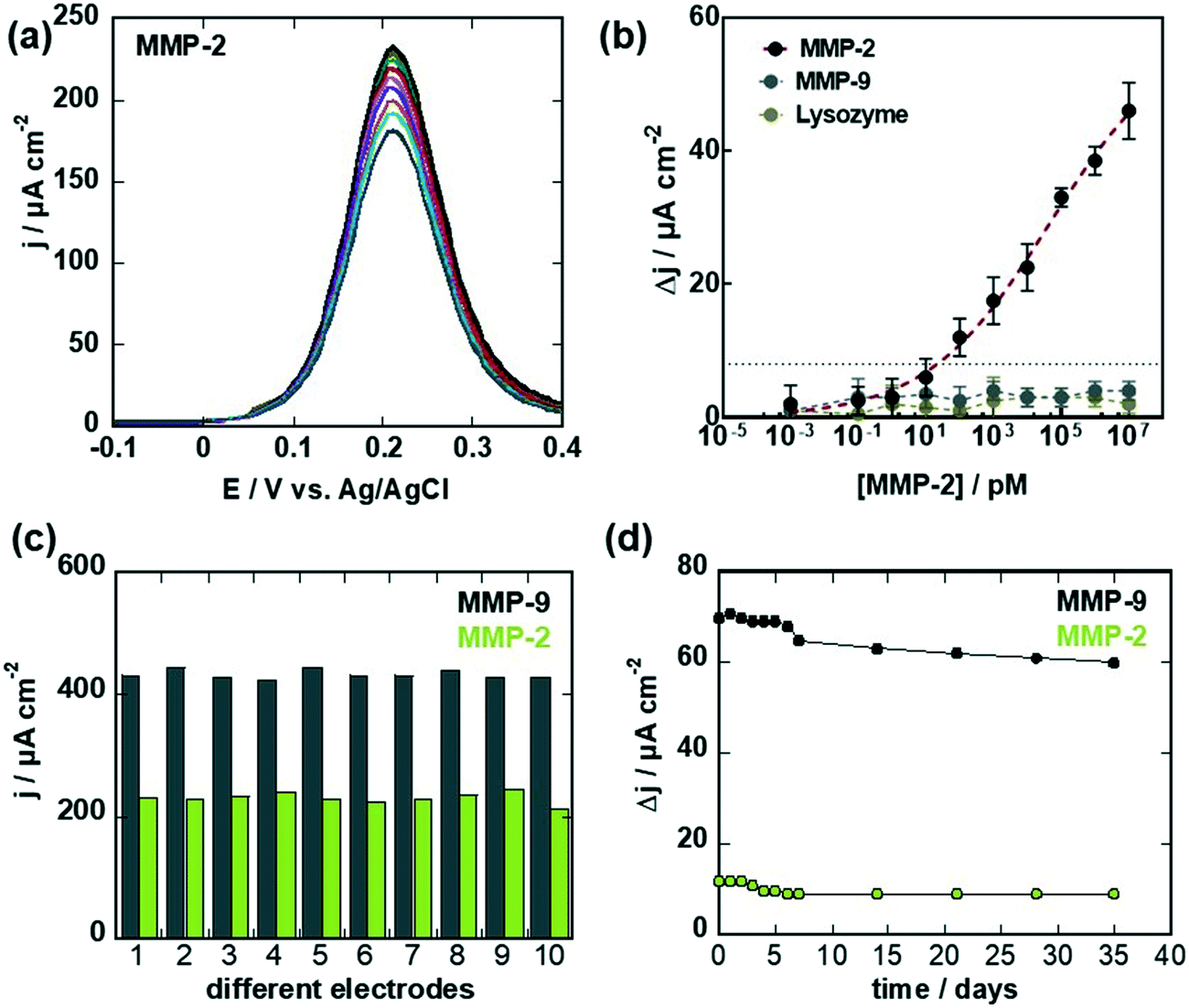 | ||
| Fig. 6 Electrochemical sensing of MMP-2: (a) differential pulse voltammograms (DPVs) of G-aptamer in ferrocenemethanol (1 mM in 0.1 M PBS, pH 7.4) before (black) and after addition of different MMP-2 concentrations. (b) Dose–response curves of MMP-2 (black) MMP-9 (grey) and lysozyme (blue). Data were fitted to a Langmuir adsorption isotherm assuming a 1:1 complex. The results are expressed as the mean ± SEM of at least 3 independent samples for each group. (c) Reproducibility of the MMP-9 (grey) and MMP-2 (green) aptamer sensor determined by differential pulse voltammograms in ferrocenemethanol (1 mM in 0.1 M PBS, pH 7.4). (d) Long-term stability of the aptamer-based sensors tested over 35 days using 100 pg mL−1 of MMP-9 and MMP-2. Sensor was stored at 4 °C in PBS during each measurement. | ||
Before testing the relevance of these sensors for detection of MMP-9 and MPP-2 biomarkers in wound fluids, the stability of the aptamer sensors as well as the reproducibly of the sensor fabrication were investigated. The reproducibility of the aptamer sensor construct (Fig. 6c) was determined by comparing the current density via differential pulse voltammograms in ferrocenemethanol (1 mM) for 10 different devices. The current density variations were 3.8% for MMP-9 aptamer sensor and 9.8% for the MMP-2 sensor. The long-term stability of the aptamer-based MMP sensor showed a loss of 3.4% and 4.6% when tested using 100 pg mL−1 of MMP-9 and MMP-2, respectively when stored at 4 °C for 1 month (Fig. 6d).
2.4. Relevance to targeting MMP-9 and MMP-2 in diabetic foot ulcers
Matrix metalloproteinases (MMPs) play important roles in the pathology of wounds and in the wound healing process of diabetic foot ulcers (DFUs), significant complications of diabetes.25,35,36 Chronic wounds typically stall in the inflammation stage of wound healing, where it is believed that excessive MMP activity plays a role in preventing the wound from healing, the increase believed to be responsible for the increased extracellular matrix destruction observed in chronic wounds. Wound liquids are complex fluids containing next to ions such as sodium, potassium, chloride, and urea, proteins notably lysozyme and matrix metalloproteinases as well as cytokines, neutrophils, macrophages and leucocytes.37 The selective sensing of MMP over lysozyme is clearly seen in Fig. 6b. To understand the concentration of MMP-9 and MMP-2 in non-healing wound, wound fluids were collected from open wound with an absorbent cotton pad, as it is a local noninvasive manner for sample collection, which reflects well the microenvironment of the wound bed. The total protein level of the sample was determined without differentiation of the different proteins present (Table 2).18| Samplea | Protein content [μg mL−1] | ELISA | Aptasensor | ||
|---|---|---|---|---|---|
| MMP-9 pM (ng mL−1) | MMP-2 pM (ng mL−1) | MMP-9 pM (ng mL−1) | MMP-2 pM (ng mL−1) | ||
| a Details of patient samples: sample 1: female, 48 years, type 2 diabetics, neuropathic wound, wound duration: 2 weeks, dermal infections, HBA1c = 6.6%. Sample 2: male, 76 years, type 2 diabetics, neuropathic wound, wound duration: 12 months, osteitis, HBA1c = 8.2.0%. Sample 3: male, 45 years, type 1 diabetics, neuropathic wound, wound duration: 1-month, dermal infectionHBA1c ≥ 11.0%. Sample 4: male, 47 years, secondary pancreatitis type 1 diabetics, neuropathic wound, wound duration: 12 months, osteitis, HBA1c = 6.0%. Sample 5: male, 71 years, type 2 diabetics, post operation wound, wound duration: 2 months, osteitis, HBA1c = 8.7%. | |||||
| 1 | 0 | 0 | 0 | 0 | <LoD |
| 2 | 5 | 1.5 ± 1.5 (0.1 ± 0.1) | 0.06 ± 0.03 (1.0 ± 0.5) | 1.2 ± 0.6 (0.08 ± 0.01) | <LoD |
| 3 | 10 | 4.50 ± 1.5 (0.3 ± 0.1) | 0.27 ± 0.1 (4.5 ± 1.6) | 5.2 ± 0.9 (0.34 ± 0.01) | 5.5 ± 0.8 (0.38 ± 0.01) |
| 4 | 134 | 160 ± 3.0 (8.7 ± 0.2) | 38.3 ± 2.8 (2.3 ± 0.2) | 203 ± 52 (11.0 ± 2.9) | 40 ± 3 (2.2 ± 0.2) |
| 5 | 625 | 912 ± 3.0 (60.8 ± 0.2) | 180 ± 1.6 (10.8 ± 0.1) | >10000 (>667) |
182 ± 3 (9.8 ± 0.2) |
From the current density changes obtained for the different samples for MMP-9 and MMP-2 (Fig. 7a–c) and using the calibration curves from Fig. 5d and 6b for MMP-9 and MMP-2, respectively, concentrations between 0–912 pM were obtained for MMP-9 and 0–180 pM for MMP-2 by ELISA as listed in Table 2. In the case of MMP-2, the concentrations obtained with the aptasensor are comparable for samples 3–5, with samples 1 and 2 showing a current density change blow the threshold (Fig. 7c) and could thus not be validated with accuracy. In the case of MMP-9, at low MMP-9 concentrations, agreement between ELISA and the aptasensor is obtained, while at larger concentrations, deviations were observed. This might be due to nonspecific interactions with the surface of the high protein load and the limit of the linear sensing range of the sensor.
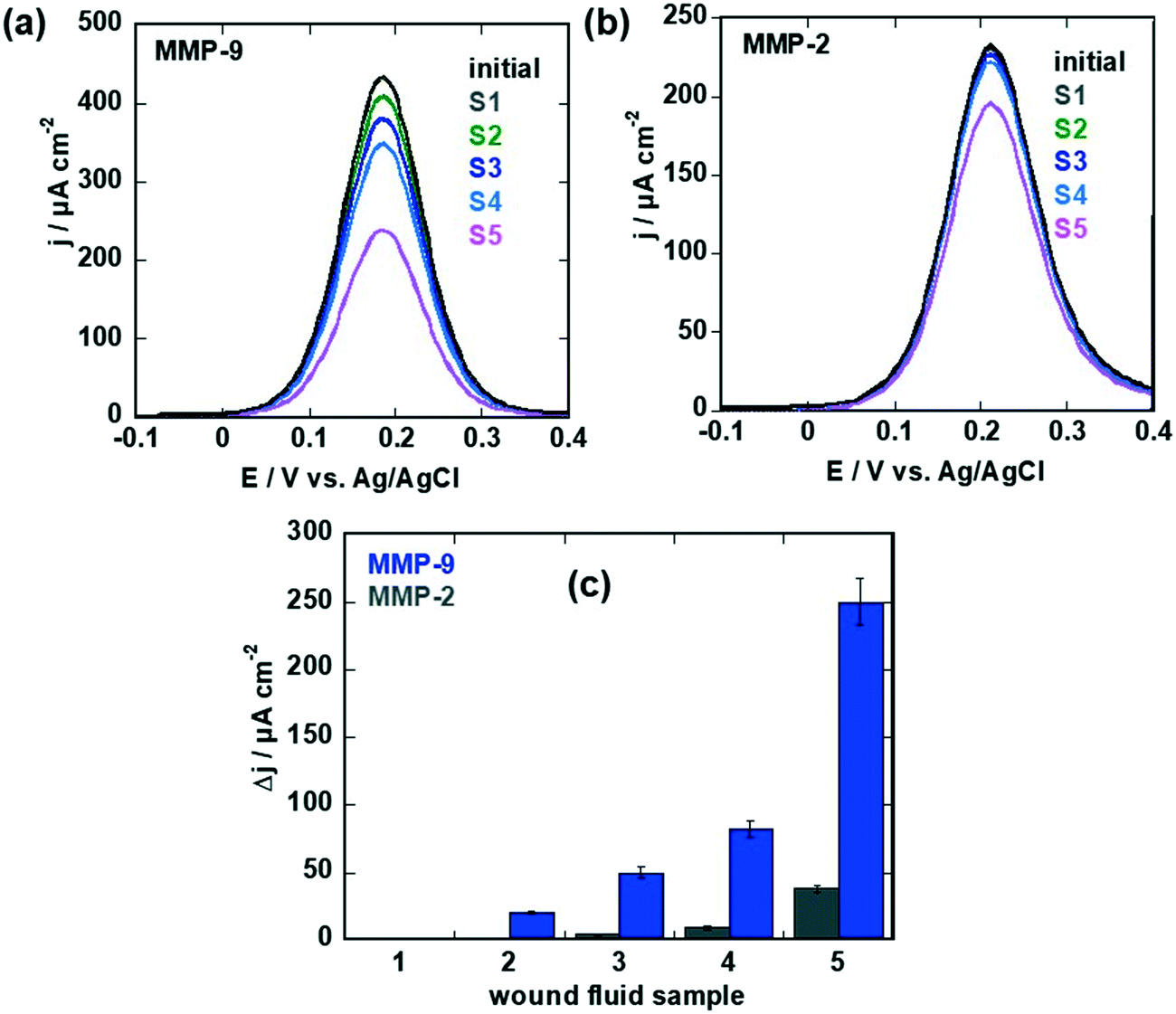 | ||
| Fig. 7 Sensing of MMP-9 and MMP-2 levels in wound fluids: (a) differential pulse voltammograms (DPVs) of MMP-9 specific aptasensor in ferrocenemethanol (1 mM in 0.1 M PBS, pH 7.4) before (black) and after sensing of samples 1–5 (after each measurement the sensor was regenerated by immersion into 0.1 M NaOH for 15 min). (b) Differential pulse voltammograms (DPVs) of MMP-2 specific aptasensor in ferrocenemethanol (1 mM in 0.1 M PBS, pH 7.4) before (black) and after sensing of samples 1–5 (after each measurement the sensor was regenerated by immersion into 0.1 M NaOH for 15 min). (c) Change of current on MMP-9 (blue) and MMP-2 (grey) specific sensors for samples 1–5 (Table 2). The results are expressed as the mean ± SEM of at least 3 different sensors for each group. | ||
3. Conclusion
In conclusion, the first electrochemical sensor for the sensing of matrix metalloproteases in wound fluids of diabetic patients was developed and compared to ELISA-based assay. The electrochemical aptasensor showed different sensitivities to MMP-9 and MMP-2 of 0.1 pM (9.2 pg mL−1) and 100 pM (920 pg mL−1), respectively. We believe this is mostly due to the difference in affinity constants being in the picomolar range for MMP-9, but in the nanomolar range for MMP-2. The possibility of the same sensor for sensing MMP-9 and MMP-2 sequentially was achieved using an innovative surface ligand, 1-pyrene pyridyl disulphide (PDS), engineered to allow the regeneration of the whole ligand/analyte construct. While such sensor concepts have to be now integrated into real wound healing bandages to test their efficiency, this work provides one of the first demonstrator of the feasibility of an intelligent wound healing bandage, which can be used as early warning system for lack of wound closer and the occurrence of non-healing wounds. It has to be noted, in addition, that the ELISA technique detects both active and inactive forms of the enzyme and does not reflect actual enzyme activity in the wounds. The aptamer interacts only with the active form of the MMP, being thus clinically more relevant. Finally, a major issue in the clinical utility of MMP assays is the detection of MMP activity following treatment with agents inhibiting activity, as recently outlined by Fields et al.38 The effect of different MMP-9 and MMP-2 inhibitors on the efficiency of the sensor to detect such changes is one of the follow-up studies to be carried out. Such considerations will greatly enhance the clinical utility of MMP biosensor and will allow bridging the utility of such diagnostic devices for obtaining clinical responses to treatment choices for non-healing wounds.4. Experimental section
Materials
4-Pyrenebutyric acid was purchased from Acros Organics. 1-(3-Dimethyl-aminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI) and 4-(dimethylamino)pyridine (DMAP) were purchased from Alfa Aesar. 2,2′-Dipyridyl disulfide was obtained from TCI Chemicals. Anhydrous dichloromethane (CH2Cl2) was procured from SciMatCo purification system. 11-Mercaptoundecanoic acid, N-hydroxysuccinimide (NHS), 3-(N-morpholino)propanesulfonic acid, sodium chloride (NaCl), calcium chloride (CaCl2), Tween 20, tris(2-carboxyethyl)phosphine hydrochloride (TCEP) and recombinant human MMP-2 protein (Mw = 62 kDa) were all obtained from Sigma-Aldrich. Maleimide-poly(ethylene glycol)-amine (Mw = 1 kDa) was purchased from Interchim. Glycine-HCl buffer (pH = 2.5) was obtained from Cytiva. Native human MMP-9 protein (Mw = 92 kDa) was obtained from Abcam (Cambridge, UK). Recombinant human MMP-2 protein (Mw = 62 kDa) was obtained from Sigma-Aldrich.Graphene coated electrodes
Monolayer graphene is grown by chemical vapor deposition (CVD) on commercial Cu foil from Alpha Aesar (high purity – 99.9999%) and transferred to the interdigitated microelectrodes (ED-IDE1-Au w/o SU8, Micrux Technologies), as described recently by us.3Surface modification with PSE
Graphene coated electrodes were immersed in PSE (5 mM) in dry DMF under constant agitation in ambient conditions for different time intervals. After that, the interfaces were withdrawn, copiously washed with DMF and water and dried under nitrogen flow.Biosensor
Stock solutions of the MMP-9 and MMP-9 DNA aptamers (880 μg) were suspended in 1.1 mL 1× MOPS (20 mM MOPS + 150 mM NaCl + 1 mM CaCl2), resulting in an aptamer concentration of 55 μM. An aptamer solution (100 μg mL−1) was then heated at 90 °C for 5 min and cooled down slowly to room temperature to insure proper folding. Thiolated-PEG4-methyl was used to prevent non-specific adsorption on graphene. The PEG solution was prepared in the same buffer used for the aptamer with an aptamer/PEG molar ratio of 2:1. Both aptamer and PEG solutions were diluted in a TCEP solution (1:1 volume ratio, 100 molar excess) for 20 min right before use in order to reduce eventual disulphide bond formation. The graphene electrodes were functionalized by dropping both solutions onto the graphene surface, incubated for 2 h and rinsed with 1× MOPS buffer before electrochemical measurements. MMP-9 and MMP-2 protein solutions were prepared in 1× MOPS equally.
Characterization
Scanning electron microscopy (SEM) images were recorded on an electron microscope ULTRA 55 (Zeiss, France). Raman spectroscopy measurements were performed on a LabRam HR Micro-Raman system (Horiba Jobin Yvon, France) combined with a 473 nm laser diode as excitation source. Visible light was focused by a 100× objective. The scattered light was collected by the same objective in backscattering configuration, dispersed by an 1800 mm focal length monochromator and detected by a CCD. X-ray photoelectron spectra (XPS) were recorded with an SPECSLAB II (Phoibos-Hsa 3500 150, 9 channels) spectrometer using an Al Kα source (E = 1486.6 eV) operating at 12 kV, pass energy of 40 eV, 0.1 eV energy step and acquisition time of 1 s per point. The residual pressure inside the analysis chamber was ∼1 × 10−8 Torr. All XPS peaks were referenced according to the adventitious C1s peak at 284.5 eV.Surface plasmon resonance
SPR measurements were carried out with a BIACORE T200 (Cytiva Life science). Gold-based SPR interfaces were first immersed in 11-mercaptoundecanoic acid (25 mM in ethanol) for 30 min, rinsed with ethanol and Milli-Q water before loading in the SPR. The formed Au–COOH surface was then exposed to a flow (20 μL min−1) of an equimolar mixture of EDC/NHS (15 mM/15 mM) prepared in MOPS buffer (3-(N-morpholino)propanesulfonic acid (20 mM), NaCl (150 mM), CaCl2 (1 mM) and 0.05% in volume of Tween-20) for 20 min to activate the surface with an ester chemical functionality. The surface was then exposed to a flow (10 μL min−1) of maleimide-poly(ethylene glycol)-amine (100 μL min−1) for 2 h to provide a maleimide terminal functionality necessary for binding the thiolated aptamer. Before injection, the MMP-9 aptamer solution (2 μL min−1, 160 nM diluted in MOPS buffer) was first heated at 90 °C for 5 min and cooled down to room temperature to insure a proper folding. The aptamer solution was then diluted in a TCEP solution (16 μM in MOPS buffer) for 20 min right before use in order to reduce eventual disulphide bond formation. Finally, the aptamer solution was injected at a low flow rate (1 μL min−1) for 2 h to insure the aptamer binding to the chip surface. Association/dissociation cycles consisted in first injecting for 5 min a solution of MMP-9 diluted to the working concentration in the MOPS running buffer, followed by pure buffer injection for the dissociation step and finally surface regeneration using an aqueous buffer (10 mM, glycine-HCl at pH 2.5).Electrochemical measurements
Electrochemical measurements were performed on a potentiostat/galvanostat/impedance analyzer (PalmSens4, PalmSens, The Netherlands). A conventional three-electrode cell (Micrux, Spain) was employed using a silver wire covered with silver chloride (Ag/AgCl) and a platinum mesh as the reference and auxiliary electrodes, respectively. The IDEs were acting as a working electrode. Cyclic voltammograms during the electrochemical grafting were recorded at a scan rate of 50 mV s−1 and potential step of 0.01 V. Differential pulse voltammograms for the sensing were acquired using a potential step of 0.005 V, a potential pulse of 0.06 V and a pulse time of 0.02 s at a scan rate of 100 mV s−1.Biological/clinical assays
Five patients with type 2 diabetes were recruited in a preclinical pilot study at the CHU Lille, each patient voluntarily gave informed consent to participate in the study. All patients had a chronic wound with a duration ranging from 1 month to 6 months and with an area of at least 0.25 cm2. Wound fluid samples were collected at CHU Lille by applying a sterile absorbent cotton swab (Distrimed, France, ref. 865461) over the wound for 1 min. The cotton swab was placed into 1 ml PBS buffer, and the proteins were eluted from the cotton swab by shaking for about 1 min manually.MMP-9 and MMP-2 levels were measured using an enzyme-linked immunosorbent assay (ELISA, Human MMP9 ELISA kit, Abcam UK). To avoid any possible variations, caused by the amount of fluid collected, the total protein concentrations were measured using an enzymatic method (BCA protein Assay Kit, Merck Millipore Germany) in addition.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the Centre National de la Recherche Scientifique (CNRS), the University of Lille, the CPER “Photonics for Society”, and the ANR PADISC (ANR-20-CE19-0021) is acknowledged. This project has received funding from the European Union's Horizon 2020 Research and Innovation Staff Exchange (RISE) Marie Skłodowska-Curie Actions under grant agreement no. 690836.References
- Q. Han, J. Pang, Y. Li, B. Sun, B. Ibarlucea, X. Liu, T. Gemming, Q. Cheng, S. Zhang, H. Liu, J. Wang, W. Zhou, G. Cuniberti and H. M. Rümmeli, ACS Sens., 2021, 6, 3841–3881 CrossRef CAS PubMed.
- A. Beraud, M. Sauvage, C. M. Bazan, M. Tie, A. Bencherif and D. Bouilly, Analyst, 2021, 146, 403–428 RSC.
- V. Mishyn, A. Hugo, T. Rodrigues, P. Aspermair, H. Happy, L. Marques, C. Hurot, O. Othmen, V. Bouchiat, R. Boukherroub, W. Knoll and S. Szunerits, Sens. Diagn., 2021, 1, 235–244 RSC.
- D. M. Beaupre and R. G. Weiss, Molecules, 2021, 26, 3332 CrossRef CAS PubMed.
- T. N. Gevrek, M. Cosar, D. Aydin, E. Kaga, M. Arslan, R. Sanyal and A. Sanyal, ACS Appl. Mater. Interfaces, 2018, 10, 14399–14409 CrossRef CAS PubMed.
- D. Meziane, A. Barras, A. Kromka, J. Houdkova, R. Boukherroub and S. Szunerits, Anal. Chem., 2012, 84, 194–200 CrossRef CAS PubMed.
- N. Cengiz, T. N. Gevrek, R. Sanyal and A. Sanyal, Bioconjugate Chem., 202, 31, 1382–1391 CrossRef PubMed.
- J. M. Ravasco, H. Faustino, A. Trindade and P. M. P. Gois, Chem. – Eur. J., 2019, 25, 43–59 CrossRef CAS PubMed.
- D. P. Nair, M. Podgorski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2014, 26, 724–744 CrossRef CAS.
- L. Zhang, N. Vila, T. Klein, G.-W. Kohring, I. Mazurenko, A. Wilcarius and M. Etienne, ACS Appl. Mater. Interfaces, 2016, 7, 17591–17598 CrossRef PubMed.
- I. Altinbasak, M. Arslan, R. Sanyal and A. Sanyal, Polym. Chem., 2020, 11, 7603–7624 RSC.
- K.-M. Song, S. Lee and C. Ban, Sensors, 2012, 12, 612 CrossRef PubMed.
- Y. Ohno, K. Maehashi and K. Matsumoto, J. Am. Chem. Soc., 2010, 132, 18012–18013 CrossRef CAS PubMed.
- N. I. Khan, M. Mousazadehkasin, S. Ghosh, J. G. Tsavalas and E. Song, Analyst, 2020, 145, 4494–4503 RSC.
- Z. Hao, Y. Zhu, X. Wang, P. G. Rotti, C. DiMarco, S. R. Tyler, X. Zhao, J. F. Engelhardt, J. Hone and Q. Lin, ACS Appl. Mater. Interfaces, 2017, 9, 27504–27511 CrossRef CAS PubMed.
- P. Aspermair, P. Aspermair, V. Mishyn, J. Bintinger, H. Happy, K. Bagga, P. Subramanian, W. Knoll, R. Boukherroub and S. Szunerits, Anal. Bioanal. Chem., 2020, 20, 1–9 Search PubMed.
- F. Sabino and U. auf dem Keller, Metalloproteinases Med., 2015, 2, 1–8 Search PubMed.
- P. Jindatanmanusan, S. Luanraksa, T. Boonsiri, T. Nimmanon and P. Arnutti, Patholog. Res. Int., 2018, 1631325 Search PubMed.
- M. P. Caley, V. L. C. Martins and E. A. O'Toole, Adv. Wound Care, 2015, 4, 225–234 CrossRef PubMed.
- J. L. Lazaro, V. Izzo, S. Meaume, A. H. Davies, R. Lobmann and L. Uccioli, J. Wound Care, 2016, 25, 277–287 CrossRef CAS PubMed.
- J. Dissemond, M. Augustin, M. Dietlein, U. Faust, W. Keuthage, R. Lobmann, K.-C. Münter, R. Strohal, M. Stücker, J. Traber, W. Vanscheidt and S. Läuchli, J. Wound Care, 2020, 26, 102–118 CrossRef PubMed.
- G. P. Ladwig, M. C. Robson, R. Liu, M. A. Kuhn, D. F. Muir and G. S. Schultz, Wound Repair Regen., 2002, 10, 26–37 CrossRef PubMed.
- M. Vaalamo, M. Weckroth, P. Puolakkainen, J. Kere, P. Saarinen and J. Lauharanta, et al. , Br. J. Dermatol., 1996, 135, 52–59 CrossRef CAS PubMed.
- P. K. Young and F. Grinnell, J. Invest. Dermatol., 1994, 103, 660–664 CrossRef CAS PubMed.
- T. E. Serena, B. M. Cullen, S. W. Bayliff, M. C. Gibson, M. J. Carter, L. Chen, R. Yaakov, J. Samies, M. Sabo, D. DeMarco, N. Le and J. Galbraith, Wound Repair Regen., 2016, 24, 589–595 CrossRef PubMed.
- S. Cao, Z. Li, J. Zhao, M. Chen and N. Ma, ACS Sens., 2018, 3, 1522–1530 CrossRef CAS PubMed.
- X.-A. Yu, Y. Hu, Y. Zhang, R. Zhang, X. Bai, L. Gu, H. Gao, R. Li, J. Tian and B.-Y. Yu, ACS Sens., 2020, 5, 1119–1125 CrossRef CAS PubMed.
- B.-B. Kou, Y.-Q. Chai, Y.-L. Yuaon and R. Yuan, Anal. Chem., 2017, 89, 9383–9387 CrossRef CAS PubMed.
- G. Ruiz-Vega, A. Garcia-Robaina, M. B. Ismail, H. Pasamar, T. Garcia-Berrocoso, J. Montaner, M. Zourob, A. Othmane, F. J. del Campo and E. Baldrich, Biosens. Bioelectron., 2018, 115, 45–52 CrossRef CAS PubMed.
- V. Mishyn, T. Rodrigues, Y. R. Leroux, P. Aspermair, H. Happy, J. Bintinger, C. Kleber, R. Boukherroub, W. Knoll and S. Szunerits, Nanoscale Horiz., 2021, 6, 819–829 RSC.
- M.-E. Han, S. Baek, H.-J. Kim, J. H. Lee, S.-H. Ryu and S.-O. Oh, Nanoscale Res. Lett., 2014, 9, 10 CrossRef PubMed.
- S. Scarano, E. Dausse, F. Crispo, J.-J. Toulmé and M. Minunni, Anal. Chim. Acta, 2015, 897, 1–9 CrossRef CAS PubMed.
- S. W. Han and W.-G. Koh, Anal. Chem., 2016, 88, 6247–6253 CrossRef CAS PubMed.
- Q. Palomar, X. X. Xu, R. Selegard, D. Aili and Z. Zhang, Sens. Actuators, B, 2020, 325, 128789 CrossRef CAS.
- J. I. Jones, T. T. Nguyen and M. Chang, Pharmaceuticals, 2019, 12, 79 CrossRef CAS PubMed.
- P. Jindatanmanusan, S. Luanraksa, T. Boonsiri, T. Nimmanon and P. Arnutt, Patholog. Res. Int., 2018, 2018, 1631325 Search PubMed.
- G. Power, Z. Moore and T. O'Connor, J. Wound Care, 2017, 26, 1–9 CrossRef.
- G. B. Fields, Front. Immunol., 2019, 10, 1278 CrossRef CAS PubMed.
- R. Bej, J. Sarkar and S. Ghosh, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 194–202 CrossRef CAS.
- S. Ghosh, S. Basu and S. Thayumanavan, Macromolecules, 2006, 39, 5595–5597 CrossRef CAS.
Footnote |
| † Part of special issue devoted to Ukrainian scientists. |
| This journal is © The Royal Society of Chemistry 2022 |