
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal complexes for the visualisation of amyloid peptides
Jean-François
Morfin
a,
Sara
Lacerda
 a,
Carlos F. G. C.
Geraldes
bc and
Éva
Tóth
*a
a,
Carlos F. G. C.
Geraldes
bc and
Éva
Tóth
*a
aCentre de Biophysique Moléculaire, CNRS, UPR 4301, Université d'Orléans, Rue Charles Sadron, 45071 Orléans Cedex 2, France. E-mail: Eva.JAKABTOTH@cnrs.fr
bDepartment of Life Sciences and Coimbra Chemistry Centre, Faculty of Science and Technology, University of Coimbra, Calçada Martim de Freitas, 3000-456 Coimbra, Portugal
cCIBIT/ICNAS, University of Coimbra, Azinhaga de Santa Comba, 3000-548 Coimbra, Portugal
First published on 5th April 2022
Abstract
Amyloid forms of many different proteins have been long identified as relevant biomarkers in important pathologies with high societal impact, including Aβ in Alzheimer's disease, amylin in type 2 diabetes, α-synuclein in Parkinson's disease, etc. With over eighty novel complexes reported in the last six years, metal-based agents designed for the detection of such amyloid fibrils represent a rapidly growing field in molecular imaging. While the majority of these examples are radiocomplexes for nuclear imaging and focus on the detection of Aβ in Alzheimer's disease, there is increasing interest in other peptides and pathologies, and few studies are also directed at magnetic resonance imaging probes. In this review, we survey the recent literature according to the chemical nature of the amyloid recognition moieties, most of which are derived from a few basic structures, including benzothiazole, benzofuran or stilbene. Relationships between chemical structure and amyloid binding properties and biodistribution, in particular brain delivery and brain clearance, are outlined in order to help further work in this emerging area of research.
 Jean-François Morfin | Jean-François Morfin obtained his PhD in 2008 from the Université de Bretagne Occidentale, Brest, France, on the synthesis of macrocyclic polyamine derivatives as bismuth(III) chelators for alpha-immunotherapy applications. Then he moved as a postdoc to the Centre of Molecular Biophysics (CBM) in Orléans (2009–2011), where he studied the kinetics of gallium(III) complexation with NOTA and developed multimodal probes for amyloid visualization. He finally joined CBM as a CNRS research engineer in 2011 and is currently working on the development of molecular imaging probes based on metal complexes for biomedical applications. |
 Sara Lacerda | Sara Lacerda received her PhD from the University of Lisboa (2009), during which she developed and studied in vitro and in vivo theranostic agents. Later, she did a short postdoc at the University of Lübeck, Germany, on Fragment-Based Drug Discovery. In 2010–2012, her research focused on bimodal optical/MRI lanthanide-based lipo-nanoparticle contrast agents, at Centre de Biophysique Moléculaire (CBM), Orléans, France. From 2013 to 2015, she worked as a Research Associate at KCL's Imaging Sciences Division, London, UK, on new PET/MRI targeted probes for cardiovascular diseases. In 2016 she rejoined CBM as a CNRS Researcher. Her current project focuses on multimodal peptide-based contrast agents. |
 Carlos F. G. C. Geraldes | Carlos Geraldes has been Full Professor at the University of Coimbra, Portugal, since 1988, retired and invited Professor since 2018. He received a PhD degree in Inorganic Chemistry at the University of Oxford, UK. He was a Fulbright scholar in the US, is a Fellow of the Lisbon Academy of Sciences and received several scientific prizes. His current research interests include inorganic contrast materials for MRI and molecular imaging. He co-authored 310 articles and 15 book chapters (h factor of 50), which have received over 9000 citations. |
 Éva Tóth | Éva Tóth is an expert in the chemistry of metal complexes for imaging applications. After a PhD from the University of Debrecen, Hungary, she occupied research positions at EPF Lausanne, Switzerland. In 2005, she became CNRS research director in the Centre of Molecular Biophysics, Orléans, France. She published over 180 papers, 16 book chapters and edited The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging (Wiley). In 2019, she became an external member of the Hungarian Academy of Sciences. Her recent work focuses on the design of responsive MRI probes and Mn alternatives to clinical Gd-based contrast agents. |
Introduction
In living systems, proteins can adopt various interconverting conformational states. Among these, amyloids refer to protein aggregates characterized by the presence of β-sheets and a fibrillar morphology. Amyloid formation of proteins has been first associated with a number of pathologies, like Alzheimer's and Parkinson's diseases or type 2 diabetes, involving amyloid Aβ, α-synuclein (α-syn) and islet amyloid polypeptide (IAPP or amylin), respectively. While in these pathological cases, amyloid formation is detrimental to protein function, amyloid structures are also observed under normal physiological conditions and fulfil functional roles in biology.1 Today it is thus accepted that the ability to form amyloid fibrils can be considered as an inherent property of polypeptide chains.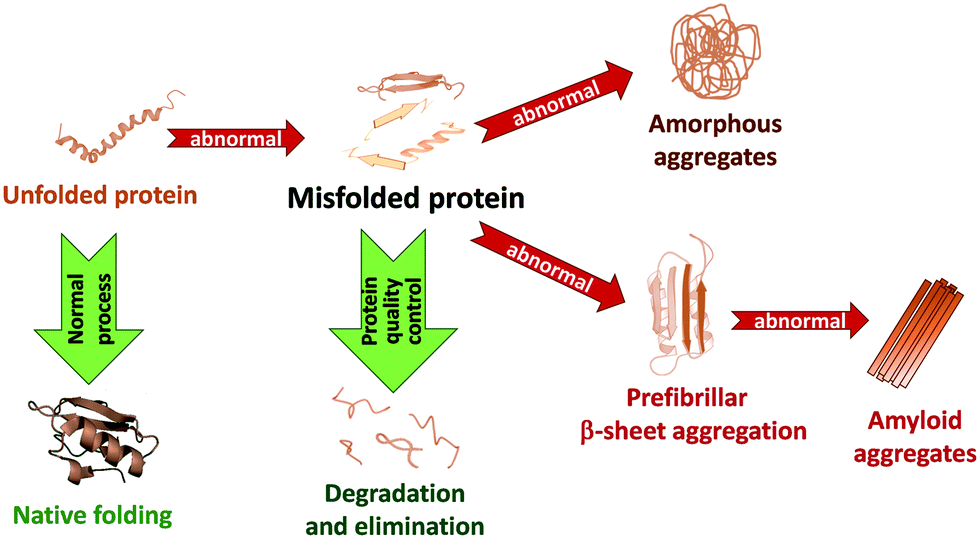 | ||
| Fig. 1 Main processes of amyloidogenesis. | ||
Amyloidogenesis, as depicted in Fig. 1, refers to the process of amyloid formation, when correctly folded proteins unfold or misfold, and in the absence of appropriate recognition of this state by protein quality control and degradation mechanisms, they misassemble into soluble and insoluble toxic oligomer/polymer cross-β-sheet fibrillary aggregates. Natively unfolded or intrinsically disordered proteins can also undergo aggregation. Amyloidogenesis is a multi-step process.1,2 Early aggregates involve a limited number of molecules which still partially retain their original conformation and are kept together by weak intermolecular interactions. The internal reorganization of these small clusters can then lead to larger and more compact aggregates, characterized by extended β-sheets conferring higher stability to them. Their further growth via self-association or monomer addition gives rise to large, ordered, fibrillary structures. In other cases, amorphous or native-like aggregates can form respectively from disordered or native-like assemblies. Monomers, oligomers and fibrils typically coexist in equilibrium. These are further complexified by the possible fragmentation of the already formed fibrils into smaller units which can in turn initiate secondary nucleation.
In amyloid-related pathologies, amyloidogenesis is an important molecular signature of the disease which is directly associated with cell/organ dysfunction. It triggers lethal effects, such as ubiquitin proteasome system failure, lipid membrane permeability, oxidative stress, endoplasmic reticulum stress and mitochondrial dysfunction, leading to cell death. While fibrils are the end product of this process, they do not necessarily represent the most toxic form.
Molecular imaging aims at visualizing molecules, molecular processes and physical–chemical parameters in tissues or cells, in order to assess physiological or pathological phenomena or to diagnose diseases and monitor their evolution. In the context of amyloid-related diseases, much attention has been devoted to the detection of amyloid protein structures. Indeed, as amyloidogenesis occurs before the earliest clinical signs of the diseases, amyloids are generally considered as highly relevant biomarkers. In the past two decades, important efforts have been made to develop molecular imaging agents that specifically recognize amyloid peptides.
The scope of this review is to present the most recent developments in the field of molecular imaging of amyloid-related pathologies. After a short description of relevant amyloid-forming peptides, we will specifically focus on the design of metal-based agents which are highly versatile as they can provide detection capability in different imaging modalities as a function of the metal ion selected. The same chelating ligand can be complexed to different radioisotopes or other metal ions and provide probes detectable in various modalities (e.g.64Cu2+ and 68Ga3+ for positron emission tomography (PET), 111In3+ for single-photon emission tomography (SPECT), Gd3+ for magnetic resonance imaging (MRI), etc.). On the other hand, most of the examples are derived from a few basic amyloid-targeting moieties such as benzothiazole, benzofuran or stilbene, and there are relatively scarce data with other types of recognition units. The majority of the agents target amyloid Aβ in the context of Alzheimer's disease or more recently amylin in diabetes, while very few examples treat other amyloid peptides. Since the chemical design and the nature of the recognition moieties are essentially independent of the peptide targeted, we propose here to survey the literature, from the past six years, by classifying the most representative examples according to the chemical structure of their targeting function. While our review does not intend to be fully exhaustive, we want to illustrate advances specifically in the following areas: (i) improving the affinity to amyloid peptides via structural optimization and understanding how affinity is affected by the solution behaviour of the amphiphilic chelates; (ii) relating organ (brain and pancreas) uptake and clearance to the molecular structure; and (iii) in vivo preclinical validation to achieve best contrast between diseased and healthy tissue.
Amyloids, related human diseases and their detection by molecular imaging
Basic properties of amyloid peptides and proteins and related diseases
Amyloidogenesis of normally soluble proteins has so far been found to be a hallmark of about 30 highly debilitating human diseases. Modern structural biology techniques have contributed to a dramatic increase in detailed structural information on amyloid fibrils. This knowledge helps understand the molecular basis of the diseases and enables the rational design of new, more specific molecular imaging probes. Although amyloid beta (Aβ) peptide and amylin have been the only targets in molecular imaging, hopefully, others will also attract attention in the future.The amyloid precursor protein (APP) is a 100–140 kDa transmembrane glycoprotein that plays a major role in the pathogenesis of Alzheimer's disease (AD), the most common neurodegenerative disease. In healthy individuals, APP cleavage by α-secretase generates a C83 carboxy-terminal fragment and soluble APP, which is associated with normal synaptic transmission. In the diseased state, APP is abnormally cleaved first by β-secretase and then γ-secretase, releasing from the cell membrane amyloid beta (Aβ) peptides containing predominantly 39–42 amino acid residues. The longer Aβ species, particularly Aβ1–40 and Aβ1–42, aggregate into neurotoxic soluble oligomers in the extracellular space. Accumulation of Aβ40/42 inhibits ion channels, impedes calcium homeostasis, and impairs neuronal energy metabolism, eventually leading to neuronal cell death. These oligomers aggregate in turn into larger insoluble β-sheet fibrils (8–10 nm) and ultimately into dense fibrillary amyloid plaques, the main hallmark of AD.3 The deposition of amyloid beta peptide on the walls of small to medium blood vessels of the central nervous system and meninges leads to a form of angiopathy called cerebral amyloid angiopathy (CAA), which is a major factor in intracerebral haemorrhage and vascular cognitive impairment.4
Amylin, or islet amyloid polypeptide (IAPP), is a peptide hormone co-secreted with insulin from pancreatic β-cells in response to food intake. It has an important role in the regulation of glycemia, preventing spikes in blood glucose levels after meals by slowing gastric emptying and promoting satiety. Amylin is produced by post-translational modifications, including protease cleavage, from the 67 amino acid pro-IAPP produced in the pancreatic β-cells. Human IAPP has 37 amino acid residues, with a disulfide bridge between cysteines 2 and 7. IAPP can adopt an α-helical structure at residues 8–18 and 22–27 and can form dimers, which is an early step in fibril formation. IAPP can also form heterodimers with insulin. While dimerization of IAPP accelerates fibril formation, insulin blocks it by blocking the IAPP dimerization interface. The early prefibrillar structures are more toxic to β-cells than the later ones. Amyloid deposition of amylin in pancreatic islets (islet amyloid) is characteristic of type 2 diabetes (T2D), the most common form of diabetes mellitus (about 90%), which is closely associated with number reduction and dysfunction of β-cells.5
The tau (τ) protein is a microtubule-associated, intrinsically disordered protein (IDP) which is mainly expressed in the axons of central nervous system neurons. Tau protein monomers, with 352–441 amino acid residues, have an N-terminal projection domain and a C-terminal microtubule-binding domain, which share a central proline-rich region. In healthy neurons, tau promotes the assembly and the stabilization of microtubules through tubulin-binding motifs in the microtubule-binding domain. Hyperphosphorylated tau protein dissociates from the neuronal microtubules, misfolds and aggregates, forming highly structured amyloid fibrils, which lead to the formation of intracellular insoluble aggregates called neurofibrillary tangles (NFTs). These intracellular NFT aggregates of tau define a group of neurological disorders known as tauopathies, such as AD, but are also present in amyotrophic lateral sclerosis (ALS) and Pick's disease, among others.6
α-Synuclein is a small (140 amino acid residues) soluble protein that normally exists in a natively unfolded state, localized in the nucleus of brain neurons. In Parkinson's disease (PD), the second most common neurodegenerative disorder, abnormal α-syn accumulation occurs in intra- and inter-neuronal inclusions (Lewy bodies and Lewy neurites, respectively), which leads to degeneration of substantia nigra dopaminergic neurons. In PD, the conformation of α-syn changes to α-helical fibrils and oligomers that form β-sheet rich fibrils. Such protein aggregation is the basis for the “misfolded hypothesis” of PD. α-Syn has also been shown to be autoproteolytic, generating a variety of low molecular weight fragments. Those containing the NAC region (non-amyloid β component, residues 61–95) undergo faster aggregation than α-syn and may play a role as intermediates or cofactors in the in vivo aggregation of α-syn. α-Syn aggregates can also migrate from affected to unaffected neurons, causing further misfolding.7
Human Cu–Zn superoxide dismutase (SOD1) is a 16 kDa protein, normally forming a 32 kDa homodimer. The structure of each SOD1 subunit consists of a β-barrel core and seven loops which are held together by an intramolecular disulfide bond, a binuclear Cu2+/Zn2+ metal binding site and a global hydrogen bond network. SOD1 is mainly distributed in the cytoplasm but is also found in the nucleus, lysosomes and mitochondria. Its main function is to dismutate superoxide radicals (O2˙−) into molecular oxygen (O2) and the less reactive hydrogen peroxide (H2O2), thus eliminating the free radicals that cause oxidative stress. Mutant SOD1 proteins, some of which originate from oxidative stress, aggregate more extensively than the normal wild-type (wt) SOD1 protein. Amyotrophic lateral sclerosis (ALS) is a fatal adult-onset neurodegenerative disorder characterized by progressive degeneration of upper motor neurons in the motor cortex and lower motor neurons in the spinal cord and brainstem. Hallmarks of ALS include mutant SOD1 protein aggregates, mainly found in spinal motor neurons. Increased aggregation, dimer destabilization and oligomerization are mechanisms proposed for mutant SOD1 toxicity.8
The TAR DNA-binding protein 43 (transactive response DNA-binding protein 43 kDa, TDP-43) in its full-length form is a dimer formed by interaction between two N-terminal domains, and this dimerisation can be propagated to form higher-order oligomers. Protein mutations located in the glycine-rich region of TDP-43 and their accumulation contribute to the development of ALS. A hyper-phosphorylated, ubiquitinated and cleaved form of TDP-43, known as pathologic TDP-43, mislocalized in the cytoplasm, is also a key pathological feature of other major neurodegenerative diseases, including AD, PD and Huntington's disease (HD).9
Beta-2 microglobulin (B2M) is a 17 kDa ubiquitously expressed protein present on all nucleated cells. It is organised in a β-sandwich fold with seven antiparallel β-strands. pH-dependent aggregation (more extensive at acidic pH) of this protein is often a problem during kidney dialysis. In patients undergoing long-term haemodialysis, B2M can aggregate into amyloid fibers that damage the kidneys' glomeruli, which become unable to filter out B2M, leading to high blood levels and dialysis-related amyloidosis (DRA).10
In its wild-type form, the huntingtin (HTT) protein (348 kDa) contains 6–35 glutamine residues. The isolated protein is believed to be intrinsically disordered. Studies of the HTT-associated protein 40 (HAP40) complex showed that HTT is largely α-helical and consists of three major domains.11 The amino- and carboxy-terminal domains contain multiple HEAT (huntingtin, elongation factor 3, protein phosphatase 2A and lipid kinase TOR) repeats arranged in a solenoid fashion. HTT is essential for embryonic development and is involved in diverse cellular activities such as vesicular transport, endocytosis, autophagy and the regulation of transcription, ensuring normal development before birth. It is expressed in many tissues, with the highest extent in the brain.12 In Huntington's disease (HD), mutated HTT contains more, up to 250, glutamine residues, leading to abnormal neuronal aggregation. Recently, HD has also been associated to tau aggregation.13
The prion protein (PrP) is mostly expressed in the nervous system. Human PrP (PrPC) is possibly involved in Cu2+ transport to cells, in cell signalling or in the formation of synapses. The protein can exist in multiple isoforms, the normal PrPC and misfolded protease-resistant forms, such as the disease-causing PrPSc (scrapie). Normal PrPC has 43% α-helical and 3% β-sheet content, whereas PrPSc possibly has a total β-sheet composition. Brain accumulation of the misfolded PrPSc is associated with cognitive disorders and neurodegenerative diseases such as ovine scrapie, bovine spongiform encephalopathy (BSE, mad cow disease) and human Creutzfeldt–Jakob disease (CJD). The predominant hypothesis for the propagation of PrPSc is that the change from normal PrPC is caused by the presence of and interaction with PrPSc.14
Transthyretin (TTR) is a transport protein found in the serum and cerebrospinal fluid that transports the thyroid hormone thyroxine (T4) and retinol-binding protein bound to retinol. TTR is a homo-tetramer where each monomer is a 127-residue polypeptide rich in β-sheet structures. Two monomers associate via their edge β-strands to form an extended β-sandwich and two of these dimers associate face-to-face to produce the homo-tetrameric structure and create two thyroxine binding sites per tetramer. TTR misfolding and aggregation into amyloid fibrils is associated with the slowly progressing, adult-onset diseases senile systemic amyloidosis (SSA), familial amyloid polyneuropathy (FAP) and familial amyloid cardiomyopathy (FAC). TTR point mutations destabilize the tetramer with mutant and wt-TTR subunits and facilitate misfolding and amyloidogenesis, leading to TTR amyloid deposition in the extracellular space. The severity of the disease varies greatly with the mutation site.15
Molecular imaging modalities for in vivo amyloid detection
Molecular imaging can play a central role in the early in vivo detection of misfolded protein aggregates considered as specific disease biomarkers. This is important for accurate diagnosis, for improved understanding of molecular and cellular mechanisms underlying the diseases and for the monitoring of novel therapeutic approaches. The currently available preclinical and clinical imaging modalities for amyloid detection include magnetic resonance imaging (MRI), positron emission tomography (PET), X-ray computed tomography (CT), single-photon emission CT (SPECT), optical imaging (OI), ultrasound (US) imaging and photoacoustic imaging (PAI). Among these, MRI displays high resolution and has the advantage of being non-invasive and non-ionising, but has poor sensitivity. PET, SPECT and OI, on the other hand, provide high sensitivity. Two or more modalities can be combined in multimodal imaging to potentiate their advantages and overcome their limitations, providing more accurate diagnostics. Modalities with high spatial resolution (MRI/CT) are ideally combined with highly sensitive modalities (OI/nuclear imaging) to complement anatomical with molecular/functional information.16,17 Detailed descriptions of the imaging techniques can be found elsewhere.16,18 Amyloid-targeted probes have been developed for most of these imaging modalities and surveyed in previous reviews by us19 and others.20,21 Among all, PET has been the most widely used technique for both clinical and pre-clinical imaging of amyloid plaques, especially in the framework of AD, mainly thanks to the successful use of 18F- or 11C-labelled probes based on Pittsburgh compound B (PiB) or other small molecules (Scheme 1).20 In addition to their good amyloid binding properties, their small size and lipophilic character are important for sufficient passage via the blood–brain barrier (BBB), which constitutes an important obstacle in brain imaging.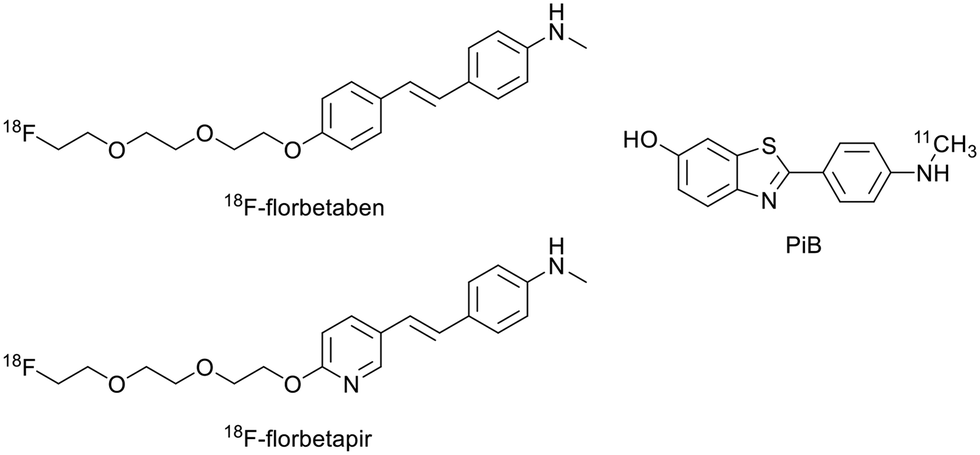 | ||
| Scheme 1 18F- or 11C-labelled PET probes. | ||
Targeted probes for amyloid detection
Probes based on benzothiazole derivatives (Table 1)
Building on the early success of PiB in targeting Aβ plaques, a vast majority of metal-based imaging probes are based on benzothiazole derivatives. Benzothiazole moieties are inspired from thioflavin S and T, well known to bind amyloid deposits. Their structure can be easily modulated.| Ligand number | Molecular weight of ligand | Metal | log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of complex P of complex |
Targeted protein | Affinity to proteina (Ki, nM) | Brain uptake at 2 minb (% ID per g) | Ref. |
|---|---|---|---|---|---|---|---|
| a Metal complexes or * ligand; $Kd. b p = pancreas; AD = murine or rat model of Alzheimer's disease. | |||||||
| 1 | 405.5 | Re/99mTc | 2.69 | Aβ1–42 | 18.8 | 1.06/p: 4.04 | 22 |
| 2 | 433.6 | Re/99mTc | 2.84 | Aβ1–42 | 22.75 | — | 22 |
| 3 | 419.5 | Re/99mTc | 2.99 | Aβ1–42 | 12.39 | 0.70/p: 4.03 | 22 |
| 4 | 447.6 | Re/99mTc | 2.79 | Aβ1–42 | 22.26 | — | 22 |
| 5 | 406.5 | Re/99mTc | 2.81 | Aβ1–42 | 204.10 | — | 22 |
| 6 | 434.5 | Re/99mTc | 2.79 | Aβ1–42 | 42.04 | 0.69/p: 3.77 | 22 |
| 7 | 420.5 | Re/99mTc | 2.81 | Aβ1–42 | 130.63 | — | 22 |
| 8 | 448.6 | Re/99mTc | 2.69 | Aβ1–42 | 15.10 | 0.54/p: 4.11 | 22 |
| 9 | 317.4 | Re/99mTc | 2.35 | Aβ1–42 | 13.6 | 0.53/p: 1.70 | 23 |
| AD: 0.52/p: 1.96 | |||||||
| 10 | 198.3 | 99mTc | 2.52 | Aβ1–42 | 65.8 | 7.94/p: 6.99 | 24 |
| 11 | 494.6 | Re | 0.15 | Aβ1–40 | — | — | 25 |
| 12 | 518.8 | Re/99mTc | 2.88 | Aβ1–42 | 30.9 | 1.57 | 26 |
| 13 | 519.8 | Re/99mTc | 2.48 | Aβ1–42 | 102.6 | 0.92 | 26 |
| 14 | 509.6 | 64Cu | 0.97 | Aβ1–40 | 30*/275 | 0.17 | 28 |
| 15 | 523.7 | 64Cu | 0.72 | Aβ1–40 | 40*/325 | 1.33 | 28 |
| 16 | 567.9 | 64Cu | 0.64 | Aβ1–40 | 320*/2350 | 0.49 | 28 |
| 17 | 779.0 | 64Cu | 0.82 | Aβ1–40 | 580*/142 | 0.61 | 28 |
| 18 | 426.6 | 64Cu | 0.92 | Aβ1–40 | 170*/795 | 0.75 | 28 |
| 19 | 439.6 | 64Cu | 1.30 | Aβ1–42 | — | 0.99 | 29 |
| 20 | 439.6 | 64Cu | 1.08 | Aβ1–42 | — | — | 29 |
| 21 | 425.6 | 64Cu | 1.29 | Aβ1–42 | — | 0.16 | 29 |
| 22 | 440.6 | 64Cu | 0.58 | Aβ1–42 | — | — | 29 |
| 23 | 426.6 | 64Cu | 0.56 | Aβ1–42 | — | — | 29 |
| 24 | 679.9 | 64Cu | 1.09 | Aβ1–42 | — | — | 29 |
| 25 | 526.7 | 64Cu | 1.26 | Aβ | — | 0.35 | 30 |
| 26 | 512.7 | 64Cu | 1.07 | Aβ | — | 0.23 | 30 |
| 27 | 498.6 | 64Cu | 1.01 | Aβ | — | 0.32 | 30 |
| 28 | 484.6 | 64Cu | 0.91 | Aβ | — | 0.46 | 30 |
| 29 | 470.6 | 64Cu | 1.15 | Aβ | — | 0.23 | 30 |
| 30 | 642.7 | Gd/111In | −0.15 | Aβ1–40/amylin | 1.8 × 105$/1.54 × 105$ | 0.86 | 31 |
| 31 | 755.9 | Gd/111In | 0.03 | Aβ1–40/amylin | 6.7 × 104$/8.3 × 104$ | — | 31 |
| 32 | 714.8 | Gd | — | Aβ1–40 | 1.94 × 105$/— | — | 31 |
| 33 | 761.9 | Gd | 0.32 | Aβ1–40 | — | — | 31 |
| 34 | 755.9 | Gd/111In | 0.63 | Aβ1–40/amylin | 5.1 × 103$/3.7 × 103$ | 0.3/p: 2.9 | 34 |
| 35 | 812.0 | Gd | 1.46 | Aβ1–40/amylin | 4.4$/4.5$ | 0.3/p: 3.7 | 34 |
A first class of benzothiazole-based probes involves cyclopentadienyl Re/99mTc tricarbonyl complexes. Their low molecular weight, small size and high stability are interesting features for BBB crossing and thus brain imaging, but these complexes present only moderate affinities to Aβ plaques.
In 2016, J. Jia et al. synthetized a series of 99mTc/Re-labeled cyclopentadienyl tricarbonyl derivatives linked to 2-phenyl/pyridylbenzothiazole via an ester function and an alkyl chain (ML1–8, Scheme 2).22 In these structures, the ester function was described to facilitate BBB crossing, as compared to an amide, by limiting hydrogen bonds. The lipophilicity could be modulated through the alkyl chain length and the pyridyl/phenyl substitution. Surprisingly, these structural modifications did not prove to have a major impact on the lipophilicity, since all the 99mTc complexes had logPoct/PBS between 2.69 and 2.99, but showed different affinities to Aβ1–42 aggregates. The affinity of the phenylbenzothiazole derivatives ML1–4 was higher (in the 12–25 nM range) than those of the pyridylbenzothiazole analogues ML5–7 (in the 42–204 nM range), except in the case of the pentyloxy linker with the N,N-dimethylated form ML8 (15 nM) (Table 1). The general tendency indicated that higher affinity was achieved with the longest linker and the phenyl group. Unfortunately, in vivo biodistribution in normal mice demonstrated that despite their small size, these probes were not able to cross effectively the BBB.
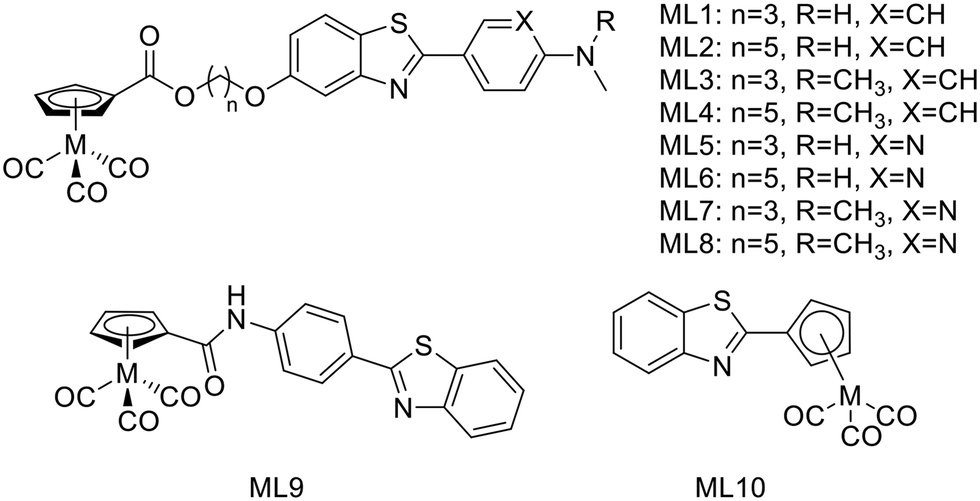 | ||
| Scheme 2 99mTc/Re-labeled cyclopentadienyl tricarbonyl derivatives linked to benzothiazole derivatives. | ||
Kiritsis et al. also designed a 99mTc cyclopentadienyl complex (99mTcL9) for SPECT imaging of Aβ plaques (Scheme 2).23 The complexing unit was directly linked to a 2-(4′-aminophenyl)-benzothiazole through an amide function, reducing the molecular size compared to those of Jia et al. These complexes (99mTc and its Re analogue) demonstrated suitable lipophilicity for BBB crossing (logPoct/water = 2.35) and also bound Aβ1–42 aggregates with high affinity (Ki = 13.6 nM). Despite these properties, the brain uptake measured in biodistribution studies in normal mice was moderate (0.53% ID per g at 2 min) with a clearance ratio 2/90 min of 2.1. In 7 month old transgenic mice (5xFAD), even if the brain uptake at 2 min was the same, a significant increase (1.94% ID per g) was observed at 90 min due to the presence of β-amyloid plaques, thus probe retention.
In 2019, the same group developed 99mTcL10 wherein the cyclopentadienyl complexing unit was directly linked to the benzothiazole part (Scheme 2).24 This radiocomplex presented a high stability in biological medium for 90 min and possessed improved properties for BBB crossing compared to 99mTcL9: a smaller size and a higher lipophilicity (logPoct/water = 2.52). It was confirmed in biodistribution studies in healthy mice by a very high brain uptake at 2 min (7.94% ID per g), respectively similar to and higher than that for the clinically used PET agents 18F-florbetapir and 18F-florbetaben.20 Besides, the brain clearance ratio 2/90 min of 39.7 indicated no unspecific retention in brain tissues, which is an essential condition for an AD imaging probe. The authors also did biodistribution studies in transgenic 5xFAD mice and compared the results at 15 min to those obtained for wild-type animals. They noted an increase of the radioactivity detected (3.90% ID per g vs. 2.68% ID per g for the wild-type mice), attributed to the presence of Aβ plaques, which was confirmed by fluorescent staining with thioflavin S. The rhenium analogue bound selectively plaques with a good affinity (Ki = 65.8 nM) and also exhibited theranostic properties such as inhibition of Aβ fibril formation and antioxidant activity.
Two other 99mTc complexes have been recently described with an N-heterocyclic carbene (NHC) (99mTcL11) or bis(aminoethanethiol) (BAT) (99mTcL12–13) scaffold by Wiratpruk et al.25 and Zhang et al.,26 respectively (Scheme 3). The first series of highly stable Re(CO)3 complexes of bifunctional bis(NHC)-amine ligands coupled to benzothiazole had been synthetized before, with promising results of selective binding to amyloid plaques from human brain tissues, but potentially low BBB permeability because of their positive charge.27 In the following work, the molecular design was improved by introducing a carboxylate function to form a neutral complex. However, the rhenium complex ReL11 became too amphiphilic (logPoct/water = 0.15), which was not optimal for brain penetration and also reduced amyloid plaque binding in AD patient brain slices. This ligand system provides a good basis for further improvements, such as the replacement of the NHC methyl substituent by a more lipophilic tert-butyl or cyclohexyl group.
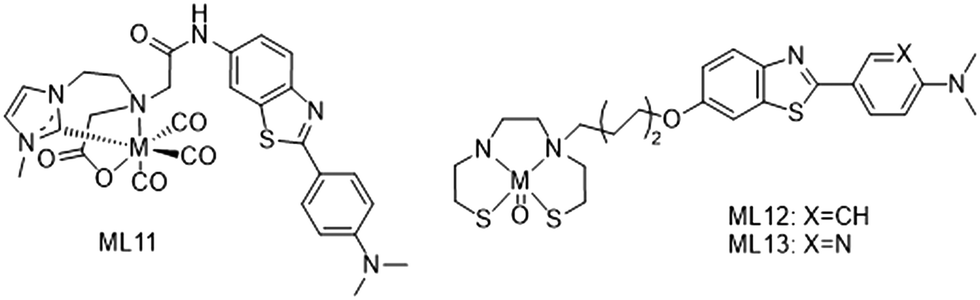 | ||
| Scheme 3 99mTc complexes with an N-heterocyclic carbene (99mTcL11) or bis(aminoethanethiol) (99mTcL12–13) scaffold. | ||
Zhang et al. linked the BAT chelator to a 2-phenyl/pyridylbenzothiazole through an oligoethyleneoxy spacer. The introduction of this pegylated moiety, instead of the alkyl chain in previously described analogues, was expected to reduce the lipophilicity of the Re/99mTc complexes and improve brain pharmacokinetics, namely the initial brain uptake and the clearance ratio from healthy brain. Both ReL12 and ReL13 presented specific and efficient binding to Aβ plaques from AD patients, and as observed by Jia et al.,22 the presence of the phenyl in ReL12 instead of the pyridyl group in ReL13 led to greater affinity to Aβ1–42 aggregates, with Ki = 30.9 nM and Ki = 102.6 nM, respectively. Biodistribution data in normal mice confirmed the benefit of the pegylated linker. Indeed, they showed increased initial brain uptake at 2 min as compared to the alkyl analogues as well as a good 2/60 min clearance ratio.
Among benzothiazole derivatives, a second class of β-amyloid imaging probes is composed of macrocyclic polyamine ligands. These chelators have the advantage of forming highly stable and kinetically inert complexes with various metals such as copper(II), gallium(III) or lanthanides(III), and can be easily functionalized to introduce an amyloid targeting unit.
For 64Cu coordination, in 2017, Bandara et al. proposed a series of five new ligands L14–18, wherein a 2-(4-hydroxy)-3-methoxyphenyl-benzothiazole is linked to 2,4-dimethyl-1,4,7-triazacyclononane (TACN) or 2,11-diaza[3.3](2,6)-pyridinophane derivatives (Scheme 4).28 The targeting unit was directly linked to the macrocycles through a Mannich reaction, and for the tetraamine derivatives, lipophilicity and charge were modulated via the R substituent (L14–16) or by adding a second benzothiazole unit (L17). The interactions between the ligands or the Cu complexes and the Aβ aggregates were studied in vitro and did not show dramatic variation upon metal complexation: all affinities remained in the nanomolar range, even if the Cu complexes of L14–16 and L18 exhibited 4–8 times lower affinities as compared to the corresponding ligand. The affinity decrease was the most visible for CuL16 that showed micromolar affinity (2.33 μM) due to the less positive charge of the metal complex via the deprotonation of the carboxylic acid function. For CuL17, an opposite behavior was observed. The 4 times higher affinity was attributed to a rearrangement of the two targeting units upon copper(II) coordination to allow for better interaction with the Aβ fibrils. Ex vivo experiments on brain slices of AD transgenic mice confirmed the specific binding of these probes to Aβ fibrils. Despite the logPoct/water values inferior to 1, the complexes, in particular CuL14, were able to cross the BBB, exhibited promising initial brain uptake and rapid clearance in biodistribution studies in wild-type mice. MicroPET imaging studies on transgenic mice also confirmed the promising properties of these compounds as diagnostic tools for AD, even if their affinity and lipophilicity might be further improved.
 | ||
| Scheme 4 Benzothiazole derivatives linked to macrocyclic ligands (2,11-diaza[3.3](2,6)-pyridinophane or TACN scaffolds) for 64Cu coordination. | ||
In a follow-up work published in 2020, the same authors reported on another series of benzothiazole derivatives L19–24 as theranostic agents, wherein the targeting unit has been modified (Scheme 4).29 They used PiB derivatives with a mono- or a dimethylamine group and introduced an additional hydroxyl group on the benzothiazole part. The replacement of the phenyl by a pyridyl group and the introduction of a hydrogen bond acceptor were also studied. These units were thus linked to methylated TACN macrocycle via the benzothiazole aromatic ring. These structural variations allowed for several conclusions. First, the affinity studies outlined the ability of all compounds to efficiently bind Aβ aggregates, but the monomethylated derivatives had higher specificity over the dimethylated analogues as well as reduced cytotoxicity. The pyridine derivative L22 and L23 ligands presented lower affinity but had improved antioxidant properties. Interestingly, L23 was able to strongly reduce neurotoxicity by accelerating the aggregation of toxic Aβ1–42 oligomers into non-toxic Aβ1–42 aggregates. All these ligands could be radiolabeled with 64Cu for PET imaging, but among all complexes, only CuL19–21 and CuL24 showed suitable lipophilicity and amyloid binding in transgenic mice brain sections. CuL20 had poorer amyloid binding specificity. In the case of CuL24, the second benzothiazole unit did not improve the binding properties, and the reduction of lipophilicity and the increase of the molecular weight were not favorable for BBB crossing. CuL19 and CuL21 were finally included in an in vivo biodistribution study in normal mice and showed very different behavior. The initial brain uptake was good for CuL19 (0.99% ID per g at 2 min) but much lower for CuL21 (0.16% ID per g at 2 min). The authors related this to the different position of the hydroxyl group on the benzothiazole ring (position 4 vs. 5). Overall, these probes were considered as very promising for combined therapy and PET imaging in Alzheimer's disease. Fig. 2 illustrates the autoradiography of the uptake of ML19–21 and ML24 in WT and AD transgenic mice and in transgenic mice in the presence of a known Aβ blocking agent.
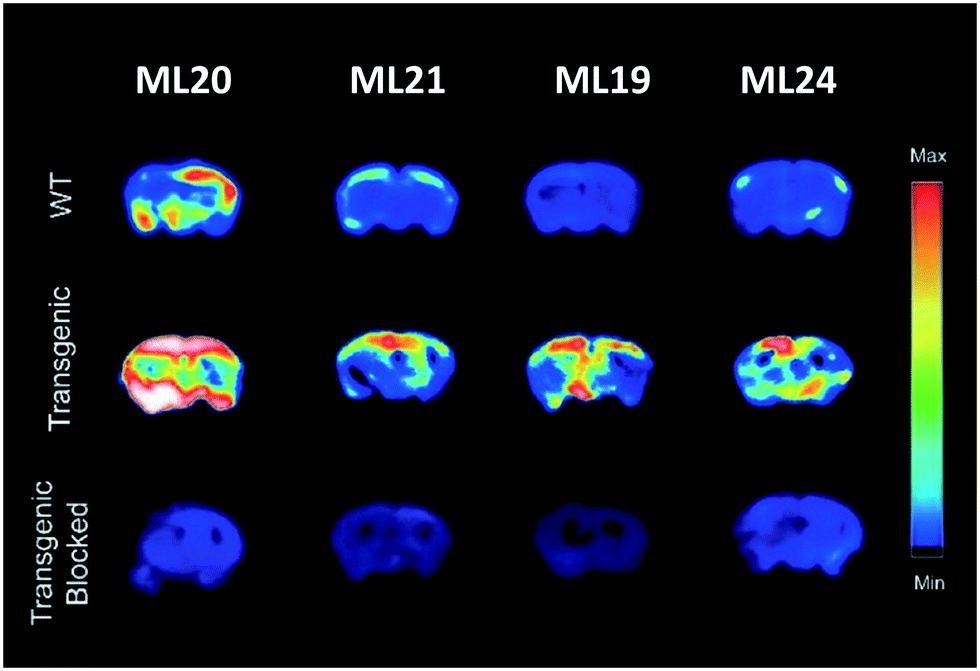 | ||
| Fig. 2 Autoradiography of brains from WT and AD transgenic mice upon injection of ML19–21 and ML24, in the absence and presence of a known Aβ blocking agent. Reproduced from ref. 29 with permission from RSC. Copyright 2020. | ||
Lately, the same group published a novel series of potential 64Cu PET imaging probes derived from L18 by substituting one methyl group on the TACN macrocycle with an acetic acid (L29) or with various alkyl carboxylate esters (L25–28) (Scheme 4).30 These structural modifications allowed for increased stability of the Cu2+ complex without affecting the amyloid binding properties. Indeed, CuL25 and CuL28 presented remarkable affinities and high specificity for amyloid plaques. The lipophilicity of these 64Cu complexes was in the same range as those of the previous compounds (logPoct/PBS between 0.9 and 1.3), while the biodistribution studies showed moderate initial brain uptake (0.23–0.46% ID per g) and rapid clearance for all complexes, indicating that free copper(II) was not released in the brain.
For a decade, our group has been developing metal complexes as potential imaging agents for amyloid Aβ or amylin detection. These probes consist of 3 parts: (i) a macrocyclic chelator based on DOTA providing high affinity for lanthanides and strong kinetic inertness, (ii) a PiB derivative as the targeting unit and (iii) a neutral spacer. Such ligands can be suitable for different imaging techniques by complexing the appropriate metallic ion (Gd3+ for MRI, In3+ for SPECT and Ga3+ for PET).31–34
We modulated the length and the nature of the linker, the orientation of the PiB unit and the global charge of the Ln3+ complexes by using DO3A monoamide or DOTAGA derivatives (Scheme 5). The first generation of these complexes (L30–33) presented an affinity to Aβ1–40 aggregates of Kd = 67–194 μM, determined by SPR. The slightly weaker affinity of LnL32 indicated that a negative global charge is less favorable for binding. The presence of the metal complex drastically deteriorated the binding properties of the PiB unit, since the dissociation constants were five orders of magnitude lower than for the PiB itself.
 | ||
| Scheme 5 PiB-conjugated DO3A monoamide or DOTAGA derivatives for Ln3+ and 111In3+ complexation. | ||
The influence of these first-generation probes on Aβ1–40 aggregation was studied by several techniques (UV-vis spectroscopy, circular dichroism (CD), dynamic light scattering (DLS) and transmission electron microscopy (TEM)). We could conclude that even small structural variations between LnL30, LnL31 and LnL32 were important, as they could either promote (LnL30 and LnL32) or inhibit Aβ1–40 fibril formation (LnL31). Regardless of the low lipophilicities, the initial brain uptake (0.36 and 0.5% ID per g in the cortex and in the cerebellum at 2 min) and the rapid clearance observed for 111InL30 in adult male WT mice were quite promising for potential nuclear imaging.
Despite these interesting initial results, the lipophilicity of the probes for better BBB permeability as well as their amyloid binding affinity needed improvement. We therefore developed a second generation of ligands wherein we changed the orientation of the PiB unit and used longer alkyl spacers (with six or ten carbons). To further increase the lipophilic character, we removed an amide function and dimethylated the amine in the PiB moiety (Scheme 5).
The first study using 111InL31 and 111InL34 complexes confirmed the gain in lipophilicity.33 The neutral complex GdL34 exhibits a more favorable logPoct/water of 0.63 but showed similarly low brain uptake to 111InL31 and 111InL34 (0.07 vs. 0.12% ID per g at 30 min post injection) in healthy mice. The increased molecular weight resulting from the longer alkyl chains was likely a limiting parameter for the BBB crossing.
Brain delivery of our Gd-complexes could be improved via conjugation to nanocarriers capable of translocating across the BBB – either per se or upon targeting to transporters expressed in the brain vasculature. Previous in vitro and in vivo proof-of-concept studies demonstrated that functionalized multi-walled carbon nanotubes (f-MWNTs) were capable of crossing an intact BBB. A significant improvement of brain accumulation was observed for 111InL31 conjugated to f-MWNTs-NH3+, with a whole-brain uptake of ∼1.16% ID per g of tissue, corresponding to a ∼10-fold increase compared to the free chelate.
Due to their large size and the hydrophilic character of the chelate part, metal complexes conjugated to amyloid targeting units exhibit poor brain penetration, which prevents their efficient use for MRI imaging of Alzheimer's disease. This is especially true given the low sensitivity of MRI detection which requires orders of magnitude higher probe concentrations than nuclear imaging.
Therefore, it seems important to envisage more accessible biomarkers. Amylin deposits are localized in pancreatic islets, where Gd3+ complexes could reach the target more easily and in larger quantities. In this regard, we have recently investigated the binding properties of GdL30–31 and GdL34–35 towards amylin in comparison with Aβ1–40 aggregates.34 Their binding affinities were characterized by SPR with aggregated Aβ1–40 and amylin. With a standard level of peptide immobilization on the SPR chips (∼4000 RU), favorable to minimize steric effects and non-specific interactions, the dissociation constants of each complex were in the μM range. No peptide selectivity was observed for any of the complexes; the Kd values were similar for Aβ1–40 and amylin fibrils. The results showed a clear tendency with respect to structural modifications of the ligand: as expected, the affinity becomes higher when the PiB unit is farther away from the metal complex. This was further confirmed by SPR using very high peptide immobilization levels (∼9000 RU) which can allow for better SPR sensitivity and the assessment of interactions at very low GdL concentrations. Under these conditions, an additional interaction in the nM concentration range was identified for GdL35 (Kd = 4.4 nM and 4.5 nM for Aβ1–40 and amylin, respectively). This finding clearly indicated the importance of a sufficiently long and hydrophobic spacer (C10) between the chelate and the targeting unit in order to achieve an affinity similar to that of the PiB unit itself.
The influence of GdL31 and GdL34–35 on the Aβ1–40 aggregation was investigated using thioflavin-T (Th-T) fluorescence measurements. Th-T is a fluorescent probe with a “turn on” response when it intercalates in β-sheets of amyloid fibrils. According to the orientation of the PiB unit, the Gd3+ complexes can either accelerate or slow down the Aβ1–40 aggregation process. GdL34 and GdL35 had a higher impact since they were able to delay the peptide aggregation. The decrease in the Th-T fluorescence signal intensity observed in the presence of the complexes indicated a competition for a common binding site. This phenomenon was more obvious for GdL35 and might result from a stronger interaction of the dimethylated aniline part with β-sheets.
Another major conclusion of this work was that the interactions of the Gd3+ complexes with the peptide fibrils are concentration-dependent. Indeed, these compounds, composed of a rather hydrophilic metal-coordinating unit and a hydrophobic amyloid-targeting moiety, are amphiphilic. Depending on the concentration and their environment, they tend to form micelle-like aggregates in which the targeting unit can be more or less accessible. The critical micellar concentrations (cmc) from which GdLs form micelles were previously determined for the paramagnetic complexes GdL30–32 (in the 0.5–1.5 mM range) by a relaxometric method, exploiting the change of motional dynamics and thus the relaxation rate between the monomeric and micellar forms. This method was not applicable for GdL34–35 because these complexes are already in a micellar state at lower concentrations. The optical properties of the PiB unit were thus used to measure the cmc of GdL34 and GdL35 (30 and 5 μM, respectively) as well as a pre-micellar concentration for GdL30 and GdL31 (53 and 13 μM, respectively). These data confirmed that different micellization regimes can exist in aqueous solutions depending on GdL concentrations.
These micellization regimes could be then related to the amyloid binding properties of the complexes. Scatchard linearization of the SPR data evidenced different interaction regimes and this was independent of the nature of the peptide (Fig. 3). For each system, these regimes were defined by straight lines with well-defined breakpoints which corresponded well to the cmc or pre-cmc values of the given Gd3+ complex as determined above. All these findings seem to indicate that the different aggregated forms of the GdL complexes interact with different affinities with the amyloid peptides.
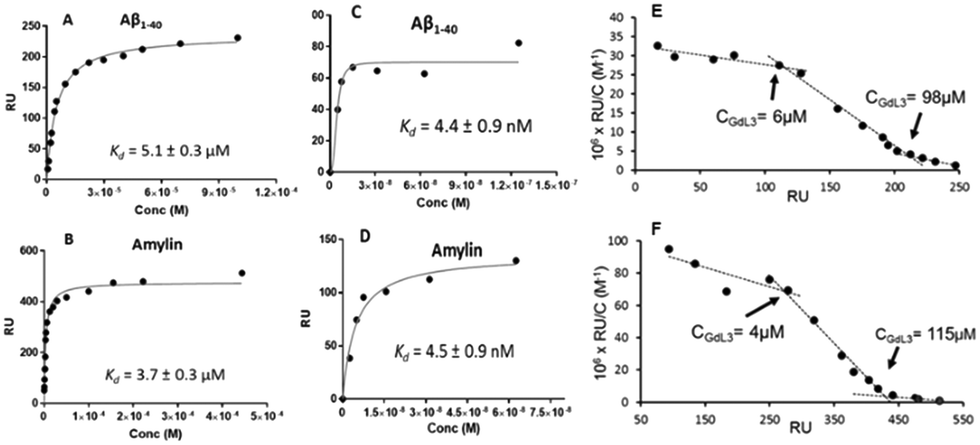 | ||
| Fig. 3 SPR binding plots fitted as Langmuir isotherm functions for the interaction of GdL35 with Aβ1–40 (A and C) and amylin (B and D). Peptide immobilization was 4000 RU (A and B) and 9000 RU (C and D). For C and D, points recorded only at low concentrations are represented. E and F are respectively the Scatchard linearization plots of A and B. | ||
To characterize their biological behavior, radiolabeled 111InL34 and 111InL35 were injected to wild-type mice. As expected, the radiocomplexes had low initial brain uptake, at the same level as that of 111InL30, but fast clearance and no specific organ retention. Nevertheless, the pancreas uptake at 2 min was significant with 2.9% ID per g and 3.7% ID per g, respectively, for 111InL34 and 111InL35. These values are promising for the perspective of amylin imaging, amylin being overexpressed in diabetic animals.
Overall, all these analyses revealed a large complexity of the systems with concentration-dependent binding affinities, which should be always kept in mind when interactions between amphiphilic metal complexes and amyloid peptides are considered.
Probes based on benzofuran derivatives (Table 2)
Benzofuran and benzothiazole are isosteric structures. With respect to benzothiazole, benzofuran has the advantage of easier preparation, since the key step of ring formation can be realized from readily available and less expensive chemicals.35 It can also be linked to aryl, pyridyl or furanyl groups to enhance amyloid binding properties.| Ligand number | Molecular weight of ligand | Metal | logP of complex |
Targeted protein | Affinity to proteina (Ki, nM) | Brain uptake at 2 minb (% ID per g) | Ref. |
|---|---|---|---|---|---|---|---|
| a Metal complexes. b p = pancreas; AD = murine or rat model of Alzheimer's disease. | |||||||
| 36 | 329.4 | Re/99mTc | — | Aβ1–42 | — | — | 36 |
| 37 | 386.5 | Re/99mTc | — | Aβ1–42 | — | — | 36 |
| 38 | 315.4 | Re/99mTc | — | Aβ1–42 | — | — | 36 |
| 39 | 372.5 | Re/99mTc | — | Aβ1–42 | — | — | 36 |
| 40 | 394.5 | 64Cu | 1.84 | Aβ1–42 | — | 1.39 | 37 |
| 41 | 408.5 | 64Cu | 1.81 | Aβ1–42 | — | 1.06 | 37 |
| 42 | 422.5 | 64Cu | 1.88 | Aβ1–42 | — | 0.77 | 37 |
| 43 | 436.5 | 64Cu | 1.48 | Aβ1–42 | — | 1.54 | 37 |
| 44 | 449.6 | 64Cu | −0.74 | Aβ1–42 | 33.7 | 0.33/p: 1.66 | 39 |
| 45 | 623.8 | 64Cu | −0.73 | Aβ1–42 | 243.5 | 0.36/p: 3.32 | 39 |
| 46 | 501.5 | Re/99mTc | 1.74 | Amylin | 146 | 0.08/p: 0.74 | 41 |
| 47 | 567.7 | Re/99mTc | 2.18 | Amylin | 3430 | 0.19/p: 1.37 | 41 |
| 48 | 683.8 | 67Ga | — | Amylin | 3183 | 0.35/p: 4.11 | 42 |
| 49 | 572.6 | 64Cu | −1.02 | Aβ | — | — | 44 |
| 50 | 600.7 | 64Cu | −0.37 | Aβ | — | — | 44 |
| 51 | 628.7 | 64Cu | 0.45 | Aβ | — | — | 44 |
| 52 | 616.7 | 64Cu | −0.93 | Aβ | — | — | 44 |
| 53 | 837.9 | 64Cu | 1.17 | Aβ | — | 0.65 | 44 |
| 54 | 894.0 | 64Cu | 1.32 | Aβ | — | 0.76 | 44 |
| 55 | 950.1 | 64Cu | 1.77 | Aβ | — | 0.38 | 44 |
| 56 | 926.0 | 64Cu | 1.02 | Aβ | — | 2.34 | 44 |
In 2018, Donnelly's group reported new technetium complexes for diagnostic SPECT imaging of cerebral amyloid angiopathy as well as their rhenium analogues (Scheme 6).36 In the complexes, the metal was coordinated by two pyridylthiosemicarbazide units, which were each conjugated to a functionalized benzofuran group (L36–39). To increase aqueous solubility, for ligands L37 and L39, the methyl substituent in the R2 position was replaced by an N,N-dimethylaminoethyl functional group.
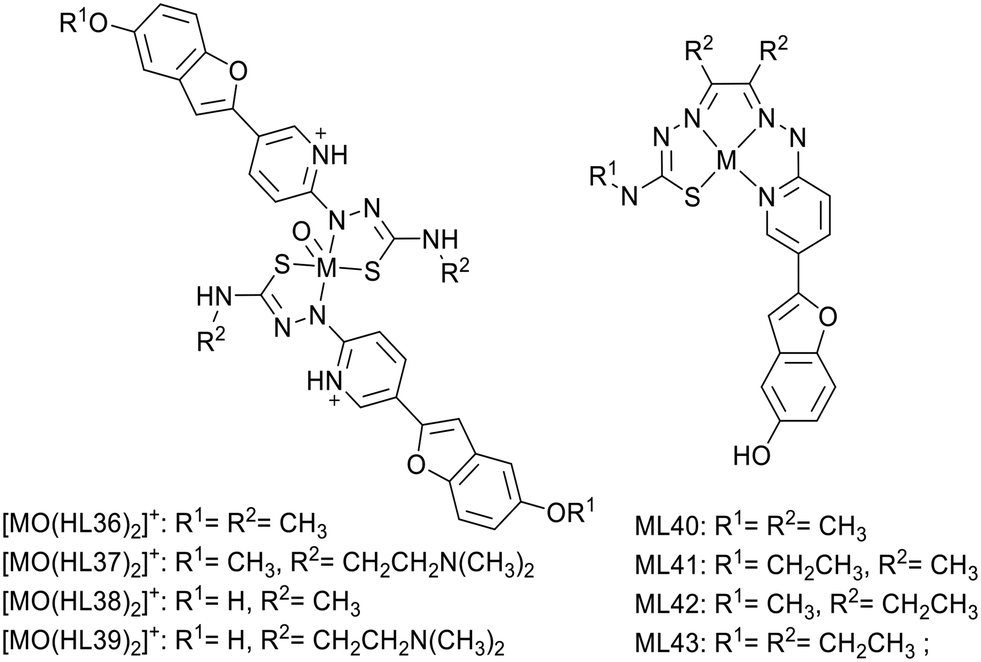 | ||
| Scheme 6 Pyridyl-benzofuran-derivative technetium complexes reported for diagnostic SPECT imaging of cerebral amyloid angiopathy and copper(II)+ complexes containing a coordinating pyridyl-benzofuran unit for PET applications. | ||
The interaction with synthetic Aβ1–42 was investigated using the well-established Th-T fluorescent probe as a competitor. The rhenium complex [ReO(HL36)2]+ induced a modest decrease of the Th-T fluorescence signal, considered as not significant, whereas the other complexes produced a reduction of >80%. The most important interaction was observed with [ReO(HL37)2]+ and [ReO(HL39)2]+ bearing the N,N-dimethylaminoethyl group. The authors did not go further with these complexes because the weak benzofuran fluorescence prevented tests on post-mortem brain tissues.
In 2020, the same group reported analogous ligands L40–43 containing the identical pyridyl-benzofuran as a targeting unit and a thiosemicarbazone–pyridylhydrazone structure for copper-64 complexation, thus potential PET application (Scheme 6).37 The ligand design was inspired by previous work where a styrylpyridyl unit was used for amyloid binding.38 Here, the choice of a pyridyl benzofuran group was justified by less non-specific binding as compared to styrylpyridine- or benzothiazole-based ligands. Further, to minimize the molecular weight, the pyridyl part also serves for metal coordination, and to modulate the in vivo behavior and Aβ interactions, methyl or ethyl groups were introduced to the ligand backbone.
The Cu2+ complexes were stable with respect to metal dissociation (Kd (7.4) ∼ 10−18 M) and resistant to reduction but had different metabolic stabilities. This was tested by incubating the Cu2+ complexes with human or murine liver microsomes which contain cytochrome P450, the major enzyme involved in drug metabolism. CuL43, which bears 3 ethyl groups, exhibited the longest half-life, 7–8 times longer than that of CuL40 which has 3 methyl groups (29 vs. 4 min in human liver microsomes). CuL41 and CuL42 with 1 or 2 ethyl groups presented intermediate and almost similar stabilities (12 and 11 min half-life, respectively). These results nicely indicated how subtle modifications on the ligand backbone imply metabolic stability alterations of the Cu2+ complexes.
All the 64Cu complexes displayed suitable lipophilicity to cross the BBB, and their brain delivery was assessed in wild-type mice. 64CuL40 and 64CuL43 possessed an initial brain uptake of 1.39 and 1.54% ID per g, respectively, but 64CuL43 was more promising because of its faster clearance (2/30 min ratio of 2.0 vs. 1.3), which is a required feature in healthy mice without amyloid plaques. Its interaction with synthetic Aβ amyloid peptide was thus studied first by TEM, resulting in a dramatic change in the morphology of the amyloid fibrils. Analysis of post mortem human AD brain tissue demonstrated that CuL43 preferentially binds to brain areas identified as being rich in Aβ plaques (measured by immunohistochemistry in the contiguous section of brain tissue).
H. Watanabe and M. Ono have developed 2 macrocyclic ligands where either a cyclen (L44) or a DO3A (L45) is conjugated to a phenylbenzofuran group through a propyl linker (Scheme 7).39 These macrocycles are known to form stable Cu2+ complexes and thus the corresponding 64Cu2+ chelates have been investigated as PET imaging probes for Aβ fibril detection.
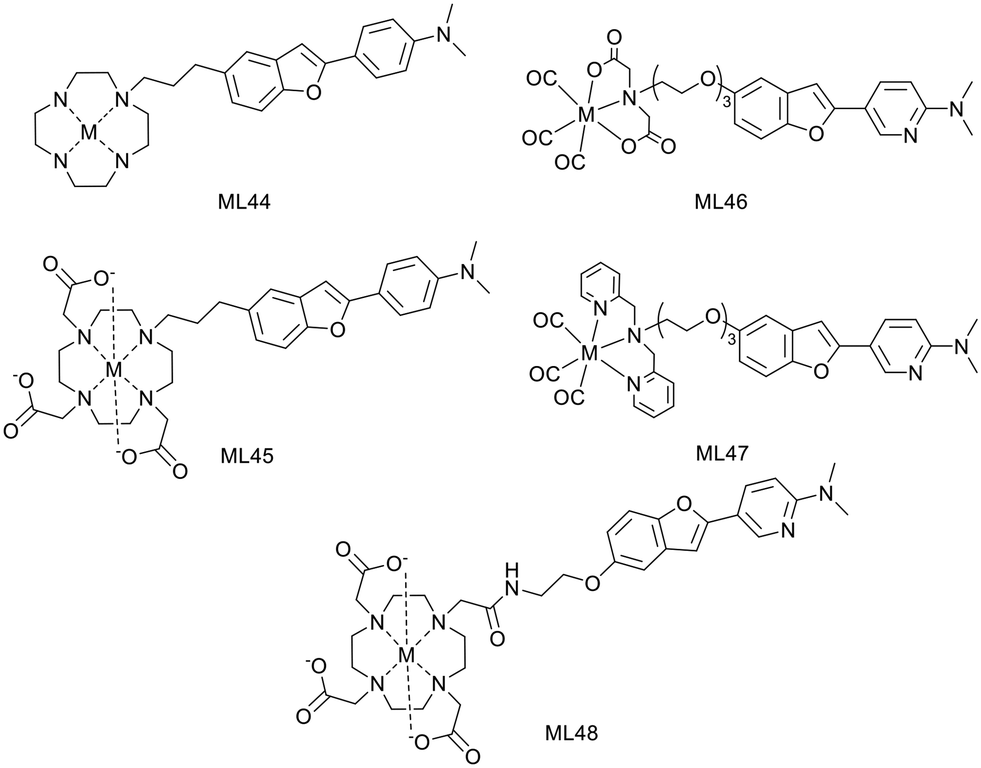 | ||
| Scheme 7 Metal-based probes containing a phenyl or a pyridyl-benzofuran unit. | ||
In vitro competitive assays were carried out with the standard radiotracer competitor [125I]IMPY to determine amyloid binding affinities of the cold Cu2+ complexes. CuL44 and CuL45 displayed inhibition constants of 33.7 and 243.5 nM, respectively, which were higher than that for other phenylbenzofuran conjugated probes (10.8–24.4 nM) previously reported by the authors or for IMPY itself (14.8 nM).35,40 The significant difference between the two complexes was related to the bulkier structure of DO3A and to their different charge. While CuL45 is anionic, CuL44 is cationic and had stronger interaction with the anionic Aβ peptide.
Both complexes remained stable in mouse plasma for 1 hour and stained Aβ plaques in brain tissue sections from Tg2576 mice, even if the behavior of CuL44 was closer to that of the Aβ staining dye Th-S. Biodistribution in normal mice revealed a low initial brain uptake at 2 min for both complexes (0.33–0.36% ID per g), which could be due to the steric bulkiness and the hydrophilic nature of the probes (logPoct/water = −0.74 and −0.73). Interestingly, a higher uptake was observed in pancreas, especially for CuL45 (3.32% ID per g), which also exhibited a good clearance. These results suggest that CuL44 and CuL45 do not have the best properties for brain imaging but may be suitable for amylin detection or for cerebral amyloid angiopathy imaging.
H. Watanabe and M. Ono reported three metal-based probes containing a pyridylbenzofuran (PBF) derivative to detect amylin by SPECT imaging. PBF was described to show high affinity for amylin aggregates (Scheme 7). The first report in 2016 proposed two technetium complexes where the pyridylbenzofuran is conjugated, via a triethylene glycol linker, to an iminodiacetic acid (IDA, L46) or to N,N-dipicolylamine (DPA, L47).41 Later, the same targeting unit was linked to 67GaDO3A through a propylacetamide coordinating spacer (L48).42
In vitro competitive inhibition assays were performed with rhenium analogues and the cold Ga3+ complex, using the 125I-labeled pyridylbenzofuran derivative [125I]IPBF, which demonstrated high binding affinity for amylin (Kd = 8.3 nM). While ReL46 displayed moderate affinity (Ki = 146 nM), ReL47 and GaL48 presented low affinities (Ki = 3430 and 3183 nM, respectively). The difference between the two rhenium complexes was attributed to their different size and charge. The steric hindrance of the bulkier DPA unit may decrease affinity. IDA forms negatively charged complexes with 99mTc, while DPA complexes are positively charged.43 This may also affect the affinity for amylin aggregates since this peptide displays a positive charge. Concerning GaL48, the bulky DOTA linked via a short alkyl chain to PBF could cause steric hindrance; a longer linker may improve the amylin binding properties.
In vitro autoradiography on healthy and T2D human pancreas sections confirmed the amylin binding properties of the 99mTc-labeled probes. 99mTcL46 specifically binds to islet amyloid in T2D pancreatic tissue section, with little nonspecific accumulation, while 99mTcL47 does not show specific binding to amylin. Despite its high inhibition constant, 67GaL48 displayed specific binding to amylin aggregates on T2D pancreas, which was explained by possible differences in the structure of amylin aggregates formed in vitro and in vivo.
It is important to note that the same authors also developed a novel, easy to prepare and ready to use islet amyloid mouse model in which human amylin aggregates are directly transplanted in the pancreas. Ex vivo autoradiography experiments with this model corroborated the poor amylin binding ability of 99mTcL47 and the good labelling potential of 99mTcL46 and 67GaL48. In contrast to the previous in vitro experiments, some nonspecific binding was identified for both complexes, suggesting their more important retention in normal tissue in the living pancreas than in tissue sections.
Biodistribution studies in normal mice indicated different pancreatic uptake for 99mTcL46 and 67GaL48. Whereas 67GaL48 presented a high initial uptake but a slow wash-out (4.1% ID per g at 2 min and 2.5% ID per g at 60 min), 99mTcL46 exhibited only 0.74% ID per g at 2 min. The uptake remained significant over 60 min in the pancreas and also in the liver (and in the spleen for 67GaL48). Such nonspecific binding in these organs may make it difficult to visualize pancreatic islet amyloids in vivo. 99mTcL46 was also injected in the mouse model with transplanted amylin aggregates. Higher pancreas accumulation of the radiotracer was observed as compared to normal mice (1.6 fold at 1 h post-injection), demonstrating both the binding ability of 99mTcL46 and the value of this model for the validation of amylin imaging probes.
L. M. Mirica published, in 2020, a series of multivalent ligands (L53–L56) containing two new furanyl-hydroxymethylbenzofuran derivatives for copper(II) complexation (Scheme 8).44 The Aβ-recognition fragment was conjugated to the macrocyclic chelator NOTA via four different spacers (ethyl, butyl, hexyl and ethylene glycol). Analogues (L49–L52) with a single targeting unit were also prepared for comparison.
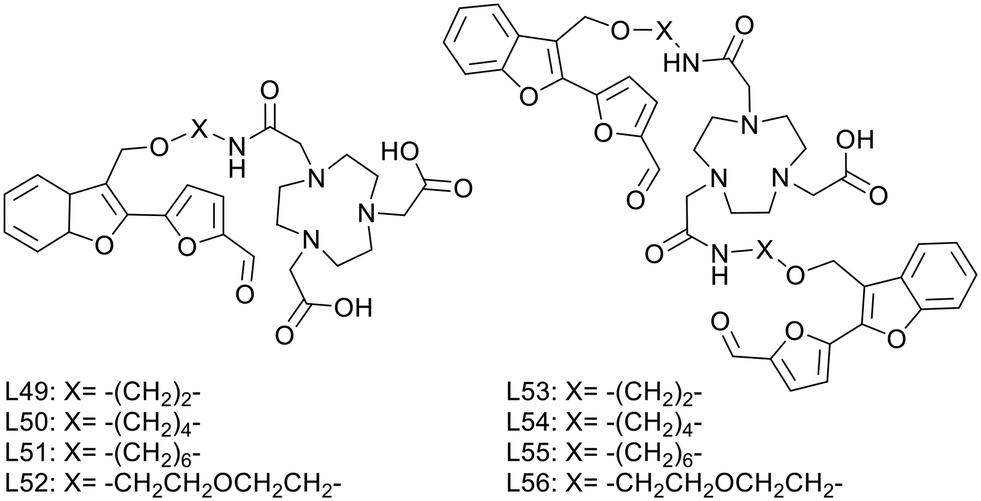 | ||
| Scheme 8 Furanyl-hydroxymethylbenzofuran derivatives for copper(II) complexation. | ||
Brain section staining from 3-month-old 5xFAD mice with the divalent complexes CuL53–56 showed specific binding to amyloid plaques. CuL54 and CuL55, with butyl and hexyl linkers, exhibited the strongest fluorescence signals and the closest co-localization with the amyloid fibril specific AF594-HJ3.4 antibody. Ex vivo data on WT and transgenic mice confirmed specific binding to Aβ plaques and negligible nonspecific interaction with other biomolecules in the brain. 64CuL54 and 64CuL55 led to the highest autoradiography contrast between 5xFAD and WT. In similar experiments, the monovalent 64CuL50 and 64CuL51 analogues yielded 1.5–1.6 lower signal intensities, confirming the existence of a multivalent binding effect.
An additional benefit of the divalent targeting approach was the increased lipophilicity. While logPoct/PBS ranged between −1.02 and 0.45 for the monovalent complexes, the divalent ones had optimal lipophilicity for potential in vivo brain imaging (logPoct/PBS of 1.02–1.77), which could be finely modulated by the nature and the length of the linker (ethylene glycol ∼ ethyl < butyl < hexyl).
In vivo biodistribution of these 64Cu complexes in WT mice showed high initial uptake and fast clearance in each organ. Surprisingly, the most lipophilic 64CuL55 compound had the lowest initial brain uptake and was mostly found in the lungs over 4 hours, whereas 64CuL54 and 64CuL56 had the best BBB permeability and were thus selected for in vivo PET imaging with age-matched WT and 5xFAD mice. Post imaging analysis showed a significantly higher brain retention in transgenic vs. WT mice for 64CuL54, which thus holds promise as a diagnostic PET imaging agent for AD.
Probes based on stilbene derivatives (Table 3)
Stilbene derivatives are conjugated molecules that were identified to selectively bind Aβ aggregates through hydrophobic non-covalent interactions.| Ligand number | Molecular weight of ligand | Metal | logP of complex |
Targeted protein | Affinity to proteina (Ki, nM) | Brain uptake at 2 minb (% ID per g) | Ref. |
|---|---|---|---|---|---|---|---|
| a Metal complexes. b p = pancreas; AD = murine or rat model of Alzheimer's disease. | |||||||
| 57 | 490.7 | Re/99mTc | 3.04 | Aβ1–42 | 13.4 | 2.10 | 26 |
| 58 | 534.8 | Re/99mTc | 3.15 | Aβ1–42 | 142.1 | 1.10 | 26 |
| 59 | 327.5 | Re/99mTc | — | Aβ1–42 | — | — | 36 |
| 60 | 384.6 | Re/99mTc | — | Aβ1–42 | — | — | 36 |
| 61 | 463.6 | Re | 0.39 | Aβ1–40 | — | — | 25 |
| 62 | 430.5 | 99mTc | 1.87 | Aβ1–40 | 855 | — | 45 |
| 63 | 384.5 | 99mTc | 1.54 | Aβ1–40 | 260 | 0.15 | 46 |
| 64 | 370.5 | 99mTc | 2.04 | Aβ1–40 | 241 | 0.36 | 46 |
| 65 | 356.5 | 99mTc | — | Aβ1–40 | 260 | — | 46 |
| 66 | 453.6 | 64Cu | 1.60 | Aβ1–40 | — | — | 47 |
| 67 | 467.7 | 64Cu | 1.61 | Aβ1–40 | — | 2.2 | 47 |
| 68 | 481.7 | 64Cu | 1.50 | Aβ1–40 | — | — | 47 |
| 69 | 495.7 | 64Cu | 1.59 | Aβ1–40 | — | — | 47 |
| 70 | 495.7 | 64Cu | 1.44 | Aβ1–40 | — | — | 47 |
| 71 | 509.7 | 64Cu | 1.56 | Aβ1–40 | — | 1.1 | 47 |
| 72 | 540.8 | 64Cu | 1.03 | Aβ1–42 | — | 0.75 | 48 |
| AD: 0.79 | |||||||
Zhang et al. conjugated the previously described Re/99mTc-chelating BAT group (L12, L13)26 to styrylpyridine as well via one or two oligoethyleneoxy blocks (L57, L58) (Scheme 9). The Re/99mTc complexes exhibited efficient and specific binding to Aβ plaques in brain, with linker-dependent affinities: the shorter oligoethyleneoxy spacer translated to higher affinity (Ki = 13.4 nM for 99mTcL57 and 102.6 nM for 99mTcL58). The replacement of the 2-arylbenzothiazole scaffold in L12 and L13 with the styrylpyridine led to more favorable initial brain uptake especially for 99mTcL57 (2.10% ID per g at 2 min), while 99mTcL58 showed lower brain delivery (1.10% ID per g at 2 min) and faster clearance (2 min/60 min = 7.33).
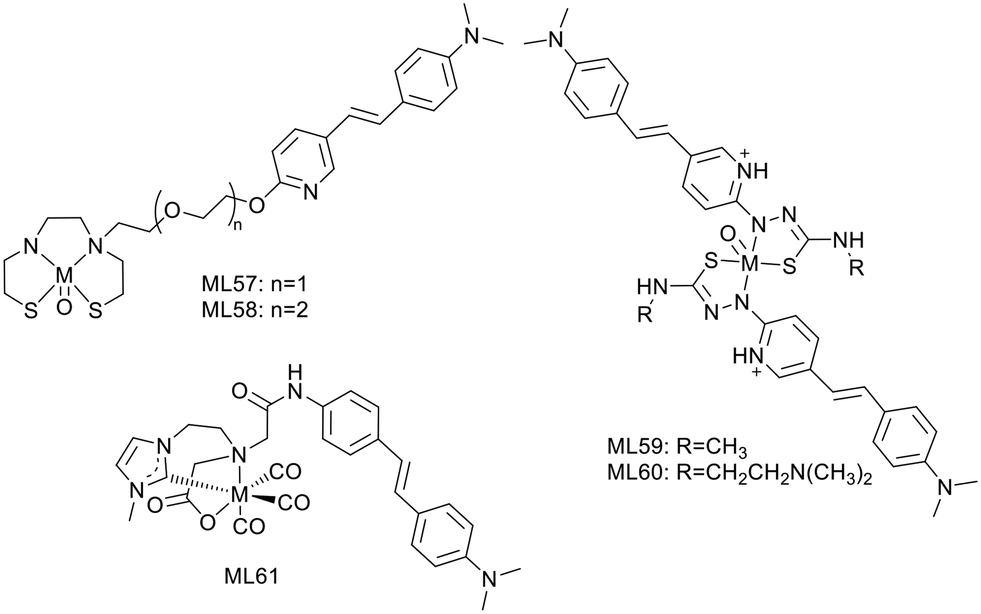 | ||
| Scheme 9 Stilbene and styrylpiridine derivative 99mTc complexes. | ||
99mTcL57, with the best overall properties, was selected for further investigations such as ex vivo autoradiography in transgenic/WT age matched mice and in vivo SPECT/CT imaging in rhesus monkeys. The good BBB permeability and the specific binding to Aβ plaques in transgenic mouse brain sections was confirmed by the excellent matching with the Th-S stained signal, while no evidence was detected in normal mice. The in vivo study in rhesus monkeys showed a significantly improved brain uptake, compared to a previous 99mTc imaging probe containing an alkyl linker and a benzothiazole unit, studied in the same animals (1.94–2.63% ID vs. 0.78–1.23% ID). Since no Aβ amyloid plaques are expected in these monkey's brains, this study did not evaluate the specific binding ability of the probe, but demonstrated its potential use for future human brain imaging.
Other examples of stilbene derivative Re/99mTc or 64Cu2+ complexes were described by P. S. Donnelly and co-workers (Scheme 9). As shown above, two substituted pyridylthiocarbazide ligands were coordinated to rhenium,36 and the authors replaced the benzofuran unit, characterized by insufficient fluorescence properties, with styrylpyridine (L59). An N,N-dimethylaminoethyl group was also added to increase solubility and modulate the binding properties (L60).
Competitive assays with Th-T in the presence of Aβ1–42 aggregates revealed strong reduction of the Th-T fluorescence, suggesting competitive binding or inhibition of fibril formation, especially with ReO(L60)2. The fluorescence of the styrylpyridine group allowed for detecting the complexes in post mortem human brain tissue using epi-fluorescence microscopy. Comparison with an Aβ specific antibody (1E8) indicated good co-localization with Re(L60)2, demonstrating significant and specific binding to Aβ plaques. The synthesis of radiolabelled 99mTc analogues was simple and gave good radiochemical yield, allowing for further investigations towards CAA diagnosis.
Another example by Wiratpruk et al. was already discussed in the benzothiazole section.25 The authors also studied the stilbene-based analogue of the neutral NHC complex ReL61 (Scheme 9). The Th-T competition assay in the presence of Aβ1–40 aggregates showed no difference between the benzothiazole and the stilbene derivative complexes. Staining of human AD brain revealed poor co-localization of ReL61 with the 1E8 antibody, which could be the consequence of the short linker in the structure. Comparison with the age-matched controls did not indicate non-specific binding in brain tissues. The logPoct/water = 0.39 indicated slightly higher lipophilicity than that of the benzothiazole derivative ReL11 (logPoct/water = 0.15), but not favourable enough for a good brain delivery.
This group also described, in 2016, a neutral oxorhenium(V) complex ReL62 based on a N3O tetradentate ligand with a metal-coordinating styrylpyridine group as an Aβ binding moiety (Scheme 10). This complex was identified to specifically bind Aβ plaques in post mortem human AD brain tissues45 and displayed suitable lipophilicity for BBB crossing, but the stability with respect to ligand exchange and radiochemical yield of the technetium analogues had to be improved. Thus, another family of oxorhenium(V) chelates was recently investigated,46 where the phenolate of ReL62 was replaced with a thiolate donor atom to lead to a N3S ligand, where the styrylpyridine part is still coordinated to the metal (Scheme 10). The amide functions (0, 1 or 2) in the ligand backbone contribute to increased stability.
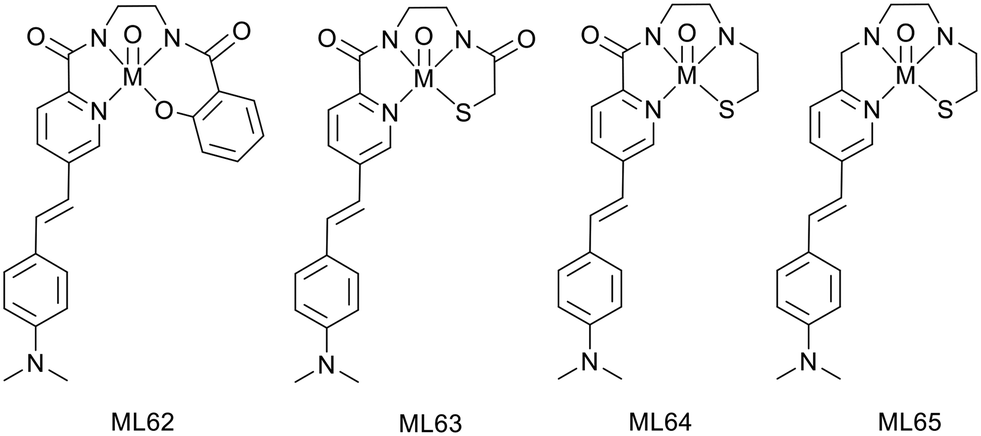 | ||
| Scheme 10 99mTc complexes with metal-coordinating styrylpyridine moiety. | ||
Inhibition constants of 240–260 nM were determined for all complexes by competition with Th-T in the presence of synthetic Aβ1–40 aggregates. Interaction with amyloid plaques in human brain tissue revealed specific binding outlined by an excellent co-localization with the 1E8 antibody. Further investigations were not possible with 99mTcL65 because of poor radiolabeling, contrary to the other two complexes 99mTcL63–64. It appeared that at least one amide function is necessary for complete radiolabeling with such ligands.
The lipophilic character of these complexes (logPoct/PBS of 1.54 and 2.04 for 99mTcL63 and 99mTcL64, respectively) might be suitable to cross the BBB. Whereas the substitution of an amide with an amine function significantly increases the lipophilicity, amides are important for complex stability with respect to ligand competition (such as histidine and cysteine).
Biodistribution in wild-type mice indicated the highest brain uptake for the monoamide–monoamine complex but still too low for further SPECT investigations (% ID per g = 0.15 and 0.36 for 99mTcL63 and 99mTcL64, respectively). This low brain delivery may be explained by the hydrogen-bond acceptor character of each amide function towards water molecules, resulting in an extensive hydrogen-bonded array which would reduce BBB permeability.
The authors expect that further structural modifications on the bis(amine) ligand, such as the introduction of geminal methyl substituents on the carbon adjacent to the sulfur donor, will improve the complex stability and the BBB crossing properties.
P. S. Donnelly and co-workers also developed, in 2020, a novel series of six Cu2+ complexes, based on bis(thiosemicarbazone) and conjugated to substituted stilbenyl functional groups (ML66–71) (Scheme 11).47 The authors already used similar bis(thiosemicarbazone) ligands as Cu2+ chelators but with benzofuran (cf. benzofuran derivatives section) or coordinating styrylpyridine amyloid recognition units and observed low brain delivery. The aim of this present work was thus to improve brain uptake.
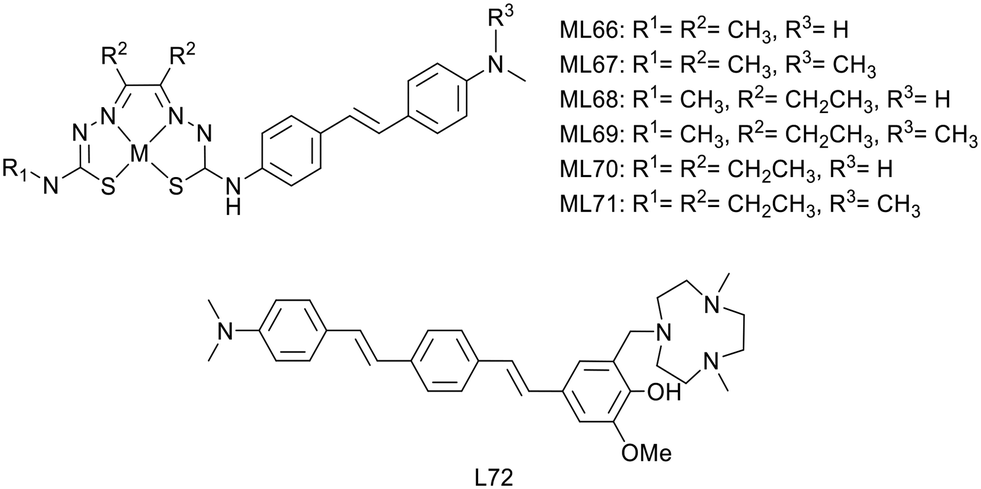 | ||
| Scheme 11 Stilbene and distyrylbenzene derivatives for copper(II) complexation. | ||
The stilbene functional group was directly attached to one thiosemicarbazone limb of the ligand through a stable N4-substituted thiosemicarbazone functional group. Electron-donating substituents are present on the stilbene unit to favor binding to amyloid aggregates. The ethyl or methyl groups on the ligand backbone are important to modulate lipophilicity, membrane permeability, retention and interaction with serum proteins as well as to retain the redox stability of the Cu2+ complexes.
The stability of the Cu2+ complexes was confirmed with respect to reduction and decomplexation in the presence of a large excess of glutathione, the most abundant biological reductant and copper binding ligand. The alkyl substituents on the ligand backbone did not induce significant changes in the distribution coefficient. Indeed, all the 64Cu complexes presented a similar lipophilic character with logPoct/PBS = 1.44–1.61, which might be appropriate for brain delivery.
Competition assays with Th-T in the presence of Aβ1–40 revealed reduced ThT fluorescence upon addition of all CuL66–71 complexes, indicative of competitive binding or inhibition of Aβ fibril formation. As all complexes had similar properties, the authors arbitrarily chose two compounds for supplementary studies: CuL67 and CuL71, with the lowest and the highest molecular weight, respectively. They both showed specific binding to Aβ plaques on post mortem human brain sections from confirmed AD patients as well as nonspecific binding on healthy tissues from age-matched control subjects.
64CuL67 and 64CuL71 crossed the BBB in wild-type mice, with initial brain uptake of 2.2 and 1.1% ID per g, respectively. These levels were similar to that of 64Cu(ATSM) already used in humans and better than that of another previously reported Cu2+-thiosemicarbazone–styrylpyridylhydrazone complex.38 Future investigations are envisaged in transgenic murine models of amyloid pathologies, and this work could also be adapted to other radioisotopes of copper such as 60Cu, 61Cu or 62Cu for imaging.
Recently, L. M. Mirica's group developed compound L72 by linking the hydrophilic triazamacrocycle (Me2TACN) to a hydrophobic distyrylbenzene derivative through a 2-methoxy-phenol fragment (Scheme 11), which is known to display antioxidant properties and to inhibit the formation of Aβ fibrils.48 Most of this work focused on the ligand itself, in terms of interaction with Aβ1–42 oligomers and aggregates, a fluorescence turn-on effect, in vivo BBB penetration and therapeutic potential. However, the authors also used the copper chelating properties of azamacrocycle and investigated the PET tracer 64CuL72. The ligand is monocationic since one amine of the macrocycle is protonated, and it is expected that the Cu2+ complex possesses the same charge due to the deprotonation of the phenolate group. Thus, both the ligand and the Cu2+ complex displayed similar turn-on fluorescence in the presence of Aβ1–42 oligomers and fibrils. Ex vivo fluorescence staining on brain sections of 7 month old 5xFAD transgenic mice indicated selective binding of CuL72 to Aβ species (fibrils and oligomers). Indeed, good co-localization has been observed with Congo red and HJ3.4 and OMAB antibodies which respectively bind to a large range of Aβ species and specifically to Aβ oligomers. Ex vivo autoradiography comparison between 10 month old 5xFAD and WT mouse brain sections further confirmed specific binding of 64CuL72 to Aβ species. The authors concluded that an amphiphilic molecule containing a hydrophilic fragment attached to a hydrophobic amyloid targeting unit is suitable to attain good binding affinity for both soluble and insoluble Aβ aggregates.
Finally, the pharmacokinetics of 64CuL72 was explored in 5xFAD and age-matched WT mice, and revealed BBB permeability and brain accumulation in AD mice. Normal and transgenic animals exhibited quite similar initial brain uptake (0.75% ID per g vs. 0.79% ID per g), but the wash-out was rapid in WT mice, in contrast to more important brain retention in the 5xFAD model.
Probes based on other small amyloid recognition moieties (Table 4)
Following their previous work on 18F fluorinated phenylquinoxaline moiety for tau targeting,49 Yang et al. explored a set of complexes based on phenylquinoxaline functionalized with a diethyl iminodiacetate group for the coordination of 99mTc and Re (ML73–76).50 The complexes varied in the position of the N,N-dimethyl amino group and in the size of the linker between the chelator and the targeting unit (Scheme 12). Variation of the position of the methyl group yielded selectivity of these probes for tau vs. Aβ, as indicated by their different affinities to those proteins.| Ligand number | Molecular weight of ligand | Metal | logP of complex |
Targeted protein | Affinity to proteina (Ki, nM) | Brain uptake at 2 minb (% ID per g) | Ref. |
|---|---|---|---|---|---|---|---|
| a Metal complexes; $Kd; §IC50. b p = pancreas; AD = murine or rat model of Alzheimer's disease. | |||||||
| 73 | 452.5 | Re/99mTc | 1.69 | Aβ1–42 | 1920 | — | 50 |
| 74 | 452.5 | Re/99mTc | 2.11 | Aβ1–42 | 1214 | 0.61/p: 2.59 | 50 |
| 75 | 438.5 | Re/99mTc | 1.17 | Aβ1–42 | 3422 | — | 50 |
| 76 | 438.5 | Re/99mTc | 1.43 | Aβ1–42 | 1475 | 0.34/p: 1.93 | 50 |
| 77 | 497.6 | Mn | — | Aβ | — | — | 51 |
| 78 | 683.8 | Mn | — | Aβ | 3190 | — | 51 |
| 79 | 652.8 | Gd | −0.32 | Aβ1–42 | 2.31 × 104$ | — | 53 |
| 80 | 448.5 | 99mTc | 1.08 | Aβ1–42 | 33.2§ | 0.78 | 54 |
| 81 | 462.5 | 99mTc | 1.1 | Aβ1–42 | 102.5§ | 0.86 | 54 |
| 82 | 959 | Gd | — | Aβ1–40/Aβ1–42 | 1–40: 9 × 104$ | — | 55 |
| 1–42: 3 × 105$ | |||||||
| 83 | 1170 | 99mTc | 1.6 | Aβ | 20.22$ | 0.35 | 56 |
| AD: 0.38 | |||||||
| 84 | 145000 |
89Zr | — | Aβ | 0.85$ | — | 58 |
| 85 | 15700 |
Gd | — | Aβ | 19§ | — | 59 |
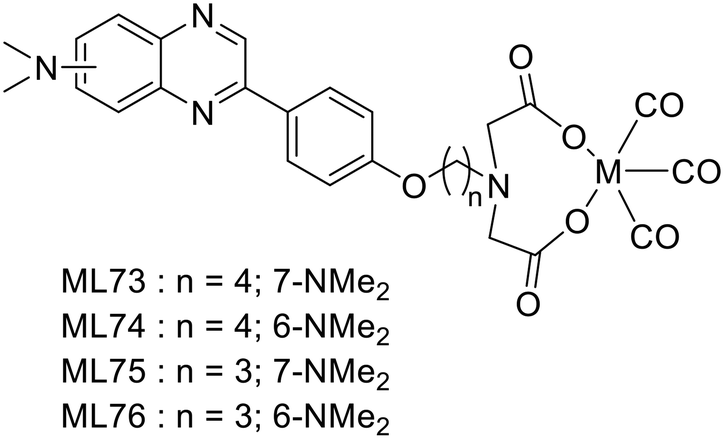 | ||
| Scheme 12 Phenylquinoxaline derivative 99mTc complexes. | ||
Indeed, Kd values ranging from 60 nM to 3.4 μM were obtained, and the ratio between the affinity to the two proteins for a given complex was 10 to 20. To further evaluate the potential of 99mTcL74 and 99mTcL76, ex vivo studies were performed in normal mice. The initial brain uptake for 99mTcL74 (0.61% ID per g, logD = 2.11), although not striking, was higher than that for 99m TcL76 (0.34% ID per g, logPoct/PBS = 1.43), probably due to the additional carbon in the linker and the subsequently increased lipophilicity of 99mTcL74. A clearance ratio 2/60 min of 3.2 and 3.8 was found for 99mTcL74 and 99mTcL76 complexes, respectively. Moreover, brain slices from human AD patients and from Tg-tau AD mice were stained with the ReL74 analogue and its tau targeting ability was validated by Gallyas–Braak staining (which detects phosphorylated tau accumulation). Overall, 99mTcL74 exhibited high and selective binding ability to neurofibrillary tangles, composed of hyperphosphorylated tau, but more structure optimizations should be performed to further improve the BBB permeability.
Tryptamine, an indole derivative, was also explored as a targeting moiety. Rastogi et al. proposed a Mn2+ complex functionalized with tryptamine as a potential T1/T2 contrast agent.51 The Mn2+ ion is 7-coordinated by the ligands (precursor MnL77 and tryptamine derivative MnL78) and the complexes hold one water molecule in their inner sphere, as confirmed by the solid-state structures (Scheme 13).
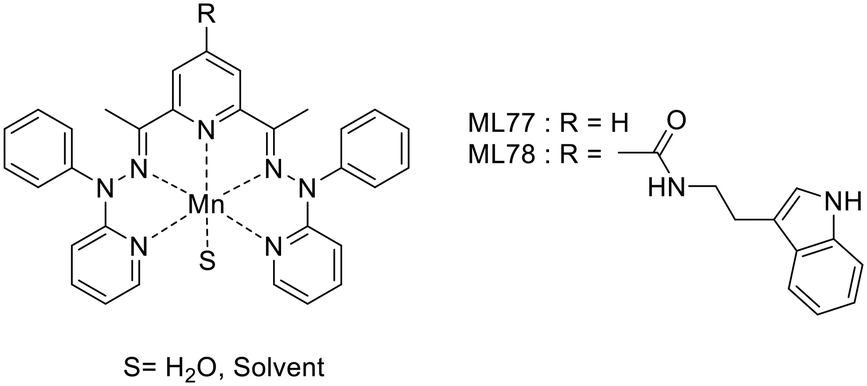 | ||
| Scheme 13 Tryptamine derivative Mn2+ complexes. | ||
The presence of the water molecule yielded surprisingly high relaxivities, r1 = 6.74 to 7.26 mM−1 s−1 and r2 = 33.38 to 23.26 mM−1 s−1 at 7 T for MnL77 and MnL78, respectively. The differences between the two chelates have been attributed to the increase in both the molecular weight and the rigidity of the complex upon tryptamine functionalization. Affinity studies towards Aβ-fibrils showed a Kd of 3.19 μM for MnL78, while MnL80 did not interact with Aβ-fibrils, highlighting the targeting properties of the indole group.
Chalcone was widely explored as a scaffold for medicinal applications. Chalcone analogues were used as inhibitors of enzymes or proteins involved in the pathogenesis of AD as well as a targeting moiety for imaging.52 A recent chalcone-based MRI probe was reported by Choi et al.53 The GdL79 complex consists of a DO3A bearing a chalcone unit functionalised with a dimethylamino group (Scheme 14). The latter is expected to improve Aβ binding as well as the fluorescence properties of the targeting moiety, enabling optical imaging. The probe had a logPoct/water of −0.32 and Kd of 23.1 μM, thus an 8-fold higher binding affinity to Aβ aggregates than the non-targeted agent Gadobutrol®.
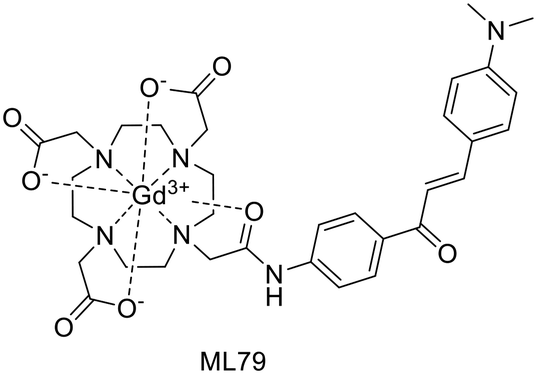 | ||
| Scheme 14 Chalcone derivative Gd3+ complex. | ||
The relaxivities of GdL79, r1 = 4.95 and r2 = 6.80 mM−1 s−1 at 3 T and r1 = 4.99 and r2 = 6.07 mM−1 s−1 at 9.4 T, were superior to those of the gold standard MRI contrast agent Gd(DOTA) and Gadobutrol®. An MRI phantom study was performed at 9.4 T to highlight the binding to Aβ and the potential of this probe for the MRI detection of low- (di-, tri-, tetramer) and high-molecular-weight (75–250 kDa) Aβ oligomers. A significant increase in MR signal intensity was observed when GdL79 was incubated with high-molecular-weight Aβ oligomers. These promising results led the authors to explore this probe in vivo, in 5XFAD transgenic mice upon IV administration of GdL79 at a dose of 0.3 mmol kg−1. Thanks to the good lipophilicity and Aβ affinity of this complex, the MR images at 4 h post injection showed signal enhancement in the brain in Aβ-containing areas which were confirmed in matched histological sections (Fig. 4).
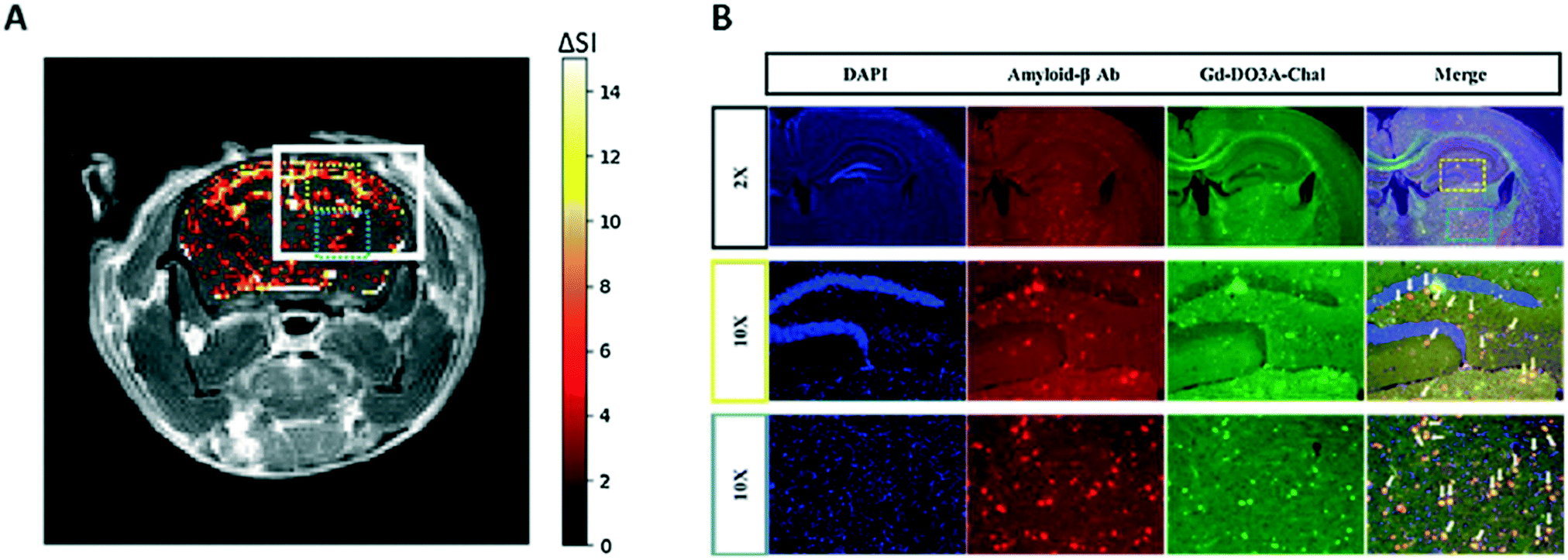 | ||
| Fig. 4 (A) Color mapped T1-weighted MR image of brain AD mouse model 4 h after intravenous administration of GdL79; (B) post mortem immunofluorescence imaging after MR acquisition, showing co-localization of GdL79 (green) and amyloid beta antibody (red). Reprinted from ref. 53. Copyright 2020 with permission from Elsevier. | ||
Molavipordanjani et al. conceived a set of 6 2-aryl-imidazo[2,1-b]benzothiazole (IBT) derivatives (Scheme 15).54 The IBT scaffold combines the benzothiazole part of PiB and the 2-arylimidazo part of IMPY. This lipophilic, planar and electron-rich conjugated heteroaromatic system is expected to improve BBB crossing and thus enable in vivo imaging of AD brains. For 99mTc radiolabelling, IBT was conjugated with 3 different units: (i) 2-thiophenemethylamine, (ii) picolylamine or (iii) ethyl glycinate, via a linker of two or three carbons. Only the latter displayed proper 99mTc labelling features (ML80–81); therefore further studies were restricted to these two complexes.
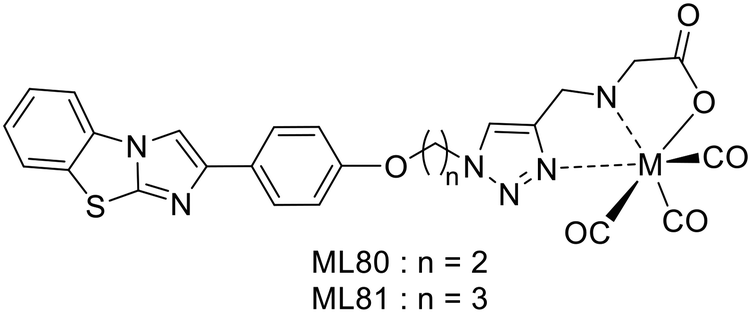 | ||
| Scheme 15 2-Aryl-imidazo[2,1-b]benzothiazole (IBT) derivative 99mTc complexes. | ||
Both radiocomplexes showed a lipophilic nature (logPoct/water = 1.08 and 1.10 for 99mTcL80 and 99mTcL81, respectively) and competition binding assays revealed that 99mTcL81 has a 3.8-fold higher affinity towards Aβ1–42, with IC50 values of 33.2 and 102.5 nM. Biodistribution studies in normal mice yielded initial brain uptake at 2 min post-injection of 0.78 and 0.86% ID per g for 99mTcL80 and 99mTcL81, respectively. The brain clearance ratios 2 to 60 min (8.67 and 7.17, respectively) indicated a good in vivo profile. The radiocomplexes were mainly eliminated via the hepatobiliary route, resulting in high radioactivity in the liver and intestines. Autoradiography was performed on brain sections of the AD rat model. 99mTcL80 and 99mTcL81 did not accumulate in control slices, while a hot spot was observed for both at the area of Aβ plaques and their co-localization was confirmed by Congo red staining experiments.
BBB-permeable and Aβ-targeted dyads obtained by conjugation of Gd(DOTA) to carbazole-based cyanine were recently reported.55 The complex GdL82 (Scheme 16) exhibited a relaxivity of 4.42 mM−1 s−1 at 3 T. The cyanine moiety could inhibit self-aggregation of Aβ, as shown by the ThT fluorescence assay, with an IC50 value of 2.83 μM. In vivo T1-weighted images (with 10 mg kg−1 injected dose, at 7 T) of 6 month-old 2xTg-AD mice led to brighter images, at different depths of the brain, at 90 min post injection than before injection, in sharp contrast to those from age-matched WT mice, demonstrating the capability of this complex to serve as an effective and sensitive T1-weighted MRI contrast agent for cerebral Aβ imaging in vivo.
 | ||
| Scheme 16 Gd(DOTA) conjugated to carbazole-based cyanine. | ||
Probes based on peptide or antibody moieties (Table 4)
To specifically target the fibril ends of Aβ1–42 deposits, LPFFD and KLVFF, two pentapeptides known to bind to Aβ, were selected and grafted on small gadolinium-based AGuIX nanoparticles modified by a PEG chain.32 The resulting nanoparticles AGuIX@PEG@LPFFD and AGuIX@PEG@KLVFF are ≈3 nm in diameter and display r1 relaxivities of 13.2 and 14.1 mM−1 s−1 and r2 of 20.1 and 23.0 mM−1 s−1 (at 60 MHz and 37 °C), respectively. Extra functionalisation with the fluorophore Cy5.5 was performed to enable the determination of the affinity of these particles towards Aβ1–42. Surprisingly, no binding has been detected under the conditions studied, while SPR measurements yielded Kd of 534 μM for AGuIX@PEG@KLVFF@Cy5.5 and of 261 μM for AGuIX@PEG@LPFFD@Cy5.5. Cytotoxicity studies showed good tolerance of the cells even for Gd3+ concentrations as high as 5 mM. Finally, incubation of these particles with AD mouse brain slices revealed their co-localization with Aβ plaques.Another probe consisted of a small lipophilic peptide (FPLIAIMA) previously reported to bind to Aβ, and a cyclopentadienyl tricarbonyl 99mTc complex.5699mTc-Cp-GABA-D-(FPLIAIMA)-NH2 (99mTcL83, Scheme 17) displayed logPoct/PBS = 1.6 and Kd = 20 μM towards Aβ1–42. In vivo studies in control and AD model rats revealed moderate initial brain uptake (0.38 and 0.35% ID per g, respectively, at 2 min post-injection). Notably, AD rats showed a high retention in the brain at 30 min (0.23% ID per g) in comparison with fast clearance in normal rat brains.
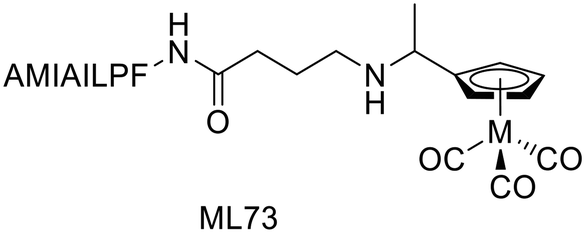 | ||
| Scheme 17 Aβ targeting with a small lipophilic peptide conjugated to a [Cp99mTc(CO)3] unit. | ||
Specific antibodies are well-known targeting moieties and have the advantage of conferring a high affinity to the imaging probes. Nevertheless, their slow pharmacokinetics is not well adapted to all imaging techniques.57 Only long-lived isotopes are appropriate for the design of antibody-based imaging probes. It is the case of 89Zr, with a half-life of 78.4 h, vs. short half-lives of currently used isotopes such as 18F (111 min), 99mTc (6 h) or 64Cu (12.7 h). The monoclonal antibody JRF/AβN/25 directed against the full-length human Aβ1–x epitope was conjugated to a Df-Bz-NCS chelator.5889Zr-Df-Bz-JRF/AβN/25 (ML84; Scheme 18) conserved its nanomolar affinity to Aβ1–40 (Kd = 0.85 nM) and biodistribution studies in healthy WT mice showed an accumulation of 0.5 and 0.36% ID per g at 1 h and 48 h post injection, respectively. This uptake was confirmed by ex vivo autoradiography performed in mice brain slices.
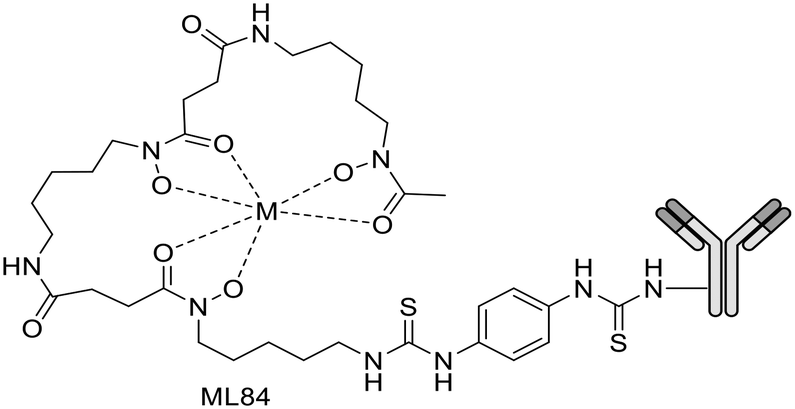 | ||
| Scheme 18 Monoclonal antibody JRF/AβN/25 conjugated to a Df-Bz-NCS chelator for 89Zr complexation. | ||
Camelid single-domain antibody fragments (VHHs), or nanobodies, are smaller (≈15 kDa), stable and highly specific. Vandesquille et al. proposed an anti-Aβ VHH, R3VQ, which was conjugated to 2 to 5 Gd(DOTA) units via an amide or a thiol bond.59 DOTA conjugation did not affect Aβ1–40 recognition, as all probes displayed IC50 in the 19–50 nM range. The potential MRI probe R3VQ-S-(Gd(DOTA))3 (ML85; Scheme 19) has an r1 of 19.2 mM−1 s−1 at 11.7 T (25 °C). Incubation of cerebral hemispheres of PS2APP mouse brains yielded a 7-fold MRI signal intensity difference between spots with amyloid plaques and the baseline and a 33-fold difference with respect to the corresponding hemisphere from WT mice. The presence of Aβ plaques in the AD model brain slices was confirmed by immunohistochemistry.
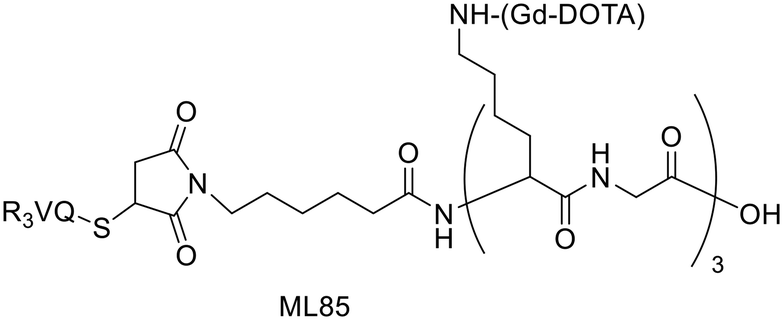 | ||
| Scheme 19 Anti-Aβ nanobody R3VQ conjugated to Gd(DOTA) units. | ||
Conclusions and perspectives
The development of metal-based probes for imaging detection of amyloid peptides has been a very active field in the past six years. However, despite the high number of amyloid peptides implicated in different pathologies, the large majority of the examples concerns the detection of Aβ fibrils in Alzheimer's disease. The success of such approaches is largely dependent on BBB permeability which remains a very important limitation. Despite the easier accessibility of the target amyloids, only a few studies have been interested so far in imaging Aβ deposits in the context of cerebral amyloid angiopathy or amylin in diabetes. Overall, most reports describe radiocomplexes, mainly with 99mTc, 64Cu, or 68Ga, for nuclear imaging applications; examples of MRI agents are very scarce. This is not surprising and is obviously related to the much lower sensitivity of MRI which makes MRI detection especially challenging in the brain.Despite the high number of novel systems investigated in recent years, the chemical versatility of the complexes, and in particular of the amyloid targeting moieties used, remains relatively limited. These are essentially restricted to a few basic structures, mainly derived from benzothiazole, benzofuran or stilbene, while targeting with biological ligands, such as peptides, antibodies or nanobodies, is rather rare. By comparing data on the various small-molecular targeting functions, none of these seems to really emerge in terms of amyloid Aβ binding affinity or promotion of BBB crossing. In vitro binding affinity to the fibrils, as mostly assessed by competition assays using radioactive or fluorescent competitors, is typically (slightly) reduced by the presence of the metal chelate, but this influence is governed to a large extent by the length and the chemical nature of the spacer between the chelate and the amyloid recognition moiety. Depending also on the peptide's charge, the charge of the chelate becomes also important, though complete and systematic studies are still lacking. A few divalent probes have been investigated as well; however, these first data are not fully conclusive on a clear benefit of two targeting units for amyloid recognition. Finally, the concentration-dependent aggregation state of the amphiphilic complex itself is also a very important element that affects the amyloid-binding affinity. Such micellization processes are likely non-existing in the concentration range where nuclear imaging probes are used, but they clearly prevail at MRI-related concentrations (from a few μM to mM range).
While it is now standard to evaluate amyloid binding of the probes on tissue, in particular brain sections, originating from pathological mouse models as well as from post mortem human patients, an increasingly higher number of in vivo data also become available. These do not always confirm the ex vivo findings, highlighting the complexity of real biological conditions.
Today, very few comparative data exist on binding constants with different amyloid peptides, and these do not show real selectivity for any of the targeting units investigated. Selectivity will likely become interesting in the future to distinguish between different fibrils, especially knowing that co-localization of amylin and Aβ has been reported and is thought to be responsible for the cross-talk between diabetes and Alzheimer's disease. In addition, affinity of the probes to other abundant proteins can also be important as it influences their biological fate. This aspect is generally overlooked today but should receive more attention in the future. Potential binding to serum albumin is essential for blood retention, and interaction with other proteins can be responsible for non-specific staining often detected in ex vivo experiments.
As for BBB permeability, the common relationships between molecular structure and brain delivery are also validated for metal complexes: smaller size and amphiphilic character promote brain delivery. Structural elements important for amyloid binding, like long and hydrophobic spacers, can also contribute to more adapted lipophilicity, but they also increase the size, which in turn can be detrimental for BBB crossing. Interestingly, several examples exist where one donor atom from the targeting moiety also participates in metal binding in order to limit molecular size, and this does not seem to severely reduce amyloid recognition.
Beyond these general trends, many examples show that even small structural modifications on the ligand can have dramatic consequences on the amyloid-binding properties or the biodistribution of the metal chelates which makes it difficult to fully predict their behaviour.
Future efforts will be increasingly focusing on in vivo translation of the imaging agents, and in this respect, pathologies which do not concern the brain are expected to experience more success. In the choice of the biomarker, extracellular localization of the fibrils is also important to facilitate target accessibility. The availability of appropriate preclinical animal models will be a key issue for successful assessment of the novel probes on pathologies other than Alzheimer's disease.
Finally, we should also note that while imaging amyloid plaques remains an important objective, there is increasing evidence that mature fibrillary species might be less relevant than oligomers to assess the severity of the pathology. In AD, soluble Aβ oligomers are accepted today to be more neurotoxic than fibrillary Aβ species and responsible for memory failure, and oligomer levels correlate better with disease progression.60
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge financial support from the French National Research Agency (ANR-16-CE18-0022). CFGCG thanks the Portuguese Foundation for Science and Technology (FCT)/MCTES for funding the Coimbra Chemistry Centre through the programmes UIDB/00313/2020 and UIDP/00313/2020, also cofunded by FEDER/COMPETE 2020-EU.Notes and references
- F. Chiti and C. M. Dobson, Annu. Rev. Biochem., 2017, 86, 27 CrossRef CAS PubMed.
- C. Giacomelli, S. Daniele and C. Martini, Biochem. Pharmacol., 2017, 131, 1 CrossRef CAS PubMed; D. Eisenberg and M. Jucker, Cell, 2012, 148, 1188 CrossRef PubMed.
- L. Gremer, D. Schölzel, C. Schenk, E. Reinartz, J. Labahn, R. B. G. Ravelli, M. Tusche, C. Lopez-Iglesias, W. Hoyer, H. Heise, D. Willbold and G. F. Schröder, Science, 2017, 358, 116 CrossRef CAS PubMed.
- L. Gatti, F. Tinelli, E. Scelzo, F. Arioli, G. Di Fede, L. Obici, L. Pantoni, G. Giaccone, P. Caroppo, E. A. Parati and A. Bersano, Int. J. Mol. Sci., 2020, 21, 3435 CrossRef CAS PubMed.
- M. Press, T. Jung, J. König, T. Grune and A. Höhn, Mech. Ageing Dev., 2019, 177, 46 CrossRef CAS PubMed.
- S. Muralidar, S. V. Ambi, S. Sekaran, D. Thirumalai and B. Palaniappan, Int. J. Biol. Macromol., 2020, 163, 1599 CrossRef CAS PubMed.
- C. R. Fields, N. Bengoa-Vergniory and R. Wade-Martins, Front. Mol. Neurosci., 2019, 12, 299 CrossRef CAS PubMed.
- O. Pansarasa, M. Bordoni, L. Diamanti, D. Sproviero, S. Gagliardi and C. Cereda, Int. J. Mol. Sci., 2018, 19, 1345 CrossRef PubMed.
- D. Chhangani, A. Martín-Peña and D. E. Rincon-Limas, iScience, 2021, 24, 102459 CrossRef CAS PubMed.
- C. P. Argyropoulos, S. S. Chen, Y. H. Ng, M. E. Roumelioti, K. Shaffi, P. P. Singh and A. H. Tzamaloukas, Front. Med., 2017, 4, 73 CrossRef PubMed.
- X. Wang, X. Wang and Z. Guo, Coord. Chem. Rev., 2018, 362, 72 CrossRef CAS.
- P. A. Broderick, L. Wenning and Y. S. Li, J. Neural Transm., 2017, 124, 57 CrossRef CAS PubMed.
- U. Saeed, J. Compagnone, R. I. Aviv, A. P. Strafella, S. E. Black, A. E. Lang and M. Masellis, Transl. Neurodegener., 2017, 6, 1 CrossRef PubMed.
- N. J. Cobb and W. K. Surewicz, Biochemistry, 2009, 48, 2574 CrossRef CAS PubMed; L.-Q. Wang, K. Zhao, H.-Y. Yuan, Q. Wang, Z. Guan, J. Tao, X.-N. Li, Y. Sun, C.-W. Yi, J. Chen, D. Li, D. Zhang, P. Yin, C. Liu and Y. Liang, Nat. Struct. Mol. Biol., 2020, 27, 598 CrossRef PubMed.
- D. Adams, H. Koike, M. Slama and T. Coelho, Nat. Rev. Neurol., 2019, 15, 387 CrossRef CAS PubMed.
- B. R. Smith and S. S. Gambhir, Chem. Rev., 2017, 117, 901 CrossRef CAS PubMed.
- M. L. James and S. S. Gambhir, Physiol. Rev., 2012, 92, 897 CrossRef CAS PubMed; D.-E. Lee, H. Koo, I.-C. Sun, J. H. Ryu, K. Kim and I. C. Kwon, Chem. Soc. Rev., 2012, 41, 2656 RSC; S. Y. Lee, S. I. Jeon, S. Jung, I. J. Chung and C. H. Ahn, Adv. Drug Delivery Rev., 2014, 76, 60 CrossRef PubMed.
- R. Weissleder and M. J. Pittet, Nature, 2008, 452, 580 CrossRef CAS PubMed.
- S. Lacerda, J. F. Morfin, C. F. G. C. Geraldes and É. Tóth, Dalton Trans., 2017, 46, 14461 RSC.
- B. C. Uzuegbunam, D. Librizzi and B. Hooshyar Yousefi, Molecules, 2020, 25, 977 CrossRef CAS PubMed.
- O. Krasnovskaya, D. Spector, A. Zlobin, K. Pavlov, P. Gorelkin, A. Erofeev, E. Beloglazkina and A. Majouga, Int. J. Mol. Sci., 2020, 21, 9190 CrossRef CAS PubMed; K. Chen and M. Cui, Med. Chem. Commun., 2017, 8, 1393 RSC.
- J. Jia, K. Zhou, J. Dai, B. Liu and M. Cui, Eur. J. Med. Chem., 2016, 124, 763 CrossRef CAS PubMed.
- C. Kiritsis, B. Mavroidi, A. Shegani, L. Palamaris, G. Loudos, M. Sagnou, I. Pirmettis, M. Papadopoulos and M. Pelecanou, ACS Med. Chem. Lett., 2017, 8, 1089 CrossRef CAS PubMed.
- M. Sagnou, B. Mavroidi, A. Shegani, M. Paravatou-Petsotas, C. Raptopoulou, V. Psycharis, I. Pirmettis, M. S. Papadopoulos and M. Pelecanou, J. Med. Chem., 2019, 62, 2638 CrossRef CAS PubMed.
- N. Wiratpruk, A. Noor, C. A. McLean, P. S. Donnelly and P. J. Barnard, Dalton Trans., 2020, 49, 4559 RSC.
- X. Zhang, Y. Hou, C. Peng, C. Wang, X. Wang, Z. Liang, J. Lu, B. Chen, J. Dai, B. Liu and M. Cui, J. Med. Chem., 2018, 61, 1330 CrossRef CAS PubMed.
- B. W. Chow and C. Gu, Trends Neurosci., 2015, 38, 598 CrossRef CAS PubMed; C. Y. Chan, A. Noor, C. A. McLean, P. S. Donnelly and P. J. Barnard, Chem. Commun., 2017, 53, 2311 RSC.
- N. Bandara, A. K. Sharma, S. Krieger, J. W. Schultz, B. H. Han, B. E. Rogers and L. M. Mirica, J. Am. Chem. Soc., 2017, 139, 12550 CrossRef CAS PubMed.
- Y. Huang, H.-J. Cho, N. Bandara, L. Sun, D. Tran, B. E. Rogers and L. M. Mirica, Chem. Sci., 2020, 11, 7789 RSC.
- Y. Wang, T. T. Huynh, H.-J. Cho, Y.-C. Wang, B. E. Rogers and L. M. Mirica, Inorg. Chem., 2021, 60, 12610 CrossRef CAS PubMed.
- A. F. Martins, J.-F. Morfin, A. Kubíčková, V. Kubíček, F. Buron, F. Suzenet, M. Salerno, A. N. Lazar, C. Duyckaerts, N. Arlicot, D. Guilloteau, C. F. G. C. Geraldes and É. Tóth, ACS Med. Chem. Lett., 2013, 4, 436 CrossRef CAS PubMed; A. F. Martins, J.-F. Morfin, C. F. G. C. Geraldes and Éva Tóth, J. Biol. Inorg. Chem., 2014, 19, 281 CrossRef PubMed; A. F. Martins, D. M. Dias, J. F. Morfin, S. Lacerda, D. V. Laurents, É. Tóth and C. F. G. C. Geraldes, Chem. – Eur. J., 2015, 21, 5413 CrossRef PubMed; D. Cressier, M. Dhilly, T. T. Cao Pham, F. Fillesoye, F. Gourand, A. Maïza, A. F. Martins, J.-F. Morfin, C. F. G. C. Geraldes, É. Tóth and L. Barré, Mol. Imaging Biol., 2016, 18, 334 CrossRef PubMed; A. C. Oliveira, T. Costa, L. L. G. Justino, R. Fausto, J.-F. Morfin, É. Tóth, C. F. G. C. Geraldes and H. D. Burrows, Photochem. Photobiol. Sci., 2020, 19, 1522 CrossRef PubMed.
- M. Plissonneau, J. Pansieri, L. Heinrich-Balard, J.-F. Morfin, N. Stransky-Heilkron, P. Rivory, P. Mowat, M. Dumoulin, R. Cohen, É. Allémann, É. Tóth, M. J. Saraiva, C. Louis, O. Tillement, V. Forge, F. Lux and C. Marquette, J. Nanobiotechnol., 2016, 14, 60 CrossRef PubMed.
- P. M. Costa, J. T.-W. Wang, J.-F. Morfin, T. Khanum, W. To, J. Sosabowski, E. Tóth and K. T. Al-Jamal, NANO, 2018, 2, 168 Search PubMed.
- S. Majdoub, Z. Garda, A. C. Oliveira, I. Relich, A. Pallier, S. Lacerda, C. Hureau, C. F. G. C. Geraldes, J.-F. Morfin and É. Tóth, Chem. – Eur. J., 2021, 27, 2009 CrossRef CAS PubMed.
- M. Ono, Y. Fuchi, T. Fuchigami, N. Kobashi, H. Kimura, M. Haratake, H. Saji, M. Nakayama and ACS Med, Chem. Lett., 2010, 1, 443 CAS.
- S. P. Fletcher, A. Noor, J. L. Hickey, C. A. McLean, J. M. White and P. S. Donnelly, J. Biol. Inorg. Chem., 2018, 23, 1139 CrossRef CAS PubMed.
- L. E. McInnes, A. Noor, K. Kysenius, C. Cullinane, P. Roselt, C. A. McLean, F. C. K. Chiu, A. K. Powell, P. J. Crouch, J. M. White and P. S. Donnelly, Inorg. Chem., 2019, 58, 3382 CrossRef CAS PubMed.
- J. L. Hickey, S. Lim, D. J. Hayne, B. M. Paterson, J. M. White, V. L. Villemagne, P. Roselt, D. Binns, C. Cullinane, C. M. Jeffery, R. I. Price, K. J. Barnham and P. S. Donnelly, J. Am. Chem. Soc., 2013, 135, 16120 CrossRef CAS PubMed.
- H. Watanabe, A. Kawasaki, K. Sano, M. Ono and H. Saji, Bioorg. Med. Chem., 2016, 24, 3618 CrossRef CAS PubMed.
- H. Watanabe, M. Ono, S. Iikuni, M. Yoshimura, K. Matsumura, H. Kimura and H. Saji, Bioorg. Med. Chem. Lett., 2014, 24, 4834 CrossRef CAS PubMed.
- M. Yoshimura, M. Ono, H. Watanabe, H. Kimura and H. Saji, Bioconjugate Chem., 2016, 27, 1532 CrossRef CAS PubMed.
- H. Watanabe, K. Kawano, S. Iikuni, Y. Shimizu and M. Ono, Nucl. Med. Biol., 2020, 90-91, 93 CrossRef CAS PubMed.
- N. Harada, H. Kimura, M. Ono, D. Mori, Y. Ohmomo, T. Kajimoto, H. Kawashima and H. Saji, J. Organomet. Chem., 2011, 696, 3745 CrossRef CAS.
- H.-J. Cho, T. T. Huynh, B. E. Rogers and L. M. Mirica, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 30928 CrossRef CAS PubMed.
- D. J. Hayne, J. M. White, C. A. McLean, V. L. Villemagne, K. J. Barnham and P. S. Donnelly, Inorg. Chem., 2016, 55, 7944 CrossRef CAS PubMed.
- B. Spyrou, I. N. Hungnes, F. Mota, J. Bordoloi, P. J. Blower, J. M. White, M. T. Ma and P. S. Donnelly, Inorg. Chem., 2021, 60, 13669 CrossRef CAS PubMed.
- A. Noor, D. J. Hayne, S. Lim, J. K. Van Zuylekom, C. Cullinane, P. D. Roselt, C. A. McLean, J. M. White and P. S. Donnelly, Inorg. Chem., 2020, 59, 11658 CrossRef CAS PubMed.
- L. Sun, H.-J. Cho, S. Sen, A. S. Arango, T. T. Huynh, Y. Huang, N. Bandara, B. E. Rogers, E. Tajkhorshid and L. M. Mirica, J. Am. Chem. Soc., 2021, 143, 10462 CrossRef CAS PubMed.
- K. Zhou and M. Cui, J. Labelled Compd. Radiopharm., 2017, 60, 111 CrossRef PubMed; L. Fu, K. Zhou, X. Zhang, F. Yi, M. Cui, J. Zhang, B. Xu and J. Tian, Eur. J. Nucl. Med. Mol. Imaging, 2018, 45, S650 Search PubMed.
- F. Yang, K. Wang, K. Zhou, B. Dai, J. Dai, Y. Liang and M. Cui, Eur. J. Med. Chem., 2019, 177, 291 CrossRef CAS PubMed.
- N. Rastogi, N. Tyagi, O. Singh, B. S. Hemanth Kumar, U. P. Singh, K. Ghosh and R. Roy, J. Inorg. Biochem., 2017, 177, 76 CrossRef CAS PubMed.
- P. Thapa, S. P. Upadhyay, W. Z. Suo, V. Singh, P. Gurung, E. S. Lee, R. Sharma and M. Sharma, Bioorg. Chem., 2021, 108, 104681 CrossRef CAS PubMed.
- G. Choi, H.-K. Kim, A. R. Baek, S. Kim, M. J. Kim, M. Kim, A. E. Cho, G.-H. Lee, H. Jung, J.-U. Yang, T. Lee and Y. Chang, J. Ind. Eng. Chem., 2020, 83, 214 CrossRef CAS.
- S. Molavipordanjani, S. Emami, A. Mardanshahi, F. Talebpour Amiri, Z. Noaparast and S. J. Hosseinimehr, Eur. J. Med. Chem., 2019, 175, 149 CrossRef CAS PubMed.
- X. Wang, H. N. Chan, N. Desbois, C. P. Gros, F. Bolze, Y. Li, H. W. Li and M. S. Wong, ACS Appl. Mater. Interfaces, 2021, 13, 118525 Search PubMed.
- S. Jokar, H. Behnammanesh, M. Erfani, M. Sharifzadeh, M. Gholami, O. Sabzevari, M. Amini, P. Geramifar, M. Hajiramezanali and D. Beiki, Bioorg. Chem., 2020, 99, 103857 CrossRef CAS PubMed.
- J. T. Ryman and B. Meibohm, CPT: Pharmacometrics Syst. Pharmacol., 2017, 6, 576 CAS.
- J. Fissers, A.-M. Waldron, T. D. Vijlder, B. V. Broeck, D. J. Pemberton, M. Mercken, P. V. D. Veken, J. Joossens, K. Augustyns, S. Dedeurwaerdere, S. Stroobants, S. Staelens and L. Wyffels, Mol. Imaging Biol., 2016, 18, 598 CrossRef CAS PubMed.
- M. Vandesquille, T. Li, C. Po, C. Ganneau, P. Lenormand, C. Dudeffant, C. Czech, F. Grueninger, C. Duyckaerts, B. Delatour, M. Dhenain, P. Lafaye and S. Bay, mAbs, 2017, 9, 1016 CrossRef CAS PubMed.
- Z.-X. Wang, L. Tan, J. Liu and J.-T. Yu, Mol. Neurobiol., 2016, 53, 1905 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |